
Abstract
Folate ingestion below and above the physiologic dose has been shown to play a tumorigenic role in certain cancers. Also, excessive folate supplementation after establishment of pre-established lesions led to an advancement in the growth of a few tumors. However, such information has not yet been achieved in the case of HCC. In our study, HepG2 cells were administered with three different concentrations of folic acid i.e. folic acid normal (FN) (2.27 µM), folic acid deficient (FD) (no folic acid), folic acid oversupplementation (FO) (100 µM) for 10 days. Intracellular folate levels were assayed by Elecsys Folate III kit based method. The migratory and invasive abilities were estimated by transwell migration and matrigel invasion methods respectively. FACS was done to evaluate cell viability and apoptosis. Agarose-coated plates were used to access cancer stem cells (CSCs) number. Quantitative RT-PCR and western blotting approaches were used for gene and protein expression of certain tumor suppressor genes (TSGs), respectively. FD cells depicted increased migration, invasion, apoptosis, necrosis and decreased cell viability, CSCs. On the other hand, FO cells showed increased migration, invasion, cell viability and number of CSCs and decreased apoptosis and necrosis. TSGs revealed diminished expression with both FA modulations with respect to FN cells. Thus, FA deficiency as well as abundance enhanced the HCC progression by adapting different mechanisms.
Similar content being viewed by others


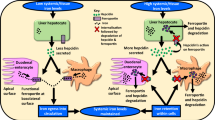
Introduction
Folate is a water soluble B9 vitamin, naturally occurring in green leafy vegetables and fruit juices. Folic acid (FA) is the oxidized form of this vitamin that is artificially added to fortified foods items1. Folate is usually considered safe and assumed to be entirely beneficial and a perfect food element helping in disease prevention. Based on its functional importance, any sort of imbalance in folate levels in the cells can lead to severe consequences. Folate deficiency is a highly prevalent deficiency all over the globe. It was the most frequent vitamin deficiency in the US, approximately affecting up to 60% of young or elderly people and10% of the general adult population belonging to low socio-economic groups2 before the food fortification program started in 1998. Folate deficiency in pregnant women has been linked with the occurrence of neural tube defects (NTDs) in neonates. It is also associated with megaloblastic anemia, cardiac problems2 atherosclerosis3, neuropsychiatric disorders4,5 and the risk of numerous cancers like colorectal, lungs, ovarian, prostate, etc. Poor dietary consumption, impairment in intestinal absorption e.g., in celiac disease, short bowel syndrome, amyloidosis and impaired renal tubular reabsorption, pregnancy, achlorhydria, hemolytic anemia, alcoholism, dialysis etc. can cause folate deficiency1. The facts from the post-fortification period have provided potential proof in decreasing NTD cases due to mandatory folic acid fortification 6. The efficacy of these programs in improving folate status proved quite striking, with a spectacular increase in blood folate concentrations in the USA and Canadian populations. Preliminary reports confirmed a reduction of about (∼15–50%) in the NTDs incidence in USA and Canada7. The circulatory folate concentrations illustrated inverse correlations with three aspects of tumor expansion: the size of the tumor8, its multiplicity and metastasis9.
In developing countries like India, FA is administered during the preconception period and through the gestation period at a much higher level than the recommended one according to WHO guidelines (600 μg vs. 5 mg)10. The consumption of FA from fortified food items (100–200 µg/d) collectively with the usage of supplements (which adds an extra 400 µg FA/standard multivitamin preparation) and ingestion of nutrition bars and drinks (which is frequently supplemented with 400 µg FA/serving) buildup a condition of folate oversupplementation in the population, indicating that folate oversupplementation conditions do prevail among humans knowingly or unknowingly11. In context to prostatic tumors, the Aspirin/Folate Polyp Prevention Trial has reported a raised risk of malignancies12 and higher circulatory folate post androgen deprivation found associated with poor prostatic tumor-specific survival13. Recent epidemiological data have depicted that FA supplementation might promote the progression of established breast malignancies in an animal model14. Also, a systematic review concluded that increased consumption of folate reduced mammary tumor risk for females consuming higher content of alcohol, but not for those having lower alcohol intake15. Studies conducted in animal models of colorectal tumors reported that FA administration prohibits tumor growth in normal tissues but encourages cancer progression if tumor lesion has been established16,17,18. Thus the time of folate intervention is significant in either providing an effectual and safe chemo-preventive role against carcinogenesis or as a promoter in favor of tumor development17.
In addition to the role of folate in nucleotide synthesis and methylation reactions, folate-dependent pathways play a major role in cell motility, including cancer cell migration as well as invasion19. A study depicted that colorectal cells when grown in inadequate FA exhibited impaired colonosphere formation20 and improved invasive properties of colon tumor cells mediated by activation of Shh signaling pathways via hypomethylation21. However, conflicting results reported in the cells of head and neck squamous cell carcinoma (HNSCC)22 and breast tumors19 which when depleted with methyl donors showed reduced growth, mobility and increased apoptosis22. Consistent with this, the dividing ability of embryonic stem cell was improved on treatment with folate23. Folate pathways are also known to promote invasiveness in prostate cancer cells24 and motility in A549 lung carcinoma cells affecting actin dynamics25 which might be due to the regulation of methylation of the genes involved26. In the case of liver, a study suggested that cells grown in folate-depleted media appeared to be more mobile and generated more number of spheres27. Apart from this, the effect of FA modulations in HCC cells has not been studied.
The molecular link between different doses of folate treatment and cancer hallmarks is also not well established. Till date, literature suggests that the effect of FA on the cancer cell is cell-type specific. So, in the present study we tried to decipher the effect of FA modulations on certain cancer hallmarks and expression of tumor suppressors in HepG2 cells, which might be help in identifying the underlying mechanism behind the HCC growth and its metastatic spread.
Results
Effect of FA deficiency and oversupplementation on cell morphology
HepG2 a liver cancer cell line possesses high proliferation rates with epithelial-like morphology. We observed that after treating these cells with different concentrations of FA, cells treated with the high FA were similar in morphology to FN cells. Cells in FA deficient media showed different morphology. The nucleus of FD cells seems to be enlarged, showing a disintegrated pattern or chromatin condensation as compared to the other two groups (Fig. 1).

Effect of folic acid deficiency and oversupplementation on the morphology of HepG2 cells under the magnifications i.e. 10×, 20× and 40×. FN-Folic acid normal, FD-Folic acid deficient, FO-Folic acid oversupplemented.
Effect of FA deficiency and oversupplementation on intracellular folate levels
In order to determine whether the treatment of FA deficiency and oversupplementation established the intracellular folate levels accordingly, we estimated folate levels in HepG2 cells. We observed that folate concentration in FD cells was significantly decreased (1.5 ± 0.6 ng/mL; P < 0.01) and in FO cells, it was significantly increased (25 ± 1.6 ng/mL; P < 0.01) when compared with FN cells (16.2 ± 2.9 ng/mL). In an intergroup comparison between FD and FO, markedly reduced (P < 0.001) folate levels in FD cells in comparison with FO cells were found (Fig. 2).

Effect of folic acid deficiency and oversupplementation on intracellular folate levels in HepG2 cells. **P < 0.01 versus FN, $$$P < 0.001 versus FO; FN-Folic acid normal, FD-Folic acid deficient, FO-Folic acid oversupplemented.
Effect of FA deficiency and oversupplementation on cell viability and apoptosis
Cell viability
The representative image of flow cytometric analysis was depicted in Fig. 3a. Viable cells were more in number in group treated with excess FA, although this increase was not statistically significant. A significantly less (P < 0.05) number of viable cells was seen in the FD group. The number of viable cells in the FD subset was observed to be reduced (P < 0.001) as compared to the FO group. Excess FA supplementation might have led to an increase in the number of proliferating cells (Fig. 3b).

Effect of folic acid deficiency and oversupplementation in HepG2 cells on (a) PI-Annexin assay by flow cytometry. (b) Cell viability (c) Early apoptosis (d) Late apoptosis (e) Necrotic cells. *P < 0.05, **P < 0.01 versus FN, $$P < 0.01, $$$P < 0.001 versus FO; FN-Folic acid normal, FD-Folic acid deficient, FO-Folic acid oversupplemented.
Apoptosis
Upon FACS, FO cells revealed that the number of cells undergoing necrotic and late apoptotic changes were significantly low (P < 0.01; P < 0.05 respectively), as was the case with cells with early apoptotic changes albeit, non-significantly. Cells grown under conditions of FA scarcity demonstrated an increase in the number of cells with early (P < 0.01) plus late (P < 0.05) apoptotic alterations. A significant increase in the number of late apoptotic (P < 0.001) and necrotic cells (P < 0.01) in the FD treated group was observed in comparison to FO. FO treatment resulted in a decrease in the number of necrotic and late apoptotic cells. Whereas with FA scarcity, cells with early and late apoptotic changes were increased, indicating more apoptosis under deficiency treatment {Fig. 3c–e}. Annexin V-positive and PI-positive cells were considered as the late apoptotic cells.
Effect of FA deficiency and oversupplementation on cell migration
On staining, the migratory HepG2 cells with crystal violet, the cell number was found significantly more (P < 0.01) for FD cells when compared with FN cells. However the number of migratory cells also increased for FO cells but this change was not statistically significant. Thus, FA deficit conditions increased the migratory activities in HepG2 cells, thus favoring metastasis {Fig. 4a(i&ii)}.

Effect of folic acid deficiency and oversupplementation in HepG2 cells. (a(i)) Representative images of migratory cells under microscope under 20× magnification (a(ii)) Quantification of migratory cells. (b(i)) Representative images of invasive cells under microscope under 20X magnification (b(ii)) Quantification of invading cells. (c(i)) Representative images of spheres under microscope under 20× magnification (c(ii)) Quantification of spheres formed. 20X = 200 µm.*P < 0.05, **P < 0.01, ***P < 0.001 versus FN, $P < 0.05, $$$P < 0.001 versus FO; FN-Folic acid normal, FD-Folic acid deficient, FO-Folic acid oversupplemented.
Effect of FA deficiency and oversupplementation on cell invasion
The matrigel invasion assay revealed that the number of invading cells in the FD group was almost similar to cells treated with normal FA concentrations. However, the invading cell count was found significantly raised (P < 0.05 each) for FO cells when compared with FN or FD, respectively. Therefore, treatment with excessive FA increased the invading properties of HepG2 cells {Fig. 4b(i&ii)}.
Effect of FA deficiency and oversupplementation on sphere formation
Cancer stem cells have the property of proliferating and surviving even in such harsh conditions. One sphere results from proliferation of single stem cell28. We observed a significantly lesser (80.7%; P < 0.05) number of spheres in the cells grown without FA compared to FN cells in sphere formation assay. A marked elevation of 63% (P < 0.001) in sphere count was observed for cells administered with an additional quantity of FA when compared with FN cells. On the evaluation of FD cells with FO cells, a considerably 14-fold lesser number of spheres were seen in the FD fraction (P < 0.001) of HepG2 cells. FA deficiency decreased and oversupplementation increased number of spheres formed, indicating beneficial effect of excessive FA for cancer stem cell survival {Fig. 4c(i&ii)}.
Effect of FA deficiency and oversupplementation on gene expression of TSGs
RNA quality was checked on 1.5% agarose gel. The intact bands of 28S and 18S rRNA species on the gel depicted a good quality of RNA. Primer sequences used in RT-PCR for all the studied gene has been represented in Table S1 in supplementary information.
In HepG2 cells, mRNA expression studies revealed a decrease in the expression of DPT gene in the cells which were allowed to grow under the conditions of no FA (FD) when compared with the cells which were grown under normal concentration of FA (FN). For FO cells, no noticeable change in the expression was observed (Fig. 5a). Relative to FO, cells of FD treatment illustrated a decline in DPT expression.

Effect of folic acid deficiency and oversupplementation on the gene expression of tumor suppressor genes: (a) DPT (b) RUNX3 (c) p16 (d) RASSF1A and (e) SOCS1. 18S was used to normalize expression of studied genes. **P < 0.01, ***P < 0.001 versus FN, $P < 0.05, $$P < 0.01 versus FO; FN-Folic acid normal, FD-Folic acid deficient, FO-Folic acid oversupplemented.
Similar to DPT gene, mRNA levels of RUNX3 depicted a significant decrease in the expression in FD cells in comparison with FO cells. No significant effects were observed in FO cells when compared to FN cells (Fig. 5b). Gene expression of p16 was found to be decreased, albeit non-significantly, in FD and unaltered in FO cells with respect to FN cells (Fig. 5c).
In contrast, RASSF1A mRNA levels revealed a significant decrease in both the treated cell groups i.e. in FD and in FO cells with respect to FN cells (Fig. 5d). Similarly, diminished gene expression in FD as well as FO groups was observed for SOCS1 gene relative to cells administered with normal FA quantity (Fig. 5e). Fold change in gene expression of TSGs is detailed in Table S2 in supplementary information.
Effect of FA deficiency and oversupplementation on protein expression of TSGs
Blot images were normalized with the expression of β-actin protein. The representative images of the β-actin, p16 and SOCS1 blots have been shown in (Fig. 6a,c). Similar to gene expression, densitometric analyses of protein levels of p16 depicted a decrease in FA deficit but the change was insignificant (Fig. 6b). SOCS1 protein levels were significantly decreased in FO cells when a comparison was made with FN cells. No considerable changes were observed in FD cells when the evaluation was done with FN cells. However in comparison with FO cells, cells of deficient fraction illustrated an increase in its protein expression (Fig. 6d). Numerical data for the same has been depicted in Table S3 in supplementary.

Effect of folic acid deficiency and oversupplementation on protein expression in HepG2 cells (a) Representative images of β-Actin & p16 blot (b) Densitometric analysis of the p16 blot. (c) Representative image of SOCS1 blot (d) Densitometric analysis of the SOCS1 blot. **P < 0.01 versus FN, $$P < 0.01 versus FO; FN-Folic acid normal, FD-Folic acid deficient, FO-Folic acid oversupplemented.
Data retrieval of TSGs from TCGA database
The survival curves from TCGA database depicted a decrease in patient survival months having an alteration in any of these genes, viz. DPT, RUNX3, P16, RASSF1, SOCS1 (Fig. 7a-e). However, the survival was significantly reduced (P < 0.015) with alterations in RASSF1A gene.

Survival curves retrieved from TCGA database for all the studied genes; (a) DPT (b) RUNX3 (c) p16 (d) RASSF1A and (e) SOCS1.
Discussion
DNA synthesis and methylation are the vital cellular processes that claim the presence of folate29. Dysregulation in folate levels has been linked with induction of base substitution, gene deletions and DNA strand breaks as well as altered gene expression30. Folate probably plays a double role in tumorogenesis, initially as a protector of DNA damage before the onset of neoplastic transformation and later behave as a promoter for cancer growth by increasing proliferation and invasion-like processes16,31. We attempted to study how modulation of folate levels in HCC cells affects the various hallmarks of cancer.
As expected, FA deficiency and it’s over-administration resulted in a significant decrease and increase in folate levels, respectively, in HepG2 cells. The previous studies also showed a similar reduction in folate levels under FD treatment32,33,34,35. Importantly, low folate levels have been linked with the risk for certain tumors36.
HepG2 cells, when grown without FA for ten days, showed enlarged nuclei relative to the other two groups, which might be due to nuclear chromatin condensation. These observations were in agreement with the previous study on the same cell type37. Moreover, a lack of correlation between nuclear dimension and ploidy has been depicted for carcinomas of the urinary bladder, colorectum, mammary glands, lung, skin, cervix, and prostate38. Morphologically FN and FO cells were indistinguishable from each other. However it was earlier seen that FO exerted a protective effect on hexagonal parenchymatous liver cells, thus improving liver morphology in aged rats39.
For HepG2 cells, we achieved a reduced number of live cells in FD when stained with PI-annexin dye. This decreased viability was reported earlier too in the same cell type37. Also experiments with FD-conditioned media with colon cells illustrated the same results40. Hepatoma cells that were supplemented with excess FA presented with increased counts of viable cells in our study. A previous finding also suggested that FO markedly enhanced the polyp number and tumour burden in the intestine41. In a contrasting observation, FA treatment (20 & 50 µM) inhibited nasopharyngeal cancer cell proliferation, suggesting that FA affects cell proliferation in a dose and cell type-specific manner42.
Our experiments with hepatic cells showed an increased migration under FD conditions, while extra FA did not significantly affect it. While studying invasion, FO cells showed a pronounced increase and decline in the invasion relative to FN and FD cells, respectively27. According to the previous findings, FD-conditioned SK-Hep1 and Mahlavu (the liver cancer cells) displayed a higher migration and invasion when compared with their respective control25. Contrasting evidences suggested that folate withdrawal inhibited migration and invasion of A549 cells43, while did not affect the migratory activity of A375 human melanoma cells. Also, Hearnden22 recently demonstrated that HNSCC cells in the absence of methyl donors showed extensively diminished cellular proliferation, impairment in cellular migration and increased apoptotic activities, indicating a less aggressive phenotype of the tumor. In line with our observation, the literature data suggested that FO can boost the growth and invasive potential of prostate carcinoma cells44 and modify the expression of genes related to cell adhesion, migration and invasion45. Folate treatment above physiological levels supports epithelial to mesenchymal transition in experimental mouse tumor cells and silencing of RhoA and Rac1, which serve as immediate sensors of folate intracellularly, inhibited the effects of folate on cellular migratory and invasive abilities46. These observations signify the cell and dose type-dependent effects of folic acid modulations.
Spheres in HepG2 were considerably reduced upon folic acid restriction and markedly increased upon its oversupplementation. A previous study on colon cells grown without adequate FA exhibited impairment in colonosphere forming ability20, similar to our results. In contrast to our findings, Sk-Hep1 and Mahlavu, under FD-conditioned media, generated a higher number of spheres, again stressing the fact that effects of folic acid deficiency are not only tissue-specific but also cancer type-specific, too27. A prior study reported the same findings that FA administration activated neural stem cell (NSC) proliferation in a dose-dependent manner and the plausible mechanisms were deciphered to be notch signaling pathway23. Thus, folic acid depletion can increase or decrease the number of tumorospheres generated, whereas folic acid oversupplementation raises the oncospheres, inferring the cell or tissue-specific role of folate modulations.
In HepG2 cells, TSGs transcript (RASSF1A and SOCS1) were found to be significantly decreased in both the dietary folate modulations relative to cells administered with normal amounts of FA. Similar to our study, dermatopontin (DPT), an extracellular matrix component47, was appreciably decreased in 202 HCC patients and can be correlated with metastasis and prognosis. Also, it inhibited cell motility in HCC mainly via α3β1 integrin, including diminished RhoA activity48. So, it could be possible that different folic acid modulations affect RhoA activity and lead to increased migration and invasion in our experiments. A study by Xirong et al., confirmed high expression of DPT in the fully developed liver and fetal life, while it decreased in HCC tissues49. Reduced DPT mRNA in our experiments was in concordance with findings in ApoE null mice, in which feeding mice with a combination of folate deficiency and a fat rich diet decreased DPT expression, which regulate differentiation, adhesion and migration abilities of the cell50. It is also among the top up-regulated genes in severe NAFLD51, NASH and fibrosis52. DPT expression was closely found associated with overall survival (OS) as well as with disease-free survival in HCC48, similar to the data we retracted from the database in September, 2020.
Also, the diminished expression of RUNX3 correlates well with the ample literature- based evidence, which states that RUNX3 is lowered in HCC tissues and cell lines53,54,55. Restoration of RUNX3 expression helps in lowering the number of stem cells by suppressing Notch signaling56 and controls metastatic spread via interacting with miR-186/E-cadherin/EMT in HCC57. Seventy percent of human HCCs revealed RUNX3 hypermethylation58. In comparison with RUNX3-positive HCC, RUNX3-negative malignancies showed poor OS and relapse related survival57. A study reported raised RUNX3 was reduced after folate repletion in keratinocytes59.
In FD-conditioned HepG2 cells, p16 expression was decreased at gene as well as protein levels relative to FN. Also, under folate oversupplemented conditions, p16 revealed non-significant changes in expression. The reduced p16 expression in FD cells was in concordance with a report which observed diminished p16 mRNA due to hypermethylation in liver malignancies60. On exposure to folate/methyl deficiency, hypermethylation was seen for p16 in hepatic tumors61. In HCC patients, high circulatory folate is associated with p16 hypomethylation in the elderly Chilean population62. Insufficient folate consumption is associated with p16 hypermethylation in an older population in colon63, head and neck64 and gastric tumors65. Folate withdrawal unaltered the methylation of p16 in HCT116 and Caco-2 cells66.
In HepG2 cultured cells, the RASSF1A transcript was diminished for both FD and FO. Previous data confirmed RASSF1A and p16 inactivation by aberrant methylation, a commonly occurring event in HCC67, while no change in RASSF1A methylation was identified for Hep3B and HepG2 cells68. The TCGA database depicted decreased survival in liver cancer patients with altered RASSF1. However, literature also evidenced the reverse findings that RASSF1A hypermethylation results in longer OS in HCC patients69. Another study depicted that the low folate diet in combination with increased alcohol intake was associated with hypermethylated RASSF1A in CRC subjects70. In a contrasting observation in lung cancer patients, blood cell folate was found associated with raised RASSF1A methylation71, whereas folate levels less actively influenced RASSF1A in breast cancer tissues72.
SOCS1, the negative regulator in cytokine signaling, showed expression in both treated groups in HepG2 cells in the present study. Prior studies have evidenced the same results that frequent CpG promoter methylation and its subsequent loss has been identified in HCC for SOCS173. Previously, an immunohistochemical study illustrated intense homogeneous/ heterogeneous staining in the non-cancerous hepatic tissue as compared to HCC tissue which presented a noteworthy decrease with heterogeneous staining74. Also, in the rodent model of HCC induced by methyl deficient diet, epigenetic silencing of the SOCS1 was observed75. Likewise, altered methylation and expression of SOCS1 was confirmed in HCC cell lines and upon restoration of the expression, suppressed expansion and anchorage-free cell growth were seen76. In line with this, a study referred to aberrant SOCS1 methylation as the main event in the transformation of cirrhotic nodules into HCC77,78. Also, high folate intake was associated with raised SOCS1 methylation in CRC in European American individuals79. Thus, both folate modulations reduced the expression of the studied TSGs, which might lead to enhanced cancer progression. The aberrant expression of these genes might be due to altered promoter methylation. However, the limitation of our study is that the promoter methylation of the studied tumor suppressor genes was not determined in response to the modulation of folate levels.
Therefore, in HepG2 cells, FA deprivation led to decreased cell proliferation, number of CSCs, expression of TSGs and increased apoptosis, migration and invasion. On the other hand, excessive FA exposure to these cells led to increased cell proliferation, number of CSCs, migration, invasion and decreased apoptosis, necrosis and generally reduced expression of TSGs. All the said hallmarks together might result in increased cancer expansion under FO treatment. Hence FA modulations do affect the development of HCC differentially; FA deficient tumors might be less aggressive while FA oversupplemented ones be more hostile tumor types by employing differential regulation of TSGs and manipulating the pathways leading to altered cell proliferation, CSCs, apoptosis, migration and invasion.
Materials and methods
Cell culture
The human liver cancer cell line HepG2 was obtained from NCCS, Pune. HepG2 cells were cultured at 37ºC in an atmosphere of 5% CO2 and 95% relative humidity in 75cm2 cell culture flasks containing minimum essential medium (MEM) supplemented with 10% fetal bovine serum (FBS), sodium bicarbonate, penicillin (100,000U/L) and streptomycin (10 mg/L). These cells were obtained at passage 30 and used experimentally between passages 35–50. They were routinely passaged at 2-3 days interval. The medium was refreshed after 2 and 3 days of treatment for FN, FO and FD cells, respectively. Cells were regularly watched in order to see for their deadherance from the flask surface. After that, cells were seeded in 75cm2 cell culture flasks at a density of 200,000 cells per flask. Based on FA content administered to the cells, these were divided into three groups i.e. FN, FD and FO. FN or control cells were maintained under the conditions mentioned above in MEM media containing 2.27 μM folate concentration with 10% FBS. FD cells were grown on a commercially available folate-deficient medium supplemented with 20% dialyzed FBS. Cells were routinely monitored for any morphological changes. FO cells were grown on MEM media with FA concentration of 100 μM and 10% FBS80. All the treatments were carried out for five generations which is equivalent to 10 days of treatment.
Estimation of intracellular folate levels in HepG2 cells by using Elecsys Folate III kit
For folate determination in cultured cells, the viability of cells was determined by trypan blue exclusion assay after harvesting. Thereafter, 104 cells were lysed by heating at 100ºC for 5 min in folate extraction buffer. Samples were cooled and clarified by centrifugation and supernatant was incubated at 37ºC following the adding 0.25volumes of rat plasma conjugase for three hours in folate extraction buffer. After incubation, samples were boiled to precipitate protein and the solution was cooled and clarified by centrifugation. Further, folate levels were measured by using elecsys folate III kit on Elecsys and Cobas e analyzer. The detailed composition of the buffers used for folate estimation has been given in supplementary information.
Cell viability and apoptosis by FACS
Annexin V/PI assay was used for measure the number of viable cells and apoptosis of HepG2 cells using FITC Annexin V Apoptosis Detection Kit II (BD, Pharmigen). After ten days of treatment, HepG2 cells were harvested by using trypsin–EDTA. Cells were suspended in 1X binding buffer at a concentration of 1 × 106cells/mL in 100μL and were transferred into a FACS tube. Two microlitres (µL) of Annexin V-FITC were added to each tube and mixed well. The samples were incubated for 15 min at room temperature in the dark. Additional 400μL of 1X binding buffer was added to each tube and 5μL of PI was freshly added into the cell suspension just before analysis and mixed. The cells were analyzed on a fluorescence-activated cell sorter and data was analysed using Cell Quest software.
Transwell migration assay
Cell migration assay was performed using 24well transwell chambers (8.0 μm pore size polycarbonate membrane 65 mm, 0.33cm2). After ten days of respective media and FBS treatment, 1X105 HepG2 cells were seeded in the upper of transwell chamber with respective media. MEM supplemented with their respective FBS was added to the lower chamber as a chemoattractant. Following incubation for 24 h at 37 ºC, non-invading cells remaining on the upper surface were removed using a wet cotton swab, whereas cells on the lower surface were fixed with 0.1% crystal violet and 20% methanol-containing solution for 30 min and washed with 1X PBS twice and kept for drying for 20 min and then placed on a glass slide. Then cells migrating across the membrane were imaged in random visual fields under an inverted microscope. After that, crystal violet dye retained on the filters was extracted using 600 μL of 30% acetic acid and then OD was taken at 590 nm. The values obtained and the percentage of invasion and migration were calculated using the following formula: (OD of extracted crystal violet from treated cells migrating through inserts minus blank)/OD of extracted crystal violet from negative control minus blank) * 100. All the experiments were repeated at least twice.
Matrigel invasion assay
For invasion assay, HepG2 cells were plated onto the top of a 30 μg/cm2 matrigel coated transwell chamber of 24-well plate in 100μL serum-free medium. The bottom well of the chamber contained 600 μL of MEM supplemented with its respective FBS. After incubating for 24 h, chambers were disassembled and stained, following the protocol as discussed above for migration assay.
Sphere formation assay
To check the ability of cancer stem cells to proliferate, a sphere formation assay was done on low attachment plates. For low attachment, 6 well plates were coated with 1% agarose, allowed to solidify and kept under UV for sterilization for at least six hours. Then HepG2 cells were seeded at a density of about 3000 cells per well with their specific MEM and FBS. Cells were further incubated at 37 ºC for about 15 days. Meanwhile after 3–4 days of treatment, additional media and FBS were added along the sides of the wells without disturbing spheres. During incubation sphere formation was observed under a microscope and images were captured. After 15 days of incubation, spheres were counted under a microscope and images were captured for each group.
Quantitative RT-PCR
From HepG2 cells, total RNA was isolated using TRIzol reagent (Ambion). Further reverse transcription was carried out by using Fermentas cDNA kit. RT-PCR was performed with gene-specific primers for tumor suppressor genes (TSGs), viz. DPT, RUNX3, p16, RASSF1A and SOCS1 on Applied Biosystems StepOnePlusTM Real-Time PCR. 18 s rRNA was used as endogenous control. Relative quantification of gene expression was done by using the comparative CT method (ΔΔCT).
Western blotting
HepG2 cells were lysed using RIPA buffer. After quantification with bicinchoninic acid assay, equivalent quantities of protein were run on SDS-PAGE and blotted using the PVDF membrane. Polyclonal primary antibodies for p16 (catalogue no. PAA794Ra01) & SOCS1 (catalogue no. PAH158Ra01) were purchased from Cloud Clone Corporation USA. The primary antibodies against RASSF1A (sc-58470) & β-actin (sc-47778) were obtained from Santa Cruz, CA, USA. An enhanced chemiluminescence detection system was used for developing. Densitometric analysis of blots was done with Image J software.
Data retrieval from TCGA database
For all the studied genes, we also tried to retrieve survival data from TCGA database in HCC patients available in this database till September 2020.
Statistical analysis
For statistical analysis, of more than two groups, we performed one-way analyses of variance ANOVA (α = 0.05) followed by the Tukey post-hoc test. GraphPad Prism (v.6.0.1) was used for statistical analysis. Data were expressed as mean ± SEM and were considered statistically significant at P < 0.05.
Abbreviations
- FA:
-
Folic acid
- FD:
-
Folic acid deficiency
- FN:
-
Folic acid normal
- FO:
-
Folic acid oversupplementation
- HCC:
-
Hepatocellular carcinoma
References
Khan, K. M. & Jialal, I. Folic Acid Deficiency (Springer, 2022).
Duthie, S. J. Folic acid deficiency and cancer: Mechanisms of DNA instability. Br. Med. Bull. 55, 578–592. https://doi.org/10.1258/0007142991902646 (1999).
Cui, S. et al. Folic acid supplementation delays atherosclerotic lesion development by modulating MCP1 and VEGF DNA methylation levels in vivo and in vitro. Int. J. Mol. Sci. 18, 990. https://doi.org/10.3390/ijms18050990 (2017).
Zou, R. et al. Maternal folate levels during pregnancy and offspring brain development in late childhood. Clin. Nutr. 40, 3391–3400. https://doi.org/10.1016/j.clnu.2020.11.025 (2021).
Ramaekers, V. T. et al. Genetic assessment and folate receptor autoantibodies in infantile-onset cerebral folate deficiency (CFD) syndrome. Mol. Genet. Metab. 124, 87–93. https://doi.org/10.1016/j.ymgme.2018.03.001 (2018).
Naderi, N. & House, J. D. Recent developments in folate nutrition. Adv. Food Nutr. Res. 83, 195–213. https://doi.org/10.1016/bs.afnr.2017.12.006 (2018).
Kim, Y. I. Will mandatory folic acid fortification prevent or promote cancer? Am. J. Clin. Nutr. 80, 1123–1128 (2004).
Hansen, M. F., Jensen, S. O., Füchtbauer, E. M. & Martensen, P. M. High folic acid diet enhances tumour growth in PyMT-induced breast cancer. Br. J. Cancer 116, 752–761. https://doi.org/10.1038/bjc.2017.11 (2017).
Kuo, C. S., Lin, C. Y., Wu, M. Y., Lu, C. L. & Huang, R. F. Relationship between folate status and tumour progression in patients with hepatocellular carcinoma. Br. J. Nutr. 100, 596–602. https://doi.org/10.1017/S0007114508911557 (2008).
Wald, N. J., Law, M. R., Morris, J. K. & Wald, D. S. Quantifying the effect of folic acid. Lancet 358, 2069–2073. https://doi.org/10.1016/s0140-6736(01)07104-5 (2001).
Ulrich, C. M. & Potter, J. D. Folate supplementation: Too much of a good thing? Cancer Epidemiol. Biomarkers Prev. 15, 189–193. https://doi.org/10.1158/1055-9965.EPI-152CO (2006).
Cole, B. F. et al. Folic acid for the prevention of colorectal adenomas: A randomized clinical trial. J. Am. Med. Assoc. 297, 2351–2359. https://doi.org/10.1001/jama.297.21.2351 (2007).
Liss, M. A., Ashcraft, K., Satsangi, A. & Bacich, D. Rise in serum folate after androgen deprivation associated with worse prostate cancer-specific survival. Urol. Oncol. 38(682), e21-682.e27. https://doi.org/10.1016/j.urolonc.2020.04.003 (2020).
Deghan, M. S. et al. Folic acid supplementation promotes mammary tumor progression in a rat model. PLoS One 9, e84635–e84635. https://doi.org/10.1371/journal.pone.0084635 (2014).
Chen, P. et al. Higher dietary folate intake reduces the breast cancer risk: A systematic review and meta-analysis. Br. J. Cancer 110, 2327–2338. https://doi.org/10.1038/bjc.2014.155 (2014).
Smith, A. D., Kim, Y. I. & Refsum, H. Is folic acid good for everyone? Am. J. Clin. Nutr. 87, 517–533. https://doi.org/10.1093/ajcn/87.3.517 (2008).
Troen, A. M. et al. Unmetabolized folic acid in plasma is associated with reduced natural killer cell cytotoxicity among postmenopausal women. J. Nutr. 136, 189–194. https://doi.org/10.1093/jn/136.1.189 (2006).
Liu, Z., Cheng, M. & Cao, M. Z. Potential targets for molecular imaging of apoptosis resistance in hepatocellular carcinoma. Biomed. Imaging Interv. J. 7, e5. https://doi.org/10.2349/biij.7.1.e5 (2011).
Ashkavand, Z. et al. Metabolic reprogramming by folate restriction leads to a less aggressive cancer phenotype. Mol. Cancer Res. 15, 189–200. https://doi.org/10.1158/1541-7786.mcr-16-0317 (2017).
Farias, N. et al. The effects of folic acid on global DNA methylation and colonosphere formation in colon cancer cell lines. J. Nutr. Biochem. 26, 818–826. https://doi.org/10.1016/j.jnutbio.2015.02.002 (2015).
Wang, T., Hsu, S. & Feng, H. Folate deprivation enhances invasiveness of human colon cancer cells mediated by activation of sonic hedgehog signaling through promoter hypomethylation and cross action with transcription nuclear factor-kappa B pathway. Carcinogenesis 33, 1158–1168. https://doi.org/10.1093/carcin/bgs138 (2012).
Hearnden, V., Powers, H. J., Elmogassabi, A., Lowe, R. & Murdoch, C. Methyl-donor depletion of head and neck cancer cells in vitro establishes a less aggressive tumour cell phenotype. Eur. J. Nutr. 57, 1321–1332. https://doi.org/10.1007/s00394-017-1411-5 (2018).
Liu, H., Huang, G. W., Zhang, X. M., Ren, D. L. & Wilson, J. X. Folic acid supplementation stimulates notch signaling and cell proliferation in embryonic neural stem cells. J. Clin. Biochem. Nutr. 47, 174–180. https://doi.org/10.3164/jcbn.10-47 (2010).
Green, T., Chen, X., Ryan, S., Asch, A. S. & Ruiz-Echevarría, M. J. TMEFF2 and SARDH cooperate to modulate one-carbon metabolism and invasion of prostate cancer cells. Prostate 73, 1561–1575. https://doi.org/10.1002/pros.22706 (2013).
Oleinik, N. V., Krupenko, N. I. & Krupenko, S. A. ALDH1L1 inhibits cell motility via dephosphorylation of cofilin by PP1 and PP2A. Oncogene 29, 6233–6244. https://doi.org/10.1038/onc.2010.356 (2010).
Björklund, N. K. & Gordon, R. A hypothesis linking low folate intake to neural tube defects due to failure of post-translation methylations of the cytoskeleton. Int J Dev Biol 50, 135–141. https://doi.org/10.1387/ijdb.052102nb (2006).
Su, Y. H. et al. Folate deficient tumor microenvironment promotes epithelial-to-mesenchymal transition and cancer stem-like phenotypes. Oncotarget 7, 33246–33256. https://doi.org/10.18632/oncotarget.8910 (2016).
Singec, I. et al. Defining the actual sensitivity and specificity of the neurosphere assay in stem cell biology. Nat. Methods 3, 801–806. https://doi.org/10.1038/nmeth926 (2006).
Weinstein, S. J. et al. Null results in brief null association between prostate cancer and serum folate, vitamin B 6, vitamin B 12, and homocysteine. Cancer Epidemiol. Biomarkers Prev. 12, 1271–1272 (2003).
De Cabo, S. F., Santos, J. & Fernández-Piqueras, J. Molecular and cytological evidence of S-adenosyl-L-homocysteine as an innocuous undermethylating agent in vivo. Cytogenet. Cell Genet. 71, 187–192. https://doi.org/10.1159/000134104 (1995).
Mason, J. B. et al. a temporal association between folic acid fortification and an increase in colorectal cancer rates may be illuminating important biological principles: A hypothesis. Cancer Epidemiol. Biomarkers Prev. 16, 1325–1330. https://doi.org/10.1158/1055-9965.EPI-07-0329 (2007).
Fu, Y. & Chung, F. Oxidative stress and hepatocarcinogenesis. Hepatoma Res. 4, 39. https://doi.org/10.20517/2394-5079.2018.29 (2018).
Yu, L. X., Ling, Y. & Wang, H. Y. Role of nonresolving inflammation in hepatocellular carcinoma development and progression. NPJ Precis. Oncol. 2, 6. https://doi.org/10.1038/s41698-018-0048-z (2018).
Mittal, S. & El-Serag, H. B. Epidemiology of hepatocellular carcinoma: Consider the population. J. Clin. Gastroenterol. 47, S2-6. https://doi.org/10.1097/MCG.0b013e3182872f29 (2013).
Tian, Y., Wong, V. W. S., Chan, H. L. Y. & Cheng, A. S. L. Epigenetic regulation of hepatocellular carcinoma in non-alcoholic fatty liver disease. Semin. Cancer Biol. 23, 471–482. https://doi.org/10.1016/j.semcancer.2013.08.010 (2013).
Lind, M. & Johansson, L. High homocysteine and low folate plasma concentrations are associated with cardiovascular events but not bleeding during warfarin treatment. Clin. Chem. Lab. Med. 54, 1981–1986. https://doi.org/10.1515/cclm-2016-0092 (2016).
Huang, R. F. S., Ho, Y. H., Lin, H. L., Wei, J. S. & Liu, T. Z. Folate deficiency induces a cell cycle-specific apoptosis in HepG2 cells. J. Nutr. 129, 25–31. https://doi.org/10.1093/jn/129.1.25 (1999).
Jevtić, P. & Levy, D. L. Mechanisms of nuclear size regulation in model systems and cancer. Adv. Exp. Med. Biol. 773, 537–569. https://doi.org/10.1007/978-1-4899-8032-8_25 (2014).
Roncales, M. et al. Folic acid supplementation for 4 weeks affects liver morphology in aged rats. J. Nutr. 134, 1130–1133. https://doi.org/10.1093/jn/134.5.1130 (2004).
Khosraviani, K., Weir, H. P., Hamilton, P., Moorehead, J. & Williamson, K. Effect of folate supplementation on mucosal cell proliferation in high risk patients for colon cancer. Gut 51, 195–199. https://doi.org/10.1136/gut.51.2.195 (2002).
Bashir, O., FitzGerald, A. J. & Goodlad, R. A. Both suboptimal and elevated vitamin intake increase intestinal neoplasia and alter crypt fission in the ApcMin/+ mouse. Carcinogenesis 25, 1507–1515. https://doi.org/10.1093/carcin/bgh137 (2004).
Liu, Z., Jin, X., Pi, W. & Liu, S. Folic acid inhibits nasopharyngeal cancer cell proliferation and invasion via activation of FRα/ERK1/2/TSLC1 pathway. Biosci. Rep. https://doi.org/10.1042/bsr20170772 (2017).
Tu, H. C. et al. One crisis, diverse impacts-tissue-specificity of folate deficiency-induced circulation defects in zebrafish larvae. PLoS ONE 12, 1–23. https://doi.org/10.1371/journal.pone.0188585 (2017).
Petersen, L. F. et al. Elevated physiological levels of folic acid can increase in vitro growth and invasiveness of prostate cancer cells. BJU Int. 109, 788–795. https://doi.org/10.1111/j.1464-410X.2011.10437.x (2012).
Glen, C. D., Mcveigh, L. E., Voutounou, M. & Dubrova, Y. E. The effects of methyl-donor deficiency on the pattern of gene expression in mice. Mol. Nutr. Food Res. 59, 501–506. https://doi.org/10.1002/mnfr.201400660 (2015).
Oleinik, N. V., Helke, K. L., Kistner-Griffin, E., Krupenko, N. I. & Krupenko, S. A. Rho GTPases rhoa and rac1 mediate effects of dietary folate on metastatic potential of A549 cancer cells through the control of cofilin phosphorylation. J. Biol. Chem. 289, 26383–26394. https://doi.org/10.1074/jbc.M114.569657 (2014).
Yamatoji, M. et al. Dermatopontin: A potential predictor for metastasis of human oral cancer. Int. J. Cancer 130, 2903–2911. https://doi.org/10.1002/ijc.26328 (2012).
Fu, Y. et al. DNA methylation-mediated silencing of matricellular protein dermatopontin promotes hepatocellular carcinoma metastasis by integrin-Rho GTPase signaling. Oncotarget https://doi.org/10.18632/oncotarget.2239 (2014).
Li, X. et al. Dermatopontin is expressed in human liver and is downregulated in hepatocellular carcinoma. Biochemistry (Mosc) 74, 979–985. https://doi.org/10.1134/s0006297909090053 (2009).
Duthie, S. J. et al. Nutritional B vitamin deficiency alters the expression of key proteins associated with vascular smooth muscle cell proliferation and migration in the aorta of atherosclerotic apolipoprotein E null mice. Genes Nutr. 10, 1–11. https://doi.org/10.1007/s12263-014-0446-y (2015).
Moylan, C. A. et al. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology 59, 471–482. https://doi.org/10.1002/hep.26661 (2014).
Lefebvre, P. et al. Interspecies NASH disease activity whole- genome profiling identifies a fibrogenic role of PPAR a -regulated dermatopontin. JCI Insight 2, e92264. https://doi.org/10.1172/jci.insight.92264 (2017).
Gómez-Angelats, M., Teeguarden, J. G., Dragan, Y. P. & Pitot, H. C. Mutational analysis of three tumor suppressor genes in two models of rat hepatocarcinogenesis. Mol. Carcinog 25, 157–163. https://doi.org/10.1002/(SICI)1098-2744(199907)25:3%3c157::AID-MC1%3e3.0.CO;2-8 (1999).
Lee, J. S. et al. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nat. Genet. 36, 1306–1311. https://doi.org/10.1038/ng1481 (2004).
Talaulikar, V. S. Folic acid in obstetric practice: A review. Obstet. Gynecol. Surv. 66, 240–247. https://doi.org/10.1097/OGX.0b013e318223614c (2011).
Nishina, S. I. et al. Restored expression of the tumor suppressor gene RUNX3 reduces cancer stem cells in hepatocellular carcinoma by suppressing Jagged1-Notch signaling. Oncol. Rep. 26, 523–531. https://doi.org/10.3892/or.2011.1336 (2011).
Gou, Y., Zhai, F., Zhang, L. & Cui, L. RUNX3 regulates hepatocellular carcinoma cell metastasis via targeting miR-186 / E-cadherin/EMT pathway. Oncotarget 8, 61475–61486. https://doi.org/10.18632/oncotarget.18424 (2017).
Wang, S. et al. Aberrant methylation of RUNX3 is present in aflatoxin B1-induced transformation of the L02R cell line. Toxicology 385, 1–9. https://doi.org/10.1016/j.tox.2017.04.011 (2017).
Savini, C. et al. Folate repletion after deficiency induces irreversible genomic and transcriptional changes in human papillomavirus type 16 (HPV16)-immortalized human keratinocytes. Int. J. Mol. Sci. 20, 1100. https://doi.org/10.3390/ijms20051100 (2019).
Esteller, M. et al. Cancer epigenetics and methylation. Science 297, 1807–1808. https://doi.org/10.1126/science.297.5588.1807d (2002).
Pogribny, I. P. & James, S. J. D. novo methylation of the p16INK4A gene in early preneoplastic liver and tumors induced by folate/methyl deficiency in rats. Cancer Lett. 187, 69–75. https://doi.org/10.1016/s0304-3835(02)00408-1 (2002).
Sanchez, H. et al. High levels of circulating folate concentrations are associated with DNA methylation of tumor suppressor and repair genes p16, MLH1, and MGMT in elderly Chileans. Clin. Epigenet. 9, 74. https://doi.org/10.1186/s13148-017-0374-y (2017).
Keyes, M. K. et al. Older age and dietary folate are determinants of genomic and p16-specific DNA methylation in mouse colon. J. Nutr. 137, 1713–1717 (2007).
Kraunz, K. S. et al. Dietary folate is associated with p16 INK4A methylation in head and neck squamous cell carcinoma. Int. J. Cancer 119, 1553–1557. https://doi.org/10.1002/ijc.22013 (2006).
Qu, Y., Dang, S. & Hou, P. Gene methylation in gastric cancer. Clin. Chim Acta 424, 53–65. https://doi.org/10.1016/j.cca.2013.05.002 (2013).
Stempak, J. M., Sohn, K. J., Chiang, E. P., Shane, B. & Kim, Y. I. Cell and stage of transformation-specific effects of folate deficiency on methionine cycle intermediates and DNA methylation in an in vitro model. Carcinogenesis 26, 981–990. https://doi.org/10.1093/carcin/bgi037 (2005).
Schagdarsurengin, U. et al. Frequent epigenetic inactivation of the RASSF1A gene in hepatocellular carcinoma. Oncogene 3, 1866–1871. https://doi.org/10.1038/sj.onc.1206338 (2003).
Zhang, Y. J. et al. High frequency of promoter hypermethylation of RASSF1A and p16 and its relationship to aflatoxin B1-DNA adduct levels in human hepatocellular carcinoma. Mol. Carcinog 35, 85–92. https://doi.org/10.1002/mc.10076 (2002).
Saelee, P., Wongkham, S., Chariyalertsak, S., Petmitr, S. & Chuensumran, U. RASSF1A promoter hypermethylation as a prognostic marker for hepatocellular carcinoma. Asian Pac. J. Cancer Prev. 11, 1677–1681 (2010).
Van Engeland, M. et al. Effects of dietary folate and alcohol intake on promoter methylation in sporadic colorectal cancer: The Netherlands cohort study on diet and cancer. Cancer Res. 63, 3133–3137 (2003).
Vineis, P. et al. DNA methylation changes associated with cancer risk factors and blood levels of vitamin metabolites in a prospective study. Epigenetics 6, 195–201. https://doi.org/10.4161/epi.6.2.13573 (2011).
Pirouzpanah, S., Taleban, F. A., Mehdipour, P. & Atri, M. Association of folate and other one-carbon related nutrients with hypermethylation status and expression of RARB, BRCA1, and RASSF1A genes in breast cancer patients. J. Mol. Med. 93, 917–934. https://doi.org/10.1007/s00109-015-1268-0 (2015).
Aravalli, R. N., Steer, C. J. & Cressman, E. N. K. Molecular mechanisms of hepatocellular carcinoma. Hepatology 48, 2047–2063. https://doi.org/10.1002/hep.22580 (2008).
Chu, P. Y. et al. Epigenetic alteration of the SOCS1 gene in hepatocellular carcinoma. Swiss Med. Wkly 140, w13065. https://doi.org/10.4414/smw.2010.13065 (2010).
Bagnyukova, T. V. et al. Epigenetic downregulation of the suppressor of cytokine signaling 1 (Socs1) gene is associated with the STAT3 activation and development of hepatocellular carcinoma induced by methyl-deficiency in rats. Cell Cycle 7, 3202–3210. https://doi.org/10.4161/cc.7.20.6816 (2008).
Yoshikawa, H. et al. SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity. Nat. Genet. 28, 29–35. https://doi.org/10.1038/88225 (2001).
Okochi, O. et al. Methylation-mediated silencing of SOCS-1 gene in hepatocellular carcinoma derived from cirrhosis. Clin. Cancer Res. 9, 5295–5298 (2003).
Miyoshi, H. et al. Methylation status of suppressor of cytokine signaling-1 gene in hepatocellular carcinoma. J. Gastroenterol. 39, 563–569. https://doi.org/10.1007/s00535-003-1343-0 (2004).
Busch, E. L., Galanko, J. A., Sandler, R. S., Goel, A. & Keku, T. O. Lifestyle factors, colorectal tumor methylation, and survival among African Americans and European Americans. Sci. Rep. 8, 9470. https://doi.org/10.1038/s41598-018-27738-x (2018).
Ashokkumar, B., Mohammed, Z. M., Vaziri, N. D. & Said, H. M. Effect of folate oversupplementation on folate uptake by human intestinal and renal epithelial cells. Am. J. Clin. Nutr. 86, 159–166. https://doi.org/10.1093/ajcn/86.1.159 (2007).
Funding
The financial assistance to R.S as fellowship by ICMR has sincerely been acknowledged.
Author information
Authors and Affiliations
Contributions
J.K. and R.S. has designed the overall study. R.S. has conducted hands on experiments. T.A. has contributed in the acquisition, and analysis of data related to blotting experiments.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sharma, R., Ali, T. & Kaur, J. Folic acid depletion as well as oversupplementation helps in the progression of hepatocarcinogenesis in HepG2 cells. Sci Rep 12, 16617 (2022). https://doi.org/10.1038/s41598-022-21084-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-21084-9
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
