
Abstract
According to molecular profiling studies, a considerable number of patients with pancreatic cancer harbor potentially actionable mutations. However, there are limited relevant data from the Korean population. We assessed the molecular profiles of patients with pancreatic cancer in Korea. This study collected molecular profiling data from patients with pancreatic cancer who visited Seoul National University Bundang Hospital between March 2018 and August 2020. Formalin-fixed, paraffin-embedded tumor specimens were sequenced using a targeted next-generation sequencing (NGS) platform. Cancer-associated mutations were analyzed, and potentially actionable mutations were identified. Potentially actionable mutations were classified into “highly actionable” and “modifies options” based on the Know Your Tumor registry study. In total, 87 patients with NGS tumor panel data were identified. Sixty-one patients (70.1%) had metastatic disease at the time of tissue acquisition. Tissues were obtained from the primary tumors and metastatic sites in 41 (47.1%) and 46 (52.9%) patients, respectively. At least one pathogenic mutation was reported in 86 patients (98.9%). The frequencies of four common mutations in our cohort were similar to those in The Cancer Genome Atlas data. Potentially actionable mutations were identified in 27 patients (31.0%). Of these, mutations categorized as highly actionable and modifies options were identified in 12 (13.8%) and 18 patients (20.7%), respectively. The most frequent highly actionable mutations were located in DNA damage response genes, such as BRCA1, BRCA2, or ATM (n = 6, 6.9%). Two patients with germline BRCA1 mutations received maintenance poly(adenosine diphosphate-ribose) polymerase inhibitor therapy. One patient has been receiving maintenance treatment for 18 months while remaining in radiologically complete remission. Mutational profiles using targeted NGS in Korean patients with pancreatic cancer were similar to those in Western patients. The present study supports the clinical potential and possible expanded clinical use of genetic profiling.
Similar content being viewed by others


Introduction
Pancreatic cancer is a lethal cancer with a 5-year survival rate of only 10% in the United States1. The incidence and death rates of pancreatic cancer are gradually increasing, and it is expected to become the second leading cause of cancer-related death in the United States by 20301,2. In Korea, in 2019, pancreatic cancer was reported to be the eighth most common cancer with more than 8000 new cases per year3. Moreover, it was reported to be the fifth most common cause of cancer-related death, and the 5-year survival rate was only 13.9% in 2019. The main obstacle in the improvement of the poor prognosis of pancreatic cancer is that less than 20% of patients have resectable lesions at diagnosis, and the median overall survival of patients with advanced stage is less than 1 year with the current combination chemotherapy1,4,5.
Recent molecular profiling studies in pancreatic cancer revealed clinically relevant recurrent molecular alterations and genomic subtypes, which are collectively termed actionable mutations6,7,8,9,10,11. They reported that approximately 25% of patients with pancreatic cancer harbor potentially actionable mutations, and mutations were most commonly present in DNA damage response (DDR) genes. Additionally, the recently reported Know Your Tumor (KYT) registry trial, which compared survival between patients who received matched therapy based on molecular testing and those who received unmatched therapies, revealed improved survival outcomes with molecular profiling-based precision medicine in pancreatic cancer12.
Although several studies have described the potential and clinical usefulness of molecular profiling in pancreatic cancer, the data related to Asian populations remain limited. Recently, a clinical next-generation sequencing (NGS) platform was implemented in daily practice, and molecular profiling data in pancreatic cancer are accumulating. Therefore, we aimed to report the results of molecular profiling for pancreatic cancers at a single tertiary center in Korea as well as to share the relevant clinical experiences.
Materials and methods
Patients and tumor specimens
This retrospective cohort study included patients pathologically diagnosed with pancreatic cancer who underwent NGS-based targeted gene mutational assays between March 2018 and August 2020 at Seoul National University Bundang Hospital (SNUBH). Formalin-fixed, paraffin-embedded tumor specimens were obtained for NGS at the time of diagnosis or when the disease recurred or progressed. Biopsies were conducted for primary pancreatic tumors or metastatic lesions. Paired germline testing using blood samples was performed at the discretion of the attending physician.
NGS panel information and data analysis
Tumor tissue specimens were sequenced on the SNUBH pan-cancer panel, a targeted sequencing platform at SNUBH. The first three patients were profiled on SNUBH ver. 1.1, which targeted 89 genes, and subsequent patients were profiled on SNUBH ver. 2.0, which targeted 544 genes (Supplementary Table S1). Microsatellite instability (MSI) and tumor mutational burden (TMB) were reported only in SNUBH ver. 2.0.
Samples were sequenced on the MiseqDx platform (Illumina, San Diego, CA, USA) for the SNUBH panel ver. 1.1 and the NextSeq 550Dx platform (Illumina) for the SNUBH panel ver. 2.0. Reads were aligned to the human reference genome hg19. Mutect2 was used to detect single nucleotide variants (SNVs) and small insertion/deletions (INDELs), and SnpEff was used to annotate the identified variants. Only SNVs/INDELs with variant allele frequencies of ≥ 2% were selected. CNVkit was used to identify copy number variation (CNV), and a mean copy number of ≥ 5 was considered gain (amplification). Gene fusions were identified using LUMPY, and read counts ≥ 3 were interpreted as positive results for the structural variations.
The MSI phenotype was detected using mSINGs, and TMB was calculated as the number of eligible variants in the effect panel size (1.41 megabases). Eligible variants were missense mutations with the following criteria: 1) variants reported in the population database with a frequency of > 1% (East Asian, gnomAD) were excluded; 2) pathogenic and likely pathogenic mutations reported in ClinVar were excluded; 3) variants with allele frequencies of less than 2% were excluded; and 4) variants with a depth of less than 200 were also excluded.
All genetic alterations were reported and classified using a tiered system according to the standardized guideline for the interpretation and reporting of sequence variants in cancer as follows: 1) Tier I, variants of strong clinical significance such as those targeted by FDA-approved, professional guideline, or well-powered study-reported therapies; 2) Tier II, variants of potential clinical significance such as those targeted by FDA-approved treatments for different tumor types or investigational therapies; 3) Tier III, variants of unknown clinical significance; and 4) Tier IV, benign or likely benign variants13.
Potentially actionable mutations
All genetic alterations were reviewed, and potentially actionable mutations were analyzed. The gene list for potentially actionable mutations was constructed according to the previous KYT registry study (Supplementary Table S2)9. Mutations were classified as “highly actionable” or “modifies options” as described in the KYT study. Specifically, mutations with clinical evidence of a high response rate in patients with relevant molecular abnormalities in any cancer type were considered highly actionable, and those possibly implicated in the response to therapy based on the underlying mechanism were classified as modifies options. In addition, patients with MSI-high status or high TMB (which was defined as > 20 mutations per megabase) were examined.
Statistical analysis
For the comparison of mutation frequency according to datasets or clinical settings, the chi-square or Fisher’s exact test was used, and Bonferroni’s correction was applied for multiple comparisons. All statistical analyses and mutational mapping in this study were performed using the open software R version 4.0.3 (R Foundation for Statistical Computing, Vienna, Austria).
Ethical approval
The Institutional Review Board of SNUBH approved this study (IRB no. B-2007–622-108) and waived the requirement for written informed consent from the participants because of the retrospective nature of this study. This study was conducted in accordance with the principles of the Declaration of Helsinki, and all study procedures were conducted following the relevant guidelines and regulations.
Results
Baseline characteristics of the patients
Between March 2018 and August 2020, 87 patients were enrolled in this study. Patient characteristics and tumor specimen information are summarized in Table 1. The median age (range) of patients at diagnosis was 64 (35–86) years, and 54 patients (62.1%) were men. Meanwhile, 61 (70.1%) and 26 (29.9%) patients had metastatic and non-metastatic pancreatic cancers at the time of tissue acquisition, respectively. Adenocarcinoma was the dominant type of cancer (n = 82, 94.3%).
Tumor specimens for NGS were obtained from primary tumors and metastases in 41 (47.1%) and 46 patients (52.9%), respectively. The liver was the most frequent metastatic site for tissue acquisition (n = 33, 37.9%). Concerning the method of tissue acquisition, ultrasound-guided percutaneous needle biopsy (n = 36, 41.4%) was most common, followed by surgical resection (n = 25, 28.7%) and fine-needle aspiration or biopsy (n = 20, 23.0%). Meanwhile, 13 patients (14.9%) underwent additional biopsy for NGS after cancer progression or recurrence.
Landscape of genetic mutations
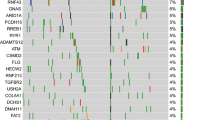
The mean depth of coverage in targeted gene sequencing ranged from 259 to 1,717. The most frequently reported mutation was KRAS mutation, which was found in 82 patients (94.3%), followed by TP53 mutations in 65 patients (74.7%), SMAD4 mutations in 26 patients (29.9%), and CDKN2A mutations in 20 patients (23.0%) (Fig. 1). The frequencies of these four mutations were similar to those reported in The Cancer Genome Atlas (TCGA) dataset (Table 2)14. Additionally, the frequencies of other significant recurrent mutations (which were previously reported in the TCGA dataset), such as RNF43 (10.3% vs. 6.0%), ARID1A (11.5% vs. 5.3%), TGFBR2 (4.6% vs. 5.3%), and GNAS (6.9% vs. 6.7%) mutations, were not significantly different from those of the TCGA dataset. CNVs were identified in 21 patients (24.1%), and KDM5A amplification (n = 5, 5.7%) was the most common amplification, followed by GATA6 (n = 4, 4.6%), RAD52 (n = 4, 4.6%), KRAS (n = 3, 3.4%), and MYC amplifications (n = 3, 3.4%) (Fig. 1). The mutation frequency was compared according to various clinical characteristics, including cancer stage, biopsy site, method of tissue acquisition, and prior chemotherapy; however, no significant differences in the frequencies of any mutations were identified (Supplementary Tables S3A and S3B).

Landscape of genomic alterations in patients with pancreatic cancer (n = 87). Distribution of recurrent single nucleotides variants (SNVs)/insertions/deletions (INDELs) and copy number variation (CNV) as well as the total percentage of patients with SNVs/INDELs or CNV for each recurrent mutated gene.
At least one Tier I or Tier II mutation was identified in 86 patients (98.9%). Other Tier I or II SNV/INDELs in oncogenes, such as BRAF (n = 1, 1.1%), GNAS (n = 1, 1.1%), and EGFR (n = 1, 1.1%), and tumor suppressor genes, such as ARID1A (n = 3, 3.4%) and RNF43 (n = 2, 2.3%), were observed, except for the four common mutations. Most common Tier I or Tier II CNVs were MYC and KRAS amplifications, where were observed in three patients (3.4%) each, followed by CDK6, CCND2, ERBB2, FGF6, FGF23, and MCL1 amplifications, which were observed in two patients (2.3%) each. Tier I or II mutations related to hereditary cancer syndrome in pancreatic cancer were identified in BRCA1 (n = 2, 2.3%), BRCA2 (n = 2, 2.3%), PRSS1 (n = 1, 1.1%), MSH6 (n = 2, 2.3%), PMS2 (n = 1, 1.1%), and APC (n = 1, 1.1%). Among them, two patients with BRCA1 mutations were confirmed to have germline pathogenic variants through paired germline testing using blood samples.
Potentially actionable mutations
In total, 27 patients (31.0%) were identified to have at least one potentially actionable mutation (Fig. 2A and Supplementary Table S4). Of these, highly actionable and modifies options mutations were found in 12 (13.8%) and 18 patients (20.7%), respectively. After excluding three patients (3.4%) who had both highly actionable and modifies options mutations, there were 15 patients with only modifies options mutations (17.2%).

(A) The percentage of patients with potentially actionable mutations (n = 87). (B) Therapeutic options according to the mutational profile (n = 87). (C) Tumor mutation burden profiling (n = 84).
Highly actionable mutations included mutations in BRCA1 (n = 2), BRCA2 (n = 2), ATM (n = 2), and BRAF (n = 1) and amplification in CDK6 (n = 2), ERBB2 (n = 2), and AKT2 (n = 1). Mutations in DDR genes such as BRCA1, BRCA2, and ATM, which were observed in six patients, were the most frequent highly actionable mutations. Concerning modifies options, mutations in ARID1A (n = 7) were the most frequent, followed by RNF43 mutations (n = 5), GNAS mutations (n = 2), and CCND2 amplification (n = 2). When classified by therapeutic options, mTOR/AKT inhibitors represented the most common drug class (n = 10, 11.5%), followed by poly(adenosine diphosphate-ribose) polymerase (PARP) inhibitors (n = 7, 8.0%), WNT inhibitors (n = 5, 5.7%), CDK inhibitors (n = 4, 4.6%), and MEK inhibitors (n = 4, 4.6%) (Fig. 2B). The MSI status and TMB were analyzed in 84 patients profiled with the SNUBH panel ver. 2.0. No patient had MSI-high tumors, but high TMB was reported in one patient (1.2%) (Fig. 2C).
Clinical implications of NGS results
In this study, three patients (3.4%) received matched therapy based on their molecular profiles. Of these, one patient was a 65-year-old man who was diagnosed with metastatic pancreatic cancer with an EGFR exon 19 deletion. He received a combination treatment, including gemcitabine and an EGFR tyrosine kinase inhibitor erlotinib (Roche, Basel, Switzerland); however, he showed rapid aggravation just after one cycle of the treatment.
The remaining two patients were diagnosed with metastatic pancreatic cancer using percutaneous needle biopsy of liver metastasis, and targeted gene profiling with the SNUBH panel ver. 2.0 was conducted at the time of diagnosis. The profiling results illustrated that both patients had tumors with Tier I BRCA1 mutations. With additional germline testing, they were confirmed to have germline pathogenic BRCA1 mutations, and they received the PARP inhibitor olaparib (AstraZeneca, Cambridge, United Kingdom) as maintenance therapy after FOLFIRINOX chemotherapy, as introduced in the POLO trial15.
The first patient who received olaparib maintenance treatment was a 54-year-old man with metastatic pancreatic cancer and a germline BRCA1 mutation (c.3412 + 1G > T). He received nine cycles of FOLFIRINOX and displayed a partial response with a 70% reduction of the tumor burden versus baseline according to RECIST ver. 1.1 (Fig. 3A). Subsequently, he received maintenance treatment with a PARP inhibitor. The tumor burden gradually decreased during maintenance treatment, and the lesion became indiscernible on computed tomography scan after 1 year of PARP inhibitor treatment. He has consistently responded to PARP inhibitor therapy, which he has received for 18 months. He had a sister with ovarian cancer and a germline BRCA1 mutation and a niece (the daughter of the sister with ovarian cancer) with breast cancer and a germline BRCA1 mutation. He had three children, and genetic testing revealed a germline BRCA1 mutation in one child.

(A) and (B) Clinical course of two patients with germline pathogenic BRCA1 mutations who were treated with PARP inhibitors. Changes in tumor burden (left y-axis) and the level of CA 19–9 (right y-axis) over time of treatment (months, x-axis) were presented together with specific computed tomography imaging.
The other patient was a 64-year-old woman with metastatic pancreatic cancer and a germline BRCA1 mutation (c.5467 + 1G > A). She received nine cycles of FOLFIRINOX and displayed a partial response with a 50% reduction of the tumor burden versus baseline according to RECIST ver. 1.1 (Fig. 3B). She received maintenance therapy with a PARP inhibitor, but the tumor progressed after 3 months of treatment. Subsequently, considering the germline BRCA1 pathogenic variant, she received third-line gemcitabine and cisplatin chemotherapy, but the tumor progressed after four cycles of treatment. She is currently receiving nanoliposomal irinotecan with fluorouracil as fourth-line therapy. She had a family history of variant cancers, including two first-degree relatives with gastric cancer (father and younger brother), two second-degree relatives with gastric cancer (father’s brother) and pancreatic cancer (mother’s brother), and a nephew (the child of a younger brother with gastric cancer) who was diagnosed with cholangiocarcinoma at the age of 28 years. Of her two children, one underwent germline BRCA1 mutation testing, and the result was negative.
Discussion
The present study reviewed the result of targeted gene profiling of patients with pancreatic cancer in a single tertiary center in Korea. Similar to the results of previous studies, which were mostly conducted in Western countries, KRAS, TP53, CDKN2A, and SMAD4 mutations were most common, and the frequencies of these recurrent mutations were comparable to those in the TCGA dataset. In addition, potentially actionable mutations were found in more than 30% of patients, versus rates of 11–50% in previous studies6,7,8,9,10,11.
There have been several mutation profiling data of patients with pancreatic cancer in Asian populations16,17,18. Hong et al. conducted whole-exome and RNA sequencing in 83 patients with pancreatic cancer in Korea, and they attempted to demonstrate the molecular profiles and find potential prognostic biomarkers using molecular aberrations16. Moreover, Yang et al. and Zhang et al. also attempted revealing genetic characteristics of pancreatic cancer in two other studies in Chinese patients with pancreatic cancer using targeted NGS panels17,18. However, the number of studies on molecular profiling of pancreatic cancer in Asian populations is still much less than that in Western populations, and it is also difficult to integrate and compare these data with our study results because of significant differences in study objectives, clinical settings, and methodology. Therefore, further large-scale and standardized genomic studies on pancreatic cancer in Asian populations are necessary.
Mutational profiles in this study were analyzed by constructing a gene list for potentially actionable mutations in reference to the KYT study and classifying the mutations into two groups (highly actionable and modifies options)9. In the KYT study, 50% of patients had potentially actionable mutations, and 27% had highly actionable mutations. However, the rates of potentially and highly actionable mutations in our study were lower than those in the KYT study (31 and 14%, respectively). The gap between the two studies may be explained by multiple factors such as differences in the NGS panel (FoundationOne® panel [Cambridge, MA, USA] vs. customized NGS panel), the database used for variant interpretation, the number of patients with KRAS mutations (87% vs. 94%), and ethnicity. In addition, the percentage of actionable mutations varied in prior studies (11–50%), and a large targeted gene profiling study of more than 3500 patients with pancreatic cancer using the FoundationOne® panel also reported that 17% of patients had therapeutically relevant alterations, which was lower than the rate in the KYT study6,7,8,9,10,11.
Similar to previous studies of actionable mutations in pancreatic cancer6,9,10,11, mutations in DDR genes, such as BRCA1, BRCA2, and ATM, were the most frequent highly actionable mutations in this study. Several studies have demonstrated the survival benefit of platinum-based chemotherapy in such patients12,19,20,21. In addition, the recent POLO trial reported the clinical benefit of the PARP inhibitor olaparib as a maintenance treatment in patients with metastatic pancreatic cancer and germline BRCA mutations15. Two patients (2.3%) in this study were confirmed to have germline pathogenic BRCA1 mutations. Both of them displayed partial responses to FOLFIRINOX, and one patient who harbored a stop-gain variant (c.3412G > T) exhibited a durable response to PARP inhibitor therapy for 18 months. Moreover, the number of patients with germline BRCA mutations who can benefit from PARP inhibitors, currently the only approved indication for targeted therapy in pancreatic cancer, might have been underestimated in this study because two other patients with BRCA2 mutations (Tier I and Tier II, respectively) did not undergo germline testing. In addition, recent studies described the possibility of expanding the indication for treatments related to the DDR pathway, such as treatments targeting non-canonical DDR gene mutations, somatic DDR gene mutations, or genomic scars associated with DDR pathway deficiency through mutational signature or genomic instability analysis19,22,23.
Another potential targeted treatment in pancreatic cancer is immunotherapy. Based on the results of the KEYNOTE-158 study, the immune checkpoint inhibitor pembrolizumab is currently approved for treating any solid cancer with high MSI24. More recently, any solid cancer with high TMB, defined as > 10 mutations per megabase in the FoundationOne® CDx panel, was also approved as an indication for immune checkpoint inhibitor therapy25. In addition, POLE and/or POLD1 mutations, which induced impairments in DNA proofreading and replication, were reported to be associated with the hypermutation phenotype and immune checkpoint inhibitor response26,27,28. As previously known that high MSI and pathogenic POLE and/or POLD1 mutations are rare in pancreatic cancer, they were not identified in our study29. High TMB was also rare in this study with only one patient (1.2%), but there remained pending issues such as variability of TMB results across different NGS panels or the proper cutoff level according to cancer types30,31. Therefore, further researches on TMB as predictive biomarker for immunotherapy in pancreatic cancer are necessary.
Our study had several limitations. First, this was a retrospective study, and selection bias was possible because the profiling test was not performed in all patients with pancreatic cancer. Second, the reliability of the SNUBH panel may be insufficient compared with other commercialized panels. However, the MSI status and ERBB2 amplification, which were confirmed by other test methods in this study, revealed results that were consistent with the SNUBH panel (Supplementary Table S5). Third, only two patients underwent germline genetic testing following the result of tumor sequencing because germline pathogenic BRCA1 and/or BRCA2 mutations are the only approved indication for targeted therapy in pancreatic cancer. Fourth, potentially actionable mutations in this study were identified according to previous KYT study protocols; however, actionability can change over time. Therefore, the frequency of actionable mutations in this study may have been overestimated or underestimated in the present circumstances. Fifth, the number of patients who received targeted therapy or therapy based on a previously reported clinical trial according to the result of gene profiling was small. Finally, the sample size was insufficient to demonstrate the difference in the mutation profile according to various clinical characteristics. However, the present study had enough strengths as it attempted to compare the potentially actionable mutations with those of previous Western datasets using the same criteria as that used in Western datasets, and it further expanded the information in Asian populations.
In summary, targeted gene profiling in Korean patients with pancreatic cancer revealed similar frequencies of common recurrent mutations and potentially actionable mutations as recorded in Western data. Considering that approximately one-third of patients had at least one potentially actionable mutation and the number of actionable mutations can expand gradually, mutational profiling is expected to have significant clinical impact in patients with pancreatic cancer.
Data availability
The datasets generated during the current study are available from the corresponding author upon reasonable request.
References
Siegel, R. L., Miller, K. D., Fuchs, H. E. & Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 71, 7–33 (2021).
Rahib, L. et al. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 74, 2913–2921 (2014).
Kang, M. J. et al. Cancer statistics in Korea: Incidence, mortality, survival, and prevalence in 2019. Cancer Res. Treat. 54, 330–344 (2022).
Conroy, T. et al. FOLFIRINOX versus Gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 364, 1817–1825 (2011).
Von Hoff, D. D. et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 369, 1691–1703 (2013).
Lowery, M. A. et al. Real-time genomic profiling of pancreatic ductal adenocarcinoma: Potential actionability and correlation with clinical phenotype. Clin. Cancer Res. 23, 6094–6100 (2017).
Aguirre, A. J. et al. Real-time genomic characterization of advanced pancreatic cancer to enable precision medicine. Cancer Discov. 8, 1096–1111 (2018).
Aung, K. L. et al. Genomics-driven precision medicine for advanced pancreatic cancer: Early results from the COMPASS trial. Clin. Cancer Res. 24, 1344–1354 (2018).
Pishvaian, M. J. et al. Molecular profiling of patients with pancreatic cancer: Initial results from the know your tumor initiative. Clin. Cancer Res. 24, 5018–5027 (2018).
Singhi, A. D. et al. Real-time targeted genome profile analysis of pancreatic ductal adenocarcinomas identifies genetic alterations that might be targeted with existing drugs or used as biomarkers. Gastroenterology 156, 2242-2253.e4 (2019).
Ding, D. et al. Challenges of the current precision medicine approach for pancreatic cancer: A single institution experience between 2013 and 2017. Cancer Lett. 497, 221–228 (2021).
Pishvaian, M. J. et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: A retrospective analysis of the know your tumor registry trial. Lancet Oncol. 21, 508–518 (2020).
Li, M. M. et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: A joint consensus recommendation of the association for molecular pathology, American Society of clinical oncology, and college of American pathologists. J. Mol. Diagn. 19, 4–23 (2017).
The Cancer Genome Atlas Research Network. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 32, 185–203 (2017).
Golan, T. et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N. Engl. J. Med. 381, 317–327 (2019).
Hong, J. Y. et al. Comprehensive molecular profiling to predict clinical outcomes in pancreatic cancer. Ther. Adv. Med. Oncol. 13, 1–14 (2021).
Yang, Y. et al. The genetic landscape of pancreatic head ductal adenocarcinoma in china and prognosis stratification. BMC Cancer 22, 186 (2022).
Zhang, X. et al. Characterization of the genomic landscape in large-scale chinese patients with pancreatic cancer. eBioMedicine. 77, 103897 (2022).
Golan, T. et al. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br. J. Cancer. 111, 1132–1138 (2014).
Park, W. et al. Genomic methods identify homologous recombination deficiency in pancreas adenocarcinoma and optimize treatment selection. Clin. Cancer Res. 26, 3239–3247 (2020).
O’Reilly, E. M. et al. Randomized, multicenter, phase II trial of gemcitabine and cisplatin with or without veliparib in patients with pancreas adenocarcinoma and a germline BRCA/PALB2 mutation. J. Clin. Oncol. 38, 1378–1388 (2020).
Davies, H. et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 23, 517–525 (2017).
Golan, T. et al. genomic features and classification of homologous recombination deficient pancreatic ductal adenocarcinoma. Gastroenterology 160, 2119-2132.e9 (2021).
Marabelle, A. et al. Efficacy of Pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair-deficient cancer: Results from the phase II KEYNOTE-158 study. J. Clin. Oncol. 38, 1–10 (2020).
Marabelle, A. et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with Pembrolizumab: Prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet. Oncol. 21, 1353–1365 (2020).
Rayner, E. et al. A panoply of errors: polymerase proofreading domain mutations in cancer. Nat Rev Cancer. 16, 71–81 (2016).
Wang, F. et al. Evaluation of POLE and POLD1 mutations as biomarkers for immunotherapy outcomes across multiple cancer types. JAMA Oncol. 5, 1504–1506 (2019).
Garmezy, B. et al. Clinical and molecular characterization of POLE mutations as predictive biomarkers of response to immune checkpoint inhibitor in advanced cancers. JCO Precis. Oncol. 6, e2100267 (2022).
Guenther, M. et al. POLE gene hotspot mutations in advanced pancreatic cancer. J. Cancer Res. Clin. Oncol. 144, 2161–2166 (2018).
Vega, D. M. et al. Aligning tumor mutational burden (TMB) quantification across diagnostic platforms: Phase 2 of the friends of cancer research TMB harmonization project. Ann. Oncol. 32, 1626–1636 (2021).
Buchhalter, I. et al. Size matters: Dissecting key parameters for panel-based tumor mutational burden analysis. Int. J. Cancer 144, 848–858 (2019).
Acknowledgements
This study was supported by Chong Kun Dang Pharm.
Author information
Authors and Affiliations
Contributions
Conception and design: S.L., H.Y.N., J.W.K., and J.K. Acquisition of data: J-W.K., J-C.L., J.H.H., J.W.K., and J.K. Analysis: K.J. and S.L. Interpretation of the data and drafting of the article: K.J. and S.L. Critical revision of the article for important intellectual content: J. W.K. and J.K. Final approval of the article: J.K. All the authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jung, K., Lee, S., Na, H.Y. et al. NGS-based targeted gene mutational profiles in Korean patients with pancreatic cancer. Sci Rep 12, 20937 (2022). https://doi.org/10.1038/s41598-022-24732-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-24732-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
