
Abstract
Epidermal growth factor receptor (EGFR)-mutated (mt) lung adenocarcinoma (LA) is refractory to immune checkpoint inhibitors (ICIs). However, the mechanisms have not been fully elucidated. CD8+ T cell infiltration was significantly lower in EGFR-mt than in EGFR-wild-type LA, which was associated with suppression of chemokine expression. Since this T cell-deserted tumor microenvironment may lead to the refractoriness of ICIs against EGFR-mt LA, we investigated the mechanism by focusing on the regulation of chemokine expression. The expression of C-X-C motif ligand (CXCL) 9, 10 and 11, which constitute a gene cluster on chromosome 4, was suppressed under EGFR signaling. The assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq) revealed open chromatin peaks near this gene cluster following EGFR-tyrosine kinase inhibitor (TKI) treatment. The histone deacetylase (HDAC) inhibitor recovered the expression of CXCL9, 10 and 11 in EGFR-mt LA. Nuclear HDAC activity, as well as histone H3 deacetylation, were dependent on oncogenic EGFR signaling. Furthermore, the Cleavage Under Targets and Tagmentation (CUT & Tag) assay revealed a histone H3K27 acetylation peak at 15 kb upstream of CXCL11 after treatment with EGFR-TKI, which corresponded to one of the open chromatin peaks detected by ATAC-seq. The data suggest that EGFR-HDAC axis mediates silencing of the chemokine gene cluster through chromatin conformational change, which might be relevant to the ICI resistance by creating T cell-deserted tumor microenvironment. Targeting this axis may develop a new therapeutic strategy to overcome the ICI resistance of EGFR-mt LA.
Similar content being viewed by others


Introduction
Since the early 2010s, the success of clinical trials using immune checkpoint inhibitors (ICIs), such as anti-programmed death-1 (PD-1) monoclonal antibodies (mAbs), has changed the clinical practices in cancer treatment1,2,3. Additionally, new clinical pieces of evidence, which reveal the efficacy of ICIs against various tumors and in different settings, are emerging4. However, some patients with cancer are resistant to ICIs. Various strategies have been devised to mitigate the resistance of cancers to ICIs.
Early clinical studies have revealed that human lung adenocarcinoma (LA) harboring oncogenic EGFR mutations is resistant to ICIs5,6,7. Hence, patients harboring EGFR mutations have been likely to be excluded from most of the ongoing ICI clinical trials on human LA. Oncogenic EGFR mutations were observed in approximately 50% of LA cases in a Japanese population8. The elucidation of the resistance mechanisms of EGFR-mutated (EGFR-mt) LA can contribute to the development of therapeutic strategies to overcome ICI resistance in EGFR-mt LA and facilitate an improved understanding of ICI mechanisms in cancer immunity. Several previous reports have suggested the potential refractory mechanisms of EGFR-mt LA for ICIs9,10,11, however, no clinical trials to mitigate the resistance have been performed. Since we noticed the significant decrease of CD8A mRNA in EGFR-mt LA from TCGA data, we further attempted to clarify the underlying mechanism.
C-X-C motif ligand (CXCL) chemokines, CXCL9, 10, and 11, are IFN-responsive and associated with the recruitment of activated Th1 cells, and have pleiotropic functions against T cells, such as migration, differentiation and activation, and play crucial roles in immune activation and tumor rejection in TME via its cognate receptor, CXCR312, and anti-tumor effect of anti-PD-1 therapy decreases in CXCR3 knock-out mice13.
We have confirmed that the number of CD8+ T cells in EGFR-mt LA was significantly lower than that in LA with wild-type EGFR (EGFR-wt). We demonstrated that the expression of CXCL9, 10 and 11, which comprises a gene cluster on chromosome 4, was significantly downregulated in EGFR-mt LA cell lines and that oncogenic EGFR signaling inhibits histone H3 acetylation near the gene cluster through the activation of histone deacetylases (HDACs), suggesting the intrinsic CXCL chemokine suppression in LA cells through an epigenetic mechanism under EGFR signaling. This study could explain one of the mechanisms of ICI resistance in EGFR-mt LA, which may contribute to the development of therapeutic strategies to mitigate ICI resistance in EGFR-mt LA.
Materials and methods
The Cancer Genome Atlas (TCGA) data analysis
The mRNA levels of CD8A were comparatively analyzed according to the mutational status of EGFR, ALK, and KRAS in LA (n = 230) cases curated in TCGA database (TCGA, Nature 2014). The mRNA levels of chemokines and chemokine receptors whose mRNA levels were positively correlated with those of CD8A were searched in the same dataset. All analyses were conducted using cBioPortal (http://www.cbioportal.org/)14,15.
Immunohistochemical (IHC) analysis
The surgical specimens of patients with LA with or without known EGFR mutations (mutant, n = 56; wild-type, n = 84) were subjected to IHC using anti-CD8a mouse mAb (1:1,000; Proteintech, 66868-1-Ig). IHC staining was performed using Ventana Discovery XT (Roche, Japan) following the manufacturer’s instructions. The IHC images were captured using a virtual slide scanner, NanoZoomer-SQ C13140 (Hamamatsu, Japan). The CD8a-positive spots were automatically counted in three areas (200 × 200 µm2) of each sample using cellSens Dimension (Olympus, Japan).
Cell lines
HCC827, NCI-H1373, A549 (purchased from American Type Culture Collection) and PC-9 (purchased from RIKEN Cell Bank) cells were cultured in Rosewell Park Memorial Institute (RPMI)-1640 (WAKO, Japan) medium supplemented with 10% fetal calf serum and penicillin/streptomycin at 37 °C and 5% CO2. All cell lines were used immediately after purchase (HCC827 and PC-9; EGFR-mt (Exon19 deletion)) or after authentication using short tandem repeat DNA typing (NCI-H1373 and A549; EGFR-wt LA).
Multiplex chemokine assay
HCC827 or PC-9 cells were seeded at 1 × 106 cells in a 60 mm dish and cultured overnight. The culture medium was replaced with 3 mL of fresh complete medium, and the cells were incubated with DMSO or AZD9291 (100 nM) (ChemScene,CS-2018) for 24 h in biological triplicates. In some experiments, the cells were stimulated with 10 ng/mL IFN-γ (PeproTech, #300-02) at 6 h post-DMSO/AZD9291 treatment. The chemokine concentrations in the culture supernatant were determined using the Bio-Plex suspension array system and Bio-Plex Pro human chemokine assays (Bio-Rad, 171AK99MR2 Japan), following the manufacturer’s instructions. The quantification was performed using biological triplicates.
Reverse transcription quantitative polymerase chain reaction (RT-qPCR)
The methods of RNA extraction, cDNA synthesis, and RT-qPCR were described previously16. The relative mRNA expression levels of CXCL9, 10, 11, HDAC1, 2, 3, and 4 in the cell lines were determined using the ΔΔCt method. The mRNA levels of the target genes were normalized to those of ACTB or 18S rRNA, as well as those in DMSO-treated cells, as a reference. Relative quantification (RQ) was calculated as (test sample mRNA level normalized by endogenous control) / (reference sample mRNA level normalized by endogenous control). Quantification was performed using biological triplicates. Primer sequences are listed in Table 1.
Immunoblotting analysis of nuclear extracts
EGFR-mt and EGFR-wt LA cells were treated with DMSO (Control) or AZD9291 (1 µM) for 24 h, then the nuclear proteins were extracted using Nuclear Extraction Kit (Active Motif, 40010) in biological triplicates. The protein concentrations were measured using DC Protein Assay Kit (Promega, 5000112JA). The methods of SDS-PAE, and immunoblotting were described previously16. The primary (anti-histone H3ac (pan-acetyl) Ab (Active Motif, 39040), anti-Lamin A/C Ab (Active Motif, 39287)) and secondary (goat anti-rabbit IgG (GE Healthcare, NA931)) antibodies were used at a dilution of 1:1000. Immunoreactivity was detected using Fusion SoloS (M&S Instruments Inc., Japan).
Nuclear HDAC activity measurement
The nuclear proteins were extracted as mentioned in Immunoblot analysis. The nuclear HDAC activity was measured in biological triplicates using HDAC Assay Kit (Active Motif, .56200), which is a biological assay to determine the HDAC activity using a short peptide substrate that contains an acetylated lysine residue. The HDAC activity is determined as the concentration of active HDAC by making a standard curve.
Assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq) analysis
HCC827 cells were treated with DMSO or AZD9291 (1 mM) in replicates for 18 h. The cells (3 × 105) were then frozen in cell stock solution (Cell Banker 1, TAKARA) and transported to DNAFORM Life Science Research Center 402 (Yokohama, Japan) to perform ATAC-seq. Next-generation sequencing was performed using 150 bp pair-end sequences at 40 million reads/sample. The quality of the FASTQ sequences was determined using Fast QC. The quality-filtered sequences were mapped to the reference genome hg38 using BWA. BAM files were used for peak calls with PePr, in which the replicate peaks were merged. The detected peaks were annotated using HOMER. The motifs within the annotated peaks were searched using FIMO (http://meme-suite.org/tools/fimo). The significantly increased open chromatin peaks in the AZD9291-treated group were visualized using the IGV genomic browser.
Cleavage under targets and tagmentation (CUT & Tag) assay
HCC827 cells were treated with DMSO (control) or AZD9291 (100 nM) for 24 h. Next, the cells were collected and analyzed using the CUT&Tag-IT Assay Kit (Active Motif, 53160) and histone H3K27ac antibody (Active Motif, 39034), following the manufacturer’s instructions. Genomic libraries containing fragments enriched by binding to anti-histone H3K27ac Ab were sequenced using an Illumina NovaSeq 6000 (Illumina, Inc.). The fastq files were uploaded to Basepair (https://activemotif.basepairtech.com/) and the differences in histone H3K27ac peaks between DMSO and AZD9291 treated samples were searched.
Statistical analysis
Student’s t-test was performed using IBM SPSS Statistics 25.0.0. Statistical significance was set at p < 0.05.
Ethics approval and consent to participate
The study using surgical specimens was approved by the Ethics Committee of Shiga University of Medical Science, and conducted in accordance with the Declaration of Helsinki. Informed consent was obtained from all patients.
Results
Infiltration of CD8+ T cells to tumor microenvironment (TME) is downregulated in EGFR-mt LA
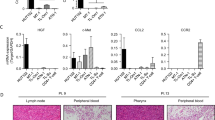
As several previous reports indicated that CD8+ T cell infiltration was reduced in EGFR-mt LA10,17,18, we tried to confirm this phenomenon. In TCGA dataset, the mRNA levels of CD8A were significantly lower in EGFR-mt than EGFR-wt LA (TCGA, Nature 2014) (p < 0.01). However, the mRNA levels of CD8A did not significantly vary according to the mutational status of ALK or KRAS in the same dataset (Fig. 1A). The detailed mutational status of these driver genes is listed on Table S1. IHC of CD8a using surgical specimens confirmed that the number of CD8a+ T cells were significantly lower in EGFR-mt than EGFR-wt LA. The number of CD8a-positive cells in LA with EGFR mutation (exon 18 G719 missense mutation, exon 19 deletion, and exon 21 (L858R) except exon 20 Ins (n = 1) were significantly lower than EGFR-wt LA (p < 0.05). (Table S2). This indicated that EGFR-mt LA was associated with a decreased infiltration of CD8+ T cells into the TME (Fig. 1B,C).

EGFR-mutated (EGFR-mt) lung adenocarcinoma (LA) exhibits a lower number of CD8+ T cells than LA with wild-type EGFR (EGFR-wt) LA. (A) Comparative analysis of the CD8A mRNA levels in human LA (TCGA, Nature 2014) according to the mutational status of EGFR, ALK, and KRAS. EGFR mt (n = 40; 17%), ALK-mt (n = 19; 8%), and KRAS-mt (n = 83; 36%). *p < 0.01. (B) Comparison of the CD8a-positive cells between EGFR-mt (n = 56) LA and EGFR-wt (n = 84) LA using immunohistochemical analysis (IHC). Mean CD8a-positive cells in three 200 × 200 µm2 areas were calculated for each sample. Vertical bars indicate the mean CD8a cells and the error bars indicate the standard deviation. *p < 0.0001. (C) Representative IHC images. Upper and lower panels indicate EGFR-mt and EGFR-wt LA, respectively. Horizontal scale bar: 200 µm.
Next, the correlation between the mRNA levels of CD8A and various chemokines or chemokine receptors was examined in the TCGA LA dataset (TCGA, Nature 2014) to speculate the chemokines possibly involved in CD8+ T cell recruitment to the TME (Table 2). Several chemokines and their receptors were significantly and positively correlated with the mRNA levels of CD8A (e.g., CXCL9, CXCL10, CXCL11, CXCR3, CCL4, CCL5, and CCR5). These chemokines may contribute to CD8+ T cell recruitment in the human LA TME.
EGFR signaling suppressed the production of several chemokines related to CD8+ T cell recruitment
Since some oncogenic signaling pathways, such as Wnt/β-catenin signaling, suppress tumor-derived chemokine expression in melanoma, which results in the reduction of tumor-infiltrating T cells (TILs)19, we speculated that EGFR signaling might also suppress some intrinsic chemokine production in human LA cells. Treatment with EGFR-TKI, AZD9291, significantly upregulated the expression levels of CXCL9, 10, and 11 in two EGFR-mt LA cell lines, PC-9 and HCC827 cells (exon 19 deleted mutation) at both protein and mRNA levels (Fig. 2A,B). We also observed the increase of CXCL10 in other EGFR-mt LA cell lines, NCI-H1975 and II-18 (L858R missense mutation), with lesser degree than exon 19 deleted LA cells (Fig. S1A,B). We addressed whether the re-expression of the chemokine CXCL10 could increase T cell migration. We conducted in vitro CD8+ T cell migration assay using the culture supernatant of HCC827 cells (EGFR-mt) with or without AZD9291 ± anti-CXCL10 mAb. AZD9291 increased T cell migration significantly, and this increase was blocked with anti-CXCL10 mAb, suggesting re-expression of CXCL10 could increase T cell migration (Fig.S1C). We also found focal CXCL10 protein expression in EGFR-wt, but not in EGFR-mt LA cells by IHC (Fig. S2). We noticed that CXCL9, 10 and 11 and their corresponding receptor, CXCR3, were included in the list of Table 2. As we stated in Introduction, these chemokines/chemokine receptor are important for Th1 immune cell recruitment/response and anti-tumor effect of anti-PD1 Ab12,13. Therefore, we speculated oncogenic EGFR signal-driven suppression of these chemokines may have important roles in the reduced TILs and refractoriness of anti-PD1/PD-L1 Ab, and tried to elucidate the mechanism.

EGFR signaling suppresses chemokine expression in EGFR-mutated (EGFR-mt) lung adenocarcinoma (LA). (A) The levels of CXCL9, CXCL10, and CXCL11 in the culture supernatant of PC-9 and HCC827 LA cells were quantified using the Bio-Plex suspension array system after treatment with dimethyl sulfoxide (DMSO) or AZD9291 and stimulation with IFN-γ. Biological triplicates were used for the analysis. *p < 0.0005, **p < 0.0001, ***p < 0.05 (Student’s t-test). (B) RT-qPCR of CXCL9, 10, and 11 of PC-9 and HCC827 LA cells treated with DMSO or AZD9291 and INF-γ stimulation. *p < 0.05, **p < 0.01, ***p < 0.005. Biological triplicates. (C) Mapping of CXCL9, CXCL10 and CXCL11 genes on chromosome 4 on UCSC genome browser (http://genome.ucsc.edu). The 3 CXCL chemokines constitute a gene cluster of 55 kb. The release date is 2018/04/11.
EGFR signaling suppresses open chromatin formation around the CXCL9, 10, and 11 gene loci in EGFR-mt LA cells
We conducted promoter analyses of CXCL10 using a luciferease vector ligated with the 0.4 and 1.2 kb DNA sequence of CXCL10 upstream from transcription start site (TSS), by focusing on IFN regulatory factor-1 (IRF-1) because an IRF-1 binding site, IFN-stimulated response element (ISRE), is located at the 5’-proximal sequence of CXCL10 (Fig. S3A), and previous reports indicated that EGFR signal downregulates IRF-1 and CXCL1010,20. However, oncogenic EGFR signal did not suppress the IRF-1 promoter activity based on our luciferase promoter assays (Fig. S3B–D). Therefore, we speculated that EGFR signal regulates the chemokine expression by suppressing the regulatory regions outside their promoters.
Since the expression of CXCL9, 10, and 11 was simultaneously upregulated upon treatment with AZD9291 (Fig. 2A,B), and these three genes make a cluster within a region of approximately 55 kb on chr4 (75,980,790–76,036,070 bp) (Fig. 2C), we hypothesized that these three genes may be simultaneously regulated from common enhancer regions, which are not located near the proximal regions of these genes. To verify this hypothesis, EGFR-mt LA cells treated with AZD9291 were subjected to ATAC-seq to determine the presence of open chromatin peaks around these three genes. One region in the first intron of CXCL9, and two regions at a distance of 15 and 19 kb from the TSS of CXCL11 were more open following EGFR-TKI treatment (Fig. 3). The large 15 kb peak comprised three merged peaks and no major peak was detected near CXCL10. The potential transcription factor (TF)-binding motifs in these loci were identified using FIMO (http://meme-suite.org/tools/fimo) (Table S3). We speculated that common enhancer regions within these peaks may contact to each promoter of these three genes simultaneously as a mechanism of a topologically associating domain (TAD)21, which is a DNA region whose DNA sequences contact each other, and that the common enhancer regions might be suppressed under oncogenic EGFR signaling.

Assay for transposase-accessible chromatin using high-throughput sequencing (ATAC-seq) analysis of the sequences near CXCL9, CXCL10, and CXCL11 in AZD9291-treated EGFR-mutated (EGFR-mt) lung adenocarcinoma (LA) cells. Treatment with AZD9291 generated three open chromatin peaks. One is in the first intron of CXCL9 (+ 159 bp from the transcription start site (TSS)), while the other two are in the 5′ region of CXCL11 (− 15,480 and − 19,130 bp from the TSS) (arrows and rectangle boxes). The large peak at the 15 kb region upstream of CXCL11 comprises three merged peaks.
Histone deacetylase (HDAC) inhibitor increased CXCL9, 10, and 11 gene expression, and EGFR-TKI decreased HDAC1,2,3 and 4 as well as nuclear HDAC activity and increased histone H3 acetylation of EGFR-mt LA cells
EGFR-mt LA cells (PC-9 and HCC827) treated with the pan-HDAC inhibitor, vorinostat (Active Motif, 14027), showed a significant increase in CXCL9, 10, and 11 mRNA expression (Fig. 4A). However, the effects of vorinostat on the expression of these chemokines were also found in EGFR-wt LA cells (Fig. S4). EGFR-wt LA cells may have other activated signaling pathways, which might induce other HDACs leading to the epigenetic silencing of these chemokines. Therefore, we determined which HDACs were specifically regulated by oncogenic EGFR signaling. RT-qPCR of HDAC1, 2, 3, 4, 5, 6, 7, 8, 9, 10, and 11 revealed that HDAC1, 2, 3, and 4 mRNA levels were significantly decreased by AZD9291 in two EGFR-mt, but not in two EGFR-wt LA cells (Fig. 4B), while the remaining HDACs did not show consistent results in the two EGFR-mt LA cells (Fig. S5). This suggests that oncogenic EGFR signaling specifically activates the expression of HDAC1, 2, 3, and 4.

Oncogenic EGFR signaling causes induction of HDAC1, HDAC2, HDAC3, and HDAC4, associated with increased nuclear HDAC activity, nuclear histone H3 deacetylation, and chemokine suppression in EGFR-mt LA cells. (A) RT-qPCR of CXCL9, CXCL10, and CXCL11 with or without 10 μM SAHA (vorinostat) in two EGFR-mt LA cells. *p < 0.05, **p < 0.005, ***p < 0.0005, ****p < 0.00005. (B) RT-qPCR of HDAC1, HDAC2, HDAC3, and HDAC4 with or without AZD9291 in two EGFR-mt and two EGFR-wt LA cells. *p < 0.01, **p < 0.05. (C) Nuclear HDAC activity with or without AZD9291 in the two EGFR-mt and two EGFR-wt LA cells. *p < 0.05. (D) Immunoblot of histone H3 acetylation of the nuclear extract with or without AZD9291 in the two EGFR-mt and two EGFR-wt LA cells. LaminA/C is a loading control. All the experiments were performed in biological triplicates.
Furthermore, nuclear HDAC activity was significantly suppressed by AZD9291 compared to DMSO in the two EGFR-mt, but not in the two EGFR-wt LA cell lines (Fig. 4C). Additionally, nuclear histone H3 acetylation increased with AZD9291 treatment in the two EGFR-mt, but not in the two EGFR-wt LA cells (Fig. 4D). These results suggest that oncogenic EGFR signaling increases HDAC activity through the induction of HDAC1, 2, 3, and 4, resulting in the suppression of histone acetylation in enhancer regions, which could cause epigenetic silencing.
Oncogenic EGFR signaling suppressed histone H3K27 acetylation at one of the ATAC-seq open chromatin peaks
To confirm whether oncogenic EGFR signaling suppressed histone H3 acetylation at the same open chromatin peaks identified by ATAC-seq, we conducted a CUT&Tag assay to identify histone H3K27 acetylation peaks after AZD9291 treatment. Compared to the control DMSO treatment, a significant increase in histone H3K27 acetylation was observed at chr4:76,050,566–76,051,369, which overlapped with one of the ATAC-seq open chromatin peaks at chr4_76,051,000_76,052,100, at 15 kb upstream of the CXCL11 TSS (Fig. 5A). The other two peaks at the 1st intron of CXCL9 and 19 kb upstream of CXCL11 TSS were not significantly different in the CUT&Tag assay (Fig. 5A,B). Because we used anti-histone H3K27 mAb as a validated Ab for CUT&Tag assay, other histone H3 or histone H4 acetylation could not be evaluated. These results suggest that oncogenic EGFR signaling suppresses histone H3K27 acetylation at the remote region of these three chemokine genes, making the chromatin conformation an inactive, closed form, which might result in the suppression of chemokine transcription.

Oncogenic EGFR signaling suppressed histone H3K27 acetylation at 15 kb upstream of CXCL11 TSS, which corresponded to the open chromatin peak by ATAC-seq. Histone H3K27 acetylation peaks are represented in Integrative genomic view (IGV). (A) Red histogram (AZD9291-treated) showed a significantly higher peak than blue histogram (DMSO-treated) at chr4:76,050,566–76,051,369 (p = 10–31, q = 10–25) (red rectangle), which overlapped to the open chromatin peak by ATAC-seq at chr4: 76,051,000–76,052,100. (B) Histone H3K27 acetylation peak in the 1st intron of CXCL9 (chr4: 76,007,200–76,007,500) did not show significant difference between AZD9291 and DMSO.
Discussion
Oncogene addiction plays a role, not only in tumorigenesis or tumor progression, but also in immune evasion in mouse and human tumors22,23. Recently, oncogenic pathway-mediated non-T cell inflamed TME has been identified as one of the causes for the resistance of cancers to ICIs24. For example, oncogenic Wnt/β-catenin signaling in melanoma decreased T cell infiltration by downregulating the expression of CCL4, which recruits the basic leucine zipper TF ATF-like 3 (BATF3)-lineage dendritic cells (DCs) that cross-present tumor-derived antigens through major histocompatibility complex class I19. Other gain-of-function (myc)25, or loss-of-function (LKB1, PTEN, and TP53)26,27,28,29 mutations also resulted in a decreased T cell infiltration in the TME. Based on these previous observations, we speculated that oncogenic EGFR signaling might also contribute to the non-T cell inflamed TME, as most of the EGFR-mt LA cases exhibited higher resistance to anti-PD1 and anti-PD-L1 mAb than EGFR-wt LA cases5,6,7. In this study, the mRNA level of CD8A and the infiltration of CD8+ T cells were confirmed to be lower in EGFR-mt than EGFR-wt LA in TCGA dataset and surgical specimens, respectively (Fig. 1A,B). Hence, this study aimed to elucidate the underlying mechanism with a focus on the regulation of chemokine gene expression.
Offin et al. reported that the tumor mutation burden (TMB) of EGFR-mt LA was significantly lower than that of EGFR-wt LA9. TMB is one of the predictive factors of ICI efficacy30,31. Hence, the low TMB in EGFR-mt LA can potentially explain the resistance to immunotherapy. However, a low TMB is not directly associated with the decreased number of CD8+ T cells in the TME, which exhibits suppressed T cell infiltration in the effector phase (but not in the induction phase) of the cancer immunity cycle32. In the effector phase, the suppression of recruitment or proliferation of effector CD8+ T cells in the TME must occur. A low TMB is closely associated with the suppression of neoantigen-specific T cells in the induction phase. Therefore, this cannot explain the formation of non-T cell inflamed TME.
Similar observations to ours have recently been reported by Sugiyama et al.10. Several differences were found between their results and ours. At first, they reported that the mRNA expression of FOXP3 in EGFR-mt was higher than that in EGFR-wt LA, which suggested that the number of regulatory T cells (Tregs) increased in EGFR-mt LA. However, the mRNA levels of FOXP3 and chemokines recruiting Treg (CCL22, CCL17, and CXCL12) were not significantly different between EGFR-mt and EGFR-wt LA in TCGA dataset (Fig. S6A). Treatment with AZD9291 significantly downregulated the expression of CCL22 in the HCC827 cells, but not in PC-9 cells, in our study (Fig. S6B). Thus, here, the relationship between oncogenic EGFR signaling and Treg recruitment was not clear. Secondly, the interpretation of the regulatory mechanism of CXCL10 suppression varied between the two studies. Sugiyama et al. claimed that EGFR downregulates CXCL10 by suppressing IRF-110. Previous studies20,33 have also suggested that IRF-1 promotes CXCL10 transcription by binding to ISRE in the CXCL10 5′-proximal region. In fact, we noticed that AZD9291 significantly increased IRF-1 expression at both mRNA and protein levels in EGFR-mt LA cells as Sugiyama et al. (Fig. S7A). We also observed that site-directed mutagenesis of ISRE of the CXCL10 promoter significantly decreased promoter activity (Fig. S7B), suggesting that IRF-1 enhances CXCL10 transcription through ISRE. However, treatment with AZD9291 decreased IRF-1 promoter activity (Figs. S3 and S7B), irrespective of the increase in the IRF-1 protein level. These results suggest that the transcriptional activity of IRF-1 is not simply regulated by its quantity, but through other complex mechanisms, such as post-translational modification of IRF-1 or formation of a complex with other transcription factors, which depends on oncogenic EGFR signaling. We speculated that the regulatory mechanism of EGFR-driven CXCL10 suppression does not exist in the 5’-proximal promoter region of CXCL10. This prompted us to examine alternative regulatory mechanisms.
In this study, the expression of three IFN-γ-responsive Th1-inducing chemokines (CXCL9, 10, and 11) was similarly increased upon treatment with AZD9291 (Fig. 2A,B). These three genetic loci were localized within a small region of approximately 55 kb. These three genes were simultaneously suppressed by histone deacetylases that were induced by oncogenic EGFR signaling. Histone deacetylation occurred at a presumed TAD near the gene cluster, which corresponded to one open chromatin peak detected by ATAC-seq. These results suggest an epigenetic suppressive mechanism of chemokine genes through the suppression of histone H3 acetylation at TAD, which was mediated by oncogenic EGFR signaling. We examined whether a DNA methyltransferase inhibitor, 5-azacytidine (5-AZA), could increase the expression of CXCL10 in EGFR-mt LA cells, but did decrease the expression level (Fig. S8), suggesting promoter methylation seems to be irrelevant. Two open chromatin peaks found by the ATAC-seq assay (the 1st intron of CXCL9 and 19 kb upstream of CXCL11) did not correspond to those in the CUT&Tag assay. We used an anti-histone H3K27 acetylation Ab because pan-histone H3 acetylation Ab was not validated for this assay. It is possible that other histone H3 acetylation or histone H4 acetylation peaks may be formed with AZD9291 and correspond to the two ATAC-seq peaks. Although our study suggests a role for TAD in the epigenetic regulation of chemokine genes, the presence of TAD must be demonstrated by more strict assays for genome-wide conformational studies, such as the chromosome conformation capture (3C) method34 or Hi-C35 in a future study. Furthermore, it remains to be elucidated how oncogenic EGFR signaling regulates HDAC1-4 expression, and whether HDAC1-4 deacetylate histone H3 near the CXCL9-11 gene cluster under oncogenic EGFR signaling, which warrant further experiments in future studies.
The significance of tumor intrinsic chemokine suppression in vivo remains unclear in our in vitro results. It remains to be demonstrated whether suppression of the 3 CXCL chemokines could cause T cell-deserted tumor microenvironment. However, it has been well documented that CXCL9, 10, and 11 play crucial roles in immune activation and tumor rejection in TME via its cognate receptor, CXCR312, and anti-tumor effect of anti-PD-1 therapy decreases in CXCR3 knock-out mice13. Enhancer of zeste homolog 2 (EZH2)-mediated histone H3K27me3 and DNMT1-mediated DNA methylation suppress the ovarian tumor production of Th1-inducing chemokine, CXCL9 and 10, and decrease TIL in TME. Inhibitors for the DNMTs could increase the TIL and improves therapeutic effects of PD-1 Ab, suggesting that epigenetic silencing of Th1-chemokine is a tumor immune evasion mechanism36. In view of the significance of the CXCL9, 10, and 11/CXCR3 axis in anti-PD-1 therapy, our findings may provide a new insight to the mechanism of EGFR-driven resistance against ICI. Further in vivo study focusing on the relationship between suppression of the 3 CXCL-chemokines and the resistance of ICIs in EGFR-mt LA, and whether HDAC inhibitor (HDACi) could overcome the ICI resistance are warranted in the future study.
In summary, this study suggested that the oncogenic EGFR signaling pathway suppresses intrinsic CXCL chemokine expression in EGFR-mt LA cells through histone deacetylation at 15 kb upstream of CXCL11 by HDACs which are induced under EGFR signaling. This finding may make a possibility for a new therapeutic strategy, a combination of ICI and HDAC inhibitors, which might be safer than combination of ICI and EGFR-TKI which increased interstitial pneumonia37. To our knowledge, this is the first report demonstrating oncogene-driven epigenetic silencing of chemokine genes, which might lead to the immune evasion of cancer, as well as the resistance to ICI, which may open a new possibility to overcome the ICI resistance of EGFR-mt LA.
Data availability
The data generated in this study are available within the article and its supplementary data files. The datasets of ATAC-seq and CUT&Tag generated during the current study are available in the DBJJ repository, DRA014913 and DRA014923, respectively.
References
Robert, C. et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa1104621 (2011).
Topalian, S. L. et al. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa1200690 (2012).
Brahmer, J. R. et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa1200694 (2012).
Salas-Benito, D. et al. Paradigms on immunotherapy combinations with chemotherapy. Cancer Discov. https://doi.org/10.1158/2159-8290.CD-20-1312 (2021).
Borghaei, H. et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa1507643 (2015).
Rittmeyer, A. et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicenter randomized controlled trial. Lancet https://doi.org/10.1016/S0140-6736(16)32517-X (2017).
Herbst, R. S. et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomized controlled trial. Lancet https://doi.org/10.1016/S0140-6736(15)01281-7 (2016).
Mitsudomi, T. Advances in target therapy for lung cancer. Jpn. J. Clin. Oncol. https://doi.org/10.1093/jjco/hyp174 (2010).
Offin, M. et al. Tumor mutation burden and efficacy of EGFR-tyrosine kinase inhibitors in patients with EGFR-mutant lung cancers. Clin. Caner Res. https://doi.org/10.1158/1078-0432.CCR-18-1102 (2019).
Sugiyama, E. et al. Blockade of EGFR improves responsiveness to PD-1 blockade in EGFR-mutated non-small cell lung cancer. Sci. Immunol. https://doi.org/10.1126/sciimmunol.aav3937 (2020).
Eric, Tu. et al. Anti-PD-L1 and anti-CD73 combination therapy promotes T cell response to EGFR-mutated NSCLC. JCI Insight https://doi.org/10.1172/jci.insight.142843 (2022).
Tokunaga, R. et al. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation—A target for novel cancer therapy. Cancer Treat Rev. 63, 40–47. https://doi.org/10.1016/j.ctrv.2017.11.007 (2018).
Zinal, S. et al. Chemoattractant receptors BLT1 and CXCR3 regulate antitumor immunity by facilitating CD8+ T cell migration into tumors. J. Immunol. 197, 2016–2026. https://doi.org/10.4049/jimmunol.1502376 (2016).
Cerami, E. et al. The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. https://doi.org/10.1158/2159-8290.CD-12-0095 (2012).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. https://doi.org/10.1126/scisignal.2004088 (2013).
Sumimoto, H., Takano, A., Teramoto, & Daigo, Y. RAS-mitogen-activated protein kinase signal is required for enhanced PD-L1 expression in human lung cancers. PLoS ONE https://doi.org/10.1371/journal.pone.0166626 (2016).
Dong, Z.-Y. et al. EGFR mutation correlates with uninflamed phenotype and weak immunogenicity, caused impaired response to PD-1 blockade in non-small cell lung cancer. Oncoimmunology 6(11), e1356145. https://doi.org/10.1080/2162402X.2017.1356145 (2017).
Zhao, C., Su, C., Li, X. & Zhou, C. Association of CD8 T cell apoptosis and EGFR mutation in non-small lung cancer patients. Thoracic Caner 11, 2130–2136. https://doi.org/10.1111/1759-7714.13504 (2020).
Spranger, S., Bao, R. & Gajewski, T. F. Melanoma-intrinsic β-catenin signaling prevents anti-tumour immunity. Nature https://doi.org/10.1038/nature14404 (2015).
Kalinowski, A. et al. EGFR activation suppresses respiratory virus-induced IRF1-dependent CXCL10 production. Am. J. Physiol. Lung Cell Mol. Physiol. https://doi.org/10.1152/ajplung.00368.2013 (2014).
Lupiáñez, D. G., Spielmann, M. & Mundlos, S. Breaking TADs: How alterations of chromatin domains result in disease. Trends Genet. https://doi.org/10.1016/j.tig.2016.01.003 (2016).
Wang, T. et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat. Med. https://doi.org/10.1038/nm976 (2004).
Sumimoto, H., Imabayashi, F., Iwata, T. & Kawakami, Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J. Exp. Med. https://doi.org/10.1084/jem.20051848 (2006).
Spranger, S. & Gajewski, T. F. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer. https://doi.org/10.1038/nrc.2017.117 (2018).
Casey, S. C. et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science https://doi.org/10.1126/science.aac9935 (2016).
Koyama, S. et al. STK11/LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T-cell activity in the lung tumor microenvironment. Cancer Res. https://doi.org/10.1158/0008-5472.CAN-20-0806 (2016).
Peng, W. et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. https://doi.org/10.1158/2159-8290.CD-15-0283 (2015).
Xue, W. et al. Senescence and tumor clearance is triggered by p53 restoration in murine liver carcinomas. Nature https://doi.org/10.1038/nature05529 (2007).
Iannello, A., Thompson, T. W., Ardolino, M., Lowe, S. W. & Raulet, D. H. P53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J. Exp. Med. https://doi.org/10.1084/jem.20130783 (2013).
Yarchoan, M., Hopkins, A. & Jaffe, E. M. Tumor mutational burden and response rate to PD-1 inhibition. N. Engl. J. Med. https://doi.org/10.1056/NEJMc1713444 (2017).
Rizvi, N. A. et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science https://doi.org/10.1126/science.aaa1348 (2015).
Chen, D. S. & Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity https://doi.org/10.1016/j.immuni.2013.07.012 (2013).
Zaheer, R. S., Koetzler, R., Holden, N. S., Wiehler, S. & Proud, D. Selective transcriptional down-regulation of human rhinovirus-induced production of CXCL10 from airway epithelial cells vai the MEK1 pathway. J. Immunol. https://doi.org/10.4049/jimmunol.0802401 (2009).
Dekker, J., Rippe, K., Dekker, M. & Kleckner, N. Capturing CHROMOSOME CONFORMATION. Science https://doi.org/10.1126/science.1067799 (2002).
Lieberman-Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science https://doi.org/10.1126/science.1181369 (2009).
Peng, D. et al. Epigenetic silencing of TH1-type chemokines shapes tumor immunity and immunotherapy. Nature 527, 249–253. https://doi.org/10.1038/nature15520 (2015).
Oxnard, G. R. et al. TATTON: A multi-arm, phase Ib trial of osimertinib combined with selumetinib, savolitinib, or durvalumab in EGFR-mutant lung cancer. Ann Oncol https://doi.org/10.1016/j.annonc.2020.01.013 (2020).
Funding
Grant-in-Aid for Scientific Research (C) (JSPS KAKENHI grant number 17K09652 (HS)) and Grant-in-Aid for Scientific Research on Innovative Areas from the Japan Society for the Promotion of Science (CoBiA: JSPS KAKENHI grant numbers 16H06277 and 22H04923 (YD)). YD is a member of the Shiga Cancer Treatment Project supported by Shiga Prefecture (Japan) and the International Joint Research Project (FY2016–2024) of the Institute of Medical Sciences (The University of Tokyo).
Author information
Authors and Affiliations
Contributions
H.S. and Y.D. conceptualized and designed the study, and developed the study methodology. T.I., J.H., K.T., and Y.D. acquired and managed patients, and provided facilities. H.S. acquired and analyzed the data (statistical analysis and biostatistics analysis). H.S., A.T., K.T., and Y.D. interpreted the data. H.S. and Y.D. wrote, reviewed, and/or revised the manuscript. A.T. and Y.D. were involved in administrative, technical, or material support. Y.D. supervised the study.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sumimoto, H., Takano, A., Igarashi, T. et al. Oncogenic epidermal growth factor receptor signal-induced histone deacetylation suppresses chemokine gene expression in human lung adenocarcinoma. Sci Rep 13, 5087 (2023). https://doi.org/10.1038/s41598-023-32177-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-32177-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
