
Abstract
In this study, 18 novel quinoline-based-benzo[d]imidazole derivatives were synthesized and screened for their α-glucosidase inhibitory potential. All compounds in the series except 9q showed a significant α-glucosidase inhibition with IC50 values in the range of 3.2 ± 0.3–185.0 ± 0.3 µM, as compared to the standard drug acarbose (IC50 = 750.0 ± 5.0 µM). A kinetic study indicated that compound 9d as the most potent derivative against α-glucosidase was a competitive type inhibitor. Furthermore, the molecular docking study revealed the effective binding interactions of 9d with the active site of the α-glucosidase enzyme. The results indicate that the designed compounds have the potential to be further studied as new anti-diabetic agents.
Similar content being viewed by others

Introduction
Diabetes mellitus (DM) is a chronic metabolic disease characterized by hyperglycemia, with the disorder in carbohydrate, fat, and protein metabolism in the body1. DM is known as an important public health threat with around 450 million cases worldwide in 2019. This number is expected to rise to 700 million by 2045 worldwide confirming further action is required in this field2,3. Long-term DM can increase the risk of various health complications including blindness, renal failure, foot amputation, as well as cardiovascular, retinopathy, and renal diseases4. Type 2 diabetes mellitus (T2DM) with around 90% of all cases is categorized as a major sub-type of DM. It was considered that glycemic control could be effective prevention and treatment for T2DM5,6,7.
α-Glucosidase (EC 3.2.1.20) is a catalytic hydrolase enzyme present on the brush border of the small intestine which hydrolyzes oligosaccharides, trisaccharides, and disaccharides to glucose and other monosaccharides at their non-reducing ends7,8,9,10. The produced monosaccharides especially glucose enter the bloodstream, resulting in postprandial hyperglycemia thus causing diabetes11,12,13. Therefore, the inhibition of α-glucosidase might reduce carbohydrate digestion, delay glucose uptake, and consequently, decrease blood sugar levels14,15. The α-glucosidase enzyme can be inhibited by acarbose, voglibose, and miglitol with sub-optimal efficacy16. Also, long-term administration of mentioned inhibitor may cause several side effects, such as abdominal pain, diarrhea, and flatulence. As a result, a need of effective inhibitors to target α-glucosidase is highly needed17,18,19.
In the last few decades, different synthetic small molecules including benzo[d]imidazole20, isatin21 benzo[b]thiophene22 pyrimidine23, xanthone24, chromene6, azole18,25 against α-glucosidase attracted increasing attention.
Regarding promising anti-diabetic properties of quinolone heterocyclic scaffold and benzo[d]imidazole moiety, in this study, the novel series of quinoline-based-benzo[d]imidazole bearing different acetamide derivatives were synthesized, and evaluated for their inhibition potential against the α-glucosidase enzyme. Also, kinetic as well as molecular docking studies of the most potent compound were performed to evaluate their inhibition pattern against α-glucosidase.
Results and discussion
Design of quinoline-based-benzo[d]imidazole derivatives
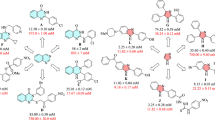
During the last years, several non-sugar-based α-glucosidase inhibitors were identified. The random screening of the in-house library resulted in introducing compound A (Fig. 1) bearing benzo[d]imidazole moiety with good potency against α-glucosidase20. The follow-up structural optimization of A resulted in a series of novel 2-phenyl-1H-benzo[d]imidazole derivatives (compound B and C) with IC50 values in the range of 0.71 to > 100 µM compared to the acarbose as a positive control with an IC50 value of 258.53 ± 1.27 µM. Preliminary structure–activity relationships (SARs) study revealed that the benzo[d]imidazole core played key role in the inhibition of α-glucosidase activity17.

Schematic illustration of previously reported α-glucosidase inhibitors and newly designed compound.
Also, recent studies demonstrated the α-glucosidase inhibitory activity of quinoline-containing compounds. The preliminary bioassay results revealed that compounds D and E (Fig. 1) had significant inhibitory potency compared to acarbose (IC50 = 66.5 ± 1.5 µg/mL)26. To further improve the α-glucosidase inhibitory activity of quinolone derivatives, the structural modification was carried out. These analogs exhibited inhibitory potential with IC50 values in the ranges between 2.60 and 102.12 μM (Compound F, Fig. 1)27.
Furthermore, it was reported that methyl-thioacetamide moiety (compounds G and H) can not only improve α-glucosidase inhibition through generating optimum structure to effectively participate within the active site but also provide a suitable site for derivatization28,29,30.
In this context, molecular hybridization as a powerful tool for drug designing was applied so that benzo[d]imidazole and quinoline as potent heterocyclic pharmacophores were conjugated to different acetamide derivatives. Novel designed compounds were synthesized and evaluated for their α-glucosidase inhibitory activities. Preliminary SAR studies were conducted. Further, kinetic study plus in silico assessments were performed to evaluate the binding of the active compound to the enzyme.
Chemistry
The synthesis of compounds 9a–r is schematically shown in Fig. 2. Briefly, phosphoryl chloride in N,N-dimethylformamide (DMF) was added dropwise to the cold N-phenylacetamide (1) under reflux conditions for 15 h to obtain 2-chloroquinoline-3-carbaldehyde (2)31. Compound 2 and sodium sulfide were then dissolved in DMF and stirred at room temperature for 2 h to achieve 3-formyl-2-mercaptoquinoline (3)32. Then, the reaction of O-phenylenediamines (4) and 3-formyl-2-mercaptoquinoline (3) in the presence of sodium metabisulfite in DMF at 150 °C for 2 h afforded the target compound 533. Synthesis of desired compounds 8a–r was performed through the reaction of aniline derivatives (6a–r) with chloroacethylchloride (7) in DMF34. Finally, the reaction of compounds 8a–r and compound 5 in acetone in presence of K2CO3 led to the formation of products 9a–r35. The structure of all compounds was confirmed using NMR and IR spectroscopy as well as elemental analysis.

Synthesis of compounds 9a–r.
Structure–activity relationship (SAR) exploration
The results of the α-glucosidase inhibitory assay are displayed in Table 1. In general, all compounds showed significant α-glucosidase inhibition with IC50 values in the range of 3.2 ± 0.3 to 185.0 ± 0.3 µM in comparison to acarbose with an IC50 value of 750.0 ± 5.0 µM. The exception come back to 9q which showed IC50 > 750.
As can be seen in Table 1, benzimidazole-thioquinoline structure bearing phenylacetamide exhibited good inhibitory activities against α-glucosidase (9a, IC50 = 30.2 ± 0.4 µM). The incorporation of a fluorine atom at the ortho position of phenylacetamide (9b) resulted in an around the twofold loss of potency compared to 9a. Furthermore, changing the position from ortho to para in compound 9c (IC50 = 13.5 ± 0.6 µM) resulted in the second potent derivative in the halogen-substituted set.
Impotently, the introduction of 3-chlorophenyl at R position, compounds 9d, displayed a significant α-glucosidase inhibition (IC50 = 3.2 ± 0.3 µM) with around 250-fold improvement in the potency compared to the positive control, acarbose. Indeed, compound 9e (R = 4-chlorophenyl, IC50 = 110.4 ± 0.2 µM) had inferior activity compared to 9d. Replacement of chlorine substitution with bromine resulted in compounds 9f and 9g. Compound 9f (R = 2-Bromophenyl) was another potent derivative in halogen-substituted set (IC50 = 23.4 ± 0.2 µM). However converting 2-Br substitution to 4-Br was not favorable (9g, IC50 = 185.0 ± 0.3 µM) compared to 9f. Additionally, 9h as the multi-substituted chlorine derivative with inferior activities compared with 9c, still exhibited promising potency compared to acarbose.
Overall, the mono-electron withdrawing group (EWG) at para position had a destructive effect against α-glucosidase while ortho and meta position seems more favorable. The exception in this trend came back to 9c bearing 4-fluorine. This could be due to the smaller size and better electronegativity compared to the rest of halogen derivatives.
The evaluations on 9j–m as the mono electron-donating-substituted group (EDG) showed overall improvement in the potency so that 9l (R = 4-methoxyphenyl) with an IC50 of 5.7 ± 0.3 μM was categorized as the top potent inhibitor in this group and second top potent entry among all derivatives followed by 9k (R = 4-methyl phenyl) and 9j (R = 2-methyl phenyl). Next, the assessment of compounds 9n and 9o were performed and, disappointingly, 9o bearing symmetric multi-substituted moiety (R = 2,6-diCH3, IC50 = 147.0 µM) recorded the reduction in the activity compared to 9n. However, 9o derivative still demonstrated around eightfold improvement in the potency compared to acarbose with IC50 of 750.0 µM.
Precise assessments on the 9j–o derivatives also indicated that the position of substitutions seems to have the most dominant role in the inhibition compared to the lipophilicity of moiety.
Ring substitution assessments were also performed in which phenyl (9a) was replaced with naphthyl (9p). An improvement in the activity showed that a bulk structure is more favorable.
Next, the investigation of SAR indicated that nitro (9i, IC50 = 19.7 ± 0.2 µM) and methoxy (9l, IC50 = 5.7 ± 0.3 µM) moieties were optimal substituents at the para position of phenylacetamide which improved the α-glucosidase inhibition. These results suggested that such substitution may probably enhance the ligand–protein interaction with the α-glucosidase active site.
9q and 9r were also synthesized to evaluate the role of elongation of the linker between aryl substitutions and thioacteamide moiety. Compound 9q with benzyl substitution exhibited dramatically reduction in the α-glucosidase inhibition compared to 9a which exhibited the destructive effect of elongation of the linker in the unsubstituted derivatives. Also, there was a similar trend in the potency in 9r bearing 4-fluorobenzyl compared to 9c (R = 4-Fluorophenyl).
The summary of the SARs to improve α-glucosidase inhibitory activity was depicted in Fig. 3. Overall, it can be understood that the most potent derivative (9d) exhibited better inhibitory activity against a-glucosidase compared to lead compounds including A to G reported in Fig. 1 concerning their positive control.

Summary of the SARs.
Enzyme kinetic studies
To gain insight into the mechanism of action of 9d as the most potent α-glucosidase inhibitor, kinetic measurements were performed. According to Fig. 4a, the Lineweaver–Burk plot showed that the Km gradually increased and Vmax remained unchanged with increasing inhibitor concentration indicating a competitive inhibition. The results show 9d bonded to the active site on the enzyme and compete with the substrate for binding to the active site. Furthermore, the plot of the Km versus different concentrations of inhibitor gave an estimate of the inhibition constant, Ki of 3.2 µM (Fig. 4b).

Kinetics of α-glucosidase inhibition by 9d. (a) The Lineweaver–Burk plot in the absence and presence of different concentrations of 9d; (b) the secondary plot between Km and various concentrations of 9d.
Docking analyses
To identify the accuracy and validation of docking procedures, the self-docking of acarbose (as a crystallographic ligand) was performed through induced fit docking of Schrödinger software. Alignment of the best pose of acarbose in the active site of α-glucosidase and crystallographic ligand recorded an RMSD value of 1.73 Å (RMSD should be less than 2 Å) which confirms the accuracy of docking. Next, the same docking procedure was repeated with all derivatives and their binding to α-glucosidase was analyzed. Results are summarized in Table 2.
The in silico studies showed the binding energy of acarbose was − 6.14 kcal/mol while the glide score value of 9a–r ranges from − 4.65 to − 6.92 kcal/mol. As can be seen, the most potent derivative in in vitro assay was 9d (IC50 = 3.2 ± 0.3) > 9l (IC50 = 5.7 ± 0.3 µM) > 9n (IC50 = 9.8 ± 0.5 µM) > 9k (IC50 = 12.3 ± 0.2 µM) exhibited the best in silico results with glide score value of − 6.92, − 6.33, − 6.90 and − 6.72 kcal/mol, respectively. Assessments on lest potent derivatives, 9q (IC50 > 750), 9g (IC50 = 185.0 ± 0.3) and 9o (IC50 = 147.0 ± 0.2) reveal low binding interaction with the targeted enzyme with binding energy of − 4.30, − 4.65 and − 4.99 value.
The docking results between α-glucosidase and compound 9d was shown in Fig. 5. Compound 9d was well inserted into the active site and recorded a Glide score of − 6.92. Compound 9d established critical hydrogen bond interaction with Trp481 and benzimidazole. Also, benzimidazole participated in pi–cation interaction with Arg600. On the other side of the molecule, 3-chlorophenylacetamide established H-bound interaction with Asp616 and halogen-bound interaction with Leu677. Notably, in most derivatives, the designed scaffold participated in the critical interactions within the active site of the enzyme and showed similar kinds of interactions to the native ligand.

3D and 2D proposed binding modes of compounds 9d (blue color) with α-glucosidase.
Conclusion
In this study, a series of novel quinoline-based-benzo[d]imidazole bearing different acetamide derivatives were designed, synthesized and their inhibitory activity against α-glucosidase was performed. Most of these derivatives showed increased activity compared to acarbose as the positive control. The analysis of the SAR indicated that meta-chlorine substitution, as well as polar group with potential hydrogen interactions at the R position, was beneficial to α-glucosidase inhibition. The most potent candidate in this series 9d (IC50 = 3.2 ± 0.3 µM) was chosen for further biological evaluation. The enzyme kinetics assessments indicated that compound 9d inhibited α-glucosidase in a competitive inhibition manner (Ki = 3.2 µM). According to the docking study, compound 9d was well fitted in the active site of α-glucosidase through both hydrophobic and hydrogen interactions. Overall, it can be understood that the most potent derivative (9d) exhibited better inhibitory activity against a-glucosidase compared to lead compounds including A, to G reported compared to positive control reported in Fig. 1. In silico assessments confirmed the critical role of benzimidazole and aryl-acetamides to participate in interactions with the binding site of an enzyme.
Regarding that T2DM is public health concern nowadays, the inhibition of α-glucosidase is considered an efficient approach to target T2DM. It was shown that quinoline-based-benzo[d]imidazole bearing different acetamides constructed a new nucleus which provided a significant role for α-glucosidase inhibition. However, to better extract the SARs of this set of compounds, in the future project, heteroaryl or aliphatic substituents at the R position will be synthesized. Also, bioisosteric replacement of benzo[d]imidazole with other heteroaromatic rings will increase our insight into the design of more potent α-glucosidase inhibitors.
Experimental
Chemistry
All the reagents were purchased from commercial sources. 1H and 13C NMR spectra were determined by a Bruker FT-400 MHz spectrometer in DMSO-d6. All the chemical shifts were reported as (δ) values ppm. The MS spectra were recorded using an Agilent 7890A spectrometer at 70 eV. CHNOS analysis was performed using ECS4010 Costech Company. IR spectra were obtained with a Nicolet, FR -IR Magna 550. Melting-point were also recorded using Kofler hot-stage apparatus.
Synthesis of 2-chloroquinoline-3-carbaldehyde (2)31
To N, N-dimethylformamide (70.0 mmol) in the round-bottomed flask, phosphorus oxychloride (120.0 mmol) was added dropwise and the reaction mixture was stirred for 1 h at 0–5 °C. To this flask, N-phenylacetamide (30.0 mmol) was added and stirred for an extra 30 min followed by refluxing for 5–4 h under N2 atmosphere. After the reaction was completed (TLC monitoring), the mixture was poured into crushed ice under constant stirring. The precipitate obtained was vacuum filtered, washed with water, air-dried, and recrystallized from EtOAc to give the 2-chloroquinoline-3-carbaldehyde.
Synthesis of 2-mercaptoquinoline-3-carbaldehyde (3)32
The reaction was initiated by stirring the mixture of 2-chloroquinoline-3-carbaldehyde 2 (1 mmol) and sodium sulfide (1 mmol) for 2 h at room temperature in dry DMF (50 mL). Then, the reaction mixture was poured into crushed ice and made acidic with acetic acid. The product was filtered off, washed with water, and dried to give the desired 2-mercaptoquinoline-3-carbaldehyde that was further purified by recrystallization in ethanol.
Synthesis of 3-(1H-benzo[d]imidazol-2-yl)quinoline-2-thiol (5)33
2-Mercaptoquinoline-3-carbaldehyde (1 mmol) and o-phenylenediamine (1.2 mmol) were dissolved in 2 mL DMF. Under stirring at room temperature, 1 mmol of sodium metabisulfite is added and allowed to react at 120 °C for about 4 h. After completion of the reaction, the mixture was precipitated in ice water, filtered, and dried at room temperature.
Synthesis of 2-chloro-N-phenylacetamide derivatives (8a–r)34
To a solution of aniline derivatives (1 mmol) in DMF (4 mL), chloroacetylchloride was added at 0 °C. The mixture was stirred at room temperature for 5 h and poured into water and then filtered to get the 8a–r. The obtained solids were then filtered, dried, and recrystallized from ethanol.
General method for synthesis of 2-((3-(1H-benzo[d]imidazol-2-yl)quinolin-2-yl)thio)-N-phenylacetamide derivatives (9a–r)35
A mixture of 3-(1H-benzo[d]imidazol-2-yl)quinoline-2-thiol (1 mmol) and potassium carbonate (1.5 mmol) in DMF were stirred at room temperature for 15–20 min. Afterward, N-chloroacetyl-aniline (1.2 mmol) was added to the above reaction mixture and stirred for an extra 4–5 h. After completion of the reaction, ice-cold water was added to the reaction mixture and stirred for 20 min. The obtained solid was filtered and washed with cold water several times. The acquired crude solid was purified by recrystallization from ethanol.
2-((3-(1H-benzo[d]imidazol-2-yl)quinolin-2-yl)thio)-N-phenylacetamide (9a)
Brown solid; Yield: 93%; MP = 180–182 °C; IR (KBr, vmax) 3310 (NH), 3025 (C–H Aromatic), 2970 (CH2 Aliphatic), 1675 (C=O) Cm−1; 1H NMR (400 MHz, DMSO-d6) δ 10.71 (s, 1H, NH), 9.07 (s, 1H, H3), 8.47 (s, 1H, H4), 8.19 (d, J = 8.00 Hz, 1H, H8), 8.10 (d, J = 8.60 Hz, 1H, H5), 7.86 (t, J = 8.40 Hz, 1H, H7), 7.81(d, J = 7.6 Hz, 2H, H2, H6), 7.65 (t, J = 8.00 Hz 1H, H4), 7.41 (t, J = 7.90 Hz, 2H, H3, H5), 7.16 (t, J = 7.40 Hz, 1H, H3) ppm. 13C NMR (100 MHz, DMSO-d6): δ 167.80, 157.98, 148.94, 147.10, 143.98, 139.79, 136.69, 135.03, 131.63, 129.24, 128.79, 127.65, 126.76, 125.08, 123.74, 123.63, 123.20, 123.40, 119.78, 119.48, 112.01, 36.55 ppm; ESI–MS (C24H18N4OS): calculated m/z 410.15 [M + H]+, observed m/z 410.10 [M + H]+ Anal. Calcd. For C24H18N4OS C, 70.22; H, 4.42; N, 13.65; Found: C, 70.39; H, 4.60; N, 13.76;
2-((3-(1H-benzo[d]imidazol-2-yl)quinolin-2-yl)thio)-N-(2-fluorophenyl)acetamide (9b)
Brown solid; Yield: 87%; MP = 183–185 °C; IR (KBr, vmax) 3325 (NH), 3060 (C–H Aromatic), 2950 (CH2 Aliphatic), 1680 (C=O) Cm−1; 1H NMR (400 MHz, DMSO-d6) δ 13.15 (s, 1H), 10.21 (s, 1H), 8.79 (s, 1H), 8.03 (d, J = 7.9 Hz, 1H), 7.98 (d, J = 8.4 Hz, 1H), 7.96–7.88 (m, 1H), 7.82 (t, J = 8.2 Hz, 1H), 7.61 (t, J = 7.4 Hz, 1H), 7.35–7.21 (m, 4H), 7.15–7.00 (m, 3H), 4.23 (s, 2H) ppm. 13C NMR (100 MHz, DMSO-d6): δ 168.45, 157.90, 154.95, 152.52 (CF, 1JCF = 243 Hz), 148.91, 147.12, 136.82, 131.63, 128.77, 127.64, 126.90, 126.84, 126.78, 125.48, 125.40, 125.14, 124.86, 124.04, 123.22, 116.00, 115.81, 35.95 ppm; ESI–MS (C24H17FN4OS): calculated m/z 428.11 [M + H]+, observed m/z 428.20 [M + H]+, Anal. Calcd. for C24H17FN4OS: C, 67.27; H, 4.00; N, 13.08; Found: C, 67.45; H, 4.18; N, 13.24;
2-((3-(1H-benzo[d]imidazol-2-yl)naphthalen-2-yl)thio)-N-(4-fluorophenyl)acetamide (9c)
Brown solid; Yield: 89%; MP = 189–191 °C; IR (KBr, vmax) 3330 (NH), 3025 (C–H Aromatic), 2915 (CH2 Aliphatic),1640 (C=O) Cm−1; 1H NMR (400 MHz, DMSO-d6) δ 13.70 (s, 1H), 10.49 (s, 1H), 8.78 (s, 1H), 8.01 (d, J = 8.00 Hz, 1H), 7.92 (d, J = 8.30 Hz, 1H), 7.79 (t, J = 7.70 Hz, 1H), 7.73–7.56 (m, 5H), 7.33–7.26 (m, 2H), 7.15 (t, J = 8.8, 2H), 4.17 (s, 2H), ppm. 13C NMR (100 MHz, DMSO-d6): δ159.57, 157.95 (CF, 1JCF = 238.1 Hz), 157.19, 148.96, 147.09, 136.68, 136.21, 131.63, 128.79, 127.3, 126.76, 125.08, 123.19, 121.25, 121.17, 115.92, 115.70, 36.49 ppm; ESI–MS (C24H17FN4OS): calculated m/z 428.11 [M + H]+, observed m/z 428.30 [M + H]+, Anal. Calcd. for C25H18FN3OS: C, 70.24; H, 4.24; N, 9.83; Found: C, 70.41; H, 4.47; N, 9.99.
2-((3-(1H-benzo[d]imidazol-2-yl)quinolin-2-yl)thio)-N-(3-chlorophenyl)acetamide (9d)
Brown solid; Yield: 89%; MP = 190–192°; IR (KBr, vmax) 3310 (NH), 3015 (C–H Aromatic), 2880 (CH2 Aliphatic), 1645 (C=O) Cm−1; 1H NMR (400 MHz, DMSO-d6) δ 13.6 (s, 1H), 10.65 (s, 1H), 8.79 (s, 1H), 8.01 (d, J = 8.10 Hz, 1H), 7.93–7.77 (m, 4H), 7.66–7.50 (m, 3H), 7.10 (d, J = 8.00 Hz, 1H), 4.18 (s, 3H) ppm. 13C NMR (100 MHz, DMSO-d6): δ 168.42, 167.60, 166.09, 162.75, 157.90, 150.04, 149.10, 147.06, 143.03, 141.29, 136.63, 136.21, 133.61, 131.56, 130.39, 128.76, 127.59, 126.73, 125.12, 123.23, 123.24, 123.02, 118.97, 117.86, 31.22 ppm; ESI–MS (C24H17ClN4OS): calculated m/z 444.08, [M + H]+, observed m/z 444.20, [M + H]+; Anal. Calcd. C24H17ClN4OS, C, 64.79; H, 3.85; N, 12.59; Found: C, 64.95; H, 4.02; N, 12.77.
2-((3-(1H-benzo[d]imidazol-2-yl)quinolin-2-yl)thio)-N-(4-chlorophenyl)acetamide (9e)
Brown solid; Yield: 90%; MP = 185–187 °C IR (KBr, vmax) 3275 (NH), 3030 (C–H Aromatic), 2980 (CH2 Aliphatic), 1680 (C=O) Cm−1; 1H NMR (400 MHz, DMSO-d6) δ 13.14 (s, 1H), 10.57 (s, 1H), 8.78 (s, 1H), 8.01 (d, J = 8.00 Hz, 1H), 7.89 (d, J = 8.40 Hz, 1H), 7.78 (t, J = 7.70 Hz, 2H), 7.68 (d, J = 8.50 Hz, 3H), 7.59 (t, J = 7.50 Hz, 1H), 7.37 (d, J = 8.50 Hz, 2H), 7.29 (d, J = 7.20 Hz, 2H) ppm. ESI–MS (C24H17ClN4OS): calculated m/z 444.08, [M + H]+, observed m/z 444.20,[M + H]+, 13C NMR (100 MHz, DMSO-d6): δ 168.06, 157.91, 148.93, 147.07, 138.77, 136.66, 131.64, 129.16, 128.79, 127.59, 127.14, 126.78, 125.05, 123.15, 120.99, 36.58 ppm; Anal. Calcd. for C24H17ClN4OS; C, 64.79; H, 3.85; N, 12.59; Found: C, 64.95; H, 3.99; N, 12.79.
2-((3-(1H-benzo[d]imidazol-2-yl)quinolin-2-yl)thio)-N-(2-bromophenyl)acetamide (9f)
Brown solid; Yield: 93%; MP = 181–183 °C; IR (KBr, vmax) 3300(NH), 3030 (C–H Aromatic), 2965 (CH2 Aliphatic), 1655 (C=O) Cm−1; 1H NMR (400 MHz, DMSO-d6) δ 13.14 (s, 1H), 10.43 (s, 1H), 8.77 (s, 1H), 8.01 (d, J = 7.90 Hz, 1H), 7.93 (d, J = 8.40 Hz, 1H), 7.80 (d, J = 5.50 Hz, 1H), 7.78 (d, J = 6.1 Hz, 1H), 7.67–7.56 (m, 3H), 7.35–7.24 (m, 4H), 7.04 (t, J = 7.3 Hz, 1H) ppm. ESI–MS (C24H17BrN4OS): calculated m/z 490.03, [M + H]+, observed m/z 488.10, [M + H]+, 13C NMR (100 MHz, DMSO-d6): δ 168.25, 157.53, 149.29, 147.09, 136.81, 136.69, 133.05, 131.53, 128.75, 128.52, 127.87, 126.91, 126.84, 125.93, 125.25, 123.60, 122.90, 116.69, 115.96, 35.84 ppm; Anal. Calcd. for C24H17BrN4OS C, 58.90; H, 3.50; N, 11.45; Found: C, 59.11; H, 3.69; N, 11.75.
2-((3-(1H-benzo[d]imidazol-2-yl)quinolin-2-yl)thio)-N-(4-bromophenyl)acetamide (9g)
Brown solid; Yield: 95%; MP = 185–187°; IR (KBr, vmax) 3340 (NH), 3025 (C–H Aromatic), 2870 (CH2 Aliphatic), 1640 (C=O) Cm−1; 1H NMR (400 MHz, DMSO-d6) δ 13.15 (s, 1H), 10.58 (s, 1H), 8.01 (d, J = 7.90 Hz, 1H), 7.90 (d, J = 8.10 Hz, 1H), 7.78 (t, J = 7.70 Hz, 2H), 7.64 (d, J = 8.50 Hz, 3H), 7.60–7.56 (m, 1H), 7.49 (d, J = 8.50 Hz 2H), 7.35–7.25 (m,2H), 4.17 (s, 2H) ppm. ESI–MS (C24H17BrN4OS): calculated m/z 490.03, [M + H]+, observed m/z 488.10, [M + H]+, 13C NMR (100 MHz, DMSO-d6): δ 168.10, 157.91, 148.94, 147.08, 143.98, 139.19, 136.66, 135.04, 132.07. 131.63, 128.78, 127.60, 126.77, 125.08, 123.76, 123.16, 122.41, 121.49, 121.40, 119.78, 115.18, 112.02, 36.62 ppm; Anal. Calcd. for C24H17BrN4OS; C, 58.90; H, 3.50; N, 11.45; Found: C, 59.05; H, 3.70; N, 11.62.
2-((3-(1H-benzo[d]imidazol-2-yl)quinolin-2-yl)thio)-N-(2,6-dichlorophenyl)acetamide (9h)
Brown solid; Yield: 86%; MP = 188–190 °C; IR (KBr, vmax) 3340 (NH), 3035 (C–H Aromatic), 2960 (CH2 Aliphatic), 1675 (C=O) Cm−1; 1H NMR (400 MHz, DMSO-d6) δ 13.15 (s, 1H), 9.92 (s, 1H), 8.81 (s, 1H), 8.03 (d, J = 8.00 Hz, 1H), 7.97 (d, J = 8.40 Hz, 1H), 7.86 (d, J = 8.80 Hz, 1H), 7.81 (t, J = 7.30 Hz, 1H), 7.75–7.57 (m, 4H), 7.37 (d, J = 8.80 Hz, 1H), 7.33–7.25 (m, 2H) 4.26 (s, 2H) ppm. ESI–MS (C24H17Cl2N4OS): calculated m/z 478.10, [M + H]+, observed m/z 444.20, [M + H]+, 13C NMR (100 MHz, DMSO-d6): δ 168.85, 157.60, 148.88, 147.10, 136.88, 134.69, 131.65, 129.30, 129.25, 128.79, 128.08, 127.77, 126.91, 126.45, 126.23, 125.19, 123.24, 35.80 ppm; Anal. Calcd. for C24H16Cl2N4OS C, 60.13; H, 3.36; N, 11.69; Found: C, 60.24; H, 3.49; N, 11.85.
2-((3-(1H-benzo[d]imidazol-2-yl)quinolin-2-yl)thio)-N-(4-nitrophenyl)acetamide (9j)
Pale yellow solid; Yield: 93%; MP = 180–182 °C; IR (KBr, vmax) 3320 (NH), 3020 (C–H Aromatic), 2965 (CH2 Aliphatic), 1670 (C=O), 1555–1350 (NO2) Cm−1; 1H NMR (400 MHz, DMSO-d6) δ 13.60 (s, 1H), 11.80 (s, 1H), 8.79(s, 1H), 8.24 (d, J = 9.30 Hz, 1H), 8.01 (d, J = 7.70 Hz, 1H), 7.91 (d, J = 9.30, 2H), 7.86–7.74 (m, 3H), 7.63 (d, J = 7.70 Hz, 1H), 7.57 (t, J = 8.00 Hz, 1H), 7.35–7.25 (m, 2H), 4.21 (s, 2H) ppm. ESI–MS (C24H17N5O3S): calculated m/z 455.11, [M + H]+, observed m/z 455.20, [M + H]+, 13C NMR (100 MHz, DMSO-d6): δ 169.12, 157.79, 148.91, 147.01, 145.99, 143.96, 142.54, 136.61, 135.04, 131.67, 128.80, 127.49, 126.81, 125.60, 125.08, 123.79, 123.04, 122.43, 119.79, 119.08, 112.02, 36.80 ppm; ESI–MS (C24H17N5O3S C): calculated m/z 455.11 [M + H]+, observed m/z 455.20 [M + H]+; Anal. Calcd. for C24H17N5O3S C, 63.29; H, 3.76; N, 15.38; N, 7.31; Found: C, 63.49; H, 3.96; N, 15.55.
2-((3-(1H-benzo[d]imidazol-2-yl)naphthalen-2-yl)thio)-N-(o-tolyl)acetamide (9k)
Brown solid; Yield: 86%; MP = 179–181 °C; IR (KBr, vmax) 3360 (NH), 3070 (C–H Aromatic), 2980 (CH2 Aliphatic), 1680 (C=O) Cm−1; 1H NMR (400 MHz, DMSO-d6) δ 13.15 (s, 1H), 10.34 (s, 1H), 8.77 (s, 1H), 8.01 (d, J = 6.60 Hz, 1H), 7.93 (d, J = 7.10 Hz, 1H), 7.80 (d, J = 7.40 Hz, 2H), 7.66–7.46 (m, 4H), 7.30 (t, J = 9.50 Hz, 2H), 7.10 (d, J = 8.10 Hz, 2H), 4.15 (s, 2H), 2.20 (s, 3H), ppm. 13C NMR (100 MHz, DMSO-d6): δ 167.54, 158.01, 148.96, 147.11, 137.30, 136.70, 132.52, 131.61, 129.60, 128.77, 127.66, 126.74, 125.08, 123.23, 119.77, 119.51, 112.02, 36.54, 20.91 ppm; Anal. Calcd. for C26H21N3OS: C, 73.73; H, 5.00; N, 9.92; Found: C, 73.92; H, 5.19; N, 10.11.
2-((3-(1H-benzo[d]imidazol-2-yl)naphthalen-2-yl)thio)-N-(p-tolyl)acetamide (9l)
Cream solid; Yield: 91%; MP = 181–183 °C; IR (KBr, vmax) 3345 (NH), 3040 (C–H Aromatic), 2900 (CH-Aliphatic), 1670 (C=O) Cm−1; 1H NMR (400 MHz, DMSO-d6) δ 13.15 (s, 1H), 10.34 (s, 1H), 8.77 (s, 1H), 8.01 (d, J = 7.30 Hz, 1H), 7.93 (d, J = 7.70 Hz, 1H), 7.79 (t, J = 7.70 Hz, 2H), 7.63–7.48 (m, 4H), 7.29 (s, 2H), 7.10 (d, J = 8.20 Hz, 2H), 4.16 (s, 2H), 2.23 (s, 3H) ppm. ESI–MS (C26H21N3OS): calculated m/z 424.14, [M + H]+, observed m/z 424.10, [M + H]+ 13C NMR (100 MHz, DMSO-d6): δ 162.6, 160.2, 147.4, 141.2, 140.1, 136.5, 133.7, 132.2, 131.0, 129.4, 128.4, 128.3, 126.2, 126.0, 124.0, 120.9, 28.1, 16.1 ppm; Anal. Calcd. for C26H21N3OS C, 73.73; H, 5.00; N, 9.92; Found: C, 73.82; H, 5.14; N, 9.99.
2-((3-(1H-benzo[d]imidazol-2-yl)quinolin-2-yl)thio)-N-(4-methoxyphenyl)acetamide (9m)
Cream solid; Yield: 93%; MP = 191–193 °C; IR (KBr, vmax) 3340 (NH), 3030 (C–H Aromatic), 2910 (CH-Aliphatic), 1680 (C=O) Cm−1; 1H NMR (400 MHz DMSO-d6) δ 13.14 (s, 1H), 10.27 (s, 1H), 8.77 (s, 1H), 8.02 (d, J = 7.70 Hz, 1H), 7.95 (d, J = 8.40 Hz, 1H), 7.80 (t, J = 8.30 Hz, 1H), 7.59 (t, J = 7.90 Hz, 1H), 7.54 (d, J = 9.00 Hz, 2H), 7.34–7.25 (m, 2H), 6.88 (d, J = 9.10 Hz, 1H), 4.15 (s, 2H), 3.71 (s, 3H) ppm. 13C NMR (100 MHz, DMSO-d6): δ 167.23, 158.02, 155.60, 148.96, 147.12, 136.71, 132.94, 131.62, 128.78, 127.68, 126.75, 125.08, 123.24, 121.03, 114.33, 55.58, 36.43 ppm; ESI–MS (C25H20N4O2S): calculated m/z 424.14 [M + H]+, observed m/z 424.10 [M + H]+; Anal. Calcd. for C25H20N4O2S; C, 68.16; H, 4.58; N, 12.72; Found C, 68.35; H, 4.76; N, 12.90.
2-((3-(1H-benzo[d]imidazol-2-yl)quinolin-2-yl)thio)-N-(4-ethylphenyl)acetamide (9n)
Brown solid;Yield:93%;MP = 178–180 °C; IR (KBr, vmax) 3300 (NH), 3020 (C–H Aromatic), 2975(CH2 Aliphatic), 1670 (C=O) Cm−1; 1H NMR (400 MHz, DMSO-d6) δ 13.12 (s, 1H), 10.34 (s, 1H), 8.78 (s, 1H), 8.01 (d, J = 8.00 Hz, 1H), 7.95 (d, J = 8.40 Hz, 1H), 7.79 (t, J = 7.30 Hz, 1H), 7.75–7.65 (m, 2H), 7.58 (t, J = 7.60 Hz, 1H), 7.54 (d, J = 8.40 Hz, 2H), 7.30 (d, J = 6.10 Hz, 2H), 7.13 (d, J = 8.30 Hz, 2H), 4.17 (s, 2H), 2.53 (d, J = 7.70 Hz, 2H), 1.14 (t, J = 7.30 Hz, 3H) ppm. ESI–MS (C26H22N4OS): calculated m/z 438.55, [M + H]+, observed m/z 438.10,[M + H]+, 13C NMR (100 MHz, DMSO-d6): δ 167.55, 158.00, 148.97, 147.11, 139.01, 137.47, 136.70, 131.61, 128.78, 128.41, 127.68, 126.75, 125.08, 123.24, 119.61, 36.52, 28.06, 16.18, 16.12 ppm; Anal. Calcd. for C26H22N4OS; C, 71.21; H, 5.06; N, 12.78; Found: C, 71.39; H, 5.26; N, 12.97.
2-((3-(1H-benzo[d]imidazol-2-yl)quinolin-2-yl)thio)-N-(2,3-dimethylphenyl)acetamide (9o)
Brown solid; Yield: 93%; MP = 185–187 °C IR; (KBr, vmax) 3300 (NH), 3020 (C–H Aromatic), 2975 (CH2 Aliphatic) 1675 (C=O) Cm−1; 1H NMR (400 MHz, DMSO-d6) δ 13.6 (s, 1H), 9.76 (s, 1H), 8.79 (s, 1H), 8.03 (d, J = 8.90 Hz, 1H), 8.01 (d, J = 7.90 Hz, 1H), 7.83 (t, J = 8.30 Hz, 1H), 7.61 (t, J = 7.90 Hz, 1H), 7.30 (d, J = 6.60, 2H), 7.15 (d, J = 7.50 Hz, 1H), 7.06–6.92 (m, 4H), 4.22 (s, 1H), 2.20 (s, 3H), 2.02 (s, 3H), ppm. 13C NMR (100 MHz, DMSO-d6): δ 167.73, 157.98, 149.00, 147.19, 137.38, 136.87, 136.66, 131.60, 131.57, 128.81, 127.77, 127.33, 126.77, 125.62, 125.15, 123.78, 123.39, 35.74, 20.59, 14.47 ppm; ESI–MS (C26H22N4OS): calculated m/z 438.15 [M + H]+, observed m/z 438.10 [M + H]+; Anal. Calcd. for C26H22N4OS C, 71.21; H, 5.06; N, 12.78; Found C, 71.39; H, 5.26; N, 12.91.
2-((3-(1H-benzo[d]imidazol-2-yl)quinolin-2-yl)thio)-N-(2,6-dimethylphenyl)acetamide (9p)
Brown solid; Yield: 93%; MP = 185–187 °C; IR (KBr, vmax) 3325 (NH), 3045 (C–H Aromatic), 2980(CH2 Aliphatic) 1665 (C=O) Cm−1; 1H NMR (400 MHz, DMSO-d6) δ 13.11 (s, 1H), 9.57 (s, 1H), 8.77 (s, 1H), 8.03 (t, J = 8.40 Hz, 2H), 7.84 (t, J = 7.70 Hz, 1H), 7.62 (t, J = 7.20 Hz, 2H), 7.35–7.25 (m, 2H), 7.07–6.98 (m, 4H), 4.24 (s, 1H), 2.05 (s, 6H) ppm. ESI–MS (C26H22N4OS): calculated m/z 438.15 [M + H]+, observed m/z 438.10 [M + H]+; 13C NMR (100 MHz, DMSO-d6): δ 162.6, 161.6, 147.4, 140.9, 139.5, 133.7, 132.7, 132.3, 131.0, 129.4, 128.9, 128.3, 127.9, 127.5, 126.3, 126.0, 123.3, 43.3 ppm; Anal. Calcd. for C26H22N4OS C, 71.21; H, 5.06; N, 12.78; Found C, 71.39; H, 5.26; N, 12.91.
2-((3-(1H-benzo[d]imidazol-2-yl)quinolin-2-yl)thio)-N-(naphthalen-2-yl)acetamide (9q)
Cream solid; Yield: 91%; MP = 182–184° C; IR (KBr, vmax) 3340 (NH), 3030 (C–H Aromatic), 2900 (CH-Aliphatic), 1670 (C=O) Cm−1; 1H NMR (400 MHz, DMSO-d6) δ 13.16 (s, 1H), 10.33 (s, 1H), 8.80 (s, 1H), 8.10 (d, J = 8.30 Hz, 1H), 8.05 (d, J = 7.90 Hz, 2H), 7.92 (d, J = 7.8 Hz, 1H), 7.86–7.80 (m, 1H), 7.79–7.66 (m, 3H), 7.63 (d, J = 7.70 Hz, 2H), 7.53–7.44 (m, 2H), 7.37 (t, J = 7.60 Hz, 1H), 7.34–7.24 (m, 2H), 4.36 (s, 2H), ppm. ESI–MS (C28H20N4OS): calculated m/z 460.14 [M + H]+, observed m/z 460.10 [M + H]+; 13C NMR (100 MHz, DMSO-d6): δ 168.50, 149.01, 147.21, 136.87, 134.23, 134.13, 131.61, 128.84, 128.54, 128.40, 127.79, 126.80, 126.46, 126.10, 126.06, 125.83, 125.18, 123.40, 122.18, 35.94 ppm; Anal. Calcd. for C28H20N4OS; C, 73.02; H, 4.38; N, 12.17; Found: C, 73.32; H, 4.55; N, 12.34.
2-((3-(1H-benzo[d]imidazol-2-yl)quinolin-2-yl)thio)-N-benzylacetamide (9r)
Brown solid; Yield: 94%; MP = 183–185 °C; IR (KBr, vmax) 3310(NH) , 3045(C-H Aromatic), 2975 (CH2 Aliphatic) 1655 (C=O) Cm−1; 1H NMR (400 MHz, DMSO-d6) δ 13.11 (s, 1H), 8.76 (s, 1H), 8.71 (t, J = 6.00 Hz, 1H), 8.03 (d, J = 8.00 Hz, 1H), 7.90 (d, J = 8.5 Hz, 1H), 7.81 (t, J = 7.70 Hz 1H), 7.62 (t, J = 7.60 Hz, 2H), 7.34–7.25 (m, 2H), 7.21–7.13 (m, 6H), 4.31 (d, J = 6.00 Hz, 2H), 4.06 (s,1H) ppm. ESI–MS (C26H22N4OS): calculated m/z 424.14 [M + H]+, observed m/z 424.10 [M + H]+; 13C NMR (100 MHz, DMSO-d6): δ 168.67, 157.83, 149.00, 147.15, 139.83, 136.79, 131.51, 128.73, 128.55, 127.90, 127.45, 127.05, 126.73, 125.10, 123.39, 42.83, 35.17 ppm; Anal. Calcd. for C25H20N4OS: C, 70.73; H, 4.75; N, 13.20; Found: C, 70.92; H, 4.90; N, 13.38.
2-((3-(1H-benzo[d]imidazol-2-yl)quinolin-2-yl)thio)-N-(4-fluorobenzyl)acetamide (9s)
Brown solid;Yield:89%; MP = 186–188 °C; IR (KBr, vmax) 3350 (NH), 3060 (C–H Aromatic), 2975 (CH2 Aliphatic), 1670 (C=O) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 8.74 (d, J = 14.20 Hz, 2H), 8.01 (d, J = 8.00 Hz, 1H), 7.90–7.75 (m, 2H), 7.70 (s, 2H), 7.60 (s, 1H), 7.39–7.17 (m, 4H), 6.95 (t, J = 8.9, 2H), 4.29 (s, 2H), 4.04 (s, 2H), ppm. ESI–MS (C25H19FN4OS): calculated m/z 442.14 [M + H]+, observed m/z 442.10 [M + H]+; 13C NMR (100 MHz, DMSO-d6): δ 168.75, 162.67, 160.27 (CF, 1JCF = 248.00 Hz), 157.81, 149.10, 147.10, 136.74, 136.04, 131.43, 129.49, 129.41, 128.71, 127.84, 126.71, 125.10, 123.45, 122.98, 115.30, 115.10, 42.20, 35.21 ppm; Anal. Calcd. for C25H19FN4OS: C, 67.86; H, 4.33; N, 12.66; Found: C, 68.04; H, 4.52; N, 12.81.
α-Glucosidase inhibitory assay
The α-glucosidase inhibitory activities of all synthesized derivatives were assayed according to the previously reported procedure9,36.
Enzyme kinetic studies
The mode of inhibition of the most active compound (9c), identified with the lowest IC50, was investigated against α-glucosidase at different concentrations of p-nitrophenyl α-d-glucopyranoside (4–16 mM) as substrate in the absence and presence of 9c at different concentrations (0, 0.8, 1.6, and 3.2 µM). A Lineweaver–Burk plot was generated to identify the type of inhibition and the Michaelis–Menten constant (Km) value was determined from the plot between reciprocal of the substrate concentration (1/[S]) and reciprocal of enzyme rate (1/V) over various inhibitor concentrations. The experimental inhibitor constant (Ki) value was constructed by secondary plots of the inhibitor concentration [I] versus Km6.
Molecular docking
To perform the molecular modeling investigations, the Maestro Molecular Modeling platform (version 10.5) by Schrödinger, LLC was used. The X-ray crystal structure of the receptor was downloaded from the PDB database (PDB ID: 5NN8). The protein is then prepared using a protein preparation wizard. At this point, all water molecules and co-crystallized ligands were removed, the missing side chains and loops were filled using the prime tool, and PROPKA assigned H-bonds at pH 7.4. To prepare the ligands, the 2D structures of the ligands were drawn in ChemDraw (ver. 16) and converted into SDF files, which were used further by the ligprep module. Ligands were prepared by OPLS_2005 force field using EPIK at a target pH of 7.0 ± 2. The grid box was generated for each binding site using entries with a box size of 25 Å, all derivatives were docked on binding sites using induced-fit docking, reporting 10 poses per ligand to form the final complex9,37.
Data availability
All data generated or analyzed during this study are included in this published article and its Supplementary Information files.
References
Roglic, G. WHO Global report on diabetes: A summary. Int. J. Noncommun. Dis. 1(1), 3 (2016).
Kondo, H., Taguchi, M., Inoue, Y., Sakamoto, F. & Tsukamoto, G. Synthesis and antibacterial activity of thiazolo-, oxazolo-, and imidazolo [3, 2-a][1, 8] naphthyridinecarboxylic acids. J. Med. Chem. 33(7), 2012–2015 (1990).
Adeghate, E., Schattner, P. & Dunn, E. An update on the etiology and epidemiology of diabetes mellitus. Ann. N. Y. Acad. Sci. 1084(1), 1–29 (2006).
Mora-Fernández, C. et al. Diabetic kidney disease: From physiology to therapeutics. J. Physiol. 592(18), 3997–4012 (2014).
Guariguata, L. et al. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res. Clin. Pract. 103(2), 137–149 (2014).
Karami, M. et al. One-pot multi-component synthesis of novel chromeno[4,3-b]pyrrol-3-yl derivatives as alpha-glucosidase inhibitors. Mol. Divers. https://doi.org/10.1007/s11030-021-10337-w (2021).
Sohrabi, M. et al. A review on α-glucosidase inhibitory activity of first row transition metal complexes: A futuristic strategy for treatment of type 2 diabetes. RSC Adv. 12(19), 12011–12052 (2022).
Seltzer, H. S., Allen, E. W., Herron, A. L. & Brennan, M. T. Insulin secretion in response to glycemic stimulus: Relation of delayed initial release to carbohydrate intolerance in mild diabetes mellitus. J. Clin. Investig. 46(3), 323–335 (1967).
Moghaddam, F. M. et al. Synthesis and characterization of 1-amidino-O-alkylureas metal complexes as α-glucosidase Inhibitors: Structure-activity relationship, molecular docking, and kinetic studies. J. Mol. Struct. 1250, 131726 (2022).
Iraji, A. et al. Cyanoacetohydrazide linked to 1, 2, 3-triazole derivatives: A new class of α-glucosidase inhibitors. Sci. Rep. 12(1), 1–15 (2022).
Tundis, R., Loizzo, M. & Menichini, F. Natural products as α-amylase and α-glucosidase inhibitors and their hypoglycaemic potential in the treatment of diabetes: An update. Mini Rev. Med. Chem. 10(4), 315–331 (2010).
Pedrood, K. et al. Design, synthesis, characterization, enzymatic inhibition evaluations, and docking study of novel quinazolinone derivatives. Int. J. Biol. Macromol. 170, 1–12 (2021).
Gulçin, İ et al. Antidiabetic and antiparasitic potentials: Inhibition effects of some natural antioxidant compounds on α-glycosidase, α-amylase and human glutathione S-transferase enzymes. Int. J. Biol. Macromol. 119, 741–746 (2018).
Kumar, S., Narwal, S., Kumar, V. & Prakash, O. α-Glucosidase inhibitors from plants: A natural approach to treat diabetes. Pharmacogn. Rev. 5(9), 19 (2011).
Abbas, G., Al Harrasi, A., Hussain, H., Hamaed, A. & Supuran, C. T. The management of diabetes mellitus-imperative role of natural products against dipeptidyl peptidase-4, α-glucosidase and sodium-dependent glucose co-transporter 2 (SGLT2). Bioorg. Chem. 86, 305–315 (2019).
Wali, S. et al. Synthesis of new clioquinol derivatives as potent α-glucosidase inhibitors; molecular docking, kinetic and structure-activity relationship studies. Bioorg. Chem. 119, 105506 (2021).
Dhameja, M. & Gupta, P. Synthetic heterocyclic candidates as promising α-glucosidase inhibitors: An overview. Eur. J. Med. Chem. 176, 343–377 (2019).
Nasli Esfahani, A. et al. Design and synthesis of phenoxymethybenzoimidazole incorporating different aryl thiazole-triazole acetamide derivatives as α-glycosidase inhibitors. Mol. Divers. 26, 1995 (2021).
Zarenezhad, E., Montazer, M. N., Tabatabaee, M., Irajie, C. & Iraji, A. New solid phase methodology for the synthesis of biscoumarin derivatives: Experimental and in silico approaches. BMC Chem. 16(1), 53 (2022).
Li, Y. et al. Discovery of new 2-phenyl-1H-benzo[d]imidazole core-based potent α-glucosidase inhibitors: Synthesis, kinetic study, molecular docking, and in vivo anti-hyperglycemic evaluation. Bioorg. Chem. 117, 105423 (2021).
Abbasi, I. et al. Isatin-hydrazide conjugates as potent α-amylase and α-glucosidase inhibitors: Synthesis, structure and invitro evaluations. Bioorg. Chem. 116, 105385 (2021).
Xie, H.-X. et al. Novel tetrahydrobenzo[b]thiophen-2-yl)urea derivatives as novel α-glucosidase inhibitors: Synthesis, kinetics study, molecular docking, and in vivo anti-hyperglycemic evaluation. Bioorg. Chem. 115, 105236 (2021).
Zarenezhad, E., Farjam, M. & Iraji, A. Synthesis and biological activity of pyrimidines-containing hybrids: Focusing on pharmacological application. J. Mol. Struct. 1230, 129833 (2021).
Santos, C. M. M., Freitas, M. & Fernandes, E. A comprehensive review on xanthone derivatives as α-glucosidase inhibitors. Eur. J. Med. Chem. 157, 1460–1479 (2018).
Sari, S., Barut, B., Özel, A. & Saraç, S. Discovery of potent α-glucosidase inhibitors through structure-based virtual screening of an in-house azole collection. Chem. Biol. Drug Des. 97, 701 (2020).
Lee, H.-W., Yang, J.-Y. & Lee, H.-S. Quinoline-2-carboxylic acid isolated from Ephedra pachyclada and its structural derivatives show inhibitory effects against α-glucosidase and α-amylase. J. Korean Soc. Appl. Biol. Chem. 57(4), 441–444 (2014).
Taha, M. et al. Novel quinoline derivatives as potent in vitro α-glucosidase inhibitors: In silico studies and SAR predictions. MedChemComm. 6(10), 1826–1836 (2015).
Mohammadi-Khanaposhtani, M. et al. Design, synthesis, docking study, α-glucosidase inhibition, and cytotoxic activities of acridine linked to thioacetamides as novel agents in treatment of type 2 diabetes. Bioorg. Chem. 80, 288–295 (2018).
Ansari, S. et al. Design, synthesis, and α-glucosidase-inhibitory activity of phenoxy-biscoumarin-N-phenylacetamide hybrids. Archiv. der Pharm. 354, e2100179 (2021).
Moghimi, S. et al. Design and synthesis of novel pyridazine N-aryl acetamides: In-vitro evaluation of α-glucosidase inhibition, docking, and kinetic studies. Bioorg. Chem. 102, 104071 (2020).
Suman, P., Patel, B. P., Kasibotla, A. V., Solano, L. N. & Jonnalagadda, S. C. Synthesis and evaluation of functionalized aminobenzoboroxoles as potential anti-cancer agents. J. Organomet. Chem. 798, 125–131 (2015).
Shashikumar, N. D., Krishnamurthy, G., Bhojyanaik, H. S., Lokesh, M. R. & Jithendrakumara, K. S. Synthesis of new biphenyl-substituted quinoline derivatives, preliminary screening and docking studies. J. Chem. Sci. 126(1), 205–212 (2014).
Celik, I., Ayhan-Kılcıgil, G., Karayel, A., Guven, B. & Onay-Besikci, A. Synthesis, molecular docking, in silico ADME, and EGFR kinase inhibitor activity studies of some new benzimidazole derivatives bearing thiosemicarbazide, triazole, and thiadiazole. J. Heterocycl. Chem. 59(2), 371–387 (2022).
Flipo, M. et al. Discovery of novel N-phenylphenoxyacetamide derivatives as EthR inhibitors and ethionamide boosters by combining high-throughput screening and synthesis. J. Med. Chem. 55(14), 6391–6402 (2012).
Kiran, B. M., Nandeshwarappa, B. P., Vaidya, V. P. & Mahadevan, K. M. Chemistry of substituted quinolines: Thieno [2, 3-b] and thiopyrano [2, 3-b] quinolines. Phosphorus Sulfur Silicon Relat. Elem. 182(5), 969–980 (2007).
Shareghi-Boroujeni, D. et al. Synthesis, in vitro evaluation, and molecular docking studies of novel hydrazineylideneindolinone linked to phenoxymethyl-1, 2, 3-triazole derivatives as potential α-glucosidase inhibitors. Bioorg. Chem. 111, 104869 (2021).
Iraji, A. et al. Cyanoacetohydrazide linked to 1, 2, 3-triazole derivatives: A new class of α-glucosidase inhibitors. Sci. Rep. 12(1), 8647 (2022).
Funding
The authors wish to thank the financial support of National Institute for Medical Research Development (NIMAD) (Grant Number: 4000379) and the Research Council of Tehran University of Medical Sciences and Health Services (Grant Number: 1400-2-411-53423), Tehran, Iran.
Author information
Authors and Affiliations
Contributions
M.N. synthesized compounds. A.D. synthesized compounds. A.I. performed docking study and contributed to the preparation of the manuscript. M.A. performed chemical analysis. N.D. and M.A. contributed to the design and characterization of compounds. M.K. performed the biological assay. M.M.K. synthesized compounds. S.M. contributed to the design and characterization of compounds. M.D. supervised the biological tests. M.A.F. supervised the biological tests. B.L. contributed to the design and characterization of compounds. M.A. supervised all phases of the study. M.M. supervised all phases of the study. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Noori, M., Davoodi, A., Iraji, A. et al. Design, synthesis, and in silico studies of quinoline-based-benzo[d]imidazole bearing different acetamide derivatives as potent α-glucosidase inhibitors. Sci Rep 12, 14019 (2022). https://doi.org/10.1038/s41598-022-18455-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-18455-7
This article is cited by
-
Coumarin linked to 2-phenylbenzimidazole derivatives as potent α-glucosidase inhibitors
Scientific Reports (2024)
-
Aryl-quinoline-4-carbonyl hydrazone bearing different 2-methoxyphenoxyacetamides as potent α-glucosidase inhibitors; molecular dynamics, kinetic and structure–activity relationship studies
Scientific Reports (2024)
-
Synthesis, in vitro potency of inhibition, enzyme kinetics and in silico studies of quinoline-based α-glucosidase inhibitors
Scientific Reports (2024)
-
Synthesis, in vitro α-glucosidase inhibitory activities, and molecular dynamic simulations of novel 4-hydroxyquinolinone-hydrazones as potential antidiabetic agents
Scientific Reports (2023)
-
Synthesis, in vitro inhibitor screening, structure–activity relationship, and molecular dynamic simulation studies of novel thioquinoline derivatives as potent α-glucosidase inhibitors
Scientific Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
