
Abstract
Glioblastomas are the most aggressive brain tumors for which therapeutic options are limited. Current therapies against glioblastoma include surgical resection, followed by radiotherapy plus concomitant treatment and maintenance with temozolomide (TMZ), however, these standard therapies are often ineffective, and average survival time for glioblastoma patients is between 12 and 18 months. We have previously reported a strong anti-glioblastoma activity of several metabolic compounds, which were synthetized based compounds, which were synthetized based on the chemical structure of a common lipid-lowering drug, fenofibrate, and share a general molecular skeleton of benzoylphenoxyacetamide (BPA). Extensive computational analyses of phenol and naphthol moieties added to the BPA skeleton were performed in this study with the objective of selecting new BPA variants for subsequent compound preparation and anti-glioblastoma testing. Initially, 81 structural variations were considered and their physical properties such as solubility (logS), blood–brain partitioning (logBB), and probability of entering the CNS calculated by the Central Nervous System—Multiparameter Optimization (MPO-CNS) algorithm were evaluated. From this initial list, 18 compounds were further evaluated for anti-glioblastoma activity in vitro. Nine compounds demonstrated desirable glioblastoma cell toxicity in cell culture, and two of them, HR51, and HR59 demonstrated significantly improved capability of crossing the model blood–brain-barrier (BBB) composed of endothelial cells, astrocytes and pericytes.
Similar content being viewed by others
Introduction
Glioblastomas are the most aggressive brain tumors for which therapeutic options are very limited1,2. Current standard of care therapies include maximal surgical resection, followed by radiotherapy plus concomitant treatment and maintenance with temozolomide (TMZ), however, these standard therapies are often ineffective, contributing to the dismal glioblastoma patient survival time of 12–18 months3. Multiple genetic and epigenetic abnormalities have been found in glioblastomas, among which p53, EGFR, PTEN, and IDH mutations are the most common4,5,6. In spite of these validated therapeutic targets, molecular, gene-therapy, and immunotherapy approaches are still ineffective7,8. Therefore, new and more effective therapies for glioblastoma patients are desperately needed.
There are several reasons why it is difficult to treat glioblastoma. First, glioblastomas are characterized by many dysregulated pathways that cannot be blocked simultaneously with a single therapy9; Second, glioblastomas are highly infiltrating and create heterogenous tumors that are very difficult to be removed by surgery without compromising the function of the surrounding brain areas10; Third, within the heterogenous tumor tissue, glioma initiating cells (GICs) have been identified as a primary mechanism involved in the development of drug resistance11,12,13,14,15. GIC’s display an embryonic stem cell phenotype11,16,17,18 and express stem-cell markers including CD133, CD44, SOX2, L1CAM, CD15, integrin α6, and BMI118,19,20,21,22,23,24. In addition, GICs express ATP binding cassette (ABC) transporter proteins, which mediate extensive drug efflux contributing to radio- and chemo-resistance25, neovascularization26, and invasiveness27; Fourth, early diagnosis of glioblastoma is rare, therefore, large highly infiltrating and vascularized tumors are often present at the time of diagnosis28; Fifth, the optimization of clinical protocols for glioblastoma treatment requires the use of a reliable preclinical model/s. Unfortunately, commonly used rodent syngeneic and xenograft models have one major problem—the experimental tumors are typically ~ 103–104 times smaller than human tumors, and therefore, drug delivery, drug retention, and effective tissue penetration by the drug, cannot be tested in a reliable manner in small animal models29; and finally Six, the blood brain barrier (BBB) prevents the majority of anticancer drugs from reaching the tumor, and current methods that enhance the BBB penetration are still not effective for glioblastoma patients30.
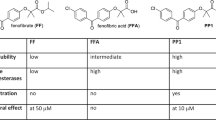
One of the drugs that readily crosses the BBB is temozolomide (TMZ). Upon oral administration, TMZ maximum plasma concentration can be reached in about one hour, and the elimination half-life is approximately 2.1 h. Importantly, penetration efficiency of TMZ into the CNS is experimentally estimated to be about 20% of the plasma levels31. Applying this estimate to calculate the logBB (Brain-Blood Distribution) for TMZ, this equation produces a value of − 0.7, which indicates sufficient capability of the compound to cross the BBB32. In spite of these positive features, TMZ-treated glioblastoma patients develop TMZ-resistance and recurrent tumors are practically incurable33. There are also several studies of the use of TMZ in combination with other drugs, which show beneficial therapeutic effects34,35. One interesting example is a combination of TMZ with lipid lowering drugs, including statins36. In addition, another class of lipid-lowering compounds, fibrates, have also attracted attention as a possible anticancer drugs37,38,39,40. We have previously reported that 50 µM fenofibrate (FF) has a strong anti-glioblastoma activity in cell culture, and in glioblastoma mouse models following intratumoral injection41 (Fig. 1). However, FF does not cross the BBB, and is quickly processed by the blood and tissue esterases to form fenofibric acid (FFA), which is no longer effective in triggering tumor cell death41,42.

Comparison between FF, FFA, and PP1 structural and anti-cancer properties. The information regarding the compounds water solubility, stability in human blood, and in vitro cytotoxicity were previously reported48. *Determined using LN229 monolayer cultures and MTT assay.
We have previously made several chemical modifications to the FF molecular skeleton, to address the FF low stability in human blood, low water solubility, and inability of penetrating the BBB. Indeed, one of the initial compounds, PP1, demonstrated improved water solubility and stability in human blood. In addition, PP1 was capable of triggering extensive glioblastoma cell death in vitro at concentrations over fourfold lower than FF43 (Fig. 1). To further improve anti-glioblastoma efficacy, we created other FF derivatives, which share the benzoyl-phenoxy-acetamide (BPA) molecular skeleton, and decided to test the addition of phenol and naphthol residues to the BPA structure due to the potential anti-cancer effects of these moieties44,45,46,47. As a result, 18 new compounds were generated and were analyzed during this study.
Results and discussion
Overall chemical design
In our previous studies we have explored the importance of a basic BPA skeleton48, and concluded that BPA could serve as a “pharmacophore”, necessary to retain anti-glioblastoma activity49,50. The amide part of the BPA skeleton can be specifically modelled to obtain a more desirable anti-tumor activity. This includes, among other properties, chemical and physical parameters (described below) that contribute to the increased BBB penetration, and possibly drug retention within the tumor tissue. In this regard, we have selected phenol and naphthol residues due to mounting evidence supporting the role of different derivatives of these compounds in health benefits51, including anti-cancer activities45,46. In this paper, three variants of BPA are discussed: a substituted phenol (Phenolic-BPA), and two naphtholic BPAs (1-Naphtholic-BPA and 2-Naphtholic-BPA) (Fig. 2) that serve as prototype molecules for further modifications.

Phenol region of BPA skeleton selected for modification (circle) in search of the optimal anti-glioblastoma drug.
The starting point for the preparation of all phenolic BPAs is fenofibric acid (FFA), and the corresponding aminophenol or aminonaphthol residues (Fig. 3) are added through amide (peptide) coupling reactions52,53. As previously reported48, due to the steric hindrance of the carboxylic group of FFA, which includes two methyl groups in the alpha position of carboxylic acid, combined with the lower amine nucleophilicity of anilines in DCC- or EDC-coupling, these reactions do not produce acceptable isolated yields. This occurs even with more reactive aminophenols and EDC or DCC, which are stronger nucleophiles compared to nonactivated anilines, which is expected to produce corresponding BPA compounds in acceptable yields54. However, we were able to detect only traces of the desirable products with these methods, and instead, decided to convert FFA into the more reactive fenofibrate chloride (FFC), followed by coupling with aminophenols or aminonaphthols (Fig. 3). We have explored several variations of this procedure and finally selected one that is very simple and can be applied to multigram and even multikilogram production scales. In particular, the FFC was prepared fresh and immediately used, for the next step of aminophenol addition (Fig. 3). The most common method of preparation of an acid chloride is by using thionyl chloride. This requires heating of thionyl chloride with the corresponding acid (in this case FFA) with appropriate traps for hydrochloric acid and sulfur dioxide, which are undesirable byproducts of the reaction55.

Schematic illustration of the procedure for preparation of hydroxylated phenyl and naphthyl derivatives of BPA.
Biological, chemical and computational testing of the compounds
Previously, we have reported that some amide derivatives of FF, including PP1, are more potent than others in eliminating glioblastoma cells43 and belong to the large family of BPA56. Further modifications of the BPA structure were designed to produce compounds with the improved ability to penetrate BBB, and superior glioblastoma cytotoxicity. This is highly relevant for glioblastoma patients, since one fundamental challenge for new drug development is the need for effective BBB penetration. So far, known BBB-permeable compounds form a very small subset of current oral drugs, and experimental models for testing BBB penetration by new drugs are complex and expensive. Therefore, an independent indicator of the BBB penetration is necessary for faster and more cost effective analyses of new anti-glioblastoma drug candidates. This is critical for the initial screening of a large number of compounds, including BPA variants, and for selection of best candidates for subsequent measurements of intracranial tumor drug penetration and anti-tumoral efficacy.
Therefore, we performed extensive molecular modeling prior to preparation of new BPA variants in order to evaluate their specific physicochemical properties considered relevant for the BBB penetration. We have applied first a weighted scoring approach named “Central Nervous System-Multiparameter Optimization” (CNS-MPO)57,58. The CNS-MPO algorithm uses 6 key physicochemical properties (clogP, clogD, MW, TPSA, HBD, and pKa) with the scores ranging between 0 and 6.0. Importantly, scores ≥ 4.0 are widely accepted for selecting compounds with high potential for CNS penetration58. The validation of this approach was based on a library of 616 compounds for which experimental distribution of the drug in CNS was determined and the corresponding parameters of the compound incorporated into CNS-MPO scores57,58. It was found that CNS-MPO scores of 1–2 (0%), 2–3 (11.6%), 3–4 (40.8%), 4–5 (53.8%) and 5–6 (81.1%) correlated with the increased probability of drugs to be found in the brain59.
In addition to CNS-MPO, other chemical and physical parameters of the Quantitative Structure Activity Relationship (QSAR) studies were also used in our calculations to further increase the probability of BBB penetration by the compound. These include: molecular polarizability (MP), minimal molecular projection area (MPA), water solubility (LogS), and lipophilicity (cLogP), to name a few. These parameters are not incorporated in the CNS-MPO score, however, they also play a role in the estimation of compound ability to penetrate the BBB60. Therefore, we have calculated and incorporated all these additional parameters when evaluating and selecting new HR compounds as potential anti-glioblastoma drugs (Figs. 4, 5, 6, 7, 8, 9); and Supplementary materials). In particular, MP is a response of electron distribution to an externally-applied static electrical field. Comparing MP values helps in understanding how different substrates may change polarization- and dispersion-type during interactions with the active sites of their interacting proteins61. It was postulated that MP values between 30 and 40 (see Supplementary data) are optimal for a molecule to bind to a biotarget62. Minimal projected area (MPA) represents another parameter that is important for drug transport and ultimately for drug activity. For instance, a distinct phenotypical pattern of drug recognition and transport for the G616N variant was reported, indicating that drug substrates with MPA over 70 Å2 are less likely to be transported compared to drugs with smaller MPA63. In addition, LogS of − 4.5 and greater are indicators of acceptable water solubility64, and the rate of passive diffusion is inversely proportional to the square root of molecular size (Graham’s law65), which are also included in our compound analysis.

Drug candidates with hydroxy substituted phenylamide moiety. (A) Cell viability (MTT assay) following exposure to modified variants of HR48 with one hydroxy groups in different positions in the phenylamide moiety (25 μM, for 72 h). (B) CV Cell viability (% of control) mean ± SD at 25 μM; ClogP calculated partitioning; HBD hydrogen bond donor at pH 7; HBA hydrogen bond acceptor at pH 7; logBB calculated blood–brain partition; PSA Polar surface area (Å2); MPA Minimal projection area (Å2); LogS Aqueous solubility (mg/mL); MPO central nervous system multiparameter optimization (CNS MPO). (C) Electrostatic potential map for H48-HR51. (D) Computed LUMO orbitals contribution with their energies. (E) Computed HOMO orbitals contribution with their energies generated by semi-empirical method PM3 as implemented in Spartan ’18 version 1.1.0.

Drug candidates with substituted 2-hydroxyphenylamide moiety. (A) Cell viability (MTT assay) following exposure to modified variants of HR48 with ortho hydroxy and ether methyl, chloro, or carboxy group in the phenylamide moiety (25 μM, for 72 h). (B) CV Cell viability (% of control) mean ± SD at 25 μM; ClogP calculated partitioning; HBD hydrogen bond donor at pH 7; HBA hydrogen bond acceptor at pH 7; logBB calculated blood–brain partition; PSA polar surface area (Å2); MPA minimal projection area (Å2); LogS aqueous solubility (mg/ml); MPO Central nervous system multiparameter optimization (CNS MPO). (C) Electrostatic potential map for H52-HR55. (D) Computed LUMO orbitals contribution with their energies. (E) Computed HOMO orbitals contribution with their energies generated by semi-empirical method PM3 as implemented in Spartan ’18 version 1.1.0.

Drug candidates with nitro-hydroxy and two hydroxy substituted phenylamide moiety. (A) Cell viability (MTT assay) following exposure to modified variants of HR48 with one hydroxy and one nitro group or with two hydroxy groups in the phenylamide moiety (25 μM, for 72 h). (B) CV cell viability (% of control) mean ± SD at 25 μM; ClogP calculated partitioning; HBD hydrogen bond donor at pH 7; HBA hydrogen bond acceptor at pH 7; logBB calculated blood–brain partition; PSA polar surface area (Å2); MPA minimal projection area (Å2); logS aqueous solubility (mg/mL); MPO Central nervous system multiparameter optimization (CNS MPO). (C) Electrostatic potential map for H56-HR59. (D) Computed LUMO + 1 orbitals contribution with their energies. (E) Computed LUMO orbitals contribution with their energies. (F) Computed HOMO orbitals contribution with their energies generated by semi-empirical method PM3 as implemented in Spartan ’18 version 1.1.0.

Drug candidates with hydroxy substituted naphthylamide moiety. (A) Cell viability (MTT assay) following exposure to modified variants of 1- and 2-naphthylamide of HR60 and HR64 with one hydroxy group in the naphthylamide moiety (25 μM, for 72 h). (B) CV Cell viability (% of control) mean ± SD at 25 μM; ClogP calculated partitioning; HBD hydrogen bond donor at pH 7; HBA hydrogen bond acceptor at pH 7; logBB calculated blood–brain partition; PSA polar surface area (Å2); MPA minimal projection area (Å2); logS aqueous solubility (mg/ml); MPO central nervous system multiparameter optimization (CNS MPO). (C) Electrostatic potential map for H60-HR65. (D) Computed LUMO orbitals contribution with their energies. (E) Computed HOMO orbitals contribution with their energies generated by semi-empirical method PM3 as implemented in Spartan ’18 version 1.1.0.

Compiled three variation of computing logBB and estimated MPO-CNS values for HR48-HR65. logP computed partition, PSA computed polar surface area; ClogP calculated partitioning; PSA polar surface area (Å2); D = hydrogen bond donor at pH 7; A = hydrogen bond acceptor at pH 7; MPO Central nervous system multiparameter optimization (CNS MPO). M1LogBB = 0.5159 × log P − 0.0277 × PSA − 0.3462. For log BB ≥ 0.332. M2logBB = 0.152logP − 0.0148PSA + 0.139. (Clark’s model). M3logBB = 0.155 × logP − 0.01 × PSA + 0.164. (Rishton’s model). M4log BB = 0.2289 × log P − 0.0326 × PSA − 0.5671 × (D + A) + 2.3420. For log BB ≥ − 132.

Cytotoxic effects of selected HR compounds. (A) Effects of 10 and 25 μM of HR51 and HR59 on the survival of human glioblastoma cell lines, LN229, U-87MG, U-118 MG, and T98G, compared to the effects of these two compounds on the survival of normal human astrocytes (NHA; LONZA/Clonetics™). Data were collected after 72 h of a continuous cell exposure to a single dose of HR51 or HR59 in the low glucose medium (1 g/L). Cells treated with DMSO (vehicle) were used as control. Data are expressed as cell viability (MTT, % of control) and represent average values with standard deviation (n = 3). (B) Dose response curves and IC50 values calculated for two most promising HR compounds, HR51 and HR59. Cell viability was evaluated by the MTT assay performed after exposure of LN229 to HR51 and HR59 for 72 h. Data represent mean values ± SD (n = 3). (C) Effects of HR51 and HR59 (25 μM each) on the sphere formation by patient-derived glioblastoma isolates (GBM12). In the sphere formation assay, low density single cell suspension of GBM12 cells was exposed to the serum-free stem-cell supporting medium (StemPro NSC SFM; Gibco: A1050901) supplemented with Recombinant Human FGF Basic and EGF (10 μg each), as previously reported66,72. The cells were allowed to form spheres for 5 days in the presence or absence of HR51 and HR59. DMSO-treated cultures (vehicle) were used as positive control for the sphere formation. In this condition, GBM12 cells grow in suspension and display stem-like phenotype—formation of multicellular gliospheres in low density cultures66. (D) Metabolic effects of HR51 and HR59 compared to the prototype drug, PP1. Metabolic responses to the drugs were evaluated in LN229 using Extracellular Flux Analyzer XF96 (Seahorse/Agilent). The oxygen consumption rate (OCR; indicative of mitochondrial respiration) was evaluated after injecting DMSO, (negative control), PP1 (positive control) and two experimental drugs, HR51 and HR59, followed by sequential injections of oilgomycin, FCCP, and rotenone (mitochondrial stress assay). Average OCR data were calculated from three independent experiments. Data represent average values ± SD. Compared to negative control (DMSO), all tested metabolic compounds (PP1, HR51 and HR59) triggered an immediate drop in OCR. In addition, the cells treated with these three compounds did not respond to FCCP injection, indicating loss of the proton gradient across the mitochondrial membrane.
Finally, the ability of the compound to penetrate the BBB can also be expressed as a decimal logarithm of brain to plasma concentration ratio (logBB), which is derived from the modified Clark’s equation: logBB = 0.152 ClogP-0.0148PSA + 0.139 30. It has been shown that compounds with logBB > 0.3 readily cross the BBB, while the compounds with logBB < − 1.0 have a problem with the CNS penetration32. Therefore, LogBB values for all HR compounds were also calculated and included (Figs. 4, 5, 6, 7, 8).
Once all above calculations were performed, initial cell viability (CV) tests were performed for all HR compounds using LN229 human glioblastoma cell line. The cells were treated with proposed HR compounds at 25 μM, and cell viability was evaluated using MTT assay, following a continuous cell exposure to a single dose of the drugs for 72 h. (Figs. 4, 5, 6, 7, 8, 9).
As a result of this initial screening, we have identified two lead drug candidates, HR51 and HR59, with phenolic moieties that contain BPA structural skeleton similar to our prototype anti-glioblastoma compound PP143. This is in addition to our recently reported BPA-based compounds (HR28, HR32, HR37, and HR46), which also demonstrated high potential as anti-glioblastoma drugs48. Anti-glioblastoma effects of HR51 and HR59 were subsequently confirmed using four different human glioblastoma cell lines, LN229, U-87 MG, U-118 MG, T98G, and the cytotoxicity data were compared to normal human astrocytes (NHA). Results in Fig. 9A demonstrate that all tested glioblastoma cells were partially responsive to 10 µM HR51 and HR59, but were almost completely eliminated following 72 h exposure to 25 µM HR51 or HR59. In contrast, these two compounds were significantly less cytotoxic to normal human astrocytes (NHA), indicating that these two new compounds may have low CNS toxicity. In addition, results in Fig. 9B show that IC50 concentrations for HR51 and HR59 are below 10 μM, which is an acceptable therapeutic concentration for clinically relevant anticancer drugs.
Another important feature shared by HR51 and HR59 is the compounds ability to interfere with the sphere formation and proliferation of GBM12—patient-derived glioblastoma cells in low density 3-D cultures designed for propagation of glioma initiating cells (GICs)66. In this sphere formation assay, low density single cell suspension cultures of GBM12 were exposed to serum-free, growth factor supplemented medium (see “Methods”). As shown in Fig. 9C, only DMSO-treated GBM12 cells (vehicle control) were able to proliferate and formed large multicellular gliosphere structures. In the presence of 25 μM HR51 or 25 μM HR59 the cells did not proliferate (not shown), and their ability to form 3-D gliospheres was severely attenuated, indicating that not only bulk tumor cells but also tumor stem-like cells are the targets for HR51 and HR59.
We have also tested if the mechanism of action of HR51 and HR59 is similar to our prototype drugs, PP1 and fenofibrate, which have been previously shown to inhibit mitochondrial respiration at the level of Complex 1 of the electron transport chain (ETC)41,43. Indeed, results in Fig. 9C confirmed that both HR51 and HR59 inhibit mitochondrial respiration by the magnitude similar to PP1.
In addition to a strong in vitro anti-glioblastoma activity (Fig. 9), HR51 and HR59 have physical properties that may contribute to the improved brain tumor penetration. Specifically, HR51 and HR59, have a minimal projection area (MPA) of 46.23 Å2 and 43.73 Å263, respectively; water solubility (LogS) of − 6.61 and − 6.1164; and brain to plasma concentration ratio (LogBB) of − 0.15 and − 0.4932, which are all considered as highly promising for compounds suspected of being capable of penetrating the brain tumor tissue.
Importantly, HR51 and HR59 can also cross the triple-coculture model of the blood brain barrier (BBB), which consists of astrocytes, pericytes and epithelial cells cultured on 24-well (3 µm pores) transwell membranes (Fig. 10A), prepared according to the protocol provided by Stone et al.67. All experiments in which the ability of HR compounds to cross the BBB were evaluated using the BBB inserts that demonstrated a significant increase of the electric resistance (Ω) in comparison to inserts without cells (ΩBBB − ΩInsert) (Fig. 10B). In particular, ΩBBB − ΩInsert values of 105.3 ± 16; 110.6 ± 14; 117.8 ± 19; 132.3 ± 23; and 129.0 ± 24 were measured for inserts used to evaluate BBB penetration of FF, caffeine, HR51 and HR59, respectively (Fig. 10B). Accordingly, trans-endothelial electric resistance (TEER = ΩBBB − ΩInsert × area of the membrane)67,68 for the inserts used for FF, caffeine, HR51 and HR59 testing are: 34.7 ± 5.3; 36.5 ± 4.6; 43.7 ± 7.6; and 42.6 ± 8.1, respectively. These TEER values are comparable to TEER values obtained from similar triple-coculture model of the BBB in which 24-well inserts with 3 μm pores were previously evaluated67, indicating effective electric resistance produced by our BBB model.

Penetration of the selected HR compounds through in vitro BBB model membrane: (A) Schematic representation of a triple-coculture model of the BBB, which consists of astrocytes, pericytes and epithelial cells cultured on 24-well transwell membranes with 3 μm pores. Trans-endothelial electric resistance (TEER) was measured using a EVOM2 meter with a STX3 electrode (World Precision Instruments). (B) Measurements of the electric resistance of inserts used for specific compounds (Ω); Difference between resistance of the insert with established BBB versus empty insert (ΩBBB − Ωinsert); and trans-endothelial electric resistance (TEER; ΩBBB − Ωinsert/cm2). * TEER coefficient = TEERcompound/TEERcaffeine. TEER coefficients were used to normalize BBB permeability (P) values to compensate for differences in TEER values between inserts selected for each compound. (C) Differences in BBB permeability (P) calculated using P = VA·CA/(t·S·CL) eqution70 and normalized by TEER coefficient. Data represent average values from 2 independent experiments in triplicates (n = 6) with standard deviation SD. *Indicates values significantly different from fenofibrate (internal negative control).
Following TEER measurements, 25 μM HR51, 25 μM HR59, as well as 50 μM caffeine (positive control69) and 25 μM fenofibrate (negative control42) were added to the corresponding insert and the plates were incubated in 37 °C and 5% CO2 for 24 h. Subsequently, aliquots of media from the corresponding inserts and from the wells were collected for HPLC measurements and to calculate BBB permeability (P = VA·CA/(t·S·CL)70. Results in Fig. 10C show that HR51, HR59 and caffeine cross the in vitro BBB at levels 4.4-fold, 3.5-fold and 22.0-fold higher compared to our internal negative control, fenofibrate, which although has a similar molecular weight and structure to the tested HR compounds (Fig. 4), its ability of crossing natural BBB is very low42.
By exploring the computed structural variations of phenolic-BPAs, and testing their cell toxicity, we have demonstrated that the addition of phenol moieties improves anti-glioblastoma activity with acceptable LogBB and LogS properties. This could be further improved by adding additional substituent(s) to the phenol moiety, including hydroxy group, halogen, alkyl, nitro on carbonyl to name a few. However, replacing the phenol residue with the larger naphthol, although it might not compromise anti-glioblastoma activity in cell culture, it may result in decreased compound bioavailability mostly due to low water solubility (Fig. 7)71. Therefore, these naphthol compounds are less likely to be considered as anti-glioblastoma candidates.
Presented in silico analyses of phenolic BPAs in combination with the in vitro BBB penetration model, represent a quick and relatively inexpensive way of testing a large number of new glioblastoma drug candidates. The observed effective penetration of the BBB model membranes by HR51 and HR59 is therapeutically promising, and it also validates our overall strategy for designing and selecting compounds with the most favorable physiochemical parameters for the BBB penetration (compare Figs. 4, 6 and 10). It should be emphasized, however, that these in silico and in vitro assays, although quite effective in narrowing down the number of potential glioblastoma drugs, cannot replace detailed pharmacokinetic analyses (tissue bioavailability and time of clearance) in clinically relevant intracranial glioblastoma animal model/s. Further experiments are necessary to confirm penetration and retention of the selected drug candidate/s in the brain tumor tissue at therapeutically-relevant concentrations.
Methods
All starting materials were reagent grade and purchased from Sigma–Aldrich, ArkPharm, and TCI America. 1H-NMR spectra were recorded on Varian Mercury 300 and Varian Mercury 400 Plus instruments in CDCl3 and DMSO-d6, using the solvent chemical shifts as an internal standard48. All computed molecular descriptors were generated by ChemAxon MarvinSketch version 19.20. Electrostatic potential maps were calculated with a PM3 semi-empirical method as implemented in Spartan ’18 v 1.1.048. 1H-NMR and 13C-NMR spectra for all HR compounds generated in this study are included in Supplementary Materials.
Method A (Larger scale preparation without extraction or crystallization)
2-(4-(4-chlorobenzoyl)phenoxy)-N-(2-hydroxyphenyl)-2-methylpropanamide (HR49). Fenofibric chloride (FFC) was freshly prepared from FFA (4.8 g; 0.015 mol) and oxalyl chloride (2.6 mL; 3.8 g; 0.03 mmol). After drying with Argon, FFC was dissolved in dichloromethane (30 mL) and mixed with a pyridine (50 mL) solution of 2-aminophenol (1.3 g; 0.012 mol). The resulting solution was stirred at room temperature for 3 h, and then at 70 °C for an additional 3 h with distillation of dichloromethane. The pyridine was removed under reduced pressure. The resulting solid substance was mixed with ethanol (50 mL) and the resulting mixture was refluxed by stirring until all of the solid was dissolved. This resulting clear alcohol solution was mixed with 3% sodium carbonate solution (300 mL) and heated with stirring at 80 °C for one hour. After cooling to room temperature, the insoluble product was separated by filtration, extensively washed with water (10 × 100 mL) and dried at 110 °C for 1 h. The isolated yield was 97% (4.77 g). 1H-NMR (DMSO-d6, 400 MHz) δ 9.95 (1H, s, OH), 9.09 (1H, s, NH), 7.93 (1H, d, J = 8.0 Hz), 7.73 (2H, d, J = 8.0 Hz), 7.70 (2H, d, J = 8.4 Hz), 7.59 (2H, d, J = 8.4 Hz), 7.11 (2H, d, J = 8.4 Hz), 6.23 (1H, t, J = 7.6 Hz), 6.83 (1H, d, J = 8.0 Hz), 6.78 (1H, t, J = 8.0 Hz), and 1.60 (6H, s) ppm. 13C-NMR (DMSO-d6) δ 193.8, 171.7, 158.9, 147.7, 137.6, 136.5, 132.2, 131.7, 131.3, 129.1, 126.1, 125.1, 121.2, 119.9, 119.6, 115.5, 82.3 and 25.3 ppm.
Method B (small scale preparation)
Preparation of 2-(4-(4-chlorobenzoyl)phenoxy)-N-(2-hydroxy-5-methylphenyl)-2-methylpropanamide (HR52). A dichloromethane (10 mL) suspension of fenofibric acid (FFA) (191 mg; 0.6 mmol) and oxalyl chloride (0.2 mL; 384 mg; 2 mmol) was stirred at room temperature for five minutes. A few drops of N,N-dimethylformamide (DMF) were then added to the suspension, which induced bubbling, and resulted in a clear reaction mixture after approximately 30 min. This solution was then stirred at 60 °C to promote slow solvent evaporation. The solvent residue and oxalyl chloride were removed by drying under an Argon flow at room temperature. The resulting yellow solid substance was dissolved in dichloromethane (10 mL) and mixed with 2-amino-4-methylphenol (62 mg; 0.5 mmol) in THF (10 mL) and Na2CO3 (1.06 g; 10 mmol) in water (10 mL). The subsequent mixture was stirred at room temperature for five hours. The solvent was then evaporated under reduced pressure and the residue was mixed with dichloromethane (50 mL) and water (50 mL). This final mixture was sonicated at room temperature until all solid was dissolved. From this bilayer solution, the water layer was discarded, and the dichloromethane layer was washed with 5% Na2CO3 (3 × 50 mL), water (50 mL), 5% HCl (3 × 50 mL), water (50 mL) and dried over anhydrous Na2CO3. After solvent evaporation, the final product was purified by crystallization from dichloromethane (~ 3 mL) and hexane (20 mL). The isolated yield was 90% (190 mg). 1H-NMR (DMSO-d6, 400 MHz) δ 9.74 (1H, broad s, OH), 9.06 (1H, s, NH), 7.81 (1H, s), 7.73 (2H, d, J = 8.8 Hz), 7.70 (2H, d, J = 8.8 Hz), 7.58 (2H, d, J = 8.8 Hz), 7.11 (2H, d, J = 8.4 Hz), 6.73 (2H, s), 2.18 (3H, s), and 1.60 (6H, s) ppm. 13C-NMR (DMSO-d6) δ 193.8, 171.6, 158.8, 145.4, 137.7, 136.5, 132.2, 131.7, 131.3, 129.1, 128.1, 125.9, 125.3, 121.5, 119.9, 115.2, 82.2, 25.3, and 20.9 ppm.
Cell culture and viability assays
All cell culture procedures used for HR compound testing were previously described43,48. Briefly, human glioblastoma cell line, LN-229 (ATCC# CRL-2611), U-87MG (ATCC# HTB-14), U-118 MG (ATCC# HTB-15) and T98G (ATCC# CRL-1690) were maintained as semi-confluent monolayer cultures in DMEM (1 g/L glucose; with sodium pyruvate and l-glutamine) supplemented with 100 U/ml penicillin, 100 μg/mL streptomycin, and 10% fetal bovine serum (FBS) at 37 °C in a 5% CO2 atmosphere. GBM12 are patient-derived human glioblastoma cells established in Dr. Sarkaria's lab33,73. GBM12 cells are routinely propagated in subcutaneous tissue of nude mice (IACUC protocol # 3750,LSUHSC, New Orleans), and were isolated from the tumor tissue for short-term monolayer and suspension cultures as previously reported41. For three-dimensional (3-D) gliosphere cultures, the cells aggregates were dissociated with Accutase (Innovative Cell Technologies: AT104), and low density single cell suspension cultures of GBM12 (1 × 104/mL) were incubated in the serum-free growth factor-supplemented stem-cell medium (StemPro NSC SFM: A1050901, which consists of KnockOut DMEM/F-12 Basal Medium (500 mL), plus StemPro NSC SFM Supplement (10 mL), 10 μg recombinant human basic FGF and 10 μg EGF)66. In the sphere-formation assay, the cells were allowed to form 3-D gliospheres, and the sphere morphology was evaluated 5 days following the plating.
Normal human astrocytes (NHA) were used to evaluate effects the selected HR compounds may have on normal non-transformed cells. NHA were cultured according to the manufacturer protocol (LONZA/Clonetics™). Prior to the treatment with HR compounds, the cells were plated in 96-well plates (BD Falcon) at the initial density of 2 × 104/well. Twenty-four hours after plating, stock solutions of the HR compounds were prepared in DMSO, diluted in cell culture medium, to the final concentration of 25 µM, and added to the cells in triplicate for every experimental condition. For the vehicle control, DMSO was used at 0.5%. After 72 h incubation, MTT assay (surrogate for cell viability) was performed according to our previous publications48. Briefly, following 1.5 h incubation with MTT, formazan crystals were dissolved in 5 mM HCl in isopropanol and the absorbance read at 540 nm. Data represent mean values expressed as % cell viability of control (DMSO) ± SD. Phase contrast images of treated cells were taken 72 h after treatment with HR compounds with a BZ-X800 fluorescence microscope (Keyence) equipped with a 20 × objective. The drug dose causing 50% inhibition in the MTT assay at 72 h time point (half maximal inhibitory concentration—IC50) was calculated using GraphPad Prism 8.
In vitro model of the blood brain barrier (BBB)
The blood brain barrier was re-created in vitro using a modified protocol provided by Stone et al.67. Briefly, 24-well transwell inserts (Falcon, catalog number 353096) were coated with 10 μg/cm2 of Collagen Type IV from human placentas (Sigma) for 24 h at 4 °C. Inserts were washed with sterile water and air-dried for 2 h. Next, the inserts were coated with 2 μg/cm2 poly-l-lysine (ScienCell) for 1 h at 37 °C, then washed twice with sterile H20 and air-dried for 2 h. 1.57 × 105 primary human astrocytes (ScienCell) and 3.125 × 104 primary human pericytes (ScienCell) were resuspended in 25 μL of astrocyte medium and pericyte medium (ScienCell), respectively, then combined in a 1:1 ratio for 50 μL total volume. Dried, coated inserts were turned upside down such that the basolateral surface was exposed at the top, and 50 μL of the cell mixture was added to the membrane, covered with the plate lid, and incubated for 2 h at 37 °C to allow cell adherence. Any medium remaining on top of the membrane was carefully removed before returning inserts to their upright position with the apical surface facing upward as they were placed in a 24-well plate containing 500 μL per well of astrocyte/pericyte medium (1:1). An additional 300 μL of medium was added to the apical compartment. Four days after plating, the apical compartment medium was removed, and 3.75 × 104 of telomerase-immortalized vain endothelial cells (TIVE; provided by Dr. Rolf Renne) in 50 μL of TIVE medium74 were added and incubated for 5 h at 37 °C to allow cell adherence, followed by the addition of an extra 250 μL of TIVE medium. Half the volume of the corresponding media in the lower and upper compartment was replaced with fresh media every third day. Ten days after initial plating, trans-endothelial electric resistance (TEER) was measured using a EVOM2 meter with a STX3 electrode (World Precision Instruments). The ability of selected HR compounds to pass through in vitro BBB was tested using inserts with effectively reconstructed BBB as confirmed by TEER67,68.
HPLC detection of selected HR compounds
Following TEER measurement, the medium from apical compartment of the in vitro BBB model (Fig. 10A) was replaced with 350 μL of fresh TIVE medium containing corresponding compounds [HR49 (PP99), HR51 (PP12), HR59 (PP171)] all used at 25 μM. In addition, 25 μM fenofibrate (FF), which does not cross the BBB42 was used as negative control, and 50 μM caffeine was used as a positive control69. Plates containing the inserts were returned to the incubator (37 °C, 5% CO2), and after 24 h of incubation conditioned media from basolateral (well) and apical (insert) compartments were collected and frozen for quantitative analyses by HPLC. 100 μL aliquots of the collected samples were subsequently mixed with 100 μL of 100% acetonitrile, samples were centrifuged at 16,000 rpm at 4 °C for 10 min and supernatants collected for HPLC analyses using UltiMate 3000 system (Thermo Scientific) equipped with analytical YMCbasic, 3 µm, 150 × 4.6 mm column (octyl silane C8; YMC America, Inc.). Isocratic elution of the compounds was performed using mobile phase composed of solvent A (50 mM acetic acid in dH2O) and solvent B (acetonitrile) mixed at ratios predetermined for each compound (Table 1). All separations were carried out with sample volume of 5 μL at flow rate of 1 mL/min, at 20 °C. Concentration of each compound was calculated using serial dilutions of the known concentration of the compound separated at the same run with experimental and control samples. After separation, integrated areas under the peak were used to prepare calibration curves and to determine concentration of the compounds.
Evaluation of metabolic parameters
Metabolic responses of human glioblastoma cells were evaluated with Extracellular Flux Analyzer XFe96 (Agilent Technologies) according to our previously established protocol43. During the day prior to each assay the cells were plated at 2 × 104 cells/well in Agilent Seahorse 96-well XF cell culture microplates with growth supporting media and incubated overnight. At the time of measurement, growth media were replaced with serum-free XF assay medium (Seahorse XF Base Medium supplemented with 1 mM sodium pyruvate, 2 mM glutamine, and 5.5 mM glucose) and cartridges equipped with oxygen-sensitive and pH-sensitive fluorescent probes were placed above the cells. The oxygen consumption rate (OCR; indicative of mitochondrial respiration) was evaluated after injecting HR compounds or PP1 (all used at 25 μM), or DMSO (0.1%; vehicle control). These initial injections were followed by sequential injections of metabolic toxins to execute mitochondrial stress assay: oligomycin (inhibitor of ATP synthase; 0.5 μM); carbonylcyanide-p-trifluoromethoxyphenylhydrazone (FCCP; uncoupling factor; 0.5 μM), rotenone (inhibitor of mitochondrial complex I; 0.3 μM), and antimycin A (inhibitor of mitochondrial complex III; 0.3 μM).
References
Nakada, M. et al. Molecular targets of glioma invasion. Cell Mol. Life Sci. 64, 458–478 (2007).
Terzis, A. J., Niclou, S. P., Rajcevic, U., Danzeisen, C. & Bjerkvig, R. Cell therapies for glioblastoma. Expert Opin. Biol. Ther. 6, 739–749 (2006).
Montemurro, N. Glioblastoma multiforme and genetic mutations: The issue is not over yet. An overview of the current literature. J. Neurol. Surg. A 81, 64–70. https://doi.org/10.1055/s-0039-1688911 (2020).
Brennan, C. W. et al. The somatic genomic landscape of glioblastoma. Cell 155, 462–477. https://doi.org/10.1016/j.cell.2013.09.034 (2013).
Network, T. C. Corrigendum: Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 494, 506. https://doi.org/10.1038/nature11903 (2013).
Wu, F. et al. Molecular classification of IDH-mutant glioblastomas based on gene expression profiles. Carcinogenesis https://doi.org/10.1093/carcin/bgz032 (2019).
Bagley, S. J. & O’Rourke, D. M. Clinical investigation of CAR T cells for solid tumors: Lessons learned and future directions. Pharmacol. Ther. 205, 107419. https://doi.org/10.1016/j.pharmthera.2019.107419 (2019).
Romani, M., Pistillo, M. P., Carosio, R., Morabito, A. & Banelli, B. Immune checkpoints and innovative therapies in glioblastoma. Front. Oncol. 8, 464. https://doi.org/10.3389/fonc.2018.00464 (2018).
Alifieris, C. & Trafalis, D. T. Glioblastoma multiforme: Pathogenesis and treatment. Pharmacol. Ther. 152, 63–82. https://doi.org/10.1016/j.pharmthera.2015.05.005 (2015).
Perry, A. & Wesseling, P. Histologic classification of gliomas. Handb. Clin. Neurol. 134, 71–95. https://doi.org/10.1016/B978-0-12-802997-8.00005-0 (2016).
Ignatova, T. N. et al. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia 39, 193–206. https://doi.org/10.1002/glia.10094 (2002).
Singh, S. K., Clarke, I. D., Hide, T. & Dirks, P. B. Cancer stem cells in nervous system tumors. Oncogene 23, 7267–7273 (2004).
Singh, S. K. et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 63, 5821–5828 (2003).
Singh, S. K. et al. Identification of human brain tumour initiating cells. Nature 432, 396–401 (2004).
Yuan, X. et al. Isolation of cancer stem cells from adult glioblastoma multiforme. Oncogene 23, 9392–9400. https://doi.org/10.1038/sj.onc.1208311 (2004).
Berger, F., Gay, E., Pelletier, L., Tropel, P. & Wion, D. Development of gliomas: Potential role of asymmetrical cell division of neural stem cells. Lancet Oncol. 5, 511–514. https://doi.org/10.1016/S1470-2045(04)01531-1 (2004).
Davis, B. et al. Comparative genomic and genetic analysis of glioblastoma-derived brain tumor-initiating cells and their parent tumors. Neuro Oncol. https://doi.org/10.1093/neuonc/nov143 (2015).
Sundar, S. J., Hsieh, J. K., Manjila, S., Lathia, J. D. & Sloan, A. The role of cancer stem cells in glioblastoma. Neurosurg. Focus 37, E6. https://doi.org/10.3171/2014.9.FOCUS14494 (2014).
Brescia, P. et al. CD133 is essential for glioblastoma stem cell maintenance. Stem cells 31, 857–869. https://doi.org/10.1002/stem.1317 (2013).
Brown, D. V. et al. Coexpression analysis of CD133 and CD44 identifies proneural and mesenchymal subtypes of glioblastoma multiforme. Oncotarget 6, 6267–6280 (2015).
Richichi, C., Brescia, P., Alberizzi, V., Fornasari, L. & Pelicci, G. Marker-independent method for isolating slow-dividing cancer stem cells in human glioblastoma. Neoplasia 15, 840–847 (2013).
Abdouh, M. et al. BMI1 sustains human glioblastoma multiforme stem cell renewal. J. Neurosci. 29, 8884–8896. https://doi.org/10.1523/JNEUROSCI.0968-09.2009 (2009).
Facchino, S., Abdouh, M., Chatoo, W. & Bernier, G. BMI1 confers radioresistance to normal and cancerous neural stem cells through recruitment of the DNA damage response machinery. J. Neurosci. 30, 10096–10111. https://doi.org/10.1523/JNEUROSCI.1634-10.2010 (2010).
Siddique, H. R. & Saleem, M. Role of BMI1, a stem cell factor, in cancer recurrence and chemoresistance: Preclinical and clinical evidences. Stem Cells 30, 372–378. https://doi.org/10.1002/stem.1035 (2012).
Eramo, A. et al. Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ. 13, 1238–1241. https://doi.org/10.1038/sj.cdd.4401872 (2006).
Bao, S. et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 66, 7843–7848. https://doi.org/10.1158/0008-5472.CAN-06-1010 (2006).
Chen, C., Xu, T., Lu, Y., Chen, J. & Wu, S. The efficacy of temozolomide for recurrent glioblastoma multiforme. Eur. J. Neurol. 20, 223–230. https://doi.org/10.1111/j.1468-1331.2012.03778.x (2013).
Rulseh, A. M. & Vymazal, J. Whole brain apparent diffusion coefficient measurements correlate with survival in glioblastoma patients. J. Neurooncol. 146, 157–162. https://doi.org/10.1007/s11060-019-03357-y (2020).
Biasibetti, E. et al. Comparison of allogeneic and syngeneic rat glioma models by using MRI and histopathologic evaluation. Comp. Med. 67, 147–156 (2017).
Harder, B. G. et al. Developments in blood-brain barrier penetrance and drug repurposing for improved treatment of glioblastoma. Front. Oncol. 8, 462. https://doi.org/10.3389/fonc.2018.00462 (2018).
Ostermann, S. et al. Plasma and cerebrospinal fluid population pharmacokinetics of temozolomide in malignant glioma patients. Clin. Cancer Res. 10, 3728–3736. https://doi.org/10.1158/1078-0432.CCR-03-0807 (2004).
Vilar, S., Chakrabarti, M. & Costanzi, S. Prediction of passive blood-brain partitioning: Straightforward and effective classification models based on in silico derived physicochemical descriptors. J. Mol. Graph. Model. 28, 899–903. https://doi.org/10.1016/j.jmgm.2010.03.010 (2010).
Kitange, G. J. et al. Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts. Neuro Oncol. 11, 281–291. https://doi.org/10.1215/15228517-2008-090 (2009).
Gao, L. et al. Suppression of glioblastoma by a drug cocktail reprogramming tumor cells into neuronal like cells. Sci. Rep. 9, 3462. https://doi.org/10.1038/s41598-019-39852-5 (2019).
Lu, G. et al. Triple-drug therapy with bevacizumab, irinotecan, and temozolomide plus tumor treating fields for recurrent glioblastoma: A retrospective study. Front. Neurol. 10, 42. https://doi.org/10.3389/fneur.2019.00042 (2019).
Maraka, S. et al. Phase 1 lead-in to a phase 2 factorial study of temozolomide plus memantine, mefloquine, and metformin as postradiation adjuvant therapy for newly diagnosed glioblastoma. Cancer 125, 424–433. https://doi.org/10.1002/cncr.31811 (2019).
Drukala, J. et al. ROS accumulation and IGF-IR inhibition contribute to fenofibrate/PPARalpha-mediated inhibition of glioma cell motility in vitro. Mol. Cancer 9, 159. https://doi.org/10.1186/1476-4598-9-159 (2010).
Grabacka, M. M. et al. Fenofibrate induces ketone body production in melanoma and glioblastoma cells. Front. Endocrinol. 7, 5. https://doi.org/10.3389/fendo.2016.00005 (2016).
Koltai, T. Fenofibrate in cancer: Mechanisms involved in anticancer activity. F1000Research 4, 1–22. https://doi.org/10.12688/f1000research.6153.1 (2015).
Wybieralska, E. et al. Fenofibrate attenuates contact-stimulated cell motility and gap junctional coupling in DU-145 human prostate cancer cell populations. Oncol. Rep. 26, 447–453. https://doi.org/10.3892/or.2011.1321 (2011).
Wilk, A. et al. Molecular mechanisms of fenofibrate-induced metabolic catastrophe and glioblastoma cell death. Mol. Cell. Biol. https://doi.org/10.1128/MCB.00562-14 (2014).
Grabacka, M. et al. Fenofibrate subcellular distribution as a rationale for the intracranial delivery through biodegradable carrier. J. Physiol. Pharmacol. 66, 233–247 (2015).
Stalinska, J. et al. Chemically modified variants of fenofibrate with antiglioblastoma potential. Transl. Oncol. 12, 895–907. https://doi.org/10.1016/j.tranon.2019.04.006 (2019).
Abotaleb, M. et al. Flavonoids in cancer and apoptosis. Cancers 11, 28. https://doi.org/10.3390/cancers11010028 (2018).
Doan, P. et al. Alkylaminophenol Induces G1/S phase cell cycle arrest in glioblastoma cells through p53 and cyclin-dependent kinase signaling pathway. Front. Pharmacol. 10, 330. https://doi.org/10.3389/fphar.2019.00330 (2019).
Rao, S., Chinkwo, K., Santhakumar, A., Johnson, S. & Blanchard, C. Apoptosis induction pathway in human colorectal cancer cell line SW480 exposed to cereal phenolic extracts. Molecules 24, 2465. https://doi.org/10.3390/molecules24132465 (2019).
Romanos-Nanclares, A. et al. Phenolic Acid subclasses, individual compounds, and breast cancer risk in a mediterranean cohort: The SUN project. J. Acad. Nutr. Diet. https://doi.org/10.1016/j.jand.2019.11.007 (2020).
Stalinska, J. et al. Exploring anticancer activity of structurally modified benzylphenoxyacetamide (BPA); I: Synthesis strategies and computational analyses of substituted BPA variants with high anti-glioblastoma potential. Sci. Rep. 9, 17021. https://doi.org/10.1038/s41598-019-53207-0 (2019).
Prinz, H. How to identify a pharmacophore. Chem. Biol. 15, 207–208. https://doi.org/10.1016/j.chembiol.2008.02.013 (2008).
Zeslawska, E. et al. Pharmacophoric features for a very potent 5-spirofluorenehydantoin inhibitor of cancer efflux pump ABCB1, based on X-ray analysis. Chem. Biol. Drug Des. 93, 844–853. https://doi.org/10.1111/cbdd.13473 (2019).
Dzialo, M. et al. The potential of plant phenolics in prevention and therapy of skin disorders. Int. J. Mol. Sci. 17, 160. https://doi.org/10.3390/ijms17020160 (2016).
Brown, D. G. & Bostrom, J. Analysis of past and present synthetic methodologies on medicinal chemistry: Where have all the new reactions gone?. J. Med. Chem. 59, 4443–4458. https://doi.org/10.1021/acs.jmedchem.5b01409 (2016).
Pattabiraman, V. R. & Bode, J. W. Rethinking amide bond synthesis. Nature 480, 471–479. https://doi.org/10.1038/nature10702 (2011).
Dunetz, J. R., Magano, J. & Weisenburger, G. A. Large-scale applications of amide coupling reagents for the synthesis of pharmaceuticals. Org. Process Res. Dev. 20, 140–177. https://doi.org/10.1021/op500305s (2016).
Greenberg, J. A. & Sammakia, T. The conversion of tert-butyl esters to acid chlorides using thionyl chloride. J. Org. Chem. 82, 3245–3251. https://doi.org/10.1021/acs.joc.6b02931 (2017).
Rani, P., Pal, D., Hegde, R. R. & Hashim, S. R. Anticancer, anti-inflammatory, and analgesic activities of synthesized 2-(substituted phenoxy) acetamide derivatives. Biomed. Res. Int. https://doi.org/10.1155/2014/386473 (2014).
Wager, T. T., Hou, X. J., Verhoest, P. R. & Villalobos, A. Moving beyond rules: The development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. Acs Chem. Neurosci. 1, 435–449. https://doi.org/10.1021/cn100008c (2010).
Wager, T. T., Hou, X. J., Verhoest, P. R. & Villalobos, A. central nervous system multiparameter optimization desirability: Application in drug discovery. Acs Chem. Neurosci. 7, 767–775. https://doi.org/10.1021/acschemneuro.6b00029 (2016).
Rankovic, Z. CNS drug design: Balancing physicochemical properties for optimal brain exposure. J. Med. Chem. 58, 2584–2608. https://doi.org/10.1021/jm501535r (2015).
Wu, Z. Y. et al. Comparison of prediction models for blood brain barrier permeability and analysis of the molecular descriptors. Pharmazie 67, 628–634 (2012).
Wang, J. M. et al. Development of polarizable models for molecular mechanical calculations I: Parameterization of atomic polarizability. J. Phys. Chem. B 115, 3091–3099. https://doi.org/10.1021/jp112133g (2011).
Naef, R. A generally applicable computer algorithm based on the group additivity method for the calculation of seven molecular descriptors: Heat of combustion, LogPO/W, LogS, refractivity, polarizability, toxicity and LogBB of organic compounds; scope and limits of applicability. Molecules 20, 18279–18351. https://doi.org/10.3390/molecules201018279 (2015).
Cha, H. J., Muller, R. T. & Pos, K. M. Switch-loop flexibility affects transport of large drugs by the promiscuous AcrB multidrug efflux transporter. Antimicrob. Agents. Chemother. 58, 4767–4772. https://doi.org/10.1128/Aac.02733-13 (2014).
Savjani, K. T., Gajjar, A. K. & Savjani, J. K. Drug solubility: Importance and enhancement techniques. ISRN Pharm. 2012, 195727. https://doi.org/10.5402/2012/195727 (2012).
Piiper, J. & Worth, H. Value and limits of Graham’s law for prediction of diffusivities of gases in gas mixtures. Respir. Physiol. 41, 233–240 (1980).
Zhou, D. et al. Distinctive epigenomes characterize glioma stem cells and their response to differentiation cues. Genome Biol. 19, 43. https://doi.org/10.1186/s13059-018-1420-6 (2018).
Stone, N. L., England, T. J. & O’Sullivan, S. E. A novel transwell blood brain barrier model using primary human cells. Front. Cell Neurosci. 13, 230. https://doi.org/10.3389/fncel.2019.00230 (2019).
Srinivasan, B. et al. TEER measurement techniques for in vitro barrier model systems. J. Lab. Autom. 20, 107–126. https://doi.org/10.1177/2211068214561025 (2015).
Garberg, P. et al. In vitro models for the blood-brain barrier. Toxicol. In Vitro 19, 299–334. https://doi.org/10.1016/j.tiv.2004.06.011 (2005).
Wang, Y. et al. An experimentally validated approach to calculate the blood-brain barrier permeability of small molecules. Sci. Rep. 9, 6117. https://doi.org/10.1038/s41598-019-42272-0 (2019).
Dahan, A. & Miller, J. M. The solubility-permeability interplay and its implications in formulation design and development for poorly soluble drugs. AAPS J. 14, 244–251. https://doi.org/10.1208/s12248-012-9337-6 (2012).
Carlson, B. L., Pokorny, J. L., Schroeder, M. A. & Sarkaria, J. N. Establishment, maintenance and in vitro and in vivo applications of primary human glioblastoma multiforme (GBM) xenograft models for translational biology studies and drug discovery. Curr. Protoc. Pharmacol. https://doi.org/10.1002/0471141755.ph1416s52 (2011).
Cen, L. et al. Efficacy of protracted temozolomide dosing is limited in MGMT unmethylated GBM xenograft models. Neuro Oncol. 15, 735–746. https://doi.org/10.1093/neuonc/not010 (2013).
An, F. Q. et al. Long-term-infected telomerase-immortalized endothelial cells: A model for Kaposi’s sarcoma-associated herpesvirus latency in vitro and in vivo. J. Virol 80, 4833–4846. https://doi.org/10.1128/JVI.80.10.4833-4846.2006 (2006).
Acknowledgements
This work was supported by P20-GM121288-01 (KR) and matching funds from the Dean, School of Medicine, LSU Health Sciences Center (KR). All chemistry and computational studies were supported by STEPFARM, LLC. (BSJ).
Author information
Authors and Affiliations
Contributions
J.S.: performed and help in design of cell culture analyses, cell toxicity tests, microscopy, imaging, data analyses and preparation of the corresponding figures; C.V.: assisted J.S. in cell culture and toxicity experiments; K.R.: helped designing cell culture experiments, authored manuscript sections related to glioblastoma, cell culture, cell toxicity, and interpretation of the in vitro studies; B.S.J.: designed chemical strategies for the development of new HR compounds, performed corresponding chemical reactions, computational analyses of the new compounds, authored the section of the manuscript related to development of chemical modifications and computational analyses; A.L. and K.B.: performed in vitro BBB penetration assay and HPLC analyses. C.F. and F.P.: designed and performed experiments and analyzed the data for all metabolic studies of glioblastoma cells treated with HR compounds. Ch.I. and S.C.C.: participated in performing specific chemical reactions designed by B.S.J., and performed IC50 analyses for some of the HR compounds; S.C.: editing and conceptual effort related to the final design of the manuscript. All Authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
Disclosure of Potential Conflict of Interest: Dr. Branko Jursic is associated with Stepharm LLC, P.O. Box 24220, New Orleans, LA; Dr. Reiss is associated with WayPath Pharma LLC. 217 Sena Dr. Metairie LA 70005. Dr. Krzysztof Reiss and Dr. Branko Jursic have LSU patent for the HR compounds presented in this manuscript ("Anticancer Composition and methods of use" WO2020/146876. Other authors do not have any competing interest in relation to this submission.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stalinska, J., Vittori, C., Ingraham IV, C.H. et al. Anti-glioblastoma effects of phenolic variants of benzoylphenoxyacetamide (BPA) with high potential for blood brain barrier penetration. Sci Rep 12, 3384 (2022). https://doi.org/10.1038/s41598-022-07247-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-07247-8
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
