
Abstract
The present article describes the design, synthesis, in vitro urease inhibition, and in silico molecular docking studies of a novel series of nitrothiazolacetamide conjugated to different thioquinazolinones. Fourteen nitrothiazolacetamide bearing thioquinazolinones derivatives (8a-n) were synthesized through the reaction of isatoic anhydride with different amine, followed by reaction with carbon disulfide and KOH in ethanol. The intermediates were then converted into final products by treating them with 2-chloro-N-(5-nitrothiazol-2-yl)acetamide in DMF. All derivatives were then characterized through different spectroscopic techniques (1H, 13C-NMR, MS, and FTIR). In vitro screening of these molecules against urease demonstrated the potent urease inhibitory potential of derivatives with IC50 values ranging between 2.22 ± 0.09 and 8.43 ± 0.61 μM when compared with the standard thiourea (IC50 = 22.50 ± 0.44 μM). Compound 8h as the most potent derivative exhibited an uncompetitive inhibition pattern against urease in the kinetic study. The high anti-ureolytic activity of 8h was confirmed against two urease-positive microorganisms. According to molecular docking study, 8h exhibited several hydrophobic interactions with Lys10, Leu11, Met44, Ala47, Ala85, Phe87, and Pro88 residues plus two hydrogen bound interactions with Thr86. According to the in silico assessment, the ADME-Toxicity and drug-likeness profile of synthesized compounds were in the acceptable range.
Similar content being viewed by others

Introduction
The urease (urea amidohydrolase EC 3.5.1.5) is a Ni-containing enzyme that catalyzes the hydrolysis of urea (CH4N2) into ammonia (NH3) and carbon dioxide (CO2). The excess release of ammonia significantly increases the pH level and contributes to pathogen-host interactions1,2. In more detail, increasing the pH by the accumulation of NH3 makes the conditions more favorable for bacterial growth and development as well as increases infections of the gastrointestinal tracts and urinary system. Additionally, severe complications may occur, such as peptic ulcers, stomach cancer, hepatic coma, hepatic encephalopathy, urinary stones, catheters blocking urolithiasis, urinary catheter encrustation, and pyelonephritis3,4.
The possible reaction mechanism for urease activities at neutral pH involves the coordination of H2O–Ni plus hydroxyl groups to other Ni. Next, the substrate (urea) is activated toward nucleophilic attack by O-coordination of Ni2+ ions, and a nickel-coordinated hydroxide ion attacks the carbonyl carbon of the coordinated substrate to form a tetrahedral intermediate. The breakdown of the tetrahedral intermediate happened to form a coordinated carbamate or carboxylate ion. Finally, the replacement of the coordinated carbamate ion or carboxylate ion by water leads to the regeneration of the enzyme5,6,7.
One of the most frequently studied bacteria related to urease is Helicobacter pylori (H. pylori), which colonizes more than half of the human population. Urease of H. pylori as a virulence factor neutralizes the acidic pH of the stomach, leading to alteration of the properties of the gastric mucous layer8 as well as providing ammonia for bacterial protein synthesis. Urease can induce destructive effects on host tissues directly by the produced ammonia and indirectly through stimulation of inflammation and immune response, including recruitment of leukocytes and triggering of the oxidative burst in neutrophils9,10. Specifically, H. pylori infection can induce and modulate the synthesis of angiogenic and invasive factors in gastric cancer cells11.
Urease inhibition can be a major strategy to target diseases associated with urease. To design effective urease inhibitors, the structure and active site of the aforementioned enzyme should be discussed. The urease consists of four domains: the N-terminal αβ domain (1–134 residue), the second αβ domain (135–285), β domain (286–401 and 702–761 located in the middle of 3D structure), and the C-terminal (αβ)8 TIM barrel domain (402–701 plus 762–840). (αβ)8 TIM barrel domain contains a flap region and an active site in which Ni1 and Ni2 are separated by a distance of less than 4 Å. Residues His519, His545, and Lys490 are connected to Ni1, while the residues His407, His409, Asp633, and Lys490 are linked to Ni212. The flap pocket modulates the entrance of urea into the active site of the enzyme13. The structures of potent inhibitors displayed the critical role of interaction with Ni (I) and Ni (II) as well as the residues of the binding site.
Several urease inhibitors with various structures have been introduced, including dihydropyrimidine thiosemicarbazones14, sulphamethazine, sulphamethoxazole15, hydroxamic acids16, thiobarbiturate17,18, bis-indole19, benzofuran20, sulfonated-coumarin21 benzimidazole22, thiazolidinone23, thiosemicarbazide24, as well as quinazoline-4(3H)-one25.
Nitrogen-containing heterocycles have attracted considerable attention due to their wide occurrence and pharmacological importance. Among these heterocycles, quinazoline and quinazolinone-based derivatives, constitute an imperative class of compounds with various methodologies for their synthesis, such as aza-reaction, metal-mediated (Pd, Zn, Cu) reaction, microwave-assisted reaction, ultrasound-promoted reaction, and phase-transfer catalysis. All these strategies provide rapid access to novel quinazoline and quinazolinone derivatives, affording the possibility of increasing structural diversity to the design and synthesis of novel agents with diverse therapeutic and pharmacological properties26,27,27,28,28,29. Considerable evidence has been found on the importance of quinazolinone derivatives in pharmaceutical chemistry as an important nucleus in the class of anticancer29, anti-inflammatory30, anticonvulsant, antihypertensive31, antidiabetic32, and antimicrobial agents33. Besides quinazolinone derivatives also exhibited significant urease inhibitory potencies34,35, but further development is required to find a lead with the quinazolinone-based structure for future advanced research. Aminonitrothiazole scaffold is also known as an important class of antibacterial agents36,37,38. By considering the rapid increase of resistance to existing drugs, a vital need for new candidates possessing urease inhibitory activity as one of the key virulence factors in the human pathogen is highly needed. Keeping this in view, in the present study, structural modifications to the previously reported quinazolinone as the elegant skeleton against urease via coupling to aminonitrothiazole were considered to evaluate the potency of newly synthesized derivatives. This manuscript describes the synthesis of nitrothiazolacetamide conjugated to thioquinazolinone 8a-n and the evaluation of their urease inhibition activities. Moreover, a structure–activity relationship (SAR) was established, followed by a mechanism of action and molecular modeling evaluations of potential inhibitors.
Results and discussion
Designing consideration
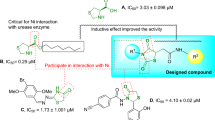
For the past few years, there are limited reports of quinazolines as urease inhibitors. New series of 2,3-disubstituted quinazolin-4(3H)-ones (Fig. 1A) were synthesized and exhibited potent urease inhibitory activity in the range of 1.55–2.65 μg/mL. The structure–activity relationship (SAR) indicated that halogen atoms on phenyl ring improved urease inhibition34. More recently, Mustafa and co-workers identified another set of quinazolinone-coumarin derivatives and the most potent compound (Fig. 1B) exhibited the IC50 values of 1.26 ± 0.07 μg/mL. In vitro results showed that the heterocyclic group substituted on N-3 position of quinazolinone ring plays an important role in the inhibitory activity39. This research group also developed and synthesized another series of quinazolin-4(3H)-ones (Fig. 1C). Most of the compounds showed excellent activity with IC50 values ranging between 1.88 ± 0.17 and 6.42 ± 0.23 μg/mL, compared to that of thiourea with an IC50 value of 15.06 μg/mL. Molecular docking interactions of compound C as the most potent derivative of this set showed key interactions with Arg439, Met637, Gln635 residues35.

Chemical structures of some biologically active agents and commercial medicine against urease.
Nitazoxanide (Fig. 1D) and nitazoxanide (Fig. 1E) are known as approved antiparasitic medications with aminonitrothiazole structure. These compounds were shown to have antibacterial activities against both metronidazole-resistant strains and sensitive clinical isolates of H. pylori pathogens. It is noteworthy that strains resistant to metronidazole were susceptible to these drugs40. Recently, thiazolbenzamide (Fig. 1F) was reported as a potential urease inhibitor. It was inferred that molecules with thio-substituted groups generally improved the urease inhibition of the target enzyme41.
Also, it would be interesting to note that the most effective inhibitors contain functional groups with electronegative atoms such as oxygen, nitrogen, or/and sulfur to form complexes with Ni ions of the enzyme as well as His residues in the active site. Stronger interaction of inhibitors with enzyme active site and higher inhibitory efficiency was observed in sulfur-containing inhibitors compared to the rest of heteroatoms 42,43.
By considering the structure of the previously reported active agents discussed herein.
-
Quinazolinone was utilized as an elegant skeleton to design urease inhibitors. Substitution at the R position of quinazolinone was performed to evaluate the type of substitution against urease.
-
To improve urease inhibitory potency, the nitrothiazole pendant with ensured anti-urease properties was incorporated into the quinazolinone ring. Nitrothiazole can improve hydrogen bonding capability within the enzyme cavity.
-
Thioacetamide is an ideal candidate to link the quinazolinone and nitrothiazol moiety with the substrate-like structure. It was assumed that sulfur atoms provide better and sometimes selective interactions with critical Ni (I) and Ni (II) coordinated with His519, His545, Lys490, His407, His409, Asp633, and Lys49045.
In continuation of our previous effort on designing urease inhibitors46,47,48, this work was aimed to report the synthesis of nitrothiazolacetamide conjugated to different thioquinazolinones. The urease inhibitory potential of all derivatives, as well as SAR and molecular docking studies, were also performed.
Chemistry
The synthetic pathway to the target compounds (8a-n) is outlined in Fig. 2. Intermediates 3a-n were synthesized by the method reported in our previous study44. Briefly, isatoic anhydride (1) was reacted with different amine (2a-n) in ethanol under reflux conditions for 3 h to obtain compound 3a-n. Carbon disulfide and KOH were added to this solution and the reaction was further refluxed for an extra 3 h. The targeted compounds (4a-n) were obtained after cooling and recrystallizing in ethanol. Compound 7 was prepared by a simple reaction of nitrothiazolamine (5) with 2-chloroacetyl chloride (6) in DMF at room temperature. The crude product was purified by recrystallization in ethanol. Compounds 8a-n were synthesized by the nucleophilic addition of thio-derivatives (4a-n) to intermediate 7 in DMF using K2CO3 as a catalyst at 50 °C. The structures of purified products were confirmed by IR, 1H NMR, 13C NMR, elemental analysis, and mass spectroscopy.

The synthetic path of the target compounds 8a-n.
Evaluation of urease inhibitory activity and structure–activity relationship
In vitro anti-urease activity of synthesized compounds, 8a-n were performed based on the calorimetric method against urease compared with thiourea as the reference inhibitor. The results of the urease inhibitory assay were shown in Table 1 in the terms of IC50. In this series, all compounds had significant inhibition against urease with IC50 values ranging from 2.22 to 8.43 µM compared with thiourea as a positive control with an IC50 value of 22.50 µM.
Based on the obtained biological results related to 8a-d, compound 8a as the unsubstituted phenyl pendant displayed an IC50 value of 4.72 µM with around fivefold improvement in the potency compared to thiourea as a standard inhibitor. Any substitution in this group including electron-withdrawing such as chlorine (8b) or bromine (8c) or even electron-donating group (8d, methoxy) improved urease inhibition and there is snot significant differences in these substituted derivatives.
The evaluations on 8e-g as the methyl-substituted group demonstrated that 8 g (R = methylpyridine) with an IC50 of 2.50 μM was categorized as the top potent urease inhibitor in this group followed by 8f (R = methyl benzyl) and 8e (R = benzyl). It seems that the presence of heteroatom in the aromatic ring could amend the interactions within the binding site of urease.
Assessments of 8a, 8e, and 8 h analogs showed the importance of the length of the alkyl chain between quinazolinone and aryl moiety. Compound 8h bearing ethyl linker demonstrated an IC50 of 2.22 μM against urease, while 8e (IC50 = 3.83 μM) possessing methyl linker was less potent compared with 8h followed by 8a with IC50 = 4.72 μM. It seems that the elongation of the alkyl linker between the quinazolinone and aryl pendant improved urease inhibitory activity.
In the case of compounds containing aliphatic chain substitution (8i-m), it can be seen that in most cases such structural modification reduced the inhibitory potency of compounds (IC50 ranging from 3.02 to 8.43 μM) compared to aromatic substituted derivatives (IC50 ranging from 2.22 to 3.85 μM). In this group, the most potent urease inhibitor was 8 k (R = n-butyl) with an IC50 value of 3.02 µM followed by 8i (R = n-propyl; IC50 = 4.26 μM), 8j (R = iso-propyl; IC50 = 6.04 μM) and 8l (R = iso-butyl; IC50 = 8.43 μM). As can be seen, the longer aliphatic chain demonstrated better inhibitory activity compared to shorter or branch one. Interestingly, compound 8n bearing cyclopentyl group as an aliphatic-ring substitute showed better activity (IC50 = 2.96 μM) compared to the rest of the aliphatic-chain group. As can be seen in this set of compounds, it seems that aliphatic-ring followed by aliphatic linear chains are more potent than aliphatic branched-chain counterparts.
Kinetic study of the most potent compound 8h
The mechanism of urease inhibition was investigated by enzyme kinetics, following the similar procedure of the urease inhibition assay. Lineweaver–Burk graphics were used to estimate the type of inhibition. Graphical analysis of the reciprocal Lineweaver–Burk plot (Fig. 3) related to compound 8h showed that Km and Vmax decreased with an increase in inhibitor concentration confirming an uncompetitive inhibition pattern against urease. Furthermore, the plot of the Km versus different concentrations of 8h gave an estimate of the inhibition constant, Ki of 1.994 µM which is in accordance with the IC50 value of 8h (Fig. 4).

The Lineweaver–Burk plot of compound 8h at different concentrations against urease of three independent experiments.

Double reciprocal Lineweaver–Burk plot of 8h against urease of three independent experiments.
Molecular docking simulation
Jack bean urease (JBU) is a T-shaped metalo-hydrolase enzyme that acts by converting urea into ammoniac within its active site. JBU monomer third structure consists of four main domains (Fig. 5). From the N-terminal of the enzyme sequence, starts by first αβ domain located in the hammer handle. The second αβ domain is located in the hammerhead which is connected through a middle β domain to the other head of the hammer which is (αβ)8 TIM barrel domain holding the active site of the enzyme12.

Schematic view of jack bean urease domains.
The enzyme kinetic study showed that the compound 8h acts as an uncompetitive inhibitor of the JBU enzyme in this type of inhibition the inhibitor interacted with the enzyme–substrate ([ES]) complex to form a final enzyme–substrate-inhibitor ([ESI]) complex; hence, the molecular docking study was performed on the [ES] complex. To make the [ES] complex the urea docked into the active site of the JBU enzyme (PDB ID: 4H9M).
In order to find the possible allosteric sites, the protein-substrate complex was treated using mastreo sitemap tool to identify the suitable sites for occupancy of hydrophobic, H-bond donor, and H-bond acceptor ligand groups. Eventually, five possible binding sites were detected which can be suitable as a drug-like binding site. As shown in Fig. 6, five binding sites were detected on the surface of the [ES] complex. Site 1 (purple) and site 2 (magenta) located in the first αβ domain which was showed the suitable size and potential interaction sites, calculated to have the best site scores (1.011 and 0.932 respectively). Site 3 (brown) nearby the canonical active site also had a plausible site score of 0.917. Site 4 (orange) and site 5 (cyan) were the smallest sites and had a few potential H-bond interactions, their site scores were calculated to be 0.724 and 0.510, respectively.

Potential binding sites of jack bean urease detected by sitemap. In each site’s magnified image the hydrophob (yellow), H-bond acceptor (red), and H-bond donor (blue) parts have been indicated.
Compound 8h as the most potent structure in the series, was docked on all of the potential binding sites of the [ES] complex to form the enzyme–substrate-inhibitor [ESI] complex. Considering the glide score (− 6.78 kcal/mol) and interactions, site 2 appeared to have the maximum affinity in comparison with other identified sites. As it is shown in Fig. 7, compound 8h well occupied the site, and the following interactions were detected: Thr86 residue acting as both H-bond acceptor and H-bond donor with amide group and quinazolinone ring nitrogen. The pi-cation interaction between Lys10 and the quinazolinone aromatic system and another pi-cation interaction between Arg48 and the thiazole ring was observed. A pi-pi stacking interactions were found among His14 and ethylbenzene moiety moreover several hydrophobic interactions were found between compound 8h and Leu11, Met44, Ala47, Ala85, Phe87, and Pro88 residues.

3D and 2D interactions of compound 8h in the [ESI] complex.
Antimicrobial and anti-ureolytic activity of tested compounds
Compounds 8c, 8g, and 8h were chosen for their antimicrobial activities against microorganisms including standard species of Cryptococcus neoformans (H99), and clinical isolate of Proteus vulgaris. The results showed that at concentrations ranging from 1 to 512 μg/ml, the examined compounds exhibited no antimicrobial activities against the tested pathogens (MIC > 512 (μg/ml).
Next, the anti-ureolytic activity of highly potent urease inhibitors (8c, 8g, and 8h) against the C. neoformans (H99) and P.vulgaris was visually and spectroscopically measured at 560 nm. Table 2 summarizes the findings. Compound 8h, like our enzymatic assay results, displayed the highest anti-ureolytic activities followed by compound 8c. Notably, 8g exhibited selective urease activity against C. neoformans but not against P. vulgaris at the tested range.
According to the findings, none of the selected derivatives had anti-microbial effects on the tested microorganisms; however, the high activity of tested compounds against ureolytic microorganisms strengthens our hypothesis that the designed pharmacophore can be an ideal candidate for targeting ureolytic microorganisms through urease enzyme inhibition.
ADME-toxicity profiles and physicochemical properties
The pkCSM server45 was used to predict the ADME-Toxicity properties of synthesized compounds. As shown in Table 3. All derivatives showed good human intestinal absorption, low clearance values, and limited toxicity.
According to the physicochemical properties predicted from the SwissADME website46, all compounds had appropriate molecular properties with no drug-likeness rules violations (Table 4).
Conclusion
In summary, fourteen new compounds with thioquinazolinone structures were designed and prepared as anti-urease agents. Among them, compound 8h exhibited the most potent inhibitory effect against urease with an IC50 value of 2.22 μM with around a ten-fold increase in the potency compared to the positive control. In addition, compound 8h possessed the uncompetitive type of inhibition in the enzymatic assay indicating that ligand bonded only to the complex formed between the enzyme and the substrate. The molecular docking study revealed that compound 8h could fit well into the binding site of urease by pi-cation, pi–pi, and H-bond interactions. 8h also demonstrated IC50 values of 129.4 ± 5.3 and 172.4 ± 8.7 µg/ml against C. neoformans and P.vulgaris on the ureolytic assay. Furthermore, in silico evaluations also found acceptable ADME-Toxicity and drug-likeness profiles.
Material and method
Chemistry
Compounds 3a-n were obtained by reaction of isatoic anhydride (compound 1, 1 mmol) with different amines (compound 2, 1.1 mmol) as the raw materials in ethanol under reflux conditions for 3 h. To the above solution carbon disulfide and KOH were added and the reaction was further refluxed for an extra 3 h to afford compounds 4a-n. Next, the intermediate 7 were synthesized by a simple reaction of nitrothiazolamine (5) with 2-chloroacetyl chloride (6) in DMF at room temperature. Finally, compounds 4a-n were reacted with 2-chloro-N-(5-nitrothiazol-2-yl)acetamide in the presence of K2CO3 to provide the crude products 8a-n which was purified by column chromatography to yield the final products.
N-(5-nitrothiazol-2-yl)-2-((4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)thio)acetamide (8a)
Brown solid; isolated yield: 80% (351 mg), mp 230–232 °C; IR (KBr) υ: 3317, 3062, 3010, 2951, 1692, 1661, 1651, 1631, 1592, 1560, 1481, 1466, 1444, 1410, 1351, 1321, 1279, 1222, 1180, 1077, 763, 719 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 13.48 (s, 0.7H, exchangeable proton), 8.66 (s, 1H), 8.08 (dd, J = 7.9, 1.6 Hz, 1H), 7.97 (s, 0.1H, exchangeable proton), 7.81 (td, J = 7.4, 1.5 Hz, 1H), 7.70–7.37 (m, 7H), 4.23 (s, 2H). 13C NMR (76 MHz, DMSO) δ 169.71, 163.49, 161.03, 157.10, 147.45, 143.63, 141.72, 136.21, 135.46, 130.58, 130.08, 129.90, 127.12, 126.61, 126.20, 120.02, 36.79. Anal.Calcd for C19H13N5O4S2: C 51.93, H 2.98, N 15.94, S 14.59; Found: C 51.72, H 3.12, N 15.70, S 14.83. MS (EI, 60 eV): m/z (%): 439 (M+, 24).
2-((3-(4-chlorophenyl)-4-oxo-3,4-dihydroquinazolin-2-yl)thio)-N-(5-nitrothiazol-2-yl)acetamide (8b)
Light brown solid; isolated yield: 82% (387 mg), mp 251–253 °C; IR (KBr) υ: 3330, 3086, 3014, 2921, 1693, 1669, 1649, 1628, 1601, 1561, 1523, 1496, 1479, 1424, 1354, 1336, 1299, 1241, 1176, 1010, 825, 751, 729 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 13.52 (s, 0.9H, exchangeable proton), 8.68 (s, 1H), 8.08 (dd, J = 8.0, 1.5 Hz, 1H), 7.97 (s, 0.1H, exchangeable proton), 7.81 (td, J = 8.2, 1.2 Hz), 7.71 (d, J = 8.7 Hz, 2H), 7.62 (d, J = 8.7 Hz, 2H), 7.52–7.37 (m, 2H), 4.25 (s, 2H). 13C NMR (76 MHz, DMSO) δ 169.30, 162.85, 160.99, 156.65, 147.39, 143.47, 142.04, 135.53, 135.35, 135.12, 131.95, 130.19, 127.12, 126.68, 126.20, 120.01, 36.53. Anal.Calcd for C19H12ClN5O4S2: C 48.15, H 2.55, N, 14.78, S 13.53; Found: 48.43, H 2.63, N, 14.49, S 13.65. MS (EI, 60 eV): m/z (%): 473 (M+, 36).
2-((3-(4-bromophenyl)-4-oxo-3,4-dihydroquinazolin-2-yl)thio)-N-(5-nitrothiazol-2-yl)acetamide (8c)
Dark brown solid; isolated yield: 84% (433 mg), mp 263–265 °C; IR (KBr) υ: 3358, 3068, 2959, 1696, 1669, 1656, 1639, 1592, 1552, 1482, 1468, 1418, 1359, 1292, 1250, 1160, 1135, 1080, 840 cm−1.
1H NMR (400 MHz, DMSO-d6) δ 13.50 (s, 0.9H, exchangeable proton), 8.67 (s, 1H), 8.06 (dd, J = 8.0, 1.5 Hz, 1H), 7.87–7.74 (m, 3H), 7.53 (d, J = 8.6 Hz, 2H), 7.46 (t, J = 7.4 Hz, 1H), 7.40 (d, J = 8.1 Hz, 1H), 4.23 (s, 2H). 13C NMR (101 MHz, DMSO) δ 169.34, 162.93, 160.93, 160.21, 156.55, 147.36, 143.50, 141.95, 135.53, 133.15, 132.20, 127.11, 126.67, 126.18, 124.01, 119.97, 36.55. Anal.Calcd for C19H12BrN5O4S2: C 44.03, H 2.33, N 13.51, S 12.37; C 44.25, H 2.18, N 13.42, S 12.51. MS (EI, 60 eV): m/z (%):) 516 (M+, 30).
2-((3-(4-methoxyphenyl)-4-oxo-3,4-dihydroquinazolin-2-yl)thio)-N-(5-nitrothiazol-2-yl)acetamide (8d)
Light brown solid; isolated yield: 78% (365 mg), mp 245–247 °C; IR (KBr) υ: 3333, 3058, 2949, 1682, 1662, 1660, 1639, 1599, 1555, 1479, 1456, 1400, 1350, 1281, 1230, 1182, 1165, 1087, 758 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 13.02 (s, 1H, exchangeable proton), 8.64 (s, 1H), 8.04–7.73 (m, 1H), 7.76 (d, J = 15.4 Hz, 1H), 7.44 (d, J = 7.7 Hz, 1H), 7.33 (t, J = 7.5 Hz, 1H), 7.18 (d, J = 8.3 Hz, 2H), 7.01 (d, J = 8.4 Hz, 2H), 4.20 (s, 2H), 3.81 (s, 3H). 13C NMR (101 MHz, DMSO) δ 169.45, 162.87, 160.66, 159.18, 157.68, 147.39, 143.44, 141.95, 139.96, 135.35, 130.42, 127.09, 126.52, 126.12, 119.95, 114.54, 55.73, 36.59. Anal.Calcd for C20H15N5O5S2: C 51.17, H 3.22, N 14.92, S 13.66; C 51.26, H 3.18, N 15.09, S 13.42. MS (EI, 60 eV): m/z (%): 469 (M+, 36).
2-((3-benzyl-4-oxo-3,4-dihydroquinazolin-2-yl)thio)-N-(5-nitrothiazol-2-yl)acetamide (8e)
Brown solid; isolated yield: 81% (366 mg), mp 241–243 °C; IR (KBr) υ: 3329, 3059, 2951, 1693, 1650, 1651, 1630, 1586, 1545, 1480, 1456, 1401, 1359, 1326, 1265, 1232, 1163, 1023, 745, 710 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 13.34 (s, 0.3 H, exchangeable proton), 8.66 (s, 1H), 8.11 (dd, J = 8.0, 1.5 Hz, 1H), 7.97 (s, J = 0.3 H, exchangeable proton), 7.79 (td, J = 8.4, 1.6 Hz, 1H), 7.47 (td, J = 8.2, 1.2 Hz, 1H), 7.42–7.23 (m, 7H), 5.37 (s, 2H), 4.35 (s, 2H). 13C NMR (76 MHz, DMSO) δ 169.77, 163.87, 161.25, 156.75, 147.06, 143.74, 141.55, 135.98, 135.48, 129.13, 128.02, 127.37, 127.16, 126.72, 126.11, 119.16, 47.60, 36.75. Anal.Calcd for C20H15N5O4S2: C 52.97, H 3.33, N 15.44, S 14.14; Found: C 53.26, H 3.56, N 15.24, S 14.44. MS (EI, 60 eV): m/z (%): 453 (M+, 33).
2-((3-(4-methylbenzyl)-4-oxo-3,4-dihydroquinazolin-2-yl)thio)-N-(5-nitrothiazol-2-yl)acetamide (8f.)
Brown solid; isolated yield: 83% (387 mg), mp 254–256 °C; IR (KBr) υ: 3326, 3050, 2941, 1689, 1652, 1656, 1632, 1590, 1552, 1488, 1463, 1404, 1349, 1316, 1291, 1230, 1182, 1087, 758, 729 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 13.07 (s, 0.4H, Exchangeable proton), 8.65 (s, 1H), 8.10 (d, J = 7.9 Hz, 1H), 7.97 (s, 0.6H, Exchangeable proton), 7.77 (t, J = 7.7 Hz, 1H), 7.45 (t, J = 7.9 Hz, 1H), 7.36 (d, J = 8.1 Hz, 1H), 7.23–7.11 (m, 5H), 5.32 (s, 2H), 4.34 (s, 2H), 2.29 (s, 3H). 13C NMR (76 MHz, DMSO) δ 169.76, 163.83, 161.23, 156.74, 147.04, 143.70, 141.57, 137.26, 135.42, 132.96, 129.65, 129.21, 127.75, 127.44, 127.14, 126.67, 119.17, 47.35, 36.73, 21.13. Anal.Calcd for C21H17N5O4S2: C 53.95, H 3.67, N 14.98, S 13.78; Found: C 53.90, H 3.75, N 14.90, S 13.65. MS (EI, 60 eV): m/z (%): 467 (M+, 32).
N-(5-nitrothiazol-2-yl)-2-((4-oxo-3-(pyridin-3-ylmethyl)-3,4-dihydroquinazolin-2-yl)thio)acetamide (8 g)
Brown solid; isolated yield: 86% (390 mg), mp 269–271 °C; IR (KBr) υ: 3335, 3120, 3096, 2863, 1691, 1652, 1651, 1630, 1586, 1584, 1545, 1489, 1456, 1406, 1340, 1401, 1366, 1359, 1326, 1265, 1232, 1191, 1028, 810, 721 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 8.66 (s, 1H), 8.65 (s, 1H), 8.53 (dd, J = 8.0, 1.6 Hz, 1H), 8.10 (dd, J = 8.0, 1.5 Hz, 1H), 7.97 (s, 0.1 H, exchangeable proton), 7.83–7.68 (m, 2H), 7.52–7.32 (m, 3H), 5.39 (s, 2H), 4.37 (s, 2H). 13C NMR (76 MHz, DMSO) δ 169.52, 163.50, 161.29, 156.32, 149.32, 149.19, 147.02, 143.62, 141.74, 135.53, 135.36, 131.87, 127.14, 126.79, 126.11, 124.22, 119.18, 45.59, 36.64. Anal.Calcd for C19H14N6O4S2: C 50.21, H 3.11, N 18.49, S 14.11; Found: C 49.89, H 3.46, N 18.18, S 14.41. MS (EI, 60 eV): m/z (%): 454 (M+, 32).
N-(5-nitrothiazol-2-yl)-2-((4-oxo-3-phenethyl-3,4-dihydroquinazolin-2-yl)thio)acetamide (8 h)
Brown solid; isolated yield: 75% (350 mg), mp 235–237 °C; IR (KBr) υ: 3320, 3071, 2922, 2893, 1690, 1655, 1652, 1622, 1585, 1559, 1482, 1453, 1414, 1357, 1326, 1281, 1220, 1180, 1126, 1079, 752, 719 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 13.51 (s, 0.4H, Exchangeable proton), 8.66 (s, 1H), 8.08 (d, J = 7.9 Hz, 1H), 7.97 (s, 0.6H, Exchangeable proton), 7.76 (t, J = 7.6 Hz, 1H), 7.56–7.11 (m, 8H), 4.40 (s, 2H), 4.28 (t, J = 8.1 Hz, 2H), 3.05 (t, J = 8.0 Hz, 2H). 13C NMR (76 MHz, DMSO) δ 169.82, 162.78, 160.75, 156.12, 146.99, 143.74, 141.58, 138.17, 135.28, 129.14, 129.13, 127.22, 126.95, 126.60, 126.04, 119.24, 46.18, 36.58, 33.91. Anal.Calcd for C21H17N5O4S2: C 53.75, H 3.55, N 14.76, S 13.43; Found: C 53.81, H 3.69, N 14.88, S 13.61. MS (EI, 60 eV): m/z (%): 467 (M+, 29).
N-(5-nitrothiazol-2-yl)-2-((4-oxo-3-propyl-3,4-dihydroquinazolin-2-yl)thio)acetamide (8i)
Light brown solid; isolated yield: 59% (238 mg), mp 179–181 °C; IR (KBr) υ: 3315, 3052, 2985, 2929, 1689, 1656, 1650, 1625, 1590, 1545, 1451, 1415, 1336, 1315, 1268, 1213, 1152, 980, 765, 741 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 13.60 (s, 0.3H, exchangeable proton), 8.67 (s, 1H), 8.06 (dd, J = 8.0, 1.5 Hz, 1H), 7.79 (s, 0.1H, exchangeable proton), 7.74 (td, J = 8.2, 1.2 Hz, 1H), 7.43 (td, J = 8.2, 1.2 Hz, 1H), 7.325 (d, J = 8.2, 1H), 4.38 (s, 2H), 4.15–3.69 (m, 2H), 1.94–1.44 (m, 2H), 0.98 (t, J = 7.4 Hz, 3H). 13C NMR (76 MHz, DMSO) δ 169.57, 163.30, 160.83, 156.23, 146.96, 143.57, 141.83, 135.21, 126.96, 126.54, 125.96, 119.21, 46.27, 36.32, 21.48, 11.59. Anal.Calcd for C16H15N5O4S2: C 47.40, H 3.73, N 17.27, S 15.81; Found: C 47.61, H 4.02, N 17.16, S 16.11. MS (EI, 60 eV): m/z (%): 405 (M+, 28).
2-((3-isopropyl-4-oxo-3,4-dihydroquinazolin-2-yl)thio)-N-(5-nitrothiazol-2-yl)acetamide (8j)
Yellow solid; isolated yield: 66% (267 mg), mp 165–167 °C; IR (KBr) υ: 3315, 3074, 2971, 2856, 1685, 1655, 1650, 1625, 1594, 1548, 1462, 1419, 1383, 1365, 1298, 1228, 1168, 1007, 759, 713 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 8.47 (s, 1H), 8.05 (td, J = 8.2, 1.2 Hz, 1H), 7.76 (td, J = 8.2, 1.2 Hz, 1H), 7.51–7.31 (m, 2H), 4.80 (s, 1H), 4.23 (s, 2H), 1.63 (d, J = 6.7 Hz, 6H). 13C NMR (76 MHz, DMSO) δ 175.25, 174.05, 161.53, 157.39, 146.84, 146.67, 136.54, 134.92, 126.54, 126.10, 120.35, 52.72, 36.26, 19.66. Anal.Calcd for C16H15N5O4S2: C 47.40, H 3.73, N 17.27, S 15.81; Found: C 47.66, H 3.87, N 17.55, S 15.59. MS (EI, 60 eV): m/z (%): 405 (M+, 23).
2-((3-butyl-4-oxo-3,4-dihydroquinazolin-2-yl)thio)-N-(5-nitrothiazol-2-yl)acetamide (8 k)
Brown solid; isolated yield: 68% (284 mg), mp 192–194 °C; IR (KBr) υ: 3310, 3042, 2996, 2921, 1687, 1658, 1651, 1628, 1592, 1547, 1456, 1412, 1333, 1309, 1274, 1223, 1175, 985, 760, 736 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 8.59 (s, 1H), 7.97 (dd, J = 8.0, 1.5 Hz, 1H), 7.65 (td, J = 8.2, 1.2 Hz, 1H), 7.34 (td, J = 7.6, 1.2 Hz, 1H), 7.23 (d, J = 7.7 Hz, 1H), 4.31 (s, 2H), 3.99 (t, , J = 7.3 Hz, 2H), 1.72–1.57 (m, 2H), 1.37–1.29 (m, 2H), 0.88 (t, J = 7.3 Hz, 3H). 13C NMR (76 MHz, DMSO) δ 169.40, 163.01, 160.78, 156.14, 146.94, 143.46, 141.97, 135.16, 126.92, 126.52, 125.94, 119.19, 44.57, 36.22, 30.07, 20.11, 14.01. Anal.Calcd for C17H17N5O4S2: C 48.68, H 4.09, N 16.70, S 15.29; Found: C 48.48, H 3.92, N 16.44, S 15.52. MS (EI, 60 eV): m/z (%): 419 (M+, 22).
2-((3-isobutyl-4-oxo-3,4-dihydroquinazolin-2-yl)thio)-N-(5-nitrothiazol-2-yl)acetamide (8 l)
Brown solid; isolated yield: 69% (285 mg), mp 183–185 °C; IR (KBr) υ: 3317, 3076, 2970, 2850, 1681, 1657, 1652, 1628, 1599, 1545, 1460, 1420, 1388, 1366, 1296, 1160, 1010, 762 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 12.94 (s, 0.5 H, exchangeable proton), 8.66 (s, 1H), 8.05 (dd, J = 8.0, 1.5 Hz, 1H), 7.95 (s, 0.5 H, exchangeable proton), 7.73 (ddt, J = 7.2, 5.1, 1.6 Hz, 1H), 7.46–7.36 (m, 1H), 7.31 (d, J = 8.1 Hz, 1H), 4.36 (s, 2H), 3.94 (d, J = 7.4 Hz, 2H), 2.38–2.14 (m, 1H), 0.94 (d, J = 6.7 Hz, 6H). 13C NMR (101 MHz, DMSO) δ 169.62, 163.38, 161.17, 156.58, 146.84, 143.61, 141.74, 135.23, 127.07, 126.56, 125.94, 119.14, 51.21, 36.50, 27.92, 20.45, 20.43. Anal.Calcd for C17H17N5O4S2: C 48.68, H 4.09, N 16.70, S 15.29; Found: C 48.73, H 4.13, N 16.52, S 15.41. MS (EI, 60 eV): m/z (%): 419 (M+, 27).
2-((3-allyl-4-oxo-3,4-dihydroquinazolin-2-yl)thio)-N-(5-nitrothiazol-2-yl)acetamide (8 m)
Brown solid; isolated yield: 74% (298 mg), mp 236–238 °C; IR (KBr) υ: 3322, 3065, 2921, 1688, 1650, 1651, 1628, 1591, 1551, 1492, 1487, 1452, 1410, 1348, 1310, 1285, 1225, 1171, 1080, 760, 730 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 13.51 (s, 0.3H, Exchangeable proton), 8.67 (s, 1H), 8.07 (dd, J = 7.9, 1.5 Hz, 1H), 7.97 (s, 0.3H, Exchangeable proton), 7.76 (td, J = 8.0, 1.5 Hz 1H), 7.45 (td, J = 7.1, 1.5 Hz 1H), 7.34 (d, J = 8.1 Hz, 1H), 5.97 (ddt, J = 17.2, 10.4, 5.1 Hz, 1H), 5.32–5.09 (m, 2H), 4.75 (d, J = 5.1 Hz, 2H), 4.37 (s, 2H). 13C NMR (76 MHz, DMSO) δ 169.56, 163.35, 160.66, 156.45, 147.01, 143.58, 141.81, 135.33, 131.76, 127.03, 126.62, 126.03, 119.16, 118.25, 46.54, 36.41. Anal.Calcd for C16H13N5O4S2: C 47.64, H 3.25, N 17.36, S 15.89; Found: C 47.86, H 3.16, N 17.41, S 15.76. MS (EI, 60 eV): m/z (%): 403 (M+, 26).
2-((3-cyclopentyl-4-oxo-3,4-dihydroquinazolin-2-yl)thio)-N-(5-nitrothiazol-2-yl)acetamide (8n)
Brown solid; isolated yield: 70% (301 mg), mp 200–202 °C; IR (KBr) υ: 3329, 3062, 2929, 2893, 1686, 1652, 1645, 1620, 1591, 1551, 1478, 1424, 1388, 1341, 1291, 1245, 1153, 1012, 763, 692 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 8.48 (s, 1H), 8.05 (dd, J = 7.9, 1.5 Hz, 1H), 7.75 (td, J = 8.2, 1.2 Hz, 1H), 7.52–7.30 (m, 2H), 5.03–4.77 (m, 1H), 4.24 (s, 2H), 2.26–2.19 (m, 2H), 2.06–1.82 (m, 5H), 1.74–1.46 (m, 2H). 13C NMR (76 MHz, DMSO) δ 175.06, 173.66, 161.02, 157.82, 146.77, 146.53, 136.77, 134.93, 126.58, 126.16, 126.11, 120.19, 60.10, 36.27, 28.74, 26.09. Anal.Calcd for C18H17N5O4S2: C 50.11, H 3.97, N 16.23, S 14.86; Found: C 49.89, H 4.16, N 16.34, S 14.59. MS (EI, 60 eV): m/z (%): 431 (M+, 29).
Urease inhibitory activity
Urease inhibition effects of the synthesized compounds were determined according to the previously reported procedure47,48,49. 100 μL of the synthesized compounds at different concentrations was added to 850 μL of urea as substrate and 15 μL urease (0.135 units dissolved in PBS, pH 7.4). After 30 min, to 100 μL of the incubated solution, 500 μL solution I (5.0 g phenol and 25.0 mg sodium nitroprusside in 500 mL water) was added followed by the addition of 500 μL of solution II (2.5 g sodium hydroxide, 4.2 mL sodium hypochlorite, and 5% chlorine in 500 mL water) which was further incubated at 37 °C for 30 min. The absorbance was determined by measuring indophenols at 625 nm. Thiourea was used as the standard inhibitor for urease. The IC50 values for all synthesized compounds were calculated using GraphPad Prism software (GraphPad Software, Inc., San Diego, CA).
Kinetic studies
The kinetic study for the inhibition of urease by compound 8h was carried out using four different concentrations of inhibitor. For the kinetic study of urease, compound 8h was used at the concentrations of 0, 1, 2, and 4 μM. The Lineweaver–Burk reciprocal plot was constructed by plotting 1/V against 1/[S] at variable concentrations of the substrate urea (3.12–100 mM). The inhibition constant Ki was calculated by the plot of slopes versus the corresponding concentrations of the compound 8h.
Molecular docking procedure
To perform the molecular modeling investigations, the Maestro Molecular Modeling platform (version 10.5) by Schrödinger, LLC has been used50,52. The X-ray crystallographic structure of the jack bean urease in complex with acetohydroxamic acid was downloaded from the protein data bank (www.rcsb.com) by the PDB ID: 4h9m. The protein is then prepared using a protein preparation wizard51. At this point, all water molecules and co-crystallised ligands were removed, the missing side chains and loops were filled using the prime tool54, and PROPKA assigned H-bonds at pH: 7.4. In order to prepare the ligands, the 2D structures of the ligands were drawn in ChemDraw (ver. 16) and converted into SDF files, which were used further by the ligprep module52. The ligand was prepared by OPLS_2005 force field using EPIK at a target pH of 7.0 ± 253.
To gain a better understanding of the active site residue conformational change in the [ES] complex, the induced fit docking method was utilized for docking the urea in the active site of the molecule54. AHA was considered as the grid center and the maximum number of 20 poses was calculated with receptor and ligand van der Waals radii of 0.7 A and 0.5 A, respectively. Structures with prime energy levels beyond 30 kcal/mol were eliminated based on standard precious glide docking. The Site map tool was used to find the possible allosteric binding sites of the [ES] complex55. The site map was tasked to report up to 5 potential binding sites with at least 15 site points per each reported site by more restrictive definition of hydrophobicity. The grid box was generated for each binding site using entries with a box size of 25 A, compound 8h was docked on binding sites using glide with extra precision and flexible ligand sampling, reporting 10 poses per ligand to form the final [ESI] complex56.
Antimicrobial activity against ureolytic microorganisms
The antimicrobial activity of compounds against the microorganisms including C. neoformans (H99), and clinical isolate of P.vulgaris was assessed using the microbroth dilution method, as recommended by the Clinical and Laboratory Standards Institute (CLSI) (M07-A9 for bacteria; M27-A3 for yeasts). The compounds were diluted, and stock solutions of 20 mg/ml in DMSO were prepared. Mueller–Hinton Broth (HiMedia) and RPMI-1640 (Sigma) were prepared as recommended for antimicrobial susceptibility testing of bacterial and fungal strains, respectively. Two-fold dilutions were made in the range of 1–512 μg/ml for tested compounds. The microbroth dilution test was accomplished using a 96-well microtiter plate, containing growth control (yeast culture in broth media) and sterility control (broth media without fungal culture). The antimicrobial susceptibility test was accomplished by adding a cell suspension adjusted to the 0.5 McFarland standard (1–2 × 108 CFU/mL for bacterial strains; 1−5 × 106 cells/ml for yeast) to different concentrations of tested compounds. Following incubation, the minimum inhibitory concentration (MIC) was established as the lowest concentration of compound that completely inhibits the growth of the organism in wells as detected visually. All experiments were performed in duplicates.
Anti-ureolytic activity against ureolytic microorganisms
The colorimetric microdilution technique using urea broth media (Merck, supplemented with glucose; pH = 6 for C. neoformans) was used to examine the urolytic activity of C. neoformans (H99), and clinical isolate of P.vulgaris treated with tested substances. Compounds in the concentration range of 1–512 μg/mL were exposed to ureolytic microorganisms, and the color of the medium was evaluated visually and spectroscopically at 560 nm after three days for C. neoformans and 24 h for P. vulgaris. The positive control, which included ureolytic bacteria but no drugs, changed color from yellow to dark pink or magenta. This shifts, allowing the determination of the inhibitory activity of compounds against urease activity of organisms even without a microtiter plate reader57,58.
In silico pharmacokinetic properties of synthesized compounds
SwissADME and pkCSM servers were used to determine the physicochemical and drug-likeness properties of the derivatives.
Data availability
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
References
Kafarski, P. & Talma, M. Recent advances in design of new urease inhibitors: A review. J. Adv. Res. 13, 101–112 (2018).
de Fátima, Â. et al. Schiff bases and their metal complexes as urease inhibitors: A brief review. J. Adv. Res. 13, 113–126 (2018).
Collins, C. M. & D’Orazio, S. E. Bacterial ureases: Structure, regulation of expression and role in pathogenesis. Mol. Microbiol. 9(5), 907–913 (1993).
Montecucco, C. & Rappuoli, R. Living dangerously: How Helicobacter pylori survives in the human stomach. Nat. Rev. Mol. Cell Biol. 2(6), 457–466 (2001).
Dixon, N. E., Riddles, P. W., Gazzola, C., Blakeley, R. L. & Zerner, B. Jack bean urease (EC 3.5.1.5). V. On the mechanism of action of urease on urea, formamide, acetamide, N-methylurea, and related compounds. Can. J. Biochem. 58(12), 1335–1344 (1980).
Carlsson, H. & Nordlander, E. Computational modeling of the mechanism of urease. Bioinorg. Chem. Appl. 2010, 364891 (2010).
Lippard, S. J. At last–the crystal structure of urease. Science (New York, N.Y.) 268(5213), 996–7 (1995).
Mobley, H. L., Mendz, G. L.& Hazell, S. L. Helicobacter Pylori: Physiology and Genetics, (2001).
Mobley, H. The role of Helicobacter pylori urease in the pathogenesis of gastritis and peptic ulceration. Aliment. Pharmacol. Ther. 10(Sup1), 57–64 (1996).
Mobley, H., Island, M. D. & Hausinger, R. P. Molecular biology of microbial ureases. Microbiol. Rev. 59(3), 451–480 (1995).
Olivera-Severo, D. et al. A new role for helicobacter pylori urease: Contributions to angiogenesis. Front. Microbiol. 8, 1883 (2017).
Balasubramanian, A. & Ponnuraj, K. Crystal structure of the first plant urease from jack bean: 83 years of journey from its first crystal to molecular structure. J. Mol. Biol. 400(3), 274–283 (2010).
Hameed, A. et al. Synthesis, biological evaluation and molecular docking of N-phenyl thiosemicarbazones as urease inhibitors. Bioorg. Chem. 61, 51–57 (2015).
Islam, M. et al. Therapeutic potential of N4-substituted thiosemicarbazones as new urease inhibitors: Biochemical and in silico approach. Bioorg. Chem. 109, 104691 (2021).
Hamad, A. et al. Probing sulphamethazine and sulphamethoxazole based Schiff bases as urease inhibitors; synthesis, characterization, molecular docking and ADME evaluation. Bioorg. Chem. 105, 104336 (2020).
Liu, Q. et al. Arylamino containing hydroxamic acids as potent urease inhibitors for the treatment of Helicobacter pylori infection. Eur. J. Med. Chem. 156, 126–136 (2018).
Rahim, F. et al. Development of bis-thiobarbiturates as successful urease inhibitors and their molecular modeling studies. Chin. Chem. Lett. 27(5), 693–697 (2016).
Abdulwahab, H. G. et al. Novel thiobarbiturates as potent urease inhibitors with potential antibacterial activity: Design, synthesis, radiolabeling and biodistribution study. Bioorg. Med. Chem. 28(23), 115759 (2020).
Uddin, I. et al. Synthesis, in vitro alpha glucosidase, urease activities and molecular docking study of bis-indole bearing Schiff base analogs. Chem. Data Collect. 28, 100396 (2020).
Taha, M. et al. Synthesis, in vitro urease inhibitory potential and molecular docking study of benzofuran-based-thiazoldinone analogues. Sci. Rep. 10(1), 1–8 (2020).
Rashid, U. et al. Synthesis of 2-acylated and sulfonated 4-hydroxycoumarins: In vitro urease inhibition and molecular docking studies. Bioorg. Chem. 66, 111–116 (2016).
Zaman, K. et al. Synthesis, in vitro urease inhibitory potential and molecular docking study of Benzimidazole analogues. Bioorg. Chem. 89, 103024 (2019).
Rahim, F. et al. Synthesis of new arylhydrazide bearing Schiff bases/thiazolidinone: α-Amylase, urease activities and their molecular docking studies. Bioorg. Chem. 91, 103112 (2019).
Taha, M. et al. Bisindolylmethane thiosemicarbazides as potential inhibitors of urease: Synthesis and molecular modeling studies. Bioorg. Med. Chem. 26(1), 152–160 (2018).
Abbasi, M. A. et al. Synthesis, in vitro and in silico studies of novel potent urease inhibitors: N-[4-({5-[(3-Un/substituted-anilino-3-oxopropyl) sulfanyl]-1, 3, 4-oxadiazol-2-yl} methyl)-1, 3-thiazol-2-yl] benzamides. Bioorg. Med. Chem. 26(13), 3791–3804 (2018).
Wang, D. & Gao, F. Quinazoline derivatives: Synthesis and bioactivities. Chem. Cent. J. 7(1), 95 (2013).
Khan, I., Ibrar, A., Ahmed, W. & Saeed, A. Synthetic approaches, functionalization and therapeutic potential of quinazoline and quinazolinone skeletons: the advances continue. Eur. J. Med. Chem. 90, 124–169 (2015).
Khan, I. et al. Quinazolines and quinazolinones as ubiquitous structural fragments in medicinal chemistry: An update on the development of synthetic methods and pharmacological diversification. Bioorg. Med. Chem. 24(11), 2361–2381 (2016).
Abuelizz, H. A., Marzouk, M., Ghabbour, H. & Al-Salahi, R. Synthesis and anticancer activity of new quinazoline derivatives. Saudi Pharm. J. 25(7), 1047–1054 (2017).
Alafeefy, A. M., Kadi, A. A., Al-Deeb, O. A., El-Tahir, K. E. & Al-Jaber, N. A. Synthesis, analgesic and anti-inflammatory evaluation of some novel quinazoline derivatives. Eur J Med Chem 45(11), 4947–4952 (2010).
Honkanen, E. et al. Synthesis and antihypertensive activity of some new quinazoline derivatives. J. Med. Chem. 26(10), 1433–1438 (1983).
Auti, P. S., George, G. & Paul, A. T. Recent advances in the pharmacological diversification of quinazoline/quinazolinone hybrids. RSC Adv. 10(68), 41353–41392 (2020).
Jafari, E., Khajouei, M. R., Hassanzadeh, F., Hakimelahi, G. H. & Khodarahmi, G. A. Quinazolinone and quinazoline derivatives: Recent structures with potent antimicrobial and cytotoxic activities. Res. Pharm. Sci. 11(1), 1–14 (2016).
Akyüz, G., Menteşe, E., Emirik, M. & Baltaş, N. Synthesis and molecular docking study of some novel 2, 3-disubstituted quinazolin-4 (3H)-one derivatives as potent inhibitors of urease. Bioorg. Chem. 80, 121–128 (2018).
Menteşe, E., Akyüz, G., Emirik, M. & Baltaş, N. Synthesis, in vitro urease inhibition and molecular docking studies of some novel quinazolin-4 (3H)-one derivatives containing triazole, thiadiazole and thiosemicarbazide functionalities. Bioorg. Chem. 83, 289–296 (2019).
Das, D., Sikdar, P. & Bairagi, M. Recent developments of 2-aminothiazoles in medicinal chemistry. Eur. J. Med. Chem. 109, 89–98 (2016).
Ballard, T. E. et al. Biological activity of modified and exchanged 2-amino-5-nitrothiazole amide analogues of nitazoxanide. Bioorg. Med. Chem. Lett. 20(12), 3537–3539 (2010).
Shrivastava, M., Shrivastava, G. & Shrivastava, G. Antimicrobial activity of schiff base of 2-Amino 5-nitrothiazole and its copper complex. PharmaTutor 6(9), 1–5 (2018).
Menteşe, E., Akyüz, G., Yılmaz, F., Baltaş, N. & Emirik, M. Synthesis of some novel quinazolin-4 (3H)-one hybrid molecules as potent urease inhibitors. Arch. Pharm. 351(12), 1800182 (2018).
Yamamoto, Y. et al. Nitazoxanide, a nitrothiazolide antiparasitic drug, is an anti-Helicobacter pylori agent with anti-vacuolating toxin activity. Chemotherapy 45(4), 303–312 (1999).
Abbasi, M. A. et al. Synthesis of novel N-(1,3-thiazol-2-yl)benzamide clubbed oxadiazole scaffolds: Urease inhibition, Lipinski rule and molecular docking analyses. Bioorg. Chem. 83, 63–75 (2019).
Rego, Y. F. et al. A review on the development of urease inhibitors as antimicrobial agents against pathogenic bacteria. J. Adv. Res. 13, 69–100 (2018).
Amtul, Z., Siddiqui, R. & Choudhary, M. Chemistry and mechanism of urease inhibition. Curr. Med. Chem. 9(14), 1323–1348 (2002).
Sepehri, N. et al. The natural-based optimization of kojic acid conjugated to different thio-quinazolinones as potential anti-melanogenesis agents with tyrosinase inhibitory activity. Bioorg. Med. Chem. 36, 116044 (2021).
Pires, D. E. V., Blundell, T. L. & Ascher, D. B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 58(9), 4066–4072 (2015).
Daina, A., Michielin, O. & Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7, 42717 (2017).
Asgari, M. S. et al. New 1,2,3-triazole-(thio)barbituric acid hybrids as urease inhibitors: Design, synthesis, in vitro urease inhibition, docking study, and molecular dynamic simulation. Arch. Pharm. (Weinheim) 353(9), e2000023 (2020).
Pedrood, K. et al. Arylmethylene hydrazine derivatives containing 1,3-dimethylbarbituric moiety as novel urease inhibitors. Sci. Rep. 11(1), 10607 (2021).
Sedaghati, S. et al. Novel (thio)barbituric-phenoxy-N-phenylacetamide derivatives as potent urease inhibitors: synthesis, in vitro urease inhibition, and in silico evaluations. Struct. Chem. 32(1), 37–48 (2021).
Maestro, Schrödinger, LLC, (2021).
Sastry, G. M., Adzhigirey, M., Day, T., Annabhimoju, R. & Sherman, W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 27(3), 221–234 (2013).
S. LigPrep, LLC (2021).
Greenwood, J. R., Calkins, D., Sullivan, A. P. & Shelley, J. C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided Mol. Des. 24(6), 591–604 (2010).
Farid, R., Day, T., Friesner, R. A. & Pearlstein, R. A. New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg. Med. Chem. 14(9), 3160–3173 (2006).
Halgren, T. A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 49(2), 377–389 (2009).
Friesner, R. A. et al. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein− ligand complexes. J. Med. Chem. 49(21), 6177–6196 (2006).
Knezevic, P., Aleksic Sabo, V., Simin, N., Lesjak, M. & Mimica-Dukic, N. A colorimetric broth microdilution method for assessment of Helicobacter pylori sensitivity to antimicrobial agents. J. Pharm. Biomed. Anal. 152, 271–278 (2018).
Nakamura, Y., Kano, R., Watanabe, S., Takahashi, H. & Hasegawa, A. Susceptibility testing of cryptococcus neoformans using the urea broth microdilution method: Empfindlichkeitsprüfung von cryptococcus neoformans in Harnstoff-Bouillon mittels Mikrodilution. Mycoses 41(1–2), 41–44 (1998).
Acknowledgements
The authors wish to thank the financial support of the Vice-Chancellor for Research of Shiraz University of Medical Sciences (Grant Number: IR.SUMS.REC.1400.529) and Iran National Science Foundation (INSF). All Figures in the article were drawn by co-authors.
Author information
Authors and Affiliations
Contributions
M.S. synthesized compounds. M.N.M. performed a docking study. S.M.F. synthesized compounds. N.T. supervised the biological tests. M.D. supervised the biological tests. A.M. performed the biological assay. K.Z. supervised and performed antimicrobial assay. S.Y. performed antimicrobial assay. M.A. performed the biological assay SH performed chemical analysis. M.B. synthesized compounds. B.L. contributed to the design and characterization of compounds. M.A. contributed to the design and characterization of compounds. M.B.T. performed in silico analysis. A.I. performed docking study and contributed to the preparation of the manuscript. M.M. supervised all phases of the study. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sohrabi, M., Nazari Montazer, M., Farid, S.M. et al. Design and synthesis of novel nitrothiazolacetamide conjugated to different thioquinazolinone derivatives as anti-urease agents. Sci Rep 12, 2003 (2022). https://doi.org/10.1038/s41598-022-05736-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-05736-4
This article is cited by
-
Synthesis, urease inhibitory and anticancer evaluation of glucosamine-sulfonylurea conjugates
Medicinal Chemistry Research (2024)
-
Design, synthesis, and biological studies of the new cysteine-N-arylacetamide derivatives as a potent urease inhibitor
Naunyn-Schmiedeberg's Archives of Pharmacology (2024)
-
New thioxothiazolidinyl-acetamides derivatives as potent urease inhibitors: design, synthesis, in vitro inhibition, and molecular dynamic simulation
Scientific Reports (2023)
-
The anti-Alzheimer potential of novel spiroindolin-1,2-diazepine derivatives as targeted cholinesterase inhibitors with modified substituents
Scientific Reports (2023)
-
Different barbiturate derivatives linked to aryl hydrazone moieties as urease inhibitors; design, synthesis, urease inhibitory evaluations, and molecular dynamic simulations
Medicinal Chemistry Research (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
