
Abstract
α-Glucosidase inhibition is an approved treatment for type 2 diabetes mellitus (T2DM). In an attempt to develop novel anti-α-glucosidase agents, two series of substituted imidazo[1,2-c]quinazolines, namely 6a–c and 11a–o, were synthesized using a simple, straightforward synthetic routes. These compounds were thoroughly characterized by IR, 1H and 13C NMR spectroscopy, as well as mass spectrometry and elemental analysis. Subsequently, the inhibitory activities of these compounds were evaluated against Saccharomyces cerevisiae α-glucosidase. In present study, acarbose was utilized as a positive control. These imidazoquinazolines exhibited excellent to great inhibitory potencies with IC50 values ranging from 12.44 ± 0.38 μM to 308.33 ± 0.06 μM, which were several times more potent than standard drug with IC50 value of 750.0 ± 1.5 μM. Representatively, compound 11j showed remarkable anti-α-glucosidase potency with IC50 = 12.44 ± 0.38 μM, which was 60.3 times more potent than positive control acarbose. To explore the potential inhibition mechanism, further evaluations including kinetic analysis, circular dichroism, fluorescence spectroscopy, and thermodynamic profile were carried out for the most potent compound 11j. Moreover, molecular docking studies and in silico ADME prediction for all imidazoquinazolines 6a–c and 11a–o were performed to reveal their important binding interactions, as well as their physicochemical and drug-likeness properties, respectively.
Similar content being viewed by others

Introduction
Diabetes mellitus (DM), mainly characterized as inadequate control of blood levels of glucose, has emerged as a remarkable health challenges over recent decades. Statics reveals that the rate of diabetes occurrence around the world was 536.6 million people in 2021, and this figure is predicted to reach 783.2 million people by 20451. Diabetes is categorized into several subtypes with various etiologies, presentations, and treatments. Moreover, this chronic disease caused various health problems including cardiovascular diseases, hypertension, obesity, kidney diseases, and blindness. Consequently, a huge financial burden on the global health system has been imposed by this illness2. Considering the alarming rate of diabetes as well as complicated, severe issues associated with it, extensive efforts have already been made to manage this disease.
Diabetes mellitus is classified into several groups, including type 1, type 2, maturity-onset diabetes of the young (MODY), gestational diabetes, neonatal diabetes, and steroid-induced diabetes. Notably, the main subtypes are type 1 diabetes mellitus (T1DM) and type 2 diabetes mellitus (T2DM)3. These two subtypes have different pathophysiology, presentation, and management strategies. T1DM is characterized by defective insulin secretion, while T2DM involves an impaired response to insulin. However, they have a potential for hyperglycemia in common. Genetic background for both types is critical as a risk factor, but T1DM tends to occur in children, whereas T2DM is prevalent among middle-aged and older adults due to prolonged hyperglycemia resulting from their poor lifestyle and dietary choices. Statistics indicate that 1 in 11 adults suffers from diabetes mellitus, and 90% of patients have T2DM. Given this high prevalence, extensive research is currently being conducted to effectively manage this particular subtype4,5.
Since T2DM is mainly identified by a high level of glucose in blood (hyperglycemia), one of the pivotal strategies to control this disease is to interfere with the digestion of dietary carbohydrates. α-Glucosidase is an enzyme located in the brush border of the small intestine, and its role is the hydrolysis of this long chain sugar to monosaccharide units, which are subsequently released to the bloodstream. Therefore, one approved approach for the treatment of T2DM and its resultant postprandial hyperglycemia is the inhibition of α-glucosidase to slow down glucose absorption, thereby reducing postprandial glucose blood concentrations. Currently, there are three commercial drugs to control T2DM through the α-glucosidase inhibitory mechanism: acarbose, voglibose, and miglitol, among which acarbose is the most widely used and studied drug6,7.
A complex oligosaccharide, acarbose, competitively and reversibly binds to the oligosaccharide site of α-glucosidase in small intestine in a dose-dependent manner. This binding prevents the breakdown of disaccharide and oligosaccharide substrates into absorbable monosaccharides. Despite the efficacy of acarbose as an α-glucosidase inhibitor, it causes several undesirable side effects for patients, the most noticeable of which are diarrhea, abdominal discomfort, as well as bloating and flatulence8. Therefore, a great deal of effort over recent decade has been made to discover and develop more potent α-glucosidase inhibitors having improved safety and pharmacological profiles to replace acarbose. To this aim, numerous heterocyclic α-glucosidase inhibitors have been reported. Among them, two valuable nitrogen-containing pharmacophores, various functionalized quinazolines9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24 and imidazoles25,26,27,28,29,30,31,32,33,34 have exhibited great inhibitory potencies compared to acarbose as the standard drug. Figure 1 summarizes some structures and IC50 values of the most active compounds from these studies. Therefore, considering the proved potency of these pharmacophores, providing novel imidazole-quinazoline analog with the hope of finding further potent α-glucosidase inhibitors could be an interesting research topic in medicinal chemistry.

α-Glucosidase inhibitors bearing substituted quinazolines A–F and substituted imidazoles G–L. The IC50 values are written in black for inhibitors and red for acarbose.
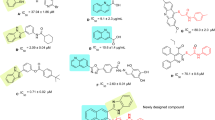
One well-stablished strategy for designing further novel and potent compounds is the hybridization of two scaffolds which have demonstrated promising inhibitory potencies. Various compounds bearing substituted quinazolines and imidazoles as potential α-glucosidase inhibitors have been already reported separately. However, there is limited investigation into the inhibitory activity of compounds containing both of these heterocycles. For example, Fig. 2 shows compounds M and N as the most potent derivatives from these studies, which were synthesized and evaluated against α-glucosidase, possessing noticeable inhibition in comparison with acarbose. Another strategy involves fusing two heterocycles to provide imidazoquinazoline backbone. Among several isomers of this skeleton, imidazo[1,2-c]quinazoline was selected for the present study. This scaffold has displayed anticancer35, antitubercular36, antifungal37, antimicrobial and antioxidant38,39,40 activities; however, its α-glucosidase inhibitory potency has yet to be explored. Therefore, this biological evaluation could be a fascinating study in medicinal chemistry (Fig. 2).

Design strategy toward two series substituted imidazo[1,2-c]quinazolines. The IC50 values are written in black for inhibitors and red for acarbose.
As part of our ongoing research to find potential α-glucosidase inhibitors41,42,43,44,45,46,47,48, imidazo[1,2-c]quinazoline was introduced as a novel inhibitor backbone in present study. To this aim, two facile, efficient synthetic protocols were employed to obtain substituted benzo[4,5]imidazo[1,2-c]quinazoline 6 and poly-substituted imidazo[1,2-c]quinazolines 11 and evaluate their in vitro potencies in comparison with acarbose as the standard drug. These compounds exhibited excellent to remarkable inhibitory activity. Subsequently, further assessments, including kinetic study, circular dichroism measurement, fluorescence quenching measurements, and thermodynamic analysis of binding to α‑glucosidase were carried out for the most active compound 11j. Finally, computational investigations, including molecular docking and in silico ADME studies were performed for imidazoquinazolines 6 and 11 to investigate the mode of their interactions with the active site of α-glucosidase and predict the compounds’ druglike properties, respectively.
Results and discussion
Chemistry
In present study, simple and efficient synthetic routes toward two series of substituted imidazoquinazolines 6 and 11 were performed. As illustrated in Scheme 1, the first step to obtain benzo[4,5]imidazo[1,2-c]quinazoline 6 was a cyclization reaction between 2-nitrobenzaldehyde 1 and benzene-1,2-diamine 2. This reaction occurred in the presence of catalytic amount of glacial acetic acid in ethanol under the reflux conditions to afford 2-(2-Nitrophenyl)-1H-benzo[d]imidazole 3. On the other hand, the protocol to obtain highly-substituted imidazo[1,2-c]quinazolines 11 was initiated through a cyclization reaction between 2-nitrobenzaldehyde 1, benzil 7, and ammonium acetate 8 under the reflux conditions in glacial acetic acid to produce 2-(2-Nitrophenyl)-4,5-diphenyl-1H-imidazole 9. Subsequent steps in the synthesis were shared for both scaffolds.

Synthesis of substituted imidazo[1,2-c]quinazolines 6a–c and 11a–o.
The nitro functionality in compounds 3 and 9 went through the reduction reaction using stannous chloride dihydrate (SnCl2∙2H2O) and hydrochloric acid in methanol to afford the amine moiety 4 and 10. Finally, a condensation-cyclization reaction between these adducts and substituted benzaldehydes 5 in glacial acetic acid at 80 °C occurred to obtain corresponding substituted imidazo[1,2-c]quinazolines 6a–c and 11a–o in great to excellent yields. The structures of the isolated compounds 3, 4, 6a–c, 9, 10, and 11a–o were deduced on the basis of their IR, 1H and 13C NMR spectroscopy, as well as mass spectrometry and elemental analysis. Partial assignments of these resonances are given in the Experimental Part.
In vitro α-glucosidase inhibitory activity
The target substituted imidazo[1,2-c]quinazolines 6a–c and 11a–o were evaluated for their in vitro Saccharomyces cerevisiae α-glucosidase inhibitory activities to investigate the role of substituents on the imidazole moiety and the phenyl ring originated from benzaldehyde moieties. In this study, acarbose was utilized as a positive control. The obtained results were summarized in Tables 1 and 2. Our studies were initiated through the synthesis of derivatives 6a–c as well as 11a, 11b, and 11g to evaluate their potencies and reveal the role of substituents on the imidazole ring. Compounds from second series exhibited superior inhibitory activities in comparison with their analogues from the first series; therefore, we followed our studies by the synthesis and investigation of other 5-(substituted aryl)-2,3-diphenylimidazo[1,2-c]quinazolines 11c–f, h–o.
As illustrated in Table 2, imidazoquinazolines 11 demonstrated good to excellent α-glucosidase inhibitory potencies, ranging from 12.44 ± 0.38 μM to 273.28 ± 0.09 μM, in comparison with acarbose (IC50 = 750.0 ± 1.5 μM). The correlations between their structures and observed activities are explained comprehensively below:
To initiate, an unsubstituted phenyl ring showed moderate inhibitory potency (compound 11a, IC50 = 209.15 ± 0.04 μM). Introducing a chlorine atom as an electron-withdrawing group at any position (compounds 11b, 11c, and 11d) caused a detrimental effect on the α-glucosidase inhibitory potencies. However, replacing this atom with electron-donating groups including methyl (Me), N,N-dimethyl (N(Me)2), and methoxy (OMe) at C-4 position improved the inhibitory activity noticeably (compounds 11e, 11f, and 11g), among which 11g exhibited better results (IC50 = 82.64 ± 0.03 μM). This led us to investigate the role of this substituent at other position of phenyl ring or the presence of additional OMe group. With this in mind, other compounds were synthesized for further evaluation. Moving the OMe from C-4 to C-3 and 2 enhanced the inhibitory activity against α-glucosidase (compound 11h with IC50 = 47.92 ± 0.18 μM and 11i with IC50 = 24.25 ± 0.13 μM).
Considering the constructive role of OMe on the phenyl ring, particularly at C-2 position, additional OMe group was introduced. This strategy led to synthesis of two valuable compounds, namely 11j and 11k, which emerged as the most potent derivatives among all the synthesized imidazoquinazolines. Compound 11j bearing two OMe groups at C-2 and 3 exhibited remarkable inhibitory potency against α-glucosidase IC50 = 12.44 ± 0.38 μM), which was 60.3 times more potent than standard inhibitor (IC50 = 750.0 ± 1.5 μM). Furthermore, compound 11k with two OMe groups at C-2 and 4 ranked as the second most potent compound in this series (IC50 = 14.32 ± 0.05 μM). Additionally, the presence of this group at C-3 and 4 showed excellent inhibitory activity (compound 11m with IC50 = 21.57 ± 0.32 μM).
Among the compounds bearing two OMe groups, 11l showed comparatively less potency, which might be related to the deteriorative effect of C-5 position. This inference could be confirmed by comparing the results of compounds 11n (IC50 = 46.73 ± 0.07 μM) and 11o (IC50 = 154.88 ± 0.36 μM) with 11m (IC50 = 21.57 ± 0.32 μM). It revealed that the presence of any substituent at C-5 position, whether electron-donating group like OMe or electron-withdrawing group like bromine, results in a moderate decrease in α-glucosidase inhibitory activity. Moreover, bromine as an electron-withdrawing group caused a detrimental effect on the inhibitory activity, which is in great agreement with earlier results in compounds 11b–d.
A statistical analysis using the T-test was performed for both series 6a–c and 11a–o. All compounds indicated a significant statistical difference (p < 0.001) between the IC50 values of each compound in comparison with acarbose as standard drug.
Comparing the IC50 values of benzo[4,5]imidazo[1,2-c]quinazolines 6 with their corresponding analogs from 5-(substituted aryl)-2,3-diphenylimidazo[1,2-c]quinazolines 11 revealed the notable influence of substituents on the imidazole moiety on the α-glucosidase inhibitory activity, as the presence of two phenyl rings on this core improved the potency. Moreover, the aforementioned SAR analysis showed that electron-donating group, particularly OMe, improved the α-glucosidase inhibitory potency, while electron-withdrawing group like chlorine or bromine caused a noticeable detrimental inhibition effect. In conclusion, imidazoquinazolines bearing two OMe groups, particularly when positioned at C-2 and 3 as seen in compound 11j, exhibited substantial inhibitory activities. Finally, compound 11j emerged as the most potent derivative having remarkable activity against α-glucosidase. Consequently, it was chosen for further evaluations.
Enzyme kinetic study
The enzyme kinetic study was performed to reveal the inhibition mode of imidazoquinazoline 11j. There are two enzyme kinetic constants: Michaelis constant (Km) and maximum velocity of the reaction (Vmax) which are calculated using initial velocity measurements at different inhibitors concentrations (for example, 0, 3.1, 6.2, and 12.4 μM in present study). As illustrated in Fig. 3A, the Lineweaver–Burk plot exhibited the Km value increased with increasing concentration of compound 11j, while Vmax did not change. The results indicated that this imidazoquinazoline bonded to the active site on the enzyme and competes with the substrate for binding to this region, indicating a competitive type of inhibition. Moreover, the plot of the Km versus different concentrations of inhibitor gave an estimate of the inhibition constant as Ki value of 11.0 µM (Fig. 3B).

Kinetics of α-glucosidase inhibition by sample 11j: (A) the Lineweaver–Burk plot in the absence and presence of different concentrations of sample 11j; (B) the secondary plot between Km and various concentrations of sample 11j.
Circular dichroism spectroscopy
The difference between the absorption of right and left circularly polarized light is measured in circular dichroism spectroscopy (CD) in order to reveal the chiral environment around amino acid residues. The CD spectrum in the far ultraviolet region ranged from 190 to 240 nm is mainly used to provide valuable information about the arrangement of protein bonds and secondary structure of the proteins in dilute solutions. There are several principal conformations like α-helix, extended β structure (or β-sheet), β-turn, and random coil (which are unordered structures). They are characterized as follow: α-helix structures by negative CD bands at 222 and 208 nm and a positive CD band at approximately 190 nm; β-sheet structures by a negative CD band in the region of 210–220 nm; β-turn structures by a negative CD band between 180 and 190 nm; and the spectra of random coil by a characteristic negative CD band in region of 200 nm49,50.
To study the impact of imidazo[1,2-c]quinazoline 11j on the secondary structure of α-glucosidase (Fig. 4b), the CD spectra (180–250 nm) was measured and analyzed using the CDNN software to be compared with the native enzyme (Fig. 4a). The percent of observed conformations are summarized in Table 3. As it can be seen, our inhibitor increased noticeably the figures for α-helix and β-turn; while random coils removed; therefore, this imidazo[1,2-c]quinazoline 11j can determine the conformation of the enzyme and fix chiral side chains in orientations. Moreover, this compound can change the secondary structure of α-glucosidase, resulting to inhibit its performance.

Circular dichroism (CD) spectra of the α-glucosidase: (A) in the absence of inhibitor (control); (B) in the presence of imidazoquinazoline 11j.
Fluorescence spectroscopy measurements
Fluorescence spectroscopy assay is a frequently used method to investigate the potential interactions between inhibitors and enzymes under physiological conditions, because binding of inhibitors changes the fluorescence characteristics and tertiary structure of the protein. Moreover, it can predict the tertiary structure of the enzyme and provide more accurate information about the binding constant, number of binding sites, and thermodynamic parameters of the studied interactions.
In present study, fluorescence spectroscopy measurement was performed between imidazo[1,2-c]quinazoline 11j and the active site of enzyme using a Synergy HTX multi-mode reader (Biotek Instruments, Winooski, VT, USA) equipped with a quartz cuvette of 10 mm. The excitation wavelength was 280 nm, and the emission spectra were reported at five different temperatures in the range from 300 to 450 nm with 10 accumulations for each collection point. The emission spectrum was adjusted for the background fluorescence from the buffer solution and for the inner filter effect promoted by the inhibitors (Fig. 5).

Fluorescence spectra of α-glucosidase: (A) in the absence of compound 11j at 20–60 ℃, (B) in the presence of compound 11j at inhibitory concentration (12.4 µM) at 20–60 ℃.
The results obtained from this evaluation revealed that the fluorescence intensity of α-glucosidase increased to 340 nm and subsequently decreased (the λmax was 340 nm). On the other hand, there are three amino acids—tryptophan, tyrosine, and phenylalanine—that play a role in the enzyme's intrinsic fluorescence property, referred to as fluorophores. Among them, the maximum intensity of tryptophan at 280 nm is about 340 nm; therefore, imidazo[1,2-c]quinazoline 11j must be located proximity to the tryptophan residues within the α-glucosidase binding site when this inhibitor bound to the enzyme and changed the tertiary structure of the enzyme.
There are two types of fluorescence quenching: dynamic and static. Dynamic quenching arises from the collisional encounter between the fluorophore (tryptophan residues) and the quencher (inhibitor). Conversely, static quenching results from the formation a ground‐state complex between fluorophore and quencher. Results revealed that the combination of fluorophores (tryptophan residues) and quencher (imidazo[1,2-c]quinazoline 11j) exhibited a static quenching mechanism. Therefore, the biding parameters can be determined as follow:
The reaction is determined as P + D → DnP in which P is the protein, D is the drug molecule (inhibitor), and DnP is the new complex molecule. The binding constant for this complex for this complex, denoted as KA, is calculated using Eq. (1). In the static quenching mechanism, the number of binding sites, which is named “n”, remains constant. Since the number of the binding site of protein and drug is n and 1, respectively, the equivalent concentration of the complex DnP is n[DnP]. Moreover, the equivalent concentration of the protein is n[P], and the equivalent concentration of the drug is [D]:
The total concentration of protein is [Pt], and the total concentration of the drug is [Dt]; therefore, [Pt] = [Pf] + [DnP] and [Df] = [Dt]−n[DnP]. Since protein (P) is the only fluorescence in present study, thus:
F and F0 are the fluorescence intensity of protein in the presence and absence of inhibitor, respectively. Therefore, the relationship between the fluorescence intensity and the total concentration of the drug could be deduced:
As the total concentration of protein was kept at a constant value (at 46 nM), while the total concentration of the drug was changed. Using the Eq. (3), a plot of F0/F Vs. [Dt] F0/(F0−F) was obtained, as depicted in Fig. 6. KA, n, and r at 20 ℃ can also be calculated, as listed in Table 4:

The plots F0/F Vs. function of [Dt] F0/(F0−F) at 20 ℃ for imidazoquinazoline 11j.
Thermodynamic analysis of binding of imidazoquinazoline 11j to α‑glucosidase
This fluorescent intensity data was plotted as a function of temperature and binding constants; therefore, thermodynamic profile including ΔG (free energy change), ΔH (enthalpy change), and ΔS (entropy change) could be computed to determine the type of non-covalent forces between imidazo[1,2-c]quinazoline 11j and binding site of α‑glucosidase. These forces between the protein and inhibitor can be categorized into four groups: hydrogen bond, van der Waals forces, electrostatic attraction, and hydrophobic interactions. To identify the type of interactions in present study, thermodynamic parameters must be calculated using following equations:
Already, there was some information including binding constants (KA2 = 36.6 × 104 and KA1 = 40.5 × 104) as well as initial and final temperatures (T1 = 20 ℃ (293 K) and T2 = 60 ℃ (333 K)). Therefore, using Eq. (4), ΔH is obtained − 2.17 (kJ mol–1) and subsequently using Eq. (5), ΔG and ΔS values are calculated − 20.6 (kJ mol–1) and 62.9 (J mol–1 K–1), respectively.
Considering the sign of these thermodynamic parameters, the type of non-covalent force could be determined as follow: (1) ΔH > 0, ΔS > 0, hydrophobic interactions; (2) ΔH < 0, ΔS > 0, van der Waals forces; (3) ΔH < 0, ΔS < 0, hydrogen bond and van der Waals interactions; and (4) ΔH < 0, ΔS > 0, electrostatic interactions. Therefore, the obtained results indicated that the acting force between imidazoquinazoline 11j and α‑glucosidase was mainly determined as electrostatic forces51,52.
Molecular docking studies
Molecular docking study was conducted using AutoDock4 and Auto Dock Tools (version 1.5.6) to explore the interaction patterns of substituted benzo[4,5]imidazo[1,2-c]quinazoline 6a–c and imidazo[1,2-c]quinazolines 11a–o within the active site of human acid-α-glucosidase (PDB ID: 5NN8). This PBD ID was also used in previous studies53. Observed interactions are listed in Table 5. The binding energy of imidazo[1,2-c]quinazolines 11a–o were found to be in range of − 7.62 to − 8.59 kcal mol−1, which is noticeably better than that of acarbose (− 3.79 kcal mol−1). The docking protocol validation involved a redocking study using the crystallized ligand with PDB ID of 5NN8. This process resulted to a low RMSD value of 1.57, confirming the reliability of our docking studies.
Additionally, the similar computational process was conducted for benzo[4,5]imidazo[1,2-c]quinazolines 6a–c, showing the binding energy scores between − 7.30 to − 7.58 kcal mol−1. These values were moderately lower than the binding energies in second series. These figures were found to be − 8.07, − 7.79, and − 8.05 kcal mol−1 for compounds 11a, 11b, and 11g, respectively; however, they were − 7.30, − 7.58, and − 7.32 kcal mol−1 for compounds 6a, 6b, and 6c, respectively. It might be related to its fewer interactions. This issue is illustrated by comparing compounds 6a with 11a as shown in Fig. 7. It must be noted no hydrogen bond formation was observed, and hydrophobic interactions are shown by dashed lines.

Interactions and structures of (a) compound 6a and (b) compound 11a in the binding pocket of human acid-α-glucosidase visualized using PyMOL 2.5.2 and PLIP online service.
As previously described in SAR analysis, derivatives bearing electron-donating groups, particularly OMe (compounds 11e–o), exhibited the excellent to great inhibitory activities. Herein, molecular docking studies (as presented in Table 6) revealed that these compounds 11e-o, except 11i, created a hydrogen bond with OMe moiety or imidazoquinazoline backbone, which might be responsible for their significant inhibitory potencies.
Imidazo[1,2-c]quinazoline 11j, the most potent compound in present study with the best IC50 value (12.44 ± 0.38 μM), exhibited a noticeable binding energy of − 8.50 kcal mol−1. This affinity could be attributed to the formation of a hydrogen bond between the OMe moiety at C-3 position and ALA-284 residue within the receptor. This similar hydrogen bond was observed in the complex of acarbose and receptor in the crystal structure. Additionally, compound 11j formed several hydrophobic interactions with different residues including TRP-481, TRP-376, LEU-678, PHE-649, and ILE-441 within the active site of α-glucosidase (Fig. 8). Similar hydrophobic interactions between acarbose and some residues in the binding site of the receptor (like TRP-481 and TRP-376) were observed in the redocking results. The presence of these interactions with tryptophan residues is consistent with the results of fluorescence spectroscopy measurements. Overall, similar interactions between both compound 11j and acarbose with active site of α-glucosidase, as well as their superimposition as depicted in Fig. 9, can confirm the accuracy of the docking procedure and the validity of results.

The interactions and structure of compound 11j in the binding pocket of human acid-α-glucosidase (it must be noted that the hydrogen bond and hydrophobic interactions are displayed in blue color and dashed lines, respectively).

Superimposition of acarbose and compound 11j in the binding pocket of human acid-α-glucosidase. Acarbose is colored in blue, and compound 11j is colored in red.
In silico ADME
The ADME parameters of benzo[4,5]imidazo[1,2-c]quinazolines 6a–c and imidazo[1,2-c]quinazolines 11a–o were calculated using SwissADME online server54. The results are summarized in Table 6. All compounds possessed favorable drug-likeliness, and they were mostly consistent with Lipinski’s Rule of 5. FDA-approved α-glucosidase inhibitors, particularly acarbose, possess low oral bioavailability and act as a competitive, reversible inhibitor of membrane-bound intestinal enzyme. Considering the presence of α-glucosidase in the lumen and its mechanism, it was assumed that low Human intestinal absorption (HIA) of imidazoquinazolines 11a–o would be a promising factor to observe minimum systemic adverse effects, while being sufficiently effective in the lumen environment55,56. However, second series, including compounds 6a–c were predicted to have high HIA, indicating their potentially lower pharmacological activities.
Since acarbose acts in the gastrointestinal tract, its low systemic absorption (below 2% of the administered dose) is crucial for optimal therapeutic efficacy57. In Fig. 10, passive gastro-intestinal absorption (HIA) and blood–brain barrier (BBB) permeation were predicted by the BOILED-Egg model58. In this figure, benzo[4,5]imidazo[1,2-c]quinazolines 6a–c were shown by blue dots. Imidazo[1,2-c]quinazolines 11a–o were shown by red dots, and some of them were overlapped by each other. Compounds which are located in the yellow region are predicted to have BBB permeability. Benzo[4,5]imidazo[1,2-c]quinazolines 6a–c were predicted to be BBB permeable, which is a negative feature for using these compounds as α‑glucosidase inhibitors. However, none of imidazo[1,2-c]quinazolines 11a–o were anticipated to be BBB-permeable, which is ideal for α‑glucosidase inhibitor safety profile.

Compounds 6a–c and 11a–o were examined by the boiled-egg method available on SWISS ADME.
Moreover, compounds which are located in the white area are predicted to have good absorption. As previously discussed, considering the enzyme’s site of action (lumen environment), high bioavailability may cause side effects without any improvement on the efficacy. Overall, benzo[4,5]imidazo[1,2-c]quinazolines 6a–c possessed higher bioavailability, which is not favorable for α‑glucosidase inhibitory activity in the gastrointestinal tract, while imidazo[1,2-c]quinazolines 11a–o were able to act locally in the lumen without getting into the bloodstream. Therefore, compounds 11a–o with low HIA and no BBB-permeation could be potential candidates for further studies.
Conclusion
In attempt to find novel and potent α‑glucosidase inhibitors, efficient synthetic approaches were performed to synthesize substituted imidazo[1,2-c]quinazolines 6 and 11. Their inhibitory potencies were evaluated, showing excellent to great potencies (ranged from 12.44 ± 0.38 μM to 308.33 ± 0.06 μM) in comparison with acarbose (IC50 = 750.0 ± 1.5 μM). Notably, compound 11j exhibited the most potent inhibitory activity, therefore, it was selected for further evaluations including kinetic analysis, circular dichroism, fluorescence spectroscopy, and thermodynamic profile. It was observed that imidazoquinazoline 11j compete with the substrate for binding to the binding site of α‑glucosidase. Moreover, circular dichroism and fluorescence spectroscopy measurements confirmed that this binding led to change the secondary and tertiary structure of enzyme and inhibit its performance. Calculation of thermodynamic parameters including ΔG (free energy change), ΔH (enthalpy change), and ΔS (entropy change) values revealed the construction of spontaneous, electrostatic forces between imidazoquinazoline 11j and α‑glucosidase. The importance of the presence of electron-donating groups such as OMe was verified during the docking procedure, since an important hydrogen bond was formed in the mentioned compounds. Also, the superiority of compounds 11 over 6 was confirmed by the low HIA figures of imidazoquinazoline 11 in ADME studies. Overall, these results showed that our target imidazoquinazolines could be considered as a promising hit for further development of α‑glucosidase inhibitors as a well-stablished diabetes treatment approach.
Experimental
All chemicals were purchased from Merck (Germany) and were used without further purification. Melting points were measured on an Electrothermal 9100 apparatus. Elemental analyses for C, H and N were performed using a Heraeus CHN-O-Rapid analyzer. Mass spectra were recorded on an Agilent Technologies (HP) 5973 mass spectrometer operating at an ionization potential of 20 eV. IR spectra were recorded on a Shimadzu IR-460 spectrometer. 1H and 13C NMR spectra were measured (DMSO-d6 solution) with Bruker DRX-300 (at 300.1 and 75.5 MHz) and Bruker DRX-500 AVANCE (at 500.1 and 125.8 MHz) instruments.
General synthetic procedures
General procedure for the preparation of 2-(2-Nitrophenyl)-1H-benzo[d]imidazole 3:
A mixture of 2-nitrobenzaldehyde 1 (8.0 mmol, 1.208 g), benzene-1,2-diamine 2 (6.7 mmol, 0.723 g), and glacial acetic acid (20 mol%, 1.34 mmol, 0.076 ml) in EtOH (15 ml) was heated reflux conditions within 10 h. As the completion of compound 3 was confirmed by TLC analysis, the reaction mixture was quenched by water, and the resulting precipitation was filtered and washed completely. Afterwards, it was recrystallized by EtOAc and n-Hexane (within the proportion of 3:1) to afford the desirable compound 3 as a pure orange solid in 78% yield.
2-(2-Nitrophenyl)-1H-benzo[d]imidazole 3: Orange solid, mp 264–267 °C, yield: 78%. IR (KBr) (νmax/cm-1): 3329 (NH), 1622, 1553, 1488, 1403, 1396, 1348, 1285, 1233, 1177, 1093, 1061, 949, 913, 880, 858, 746, 693. 1H NMR (500.1 MHz, DMSO-d6): δ 10.78 (br s., 1H, NH), 8.03 (d, J = 8.2 Hz, 1H, CH), 7.99 (d, J = 7.7 Hz, 1H, CH), 7.86 (t, J = 7.6 Hz, 1H, CH), 7.74 (t, J = 7.3 Hz, 1H, CH), 7.71–7.60 (m, 2H, 2CH), 7.30–7.20 (m, 2H, 2CH). 13C NMR (125.8 MHz, DMSO-d6): δ 150.55, 147.43, 143.38, 135.49, 133.24, 131.92, 130.98, 124.78, 123.12, 120.13, 112.78. ESI–MS m/z: 240.48 [M + 1]+. Anal. Calcd. for C13H9N3O2: C, 65.27; H, 3.79; N, 17.56.; found: C, 65.08; H, 4.05; N, 17.78%.
General procedure for the preparation of 2-(2-Nitrophenyl)-4,5-diphenyl-1H-imidazole 9
A mixture of 2-nitrobenzaldehyde 1 (36 mmol, 5.436 g), benzil 7 (30 mmol, 6.302 g), and ammonium acetate 8 (300 mmol, 23.125 g) in glacial acetic acid (75 ml) were heated under the reflux conditions for 10 h. After completion of the reaction, which was monitored by TLC, the mixture was cooled down to room temperature, and it was gradually poured into crushed ice. The yellow solid residue got to precipitate, filtered, and washed with water. Finally, it was recrystallized from EtOH (40 ml) to obtain the pure adduct 9 as yellow powder in 84% yield.
2-(2-Nitrophenyl)-4,5-diphenyl-1H-imidazole 9: Yellow solid, mp 236–240 °C, yield: 84%. IR (KBr) (νmax/cm–1): 3348 (NH), 1592, 1549, 1487, 1436, 1396, 1343, 1296, 1247, 1168, 1108, 1089, 974, 903, 849, 826, 737, 640. 1H NMR (500.1 MHz, DMSO-d6): δ 10.23 (br s., 1H, NH), 8.29 (d, J = 8.0 Hz, 1H, CH), 7.98 (d, J = 7.8 Hz, 1H, CH), 7.70 (t, J = 7.7 Hz, 1H, CH), 7.62 (t, J = 7.5 Hz, 1H, CH), 7.38–7.20 (m, 10H, 10CH). 13C NMR (125.8 MHz, DMSO-d6): δ 148.98, 146.38, 141.09, 134.75, 133.30, 132.21, 130.68, 128.45, 127.74, 125.91, 124.68. ESI–MS m/z: 342.84 [M + 1]+. Anal. Calcd. for C21H15N3O2: C, 73.89; H, 4.43; N, 12.31.; found: C, 74.12; H, 4.68; N, 12.56%.
General procedure for the reduction of nitro functionality to amine moiety (compounds 4 and 10):
The procedure was common for both series: to a mixture of 2-(2-Nitrophenyl)-1H-benzo[d]imidazole 4 (5.20 mmol, 1.243 g) in hydrochloric acid (12.48 ml) and MeOH (5 ml) in ice bath at 0 °C, stannous chloride dihydrate SnCl2.2H2O (17.16 mmol, 3.878 g) was added gradually within 1 h. Afterwards, the mixture was stirred at room temperature for almost 6 h till the yellow color of nitro moiety got disappeared. As compound 3 was completely used, and it was confirmed by TLC analysis, the reaction mixture was basified by a solution of NaOH (2N) to pH 8. Then, water (20 ml) was added to the mixture and extracted three times with EtOAc (3 × 45 ml). The combined organic extracts were washed with brine, dried over Na2SO4, and then concentrated. The precipitate was filtered and washed with Et2O to afford pure product 4 as white powder in 58% yield.
2-(1H-benzo[d]imidazol-2-yl)aniline 4: White solid, mp 208–211 °C, yield: 58%. IR (KBr) (νmax/cm–1): 3184, 3058, and 3024 (3NH), 1611, 1519, 1476, 1429, 1279, 1178, 1056, 908, 882, 795, 723, 689, 633. 1H NMR (500.1 MHz, DMSO-d6): δ 7.81 (d, J = 7.8 Hz, 1H, CH), 7.76–7.69 (m, 2H, 2CH), 7.62–7.54 (m, 2H, 2CH), 7.49 (t, J = 7.8 Hz, 1H, CH), 7.10 (t, J = 7.5 Hz, 1H, CH), 6.68 (dd, J = 7.3 and 0.9 Hz, 1H, CH), 3.84–3.42 (br. s, 3H, NH and NH2). 13C NMR (125.8 MHz, DMSO-d6): δ 153.78, 147.86, 138.56, 131.08, 127.98, 122.76, 117.66, 115.38, 114.93, 110.38. ESI–MS m/z: 209.96 [M]+. Anal. Calcd. for C13H11N3: C, 74.62; H, 5.30; N, 20.08.; found: C, 74.49; H, 5.08; N, 20.23%.
In a similar procedure, 2-(2-Nitrophenyl)-4,5-diphenyl-1H-imidazole 4 (25.2 mmol, 8.593 g) in hydrochloric acid (60.48 ml) and MeOH (30 ml) in ice bath at 0 °C, stannous chloride dihydrate SnCl2.2H2O (83.16 mmol, 18.794 g) was added slowly within 1 h. The reaction took 4 h to get finish. After workup, pure white powder compound 10 was obtained in 63% yield.
2-(4,5-diphenyl-1H-imidazol-2-yl)aniline 10: White solid, mp 218–220 °C, yield: 63%. IR (KBr) (νmax/cm–1): 3408, 3369, and 3238 (3NH), 1622, 1467, 1417, 1297, 1233, 1177, 1093, 1061, 949, 913, 869, 844, 758, 673. 1H NMR (500.1 MHz, DMSO-d6): δ 10.24 (s, 1H, NH), 7.96 (d, J = 7.8 Hz, 1H, CH), 7.53–7.20 (m, 11H, 11CH), 6.94 (t, J = 7.6 Hz, 1H, CH), 6.68 (d, J = 7.4 Hz, 1H, CH), 5.23 (s, 2H, NH2). 13C NMR (125.8 MHz, DMSO-d6): δ 147.36, 146.98, 135.87, 132.45, 129.65, 128.78, 127.65, 126.24, 116.84, 115.53, 111.64. ESI–MS m/z: 311.78 [M]+. Anal. Calcd. for C21H17N3: C, 81.00; H, 5.50; N, 13.49.; found: C, 80.82; H, 5.39; N, 13.75%.
General procedure for the preparation of poly-substituted imidazo[1,2-c]quinazolines 6a-c and 11a-o
A mixture of synthesized 2-(1H-benzo[d]imidazol-2-yl)aniline 4 or 2-(4,5-diphenyl-1H-imidazol-2-yl)aniline 10 (1 mmol, g) with corresponding substituted benzaldehyde 5 (1.5 mmol) in glacial acetic acid (5 ml) was magnetically stirred at 80 °C for almost 3 to 4 h. After completion of reaction confirmed by TLC analysis, the mixture was cooled to the room temperature and poured into water. The precipitate was filtered, washed with water, and recrystallized from EtOAc to obtain the desired imidazoquinazolines 6a–c and 11a–o as pure powder in good to excellent yields.
6-phenylbenzo[4,5]imidazo[1,2-c]quinazoline 6a: Yellow solid, mp 156–158 °C, yield: 86%. IR (KBr) (νmax/cm–1): 1596, 1498, 1433, 1378, 1294, 1263, 1182, 1123, 1082, 993, 825, 755, 685, 634. 1H NMR (500.1 MHz, DMSO-d6): δ 8.23 (d, J = 7.8 Hz, 1H, CH), 8.16 (d, J = 7.7 Hz, 1H, CH), 7.74 (d, J = 8.0 Hz, 1H, CH), 7.40–7.16 (m, 8H, 8CH), 6.95 (d, J = 7.9 Hz, 1H, CH), 6.86 (t, J = 7.4 Hz, 1H, CH). 13C NMR (125.8 MHz, DMSO-d6): δ 145.47, 144.13, 139.07, 136.66, 134.20, 130.87, 129.48, 129.05, 128.98, 126.20, 125.79, 124.57, 124.11, 118.43, 115.87, 115.22, 111.83, 107.42. ESI–MS m/z: 295.87 [M]+. Anal. Calcd. for C20H13N3: C, 81.34; H, 4.44; N, 14.23.; found: C, 81.66; H, 4.28; N, 13.99%.
6-(4-chlorophenyl)benzo[4,5]imidazo[1,2-c]quinazoline 6b: Yellow solid, mp 173–175 °C, yield: 92%. IR (KBr) (νmax/cm–1): 1526, 1487, 1423, 1385, 1348, 1343, 1288, 1225, 1173, 1074, 1042, 1013, 911, 845, 827, 780, 769, 697. 1H NMR (500.1 MHz, DMSO-d6): δ 7.94 (d, J = 7.6 Hz, 1H, CH), 7.63 (d, J = 7.9 Hz, 1H, CH), 7.52 (d, J = 7.4 Hz, 1H, CH), 7.30–6.92 (m, 7H, 7CH), 6.84 (d, J = 7.8 Hz, 1H, CH), 6.81 (t, J = 7.2 Hz, 1H, CH). 13C NMR (125.8 MHz, DMSO-d6): δ 146.91, 143.55, 143.24, 138.42, 137.44, 132.77, 129.59, 129.26, 127.41, 126.28, 124.66, 122.24, 122.08, 118.52, 118.15, 114.81, 111.70, 110.60. ESI–MS m/z: 329.43 [M]+. Anal. Calcd. for C20H12ClN3: C, 72.84; H, 3.67; N, 12.74.; found: C, 73.06; H, 3.42; N, 12.96%.
6-(4-methoxyphenyl)benzo[4,5]imidazo[1,2-c]quinazoline 6c: Yellow solid, mp 198–201 °C, yield: 79%. IR (KBr) (νmax/cm–1): 1592, 1498, 1403, 1387, 1292, 1226, 1198, 1132, 1068, 1015, 937, 847, 785, 769, 684, 623. 1H NMR (500.1 MHz, DMSO-d6): δ 7.95 (d, J = 7.6 Hz, 1H, CH), 7.63 (d, J = 7.9 Hz, 1H, CH), 7.48 (d, J = 7.8 Hz, 1H, CH), 7.32–6.90 (m, 6H, 6CH), 6.88 (d, J = 8.2 Hz, 2H, 2CH), 6.81 (t, J = 7.8 Hz, 1H, CH), 3.68 (s, 3H, OCH3). 13C NMR (125.8 MHz, DMSO-d6): δ 159.72, 146.97, 143.62, 143.39, 132.80, 132.34, 131.69, 130.42, 127.52, 124.67, 122.18, 122.03, 118.52, 118.14, 114.80, 114.09, 111.72, 110.66, 55.15. ESI–MS m/z: 310.74 [M + 1]+. Anal. Calcd. for C21H15N3O: C, 84.53; H, 4.89; N, 13.58.; found: C, 84.76; H, 5.14; N, 13.84%.
2,3,5-triphenylimidazo[1,2-c]quinazoline 11a: Milky solid, mp 133–137 °C, yield: 78%. IR (KBr) (νmax/cm–1): 1598, 1496, 1456, 1412, 1368, 1289, 1263, 1148, 1129, 1067, 1026, 989, 965, 897, 822, 752, 689, 636. 1H NMR (500.1 MHz, DMSO-d6): δ 7.88 (d, J = 7.5 Hz, 1H, CH), 7.50 (d, J = 7.2 Hz, 2H, 2CH), 7.45–7.00 (m, 14H, 14CH), 6.84 (d, J = 7.1 Hz, 1H, CH), 6.82 (t, J = 6.9 Hz, 1H, CH). 13C NMR (125.8 MHz, DMSO-d6): δ 141.37, 140.93, 137.50, 133.94, 130.32, 130.13, 129.38, 129.05, 128.91, 128.54, 128.37, 128.18, 126.42, 125.10, 124.86, 123.19, 122.82, 113.24. ESI–MS m/z: 397.26 [M]+. Anal. Calcd. for C28H19N3: C, 84.61; H, 4.82; N, 10.57.; found: C, 84.93; H, 4.62; N, 10.78%.
5-(4-chlorophenyl)-2,3-diphenylimidazo[1,2-c]quinazoline 11b: Milky solid, mp 178–181 °C, yield: 94%. IR (KBr) (νmax/cm–1): 1589, 1488, 1463, 1399, 1348, 1294, 1231, 1168, 1129, 1087, 996, 929, 886, 795, 756, 718, 675, 638. 1H NMR (500.1 MHz, DMSO-d6): δ 7.90 (d, J = 7.2 Hz, 1H, CH), 7.50 (d, J = 7.8 Hz, 2H, 2CH), 7.42–7.00 (m, 13H, 13CH), 6.96 (d, J = 7.9 Hz, 1H, CH), 6.85 (t, J = 7.6 Hz, 1H, CH). 13C NMR (125.8 MHz, DMSO-d6): δ 141.13, 140.80, 139.66, 137.17, 133.42, 133.02, 130.35, 129.10, 129.02, 128.57, 128.20, 127.10, 126.84, 126.60, 126.38, 123.33, 122.97, 112.73. ESI–MS m/z: 431.86 [M]+. Anal. Calcd. for C28H18ClN3: C, 77.86; H, 4.20; N, 9.73.; found: C, 78.04; H, 4.06; N, 9.53%.
5-(3-chlorophenyl)-2,3-diphenylimidazo[1,2-c]quinazoline 11c: Milky solid, mp 149–151 °C, yield: 83%. IR (KBr) (νmax/cm–1): 1586, 1509, 1447, 1367, 1320, 1286, 1184, 1129, 1068, 1023, 967, 834, 754, 685, 622. 1H NMR (300.1 MHz, DMSO-d6): δ 7.97 (d, J = 7.7 Hz, 1H, CH), 7.76 (d, J = 7.9 Hz, 2H, 2CH), 7.60–7.17 (m, 11H, 11CH), 7.11 (d, J = 8.0 Hz, 2H, 2CH), 6.91 (d, J = 7.2 Hz, 1H, CH), 6.79 (t, J = 7.6 Hz, 1H, CH). 13C NMR (75.1 MHz, DMSO-d6): δ 141.81, 141.10, 135.39, 131.27, 131.06, 130.80, 129.62, 129.55, 129.50, 129.10, 128.99, 128.76, 128.69, 127.15, 127.02, 126.95, 125.42, 123.67, 119.06, 115.63. ESI–MS m/z: 432.29 [M + 1]+. Anal. Calcd. for C28H18ClN3: C, 77.86; H, 4.20; N, 9.73.; found: C, 77.94; H, 4.13; N, 9.89%.
5-(2-chlorophenyl)-2,3-diphenylimidazo[1,2-c]quinazoline 11d: Milky solid, mp 164–167 °C, yield: 72%. IR (KBr) (νmax/cm–1): 1599, 1498, 1438, 1399, 1358, 1294, 1256, 1162, 1129, 1089, 1012, 991, 932, 899, 836, 763, 729, 687, 624. 1H NMR (500.1 MHz, DMSO-d6): δ 8.04 (d, J = 8.0 Hz, 1H, CH), 7.87 (dd, J = 7.2, 0.9 Hz, 1H, CH), 7.74 (t, J = 7.7 Hz, 2H, 2CH), 7.68–7.17 (m, 13H, 13CH), 6.96 (d, J = 7.4 Hz, 1H, CH), 6.83 (t, J = 7.8 Hz, 1H, CH). 13C NMR (125.8 MHz, DMSO-d6): δ 147.90, 144.90, 138.14, 137.79, 136.29, 135.96, 133.42, 131.58, 130.82, 130.11, 129.81, 129.10, 128.30, 127.74, 127.25, 126.35, 124.84, 123.57, 119.94, 113.92. ESI–MS m/z: 432.56 [M]+. Anal. Calcd. for C28H18ClN3: C, 77.86; H, 4.20; N, 9.73.; found: C, 78.06; H, 4.54; N, 9.48%.
5-(4-methylphenyl)-2,3-diphenylimidazo[1,2-c]quinazoline 11e: Milky solid, mp 155–157 °C, yield: 87%. IR (KBr) (νmax/cm–1): 1595, 1489, 1402, 1387, 1285, 1216, 1194, 1143, 1098, 1034, 995, 865, 746, 658, 624. 1H NMR (500.1 MHz, DMSO-d6): δ 7.96 (d, J = 7.8 Hz, 1H, CH), 7.84 (d, J = 7.8 Hz, 1H, CH), 7.78 (s, 1H, CH), 7.70–7.00 (m, 13H, 13CH), 6.96 (d, J = 7.8 Hz, 1H, CH), 6.82 (t, J = 7.4 Hz, 1H, CH), 2.15 (s, 3H, CH3). 13C NMR (125.8 MHz, DMSO-d6): δ 141.42, 141.04, 138.06, 137.64, 137.52, 134.05, 130.27, 130.02, 129.45, 129.05, 128.15, 126.55, 126.35, 125.04, 124.74, 123.13, 122.76, 113.47, 20.57. ESI–MS m/z: 412.37 [M + 1]+. Anal. Calcd. for C29H21N3: C, 84.64; H, 5.14; N, 10.21.; found: C, 84.78; H, 4.98; N, 10.38%.
4-(2,3-diphenylimidazo[1,2-c]quinazolin-5-yl)-N,N-dimethylaniline 11f: Milky solid, mp 208–210 °C, yield: 76%. IR (KBr) (νmax/cm–1): 1594, 1487, 1438, 1422, 1368, 1276, 1239, 1188, 1052, 1028, 985, 913, 852, 758, 695. 1H NMR (500.1 MHz, DMSO-d6): δ 8.12 (d, J = 8.1 Hz, 2H, 2CH), 7.91 (d, J = 7.8 Hz, 1H, CH), 7.53 (d, J = 7.5 Hz, 2H, 2CH), 7.48–7.00 (m, 9H, 9CH), 6.92 (d, J = 7.7 Hz, 1H, CH), 6.80 (t, J = 7.4 Hz, 1H, CH), 6.53 (d, J = 8.1 Hz, 2H, 2CH), 2.81 (s, 6H, 2 NCH3). 13C NMR (125.8 MHz, DMSO-d6): δ 154.65, 141.92, 141.32, 134.15, 130.07, 129.18, 129.09, 128.97, 128.60, 128.26, 127.20, 126.81, 126.74, 126.35, 124.96, 123.07, 112.28, 111.52, 31.44. ESI–MS m/z: 441.69 [M + 1]+. Anal. Calcd. for C30H24N4: C, 81.79; H, 5.49; N, 12.72.; found: C, 82.02; H, 5.72; N, 12.48%.
5-(4-methoxyphenyl)-2,3-diphenylimidazo[1,2-c]quinazoline 11g: Milky solid, mp 189–192 °C, yield: 79%. IR (KBr) (νmax/cm–1): 1599, 1496, 1428, 1392, 1348, 1294, 1223, 1188, 1138, 1094, 1046, 991, 952, 889, 836, 756, 733, 678, 633. 1H NMR (500.1 MHz, DMSO-d6): δ 7.97 (d, J = 7.4 Hz, 1H, CH), 7.70–7.06 (m, 13H, 13CH), 6.94 (d, J = 7.8 Hz, 1H, CH), 6.85 (t, J = 7.5 Hz, 1H, CH), 6.76 (d, J = 7.8 Hz, 2H, 2CH), 3.65 (s, 3H, OCH3). 13C NMR (125.8 MHz, DMSO-d6): δ 158.91, 148.21, 144.89, 138.18, 136.99, 136.79, 136.41, 130.27, 129.85, 129.66, 129.22, 128.64, 127.94, 127.35, 124.86, 120.02, 113.90, 111.51, 55.11. ESI–MS m/z: 427.83 [M]+. Anal. Calcd. for C29H21N3O: C, 81.48; H, 4.95; N, 9.83.; found: C, 81.34; H, 5.12; N, 10.04%.
5-(3-methoxyphenyl)-2,3-diphenylimidazo[1,2-c]quinazoline 11h: Milky solid, mp 174–176 °C, yield: 87%. IR (KBr) (νmax/cm–1): 1595, 1498, 1422, 1377, 1287, 1253, 1163, 1109, 1096, 1034, 987, 857, 763, 749, 684, 633. 1H NMR (300.1 MHz, DMSO-d6): δ 7.86 (d, J = 7.4 Hz, 1H, CH), 7.50 (d, J = 7.5 Hz, 2H, 2CH), 7.45–7.00 (m, 10H, 10CH), 6.90–6.70 (m, 3H, 3CH), 6.41 (d, J = 7.6 Hz, 1H, CH), 6.35 (s, 1H, CH), 3.58 (s, 3H, OCH3). 13C NMR (75.1 MHz, DMSO-d6): δ 159.12, 142.52, 141.44, 141.07, 137.56, 134.01, 130.30, 130.10, 129.79, 129.41, 129.09, 128.90, 128.18, 126.61, 126.41, 123.15, 122.79, 113.36, 111.52, 110.86, 54.94. ESI–MS m/z: 428.44 [M + 1]+. Anal. Calcd. for C29H21N3O: C, 81.48; H, 4.95; N, 9.83.; found: C, 81.62; H, 5.23; N, 9.68%.
5-(2-methoxyphenyl)-2,3-diphenylimidazo[1,2-c]quinazoline 11i: Milky solid, mp 148–152 °C, yield: 69%. IR (KBr) (νmax/cm–1): 1593, 1472, 1436, 1399, 1358, 1276, 1212, 1196, 1153, 1090, 1041, 1023, 975, 856, 795, 769, 713, 642, 625. 1H NMR (300.1 MHz, DMSO-d6): δ 7.88 (d, J = 7.1 Hz, 1H, CH), 7.50 (d, J = 6.9 Hz, 2H, 2CH), 7.45–6.98 (m, 10H, 10CH), 6.95–6.85 (m, 2H, 2CH), 6.78 (t, J = 7.3 Hz, 1H, CH), 6.71 (d, J = 7.6 Hz, 1H, CH), 6.37 (d, J = 7.3 Hz, 1H, CH), 3.64 (s, 3H, OCH3). 13C NMR (75.1 MHz, DMSO-d6): δ 154.87, 141.90, 141.00, 137.85, 134.34, 130.30, 130.11, 129.88, 129.58, 128.85, 128.70, 128.30, 128.12, 126.42, 126.18, 122.93, 122.58, 113.32, 111.46, 111.22, 55.54. ESI–MS m/z: 428.44 [M + 1]+. Anal. Calcd. for C29H21N3O: C, 81.48; H, 4.95; N, 9.83.; found: C, 81.34; H, 4.78; N, 10.04%.
5-(2,3-dimethoxyphenyl)-2,3-diphenylimidazo[1,2-c]quinazoline 11j: Milky solid, mp 221–224 °C, yield: 63%. IR (KBr) (νmax/cm–1): 1597, 1502, 1437, 1411, 1379, 1292, 1229, 1143, 1077, 1027, 991, 943, 899, 847, 788, 753, 706, 686, 652. 1H NMR (500.1 MHz, DMSO-d6): δ 8.06 (d, J = 8.5 Hz, 1H, CH), 7.51 (d, J = 7.4 Hz, 2H, 2CH), 7.48–7.10 (m, 10H, 10CH), 6.95 (d, J = 8.2 Hz, 1H, CH), 6.87 (d, J = 7.9 Hz, 1H, CH), 6.82 (t, J = 8.3 Hz, 1H, CH), 6.05 (d, J = 7.8 Hz, 1H, CH), 3.90 and 3.75 (2s, 6H, 2OCH3). 13C NMR (125.8 MHz, DMSO-d6): δ 152.70, 151.86, 144.72, 141.90, 141.51, 133.58, 132.15, 131.02, 129.73, 129.52, 128.78, 127.10, 126.75, 124.50, 123.90, 118.76, 117.24, 115.69, 113.90, 108.38, 60.20, 56.19. ESI–MS m/z: 457.96 [M]+. Anal. Calcd. for C30H23N3O2: C, 78.75; H, 5.07; N, 9.18.; found: C, 78.99; H, 5.23; N, 9.36%.
5-(2,4-dimethoxyphenyl)-2,3-diphenylimidazo[1,2-c]quinazoline 11k: Milky solid, mp 197–200 °C, yield: 71%. IR (KBr) (νmax/cm–1): 1589, 1456, 1398, 1361, 1273, 1237, 1198, 1104, 1036, 1021, 998, 943, 836, 759, 724, 639. 1H NMR (500.1 MHz, DMSO-d6): δ 8.06 (d, J = 8.5 Hz, 1H, CH), 7.53–7.00 (m, 11H, 11CH), 6.95–6.65 (m, 4H, 4CH), 6.05 (s, 1H, CH), 3.54 and 3.51 (2s, 6H, 2OCH3). 13C NMR (125.8 MHz, DMSO-d6): δ 153.14, 151.88, 149.77, 142.12, 141.61, 137.31, 130.84, 129.77, 129.39, 128.92, 128.60, 128.20, 127.44, 126.88, 124.11, 118.81, 115.57, 114.12, 113.12, 112.89, 56.48, 55.65. ESI–MS m/z: 457.53 [M]+. Anal. Calcd. for C30H23N3O2: C, 78.75; H, 5.07; N, 9.18.; found: C, 78.58; H, 4.96; N, 8.92%.
5-(2,5-dimethoxyphenyl)-2,3-diphenylimidazo[1,2-c]quinazoline 11l: Milky solid, mp 183–186 °C, yield: 67%. IR (KBr) (νmax/cm–1): 1597, 1512, 1489, 1423, 1395, 1335, 1295, 1258, 1183, 1124, 1075, 1031, 992, 968, 899, 831, 796, 740, 685, 673, 642. 1H NMR (500.1 MHz, DMSO-d6): δ 7.88 (d, J = 7.6 Hz, 1H, CH), 7.60–7.00 (m, 11H, 11CH), 6.05 (s, 1H, CH), 6.94 (d, J = 7.8 Hz, 1H, CH), 6.80 (t, J = 8.0 Hz, 1H, CH), 6.48 (s, 1H, CH), 6.33 (d, J = 7.5 Hz, 1H, CH), 6.29 (d, J = 7.5 Hz, 1H, CH), 3.54 and 3.51 (2s, 6H, 2OCH3). 13C NMR (125.8 MHz, DMSO-d6): δ 161.11, 156.52, 142.30, 141.66, 138.00, 132.84, 130.63, 130.30, 129.33, 129.20, 128.62, 126.81, 126.41, 123.28, 121.32, 118.75, 117.01, 115.63, 105.05, 99.04, 56.11, 55.62. ESI–MS m/z: 458.64 [M + 1]+. Anal. Calcd. for C30H23N3O2: C, 78.75; H, 5.07; N, 9.18.; found: C, 78.93; H, 5.26; N, 9.33%.
5-(3,4-dimethoxyphenyl)-2,3-diphenylimidazo[1,2-c]quinazoline 11m: Milky solid, mp 234–237 °C, yield: 84%. IR (KBr) (νmax/cm–1): 1595, 1522, 1424, 1386, 1296, 1259, 1178, 1154, 1078, 1014, 997, 947, 923, 805, 768, 743, 695, 629. 1H NMR (300.1 MHz, DMSO-d6): δ 8.04 (d, J = 7.2 Hz, 1H, CH), 7.65–7.15 (m, 11H, 11CH), 6.91 (d, J = 6.9 Hz, 1H, CH), 6.85 (t, J = 7.5 Hz, 1H, CH), 6.77 (d, J = 8.1 Hz, 1H, CH), 6.64 (s, 2H, 2CH), 6.33 (d, J = 8.1 Hz, 1H, CH), 3.64 and 3.57 (2s, 6H, 2OCH3). 13C NMR (75.8 MHz, DMSO-d6): δ 156.31, 155.22, 149.29, 148.94, 142.30, 141.30, 133.04, 131.80, 130.88, 129.66, 129.31, 129.01, 128.36, 128.04, 124.26, 119.09, 117.71, 115.73, 111.84, 109.70, 55.84, 55.82. ESI–MS m/z: 458.32 [M + 1]+. Anal. Calcd. for C30H23N3O2: C, 78.75; H, 5.07; N, 9.18.; found: C, 78.92; H, 4.87; N, 9.36%.
2,3-diphenyl-5-(3,4,5-trimethoxyphenyl)imidazo[1,2-c]quinazoline 11n: Milky solid, mp 256–260 °C, yield: 78%. IR (KBr) (νmax/cm–1): 1595, 1539, 1426, 1399, 1358, 1289, 1243, 1167, 1143, 1089, 1045, 987, 935, 829, 778, 685, 645. 1H NMR (300.1 MHz, DMSO-d6): δ 8.07 (d, J = 7.0 Hz, 1H, CH), 7.60–7.12 (m, 11H, 11CH), 6.97 (d, J = 8.1 Hz, 1H, CH), 6.87 (t, J = 7.4 Hz, 1H, CH), 6.23 (s, 2H, 2CH), 3.56 and 3.53 (2s, 9H, 3OCH3). 13C NMR (75.1 MHz, DMSO-d6): δ 156.79, 155.21, 146.36, 141.69, 139.71, 138.88, 137.07, 134.82, 130.89, 130.63, 130.39, 129.78, 129.36, 128.89, 128.42, 124.83, 120.04, 115.26, 58.36, 56.72. ESI–MS m/z: 488.26 [M + 1]+. Anal. Calcd. for C31H25N3O3: C, 76.37; H, 5.17; N, 8.62.; found: C, 76.18; H, 4.98; N, 8.48%.
5-(3-bromo-4,5-dimethoxyphenyl)-2,3-diphenylimidazo[1,2-c]quinazoline 11o: Milky solid, mp 287–289 °C, yield: 90%. IR (KBr) (νmax/cm–1): 1623, 1584, 1532, 1465, 1354, 1298, 1253, 1198, 1134, 1083, 1016, 994, 949, 896, 843, 721, 692, 658, 624. 1H NMR (300.1 MHz, DMSO-d6): δ 8.02 (d, J = 7.2 Hz, 1H, CH), 7.60–7.10 (m, 11H, 11CH), 6.94 (d, J = 7.9 Hz, 1H, CH), 6.87 (t, J = 7.5 Hz, 1H, CH), 6.67 (s, 1H, CH), 3.63 and 3.61 (2s, 6H, 2OCH3). 13C NMR (75.1 MHz, DMSO-d6): δ 153.61, 152.45, 146.11, 141.90, 141.31, 137.99, 131.70, 131.01, 129.66, 129.58, 129.04, 128.77, 127.84, 127.41, 124.15, 121.54, 119.26, 116.94, 115.58, 110.52, 60.42, 56.43. ESI–MS m/z: 536.09 [M]+. Anal. Calcd. for C30H22BrN3O2: C, 67.17; H, 4.13; N, 7.83.; found: C, 67.28; H, 4.29; N, 8.05%.
α-Glucosidase inhibition assay
α-Glucosidase enzyme ((EC3.2.1.20, Saccharomyces cerevisiae, 20 U mg−1) and substrate (p-nitrophenyl glucopyranoside) were purchased from Sigma-Aldrich. Enzyme was prepared in potassium phosphate buffer (pH 6.8, 50 mM), as well as substituted benzo[4,5]imidazo[1,2-c]quinazoline 6a-c and highly-substituted imidazo[1,2-c]quinazolines 11a–o were dissolved in DMSO (10% final concentration). The various concentrations of these compounds (20 ml), enzyme solution (20 ml) and potassium phosphate buffer (135 ml) were added in the 96-well plate and incubated at 37 °C for 10 min. Afterwards, the substrate (25 ml, 4 mM) was added to the mentioned mixture and allowed to incubate at 37 °C for 20 min. Finally, the change in absorbance was measured at 405 nm by using spectrophotometer (Gen5, Power wave xs2, BioTek, America). The percentage of enzyme inhibition was calculated using Eq. (6) and IC50 values were obtained from non-linear regression curve using the Logit method.
Kinetic studies
The kinetic analysis was performed for the most potent derivative 11j to reveal the inhibition mode against α-glucosidase. The 20 ml of enzyme solution (1U ml−1) was incubated with different concentrations (0, 3.1, 6.2, and 12.4 µM) of this compound for 15 min at 30 °C. Afterwards, various concentrations of substrate (p-nitrophenyl glucopyranoside, 1–10 mM) was added to measure the change of absorbance for 20 min at 405 nm by using spectrophotometer (Gen5, Power wave xs2, BioTek, America).
In the presence of a competitive inhibitor, Km increases while Vmax does not change. Michaelis–Menten saturation curve for an enzyme reaction shows the relation between the substrate concentration and reaction rate as bellow:
According to Michaelis–Menten graph, Kmapp is also defined as:
[I] is the concentration of inhibitor.
Lineweaver Burk plot that provides a useful graphical method for analysis of the Michaelis–Menten is represented as:
Therefore, the slope of Lineweaver Burk plot is equal to:
The Kmapp value is calculated by Eq. (6):
Therefore, from replot of Kmapp Vs. [I], Eq. (7) can be used for the calculation of KI59,60:
Fluorescence spectroscopy measurements
This assay was carried out for the most potent derivative 11j to measure the fluorescence intensity. To this aim, different solutions containing different concentrations (0 to 1.0 µM) of the inhibitor and α-glucosidase (3 ml, 0.1 U ml−1) were held for 10 min to equilibrate before measurements. Moreover, the fluorescence of the buffer containing compound 11j in the absence of the enzyme were subtracted as the background fluorescence. Afterwards, at the excitation wavelength of 280 nm, the fluorescence emission spectra were measured from 300 to 450 nm using a Synergy HTX multi-mode reader (Biotek Instruments, Winooski, VT, USA) equipped with a 1.0 cm quartz cell holder61.
Molecular docking studies
Molecular docking study using AutoDock4 and Auto Dock Tools (version 1.5.6) was performed on compounds 11a–o and 6a–c to elucidate the patterns of their interactions in the active site of the human acid-alpha-glucosidase (PDB ID: 5NN8). Receptor was prepared by removing water molecules and computing Kollman charges with BIOVIA Discovery Studio visualizer and Auto Dock Tools. To validate the docking procedure, redocking process was performed with acarbose as standard ligand, and RMSD value of 1.57 was achieved. The redocked ligand identified similar binding pose to original co-crystalized position downloaded from RCSB database (5NN8). Acarbose was extracted from the PDB file using BIOVIA Discovery Studio visualizer and saved as a separate PDB file. A possible grid box was determined using Auto Dock Tools (version 1.5.6). Furthermore, genetic algorithm was selected as the searching parameter. This procedure was carried out for different potential grid coordinates. Finally, the best grid coordinates were determined by comparing RMSD values.
Afterwards, ligands 11 and 6 were prepared by adding Gasteiger Charges using Auto Dock Tools, and the docking procedure was conducted with 100 genetic algorithm runs using AutoDock4 and AutoGrid4. The interactions were visualized by PLIP online service62 and PyMOL Molecular Graphics System, Version 2.5.2 Schrödinger, LLC.
Statistical analysis
Statistical analysis for all compounds 6a–c and 11a–o was performed using SigmaPlot version 14 (Systat-Software, USA). The experiments were replicated three times under the same conditions. Data for each compound was used as mean ± SD in T test method. A p-value lower than 0.05 was regarded as indicative of statistical significance.
Data availability
The authors confirm that the data supporting the finding of this study are available within the manuscript and supplementary file.
References:
Sun, H. et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 183, 109119. https://doi.org/10.1016/j.diabres.2021.109119 (2022).
Kshirsagar, R. P. et al. SGLT inhibitors as antidiabetic agents: A comprehensive review. RSC Adv. 10, 1733–1756. https://doi.org/10.1039/C9RA08706K (2020).
Ojebiyi, A. O. The impacts of pharmacological and other interventions for preventing the onset of diabetes. Int. J. Diabet. Metab. Disord. 8, 268–276 (2023).
Rush, T., McGeary, M., Sicignano, N. & Buryk, M. A. A plateau in new onset type 1 diabetes: Incidence of pediatric diabetes in the United States Military Health System. Pediatr. Diabetes 19, 917–922. https://doi.org/10.1111/pedi.12659 (2018).
Zheng, Y., Ley, S. H. & Hu, F. B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 14, 88–98. https://doi.org/10.1038/nrendo.2017.151 (2018).
Dowarah, J. & Singh, V. P. Anti-diabetic drugs recent approaches and advancements. Bioorg. Med. Chem. 28, 115263. https://doi.org/10.1016/j.bmc.2019.115263 (2020).
Gao, X. et al. Meta-analysis and critical review on the efficacy and safety of alpha-glucosidase inhibitors in Asian and non-Asian populations. J. Diabet. Investig. 9, 321–331. https://doi.org/10.1111/jdi.12711 (2018).
Ghani, U. Re-exploring promising α-glucosidase inhibitors for potential development into oral anti-diabetic drugs: Finding needle in the haystack. Eur. J. Med. Chem. 103, 133–162. https://doi.org/10.1016/j.ejmech.2015.08.043 (2015).
Garlapati, R. et al. Development of α-glucosidase inhibitors by room temperature C-C cross couplings of quinazolinones. Org. Biomol. Chem. 11, 4778–4791. https://doi.org/10.1039/c3ob40636a (2013).
Gurram, V. et al. Design, synthesis, and biological evaluation of quinazoline derivatives as α-glucosidase inhibitors. Med. Chem. Res. 24, 2227–2237. https://doi.org/10.1007/s00044-014-1293-5 (2015).
Javaid, K. et al. 2-Arylquinazolin-4 (3H)-ones: A new class of α-glucosidase inhibitors. Bioorg. Med. Chem. 23, 7417–7421. https://doi.org/10.1016/j.bmc.2015.10.038 (2015).
Zhang, Y. et al. Quinazoline-1-deoxynojirimycin hybrids as high active dual inhibitors of EGFR and α-glucosidase. Bioorg. Med. Chem. Lett. 27, 4309–4313. https://doi.org/10.1016/j.bmcl.2017.08.035 (2017).
Wei, M. et al. Quinazolinone derivatives: Synthesis and comparison of inhibitory mechanisms on α-glucosidase. Bioorg. Med. Chem. 25, 1303–1308. https://doi.org/10.1016/j.bmc.2016.09.042 (2017).
El-Sayed, N. N. et al. Synthesis and evaluation of anticancer, antiphospholipases, antiproteases, and antimetabolic syndrome activities of some 3 H-quinazolin-4-one derivatives. J. Enzyme Inhib. Med. Chem. 34, 672–683. https://doi.org/10.1080/14756366.2019.1574780 (2019).
Santos-Ballardo, L. et al. Synthesis, biological evaluation and molecular docking of 3-substituted quinazoline-2, 4 (1 H, 3 H)-diones. J. Chem. Sci. 132, 1–10. https://doi.org/10.1007/s12039-020-01813-1 (2020).
Babatunde, O. et al. Dihydroquinazolin-4 (1 H)-one derivatives as novel and potential leads for diabetic management. Mol. Divers. 2021, 1–20. https://doi.org/10.1007/s11030-021-10196-5 (2021).
Tokalı, F. S. et al. Design, synthesis, molecular docking, and some metabolic enzyme inhibition properties of novel quinazolinone derivatives. Arch. Pharm. 354, 2000455. https://doi.org/10.1002/ardp.202000455 (2021).
Wali, H. et al. Synthesis, in vitro, and in silico studies of newly functionalized quinazolinone analogs for the identification of potent α-glucosidase inhibitors. J. Iran. Chem. Soc. 18, 2017–2034. https://doi.org/10.1007/s13738-021-02159-2 (2021).
Ayan, E. K., Soyer, Z. & Uysal, Ş. Synthesis and enzymological characterization of some 2-(substitutedphenylamino) quinazolin-4 (3H)-one derivatives as potent α-glucosidase inhibitors in vitro. Lett. Drug Des. Discov. 18, 723–732. https://doi.org/10.2174/1570180818999201224121929 (2021).
Moheb, M. et al. Synthesis and bioactivities evaluation of quinazolin-4 (3H)-one derivatives as α-glucosidase inhibitors. BMC Chem. 16, 1–12. https://doi.org/10.1186/s13065-022-00885-z (2022).
Sinan Tokalı, F. Novel benzoic acid derivatives bearing quinazolin-4 (3H)-one Ring: Synthesis, characterization, and inhibition effects on α-glucosidase and α-amylase. ChemistrySelect 7, e202204019. https://doi.org/10.1002/slct.202204019 (2022).
Ibrahim, A., Sakr, H. M., Ayyad, R. R. & Khalifa, M. M. Design, synthesis, in-vivo anti-diabetic activity, in-vitro α-glucosidase inhibitory activity and molecular docking studies of some quinazolinone derivatives. ChemistrySelect 7, e202104590. https://doi.org/10.1002/slct.202104590 (2022).
Satyanarayana, N. et al. Synthesis of 2-styryl-quinazoline and 3-styryl-quinoxaline based sulfonate esters via sp 3 C-H activation and their evaluation for α-glucosidase inhibition. New J. Chem. 46, 5162–5170. https://doi.org/10.1039/D1NJ05644A (2022).
Khalifa, M. M., Sakr, H. M., Ibrahim, A., Mansour, A. M. & Ayyad, R. R. Design and synthesis of new benzylidene-quinazolinone hybrids as potential anti-diabetic agents: In vitro α-glucosidase inhibition, and docking studies. J. Mol. Struct. 1250, 131768. https://doi.org/10.1016/j.molstruc.2021.131768 (2022).
Yar, M. et al. Organocatalyzed solvent free an efficient novel synthesis of 2, 4, 5-trisubstituted imidazoles for α-glucosidase inhibition to treat diabetes. Bioorg. Chem. 58, 65–71. https://doi.org/10.1016/j.bioorg.2014.11.006 (2015).
Arshad, T. et al. Syntheses, in vitro evaluation and molecular docking studies of 5-bromo-2-aryl benzimidazoles as α-glucosidase inhibitors. Med. Chem. Res. 25, 2058–2069. https://doi.org/10.1007/s00044-016-1614-y (2016).
Taha, M. et al. Synthesis, α-glucosidase inhibitory, cytotoxicity and docking studies of 2-aryl-7-methylbenzimidazoles. Bioorg. Chem. 65, 100–109. https://doi.org/10.1016/j.bioorg.2016.02.004 (2016).
Arshad, T. et al. 5-Bromo-2-aryl benzimidazole derivatives as non-cytotoxic potential dual inhibitors of α-glucosidase and urease enzymes. Bioorg. Chem. 72, 21–31. https://doi.org/10.1016/j.bioorg.2017.03.007 (2017).
Zawawi, N. K. N. A. et al. Synthesis, molecular docking studies of hybrid benzimidazole as α-glucosidase inhibitor. Bioorg. Chem. 70, 184–191. https://doi.org/10.1016/j.bioorg.2016.12.009 (2017).
Özil, M., Parlak, C. & Baltaş, N. A simple and efficient synthesis of benzimidazoles containing piperazine or morpholine skeleton at C-6 position as glucosidase inhibitors with antioxidant activity. Bioorg. Chem. 76, 468–477. https://doi.org/10.1016/j.bioorg.2017.12.019 (2018).
Rahim, F. et al. Synthesis, in vitro alpha-glucosidase inhibitory potential of benzimidazole bearing bis-Schiff bases and their molecular docking study. Bioorg. Chem. 94, 103394. https://doi.org/10.1016/j.bioorg.2019.103394 (2020).
Aroua, L. M. et al. A facile approach synthesis of benzoylaryl benzimidazole as potential α-amylase and α-glucosidase inhibitor with antioxidant activity. Bioorg. Chem. 114, 105073. https://doi.org/10.1016/j.bioorg.2021.105073 (2021).
Mohammadi-Khanaposhtani, M. et al. Synthesis, in vitro and in silico enzymatic inhibition assays, and toxicity evaluations of new 4, 5-diphenylimidazole-N-phenylacetamide derivatives as potent α-glucosidase inhibitors. Med. Chem. Res. 30, 1273–1283. https://doi.org/10.1007/s00044-021-02734-5 (2021).
Li, Y. et al. Discovery of new 2-phenyl-1H-benzo [d] imidazole core-based potent α-glucosidase inhibitors: Synthesis, kinetic study, molecular docking, and in vivo anti-hyperglycemic evaluation. Bioorg. Chem. 117, 105423. https://doi.org/10.1016/j.bioorg.2021.105423 (2021).
Hasan, H. A., Salman, A. & Abdulmalek, E. Anticancer activity and high content screening of new 6-substituted-5, 6-dihydrobenzo [4, 5] imidazo [1, 2-c] quinazoline derivatives. Res. J. Pharm. Technol. 14, 2397–2405. https://doi.org/10.52711/0974-360X.2021.00423 (2021).
Jadhavar, P. S. et al. Benzimidazoquinazolines as new potent anti-TB chemotypes: Design, synthesis, and biological evaluation. Bioorg. Chem. 99, 103774. https://doi.org/10.1016/j.bioorg.2020.103774 (2020).
Li, J.-C. et al. Design, synthesis and antifungal activity evaluation of isocryptolepine derivatives. Bioorg. Chem. 92, 103266. https://doi.org/10.1016/j.bioorg.2019.103266 (2019).
Hasan, H. A. et al. Synthesis of novel 6-substituted-5, 6-Dihydrobenzo [4, 5] Imidazo [1, 2-c] quinazoline compounds and evaluation of their properties. J. Mol. Struct. 1193, 482–494. https://doi.org/10.1016/j.molstruc.2019.04.111 (2019).
Hasan, H. A. et al. Microwave synthesis, crystal structure, antioxidant, and antimicrobial study of new 6-heptyl-5, 6-dihydrobenzo [4, 5] imidazo [1, 2-c] quinazoline compound. Chem. Cent. J. 12, 1–15. https://doi.org/10.1016/j.molstruc.2019.04.111 (2018).
Domány, G. et al. Imidazo [1, 2-c] quinazolines with lipid peroxidation inhibitory effect. Eur. J. Med. Chem. 33, 181–187. https://doi.org/10.1016/S0223-5234(98)80007-X (1998).
Firoozpour, L. et al. Design, synthesis and α-glucosidase inhibition study of novel pyridazin-based derivatives. Med. Chem. Res. 32, 713–722. https://doi.org/10.1007/s00044-023-03027-9 (2023).
Sadat-Ebrahimi, S. E. et al. Design, synthesis, and biological evaluation of new indole-acrylamide-1, 2, 3-triazole derivatives as potential α-glucosidase inhibitors. Polycyclic Aromat. Compd. 42, 3157–3165. https://doi.org/10.1080/10406638.2020.1854323 (2022).
Peytam, F. et al. Design, synthesis, molecular docking, and in vitro α-glucosidase inhibitory activities of novel 3-amino-2, 4-diarylbenzo [4, 5] imidazo [1, 2-a] pyrimidines against yeast and rat α-glucosidase. Sci. Rep. 11, 11911. https://doi.org/10.1038/s41598-021-91473-z (2021).
Moghimi, S. et al. Synthesis, in-vitro evaluation, molecular docking, and kinetic studies of pyridazine-triazole hybrid system as novel α-glucosidase inhibitors. Bioorg. Chem. 109, 104670. https://doi.org/10.1016/j.bioorg.2021.104670 (2021).
Peytam, F. et al. An efficient and targeted synthetic approach towards new highly substituted 6-amino-pyrazolo [1, 5-a] pyrimidines with α-glucosidase inhibitory activity. Sci. Rep. 10, 2595. https://doi.org/10.1038/s41598-020-59079-z (2020).
Moghimi, S. et al. Design and synthesis of novel pyridazine N-aryl acetamides: In-vitro evaluation of α-glucosidase inhibition, docking, and kinetic studies. Bioorg. Chem. 102, 104071. https://doi.org/10.1016/j.bioorg.2020.104071 (2020).
Mohammadi-Khanaposhtani, M. et al. New biscoumarin derivatives as potent α-glucosidase inhibitors: Synthesis, biological evaluation, kinetic analysis, and docking study. Polycyclic Aromat. Compd. https://doi.org/10.1080/10406638.2018.1509359 (2018).
Mohammadi-Khanaposhtani, M. et al. Benzoylquinazolinone derivatives as new potential antidiabetic agents: α-Glucosidase inhibition, kinetic, and docking studies. J. Chin. Chem. Soc. 67, 856–863. https://doi.org/10.1002/jccs.201900268 (2020).
Nelson, D. L., Lehninger, A. L. & Cox, M. M. Lehninger Principles of Biochemistry (Macmillan, 2008).
Adler, A. Methods in Enzymology Vol. 27 (Academic Press, 1973).
Ross, P. D. & Subramanian, S. Thermodynamics of protein association reactions: Forces contributing to stability. Biochemistry 20, 3096–3102. https://doi.org/10.1021/bi00514a017 (1981).
Farhadian, S. et al. Insights into the molecular interaction between sucrose and α-chymotrypsin. Int. J. Biol. Macromol. 114, 950–960. https://doi.org/10.1016/j.ijbiomac.2018.03.143 (2018).
Pedrood, K. et al. Design, synthesis, and molecular docking studies of diphenylquinoxaline-6-carbohydrazide hybrids as potent α-glucosidase inhibitors. BMC Chem. 16, 1–13. https://doi.org/10.1186/s13065-022-00848-4 (2022).
SwissADME. (2022). http://www.swissadme.ch/.
Bischoff, H. Pharmacology of alpha-glucosidase inhibition. Eur. J. Clin. Invest. 24, 3–10. https://doi.org/10.1111/j.1365-2362.1994.tb02249.x (1994).
McIver, L. A. P. C. & Tripp, J. Acarbose. In StatPearls [Internet]. Treasure Island (FL) (StatPearls Publishing, 2023). https://www.ncbi.nlm.nih.gov/books/NBK493214/. Accessed 21 Sep 2022.
Ahr, H. et al. Pharmacokinetics of acarbose. Part I: Absorption, concentration in plasma, metabolism and excretion after single administration of [14C] acarbose to rats, dogs and man. Arzneimittelforschung 39, 1254–1260 (1989).
Daina, A., Michielin, O. & Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7, 42717. https://doi.org/10.1038/srep42717 (2017).
Dixon, M. The determination of enzyme inhibitor constants. Biochem. J. 55, 170. https://doi.org/10.1042/bj0550170 (1953).
Todd, M. J. & Hausinger, R. Competitive inhibitors of Klebsiella aerogenes urease: Mechanisms of interaction with the nickel active site. J. Biol. Chem. 264, 15835–15842. https://doi.org/10.1016/S0021-9258(18)71553-6 (1989).
Barker, M. K. & Rose, D. R. Specificity of processing α-glucosidase I is guided by the substrate conformation: Crystallographic and in silico studies. J. Biol. Chem. 288, 13563–13574. https://doi.org/10.1074/jbc.M113.460436 (2013).
Adasme, M. F. et al. PLIP 2021: Expanding the scope of the protein–ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 49, W530–W534. https://doi.org/10.1093/nar/gkab294 (2021).
Acknowledgements
This work was supported and funded by Department of Medicinal Chemistry, Faculty of Pharmacy, Tehran University of Medical Sciences, Tehran, Iran; Gran No. 1400-2-104-54840.
Author information
Authors and Affiliations
Contributions
A.F. and F.P. designed the study and conducted the experiments. F.P., F.S.H., and M.H. synthesized the targeted compounds. F.P., Z.E., and L.F. wrote the manuscript, analyzed the characterization data, and prepared the Supporting Information File. B.B. and M.S.M carried out the docking studies and in silico ADME. S.M. and M.A.F. performed the in vitro enzymatic analysis, kinetic study, circular dichroism spectroscopy, fluorescence spectroscopy measurements, and thermodynamic analysis. S.E.S. and M.B.T. revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Peytam, F., Hosseini, F.s., Hekmati, M. et al. Imidazo[1,2-c]quinazolines as a novel and potent scaffold of α-glucosidase inhibitors: design, synthesis, biological evaluations, and in silico studies. Sci Rep 13, 15672 (2023). https://doi.org/10.1038/s41598-023-42549-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-42549-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
