
Abstract
A new series of arylmethylene hydrazine derivatives bearing 1,3-dimethylbarbituric moiety 7a–o were designed, synthesized, and evaluated for their in vitro urease inhibitory activity. All the title compounds displayed high anti-urease activity, with IC50 values in the range of 0.61 ± 0.06–4.56 ± 0.18 µM as compared to the two standard inhibitors hydroxyurea (IC50 = 100 ± 0.15 μM) and thiourea (IC50 = 23 ± 1.7 μM). Among the synthesized compounds, compound 7h with 2-nitro benzylidene group was found to be the most potent compound. Kinetic study of this compound revealed that it is a mix-mode inhibitor against urease. Evaluation of the interaction modes of the synthesized compounds in urease active site by molecular modeling revealed that that compounds with higher urease inhibitor activity (7h, 7m, 7c, 7l, 7i, and 7o, with IC50 of 0.61, 0.86, 1.2, 1.34, 1.33, 1.94 μM, respectively) could interact with higher number of residues, specially Arg609, Cys592 (as part of urease active site flap) and showed higher computed free energy, while compounds with lower urease activity (7f, 7n, 7g, and 7a with IC50 of 3.56, 4.56, 3.62 and 4.43 μM, respectively) and could not provide the proper interaction with Arg609, and Cys592 as the key interacting residues along with lower free binding energy. MD investigation revealed compound 7h interacted with Arg609 and Cys592 which are of the key residues at the root part of mobile flap covering the active site. Interacting with the mentioned residue for a significant amount of time, affects the flexibility of the mobile flap covering the active site and causes inhibition of the ureolytic activity. Furthermore, in silico physico-chemical study of compounds 7a–o predicted that all these compounds are drug-likeness with considerable orally availability.
Similar content being viewed by others

Introduction
Helicobacter pylori (H. pylori) is one of the bacteria that has caused many problems for humans by lowering their quality of life1. This bacteria in addition to causing a variety of gastrointestinal disorders, can even cause gastric cancer2. A most common treatment for inhibition of colonization of H. pylori is use of a triple therapy containing a proton pump inhibitor and two antibiotic agents3. The use of this treatment and similar treatments, in addition to causing side effects in other organs of the body, has led to H. pylori antibiotic resistance4. Therefore, the use of methods that specifically attack to bacteria is very valuable in the treatment of this disease5. One of the most popular of these methods is the use of H. pylori urease inhibitors6. H. pylori, like many other microorganisms, uses urea for growth, thus, the enzyme that breaks down urea, urease, plays an important role in its survival7. Several urease inhibitors with various structures have been introduced that can be useful for treatment of H. pylori infection8,9,10.
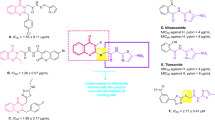
Recently, barbituric acid and its derivatives such as 1,3-dimethylbarbituric and thiobarbituric have received increasing attention in the discovery of new urease inhibitors11,12,13. Several compounds possessing barbituric acid derivatives such as compounds A display high inhibitory activity against urease (Fig. 1)14. As can be seen in Fig. 1, the later compounds have a 5-aminomethylene-1,3-dimethylbarbituric moiety. On the other hand, Saeed et al. reported the synthesis of a series of arylmethylene hydrazine derivatives containing carbothioamide B as novel urease inhibitors15. Their biological data demonstrated that the most potent compound among the compounds B showing up to 36.2-fold higher inhibitory potency than standard inhibitor thiourea (Fig. 1). By considering the potent urease inhibitors A and B, in continuing our efforts to synthesize new urease inhibitors using simple chemical reactions, here, a new series of urease inhibitors 7a–o was designed by combination of 5-aminomethylene-1,3-dimethylbarbituric moiety and arylmethylene hydrazine derivatives (Fig. 1)16,17,18,19,20. Compounds 7a–o were synthesized by a simple three-step procedure. All these compounds were evaluated for their in vitro urease inhibitory activities. Furthermore, molecular modeling and molecular dynamic studies of compounds 7a–o were also performed.

Design strategy for new arylmethylene hydrazine-1,3-dimethylbarbituric derivatives as urease inhibitors.
Results and discussion
Chemistry
Arylmethylene hydrazine-1,3-dimethylbarbituric derivatives 7a–o were synthesized by described method in Fig. 2. According to this method, reaction of 1,3-dimethylbarbituric acid 1 and trimethoxymethane 2 in ethanol under reflux condition afforded 5-(methoxymethylene)-1,3-dimethylbarbituric acid 3. The latter compound was reacted with hydrazine 4 in ethanol under reflux for produce 5-(hydrazineylmethylene)-1,3-dimethylbarbituric acid 5. In the final step, compound 5 and aromatic aldehydes 6a–o in the presence of catalytic amount of PTSA in ethanol at room temperature were converted to desired compounds 7a–o.

Synthesis of arylmethylene hydrazine-1,3-dimethylbarbituric derivatives 7a–o.
Urease inhibition
The effect of arylidenehydrazineyl-1,3-dimethylbarbituric derivatives 7a–o on urease was determined using Jack bean (JB) urease in accordance to the reported protocols18,19,20. The obtained IC50 values of title compounds against urease were presented in Table 1, in comparison with hydroxyurea and thiourea as reference inhibitors.
Generally, all newly synthesized compounds 7a–o, with IC50 values in the range of 0.61 ± 0.06–4.56 ± 0.18 μM, had significant inhibitory effect against urease. All these compounds were more potent than that of hydroxyurea (IC50 = 100 ± 0.15 μM) and thiourea (IC50 = 23 ± 1.7 μM). The 2-nitrophenyl derivative 7h and thiophen-2-yl derivative 7m with IC50 values of 0.61 ± 0.06 and 0.86 ± 0.08 μM were found to be the most active compounds. In particular, compound 7h was 37.7 times more potent than strong urease inhibitor thiourea.
Un-substituted phenyl derivative 7a, and 2,3-dichlorophenyl derivative 7j, were weaker than other phenyl derivatives against urease. Among the phenyl derivatives, the most potent compound was 2-nitro derivative 7h. The activity of this compound was 7.2-fold superior to that of parent phenyl derivative 7a. Movement of nitro substituent of 2-position into 4-position, as in compound 7i, and or replacement of 2-nitro group with 2-hydroxy substituent, as in compound 7b, decreased inhibitory activity to about two and fivefold, respectively. Moreover, introduction of 2,3-dichloro (compound 7j), 2-chloro-5-nitrophenyl (compound 7k), 3-bromo (compound 7g), 4-chloro (compound 7f), 3-phenoxy (compound 7e), and 3,4,5-trimethoxyphenyl (compound 7d) substituents instead of 2-nitro substituent dramatically decreased anti-urease activity while presence of 2,4-hydroxy (compound 7c) and 2-nitro-5-chloro (compound 7l) substituents on phenyl ring reduced inhibitory activity with less intensity in comparison to 2-nitro substituent.
The second most potent compound among the synthesized compound was thiophen-2-yl derivative 7m. As can be seen in Table 1, introduction of 5-chloro substituent on thiophen ring, as in compound 7n, led to a significant decrease in inhibitory activity. Furthermore, naphthalen-1-yl derivative 7o was one of the strongest compounds among the synthesized compounds.
Urease kinetic study
To evaluate the mechanism inhibition of new arylmethylene hydrazine-1,3-dimethylbarbituric derivatives, kinetics study was performed on the most potent urease inhibitor 7h (Fig. 3). The inhibition mode and Ki value were determined by Lineweaver–Burk plots and secondary re-plotting of the mentioned plots, respectively. As exhibited in Fig. 3a, with increasing concentrations of compound 7h, Vmax and Km increased. Therefore, this compound was a mixed-type inhibitor for urease (Fig. 3b, Ki = 0.82 µM).

Kinetic analysis of urease inhibition by compound 7h. (a) The Lineweaver–Burk plot in the absence and presence of different concentrations of compound 7h; (b) The secondary plot between 1/Vmax and various concentrations of compound 7h.
Docking study
Docking study was applied to distinguish interactions between the synthesized compounds and urease active site. The reliability of the applied docking protocol was assessed according to our previous study by re-docking of acetohydroxamic acid (AHA) into the active site of the JB urease20. This protocol was then similarly applied to all the synthesized compounds 7a–o.
The interaction mode of the top IFD scoring of all the synthesized compounds (7a–o) showed they successfully occupied in the bi-nickel active site cavity.
Figure 4a shows the top IFD pose of the compounds over JB urease. The result depicts superimposing of barbituric acid ring of all the compounds pointed toward the bi-nickel center atoms through the carbonyl group at C2 position of the barbiturate ring (Fig. 4b). The orientation of the mentioned carbonyl group is the same as the carbonyl oxygen in the AHA and thiourea as reference inhibitors. Furthermore, the arylmethylene hydrazine moieties oriented to the entering site of the active site and adapted by flexible conformation in the large hydrophobic opening of the active site flap pocket (Fig. 4a). The mentioned extended moiety in designing of the synthesized compounds has the superior inhibitory effect in comparison to the standard inhibitor (thiourea) through higher stabilization effect by implementing of various non-bonding interactions.

Representation of the compounds docking poses over the active site (a) close-up illustration of barbiturate ring relative to the bi-nuclear center (b), the active site flap colored in green color. The molecular graphic in this figure was generated using VMD 1.9.3.
In case of the most active compound 7h (Fig. 5), the carboxyl groups in C2 and C4 position of barbituric acid ring created H-bond with His492 and Arg609, the hydrazone group formed H-bond interaction through Cys592 (SH) and the ortho nitro benzylidene group was able to form salt-bridge interaction toward Arg639 (Fig. 5a). As a result, compound 7h well occupied the active pocket of urease and tightly anchoring the helix-turn-helix motif through interacting with Cys592 and Arg609 over the active-site cavity (depicted in green cartoon) (Fig. 5b), which could reduce the flexibility of flap residue (590–609) and results in the inhibition of urease activity.

Close up representation of the best energy value of the most active compound 7h complexed with JB urease (a), 90-degree anti-clockwise rotation view (b). The molecular graphic in this figure was generated using VMD 1.9.3.
In order to reveal the effect of different aromatic ring instead of phenyl ring, the docked pose complex and the gibbs free binding energy (ΔG) of the compounds 7a, 7m and 7o were compare to each other.
Figure 6a shows barbituric acid ring of compound 7a tightly coordinated along the metal bi-nickel center and further stabilized by H-bond interaction with His492. At the middle part of the molecule, the hydrazone group formed H-bond interaction through Cys592 (SH). In addition, the benzylidene group at the tail part of the compound formed hydrophobic interaction with hydrophobic pocket formed by Cys592, Met588, Val 591 and Leu589.

3D representation of ligand-residue interactions of compound 7a (a), compound 7m (b), compound 7o (c) over urease active site. Active site flap colored in green. The molecular graphic in this figure was generated using VMD 1.9.3.
Like compound 7a, compounds 7m and 7o, which have different aromatic moieties (thiophenyl and naphthyl, respectively), formed similar H-bond interactions through their barbituric acid ring and hydrazone group with His492 and Cys592, respectively. The gibbs free binding energy indicates higher value (ΔG) of − 46.47 and − 44.27, respectively as compared to compound 7a with ΔG value of − 39.52 kcal mol−1 (Table 2), which is in accordance with the experimental urease activity results (0.86, 1.94 and 4.43 μM, respectively, Table 1). In the case of compound 7m, the sulfur atom increased hydrophobic interaction by interacting with the mentioned hydrophobic pocket through sulfur atom of Met388, while in the case of compound 7o, the naphthyl ring rotated as a result of bigger size and interacted with the imidazole ring of His594 by π–π stacking interaction (Fig. 6b,c).
The result shows that the bioisoester substitution of phenyl group provides additional interactions for the compounds 7m and 7o to bind to the hydrophobic part of urease active site pocket.
This study confirms our previous investigation which outstands the key role of Cys592 and Arg609 in flexibility of mobile flap covering the active site entrance which consequently affect the inhibition activity of urease enzyme21.
Table 2 shows the interaction part of the molecule, the involving residues and the calculated gibbs free binding energies of the compound with higher and lower urease inhibition activity over JB urease active site. It indicates that compounds with higher urease inhibitor activity (7h, 7m, 7c, 7l, 7i, and 7o, with IC50 of 0.61, 0.86, 1.2, 1.34, 1.33, 1.94 μM, respectively) could interact with higher number of residues, specially Arg609, Cys592 (as part of urease active site flap), while compounds with lower urease activity (7f, 7n, 7g, and 7a with IC50 of 3.56, 4.56, 3.62 and 4.43 μM, respectively) have insufficient interaction and could not provide the proper interaction with Arg609, and Cys592 as the key interacting residues. Furthermore, this finding additionally supported by comparing the gibbs free binding energy of the mentioned compounds which are in accordance with the experimental urease inhibition activity.
Conformational analyses of the synthesized compounds over the urease active site
As the synthesized compounds hold benzylidene hydrazone moiety, they may adopt in four possible geometrical configurations including; the anti-periplanar (ap) and syn-planar rotameric (sp) forms for each Z and E isomers as shown in Fig. 7a. The superimposed structures of IFD compounds over the active site of JB urease have been shown in Fig. 7b. It represents that n-π conjugation result in a planar form of C=N–NH moiety. Considering Fig. 7a isomer E with anti-periplanar rotamer was the only configuration while no docked-structures got the Z configuration around C=N bond. This result is in accordance well with a previously reported study where there is no Z configuration around C=N of benzylidene hydrazone moiety due to the steric hindrance22,23,24. Therefore, according to the result it reveals that the E-ap form is the preferred geometrical isomer by the benzylidene hydrazone structure of barbituric acid derivatives.

Four possible configurations of benzylidene hydrazone moiety of the synthesized compounds (a). Superimposition of the docked structures in the active site of urease (b). The 2D structure representation was drawn by ChemAxon Marvin 15.10.12.030.
Molecular dynamic (MD) investigation
MD study has been investigated in order to reveal the effect of the most potent compound (compound 7h) on the urease structure and the active site environment in comparison to thiourea as the urease standard inhibitor.
Figure 8 shows the RMSD of the protein’s backbone over 100 ns MD simulation time. The RMSD simulation showed urease complexed with thiourea maintained an overall stability after 35 ns of MD simulation time with higher RMSD stabilizing at an average of 3.80 Å (Fig. 8, green line), while urease bound-state with compound 7h displayed longer equilibration time (after 15 ns of MD simulation) with obviously lower RMSD (2.4 Å) (Fig. 8, red line). In addition, the backbone RMSD of compound 7h equilibrated after about 40 ns and stabilized through the rest of the simulation time with a low RMSD fluctuation around 1.1 Å. Based on the RMSD result it is revealed that the urease-compound 7h complex obtain an equilibrium structure over the simulation time which has enough stability to investigate the structural specificity of the ligand–protein complex.

RMSD representation of the compound 7h (in yellow) and urease backbone in complex with thiourea (in green) and in complex with compound 7h (in red) for over 100 ns MD simulation time. The plot in was generated using Microsoft Excel (https://www.office.com/).
RMSF define as the fluctuation of the protein’s residues from its average position throughout the simulation, which represents the flexibility of protein structure. In this manner, helixes and sheets with organized structure depict lower RMSF value while loops with loosely organized structure have high RMSF value. Comparing RMSF values of urease-compound 7h complex shows that it has obviously lower value through the whole part of the urease structure and specially the residues 590–606 showed significantly decreased RMSF value in the urease bound-state with compound 7h than urease-thiourea complex (Fig. 9a). The mentioned conserved residues with helix-turn-helix secondary structure belong to the α-subunit part of the enzyme known as mobile flap region covering the urease active site25.

RMSF of the urease backbone in complexed with thiourea (in green) and compound 7h (in red) (a), ligand binding location for over 100 ns MD simulation time; (b) α-helical and ß-strand regions are highlighted in light red and blue backgrounds, respectively. The molecular graphic in this figure was generated using VMD 1.9.3.
Figure 9b shows that compound 7h well occupied the active pockets of urease and tightly anchoring the helix-turn-helix motif over the active-site cavity (vertical green line), which considerably reduce the flexibility of the mobile flap residue (590–609) by interacting with key amino acid residues and results in the inhibition of urease activity. The mentioned interactions could not be observed for the urease-thiourea complex which proposed the role of rigidity of the mobile flap in higher urease inhibition activity of the synthesized compounds.
Figure 10 represents different residues, types of interactions, and ligands functional group involvement during the whole MD simulation time.

The timeline representation of the interactions shows the residues interact with compound 7h in each trajectory frame (more than one specific contact with the ligand is represented by a darker shade of orange) (a). The 3D representation of urease in bound-state with compound 7h in two different orientations related to 0–14.32 ns and 14.32 to the rest of simulation time (active site flap are depicted in green cartoon mode) (b). The simulation interactions diagram panel. The stacked bar charts are normalized over the course of the trajectory: some protein residues may make multiple contacts with the ligand (c). 2D representation of ligand-residue interactions that occur at least 30% of simulation time of urease bound-state of thiurea and compound 7h (d). The molecular graphic in this figure was generated using VMD 1.9.3.
Based on result, compound 7h interacted with Arg439, Ala440, Lys490, His519, His545, Gly550, Asp633 and Ala636 for approximate the first 10 ns of the MD simulation (Fig. 10a, red dash boxes). Otherwise, after about 14.32 ns until to the end of simulation the metal-coordinated residue interactions disappeared and substituted with residues; Thr522, Gly548, Leu589, Cys592, His593, Arg609, Arg611, Ile610, and Met637 (Fig. 10a, blue dash boxes).
So, because of the shifting orientation of compound 7h as a result of different interacting residues, the MD simulation time divided in two sections; 0 ns to 14.32 ns and 14.32 ns to 100 ns. Based on the cluster analysis of compound 7h, the percent of population in cluster 1 was 86.37% in the first section and 95% in the second section in which the representative frame from cluster 1 of the section 1 (Fig. 10b, up) and the section 2 (Fig. 10b, down) were selected for investigating the 3D complex interaction.
As it is obvious in Fig. 10b (up), at the first stage of MD compound 7h tightly coordinated toward the active site bi-nickel center through its barbituric acid. In addition, the ortho nitro benzylidene moiety provided ion-bridge and π-cation interactions through Arg439. Otherwise, after about 14 ns the barbituric acid ring shifted from the bi-nickel center toward the active site flap and provided interactions with Cys592, His593 at one side and Arg609 and Arg611 at the other side of the active site flap while the ortho nitro benzylidene moiety still stabilized in its initial position faced to Arg439 through the electrostatic interaction (Fig. 10b, down) (the types of residue interactions during the MD simulation represents in different color by Fig. 10c). So, it can propose that compound 7h provides the stabilized interaction to Arg439 at the opening part of the active site through its polar nitro substitution. This finding is in accordance with our experimental results as the molecular level explanation on the higher urease activity of compound 7h and the compounds with polar substituent at the phenyl ring. Also, again the E-ap conformer, as the preferred geometrical isomer by the benzylidene hydrazone, recognized for both stages of MD simulation time.
Furthermore, Fig. 10d shows the detailed 2D-ligand atom interactions that occurred more than 30.0% of the simulation time during the equilibrated phase over urease complexed with thiourea and compound 7h. The interaction analysis suggests compound 7h stabilized by the ortho nitro group with Arg439 at the entrance of active site through salt-bridge interaction for about 43% of simulation time. Also, it formed water-mediated H-bond interaction with Arg609 and Arg611 at on side of the active site flap through the C4 carbonyl group of barbituric acid ring for 45% and 55% of simulation time, respectively. In addition, Cys592 on the other side of the active site flap provided the non-bonding interaction mostly through hydrophobic interaction for about 45% of MD simulation time (Fig. 10c, Cys592 bar chart).
It is noteworthy that Arg609 and Cys592 are of the key residues at the root part of mobile flap covering the active site. Interacting with the mentioned residue for a significant amount of time, affects the flexibility of the mobile flap covering the active site and causes inhibition of the ureolytic activity20.
Comparing the results of compound 7h interaction with thiourea indicates thiourea did not show any interaction with these key residues at the active site flap region which can proposed the reason of higher urease inhibition activity of compound 7h rather than thiourea.
In silico prediction of pharmacokinetic properties
The main physico-chemical properties of the synthesized compounds, which represent drug-likeness, partition coefficient, solubility, cell permeation, were calculated. Aiming to discuss the reliability of predictions in a consensus way, the parameters calculated with two different software. The partition coefficient (Log P) predicted by Qikpro module of schrodinger and swissADME web server while solubility (Log S), cell permeation and the predicted % human absorption (oral; %HOA and intestinal; %HIA) were measured based on Qikpro and pkCSM web server.
“Lipinski rule of five” was used to assess the drug-likeness of the synthesized compounds which includes calculating of the molecular weights, number of hydrogen bond donor, number of hydrogen bond acceptor and the predicted octanol/water partition co-efficient. According to Table 3, the number of violations of Lipinski’s rule of five (ROF violations) for all the synthesized compounds was zero (0) and therefore all compounds (7a–o) meet the drug-likeness criteria26.
Also, the bioavailability of a compound mainly depends on the absorption and metabolism process. The oral and gastrointestinal absorption procedures in turn relay on the solubility and permeability of the a compound27. The computed physico-chemical parameters used to assess absorption of the newly synthesized compounds 7a–o and hydroxyurea including; the predicted aqueous solubility (Log S), the predicted apparent Caco-2 cell permeability as a model for the gut-blood barrier (non-active transport) (PCaco-2 and LogCaco-2), the predicted % human absorption (oral; %HOA and intestinal; %HIA) which were measured based on two different methods Qikpro and pkCSM web server and presented in Table 4. Comparing the experimental solubility value of hydroxyurea which considered as 100 mg/ml (experimental Log S of 1.2 mol/L) with the calculated Log S value of 1.1 (Table 4, Qikpro method) confirm the reliability of calculated data. Based on Jorgensen’s rule of three, orally availability is determined by calculating descriptor expressed as: log Swat > − 5.7, PCaco-2 > 22 nm/s, and Primary Metabolites < 728. According to Table 4, all the titled compounds follow of Jorgensen’s rule of three. Moreover, the same result has been revealed by implementing the pkCSM web server which proved the resulted outcome. In this way, compounds 7a, 7d-g, 7j, and 7m–o exhibited high HOA and the other compounds have moderate HOA.
Conclusion
In summary, a novel series of arylmethylene hydrazine-1,3-dimethylbarbituric derivatives 7a–o have been synthesized via simple chemical reactions, and their inhibitory activities against urease were evaluated. In the enzymatic assay, all the synthesized compounds 7a–o acted as potent inhibitors against urease (IC50 values = 0.61 ± 0.06–4.56 ± 0.18 μM) and were more active than the standard inhibitors hydroxyurea and thiourea (IC50 values = 100 ± 0.15 and 23 ± 1.7 μM, respectively). Furthermore, IFD study of the synthesized compounds in the urease active site showed that compounds with higher urease inhibitor activity could interact with higher number of residues, specially Arg609, Cys592 and showed higher computed free energy, while compounds with lower urease activity could not provide the proper interaction with Arg609, and Cys592 as the key interacting residues along with lower free binding energy. MD investigation revealed compound 7h interacted with Arg609 and Cys592 which are of the key residues at the root part of mobile flap covering the active site. Interacting with the mentioned residue for a significant amount of time, affects the flexibility of the mobile flap covering the active site and causes inhibition of the ureolytic activity. Moreover, in silico pharmacokinetic study predicted that newly synthesized compounds are drug-likeness and can be orally active.
Experimental
Methods
Melting points of arylmethylene hydrazine-1,3-dimethylbarbituric derivatives 7a–o were taken on a Kofler hot‐stage apparatus. 1H spectra were recorded on Bruker FT‐500 (500 MHz), and are reported relative to DMSO-d6 (δ 2.50). 1H NMR coupling constants (J) are reported in Hertz (Hz), and multiplicities are indicated as follows: s (singlet), d (doublet), t (triplet), m (multiplet), dd (doublet of doublet), dt (doublet of triplet). Proton-decoupled 13C NMR spectra were recorded on Bruker FT‐500 (125 MHz) and reported relative to DMSO-d6 (δ 40.0). The infrared (IR) spectra of title compounds were obtained on a Nicolet Magna FT‐IR 550 spectrophotometer (KBr disks). Elemental analysis of compounds 7a–o was carried out with an Elementar Analysensysteme GmbH VarioEL CHN mode. Spectra data of arylmethylene hydrazine-1,3-dimethylbarbituric derivatives 7a–o are available in the supplementary information.
Synthesis of 5-(methoxymethylene)-1,3-dimethylbarbituric acid 3
A mixture of 1,3-dimethylbarbituric acid 1 (1 mmol) and trimethoxymethane 2 (3 mmol) in ethanol (5 ml) was refluxed for 3 h. Then, this mixture was allowed to stand overnight at room temperature for formation pure 5-(methoxymethylene)-1,3-dimethylbarbituric acid 3.
Synthesis of 5-(hydrazineylmethylene)-1,3-dimethylbarbituric acid 5
A mixture of 5-(methoxymethylene)-1,3-dimethylbarbituric acid 3 (1 mmol) and hydrazine 4 (1 mmol) in ethanol (5 ml) was refluxed for 1 h. After that, the reaction mixture was allowed to cool at room temperature and poured into water, and the pure 5-(hydrazineylmethylene)-1,3-dimethylbarbituric acid 5 were filtered off.
General procedure for the synthesis of Arylmethylene hydrazine-1,3-dimethylbarbituric derivatives 7a–o
A mixture of the compound 5 (1 mmol), aromatic aldehydes 6a–o (1.5 mmol), PTSA in ethanol (10 ml) was was stirred at room temperature for 8 h. The obtained precipitate was filtered off and washed with ethanol (2 ml) to give pure compounds 7a–o.
(E)-5-((2-benzylidenehydrazinyl)methylene)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione (7a). White solid; isolated yield: 96%, mp 172–174 °C; IR (KBr, υ): 3289, 3057, 2929, 1638 cm-1; 1H NMR (301 MHz, DMSO-d6) δ 12.71 (s, 1H, NH), 8.78 (s, 1H, CH=N), 8.44 (s, 1H, CH=C), 7.76 (dd, J = 6.6, 2.8 Hz, 2H), 7.54 – 7.48 (m, 3H), 3.20 (s, 6H, 2CH3); 13C NMR (76 MHz, DMSO-d6) δ 159.96, 154.44, 151.91, 133.23, 131.74, 129.48, 128.21, 121.88, 117.10, 90.73, 28.06 (CH3), 27.46 (CH3); Anal Calcd for C14H14N4O3, C, 58.73, H, 4.93, N, 19.57 found: C, 58.70, H, 4.98, N, 19.50.
(E)-5-((2-(2-hydroxybenzylidene)hydrazinyl)methylene)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione (7b). White solid; isolated yield: 83%, mp 173–175 °C; IR (KBr, υ): 3284, 3057, 2939, 1631 cm-1; 1H NMR (301 MHz, DMSO-d6) δ 12.62 (d, J = 12.0 Hz, 1H, NH), 10.17 (s, 1H, OH), 8.79 (s, 1H, CH=N), 8.45 (d, J = 12.0 Hz, 1H, CH=C), 8.29 (d, J = 7.8 Hz, 1H, H3), 7.47 (m, 1H, H4), 6.44 – 6.31 (m, 2H, H5 & H6), 3.19 (s, 3H, CH3), 3.18 (s, 3H, CH3); 13C NMR (76 MHz, DMSO-d6) δ 163.40, 162.41, 159.78, 153.54, 152.64, 151.90, 129.92, 110.68, 108.72, 102.92, 90.43, 89.77, 28.00 (CH3), 27.39 (CH3); Anal Calcd for C14H14N4O4, C, 55.63, H, 4.67, N, 18.53 found: C, 55.65, H, 4.64, N, 18.48.
(E)-5-((2-(2,4-dihydroxybenzylidene)hydrazinyl)methylene)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione (7c). White solid; isolated yield: 80%, mp 184–186 °C; IR (KBr, υ): 3292, 3048, 2936, 1639 cm-1; 1H NMR (301 MHz, DMSO-d6) δ 12.64 (s, 1H, NH), 10.18 (s, 1H, OH), 10.09 (s, 1H, OH), 8.80 (s, 1H, CH=N), 8.46 (s, 1H, CH=C), 7.48 (d, J = 9.1 Hz, 1H, H6), 6.42–6.30 (m, 2H), 3.19 (s, 6H, 2CH3); 13C NMR (76 MHz, DMSO-d6) δ 163.41, 162.42, 162.27, 159.79, 153.58, 152.67, 151.95, 129.93, 110.72, 108.75, 102.95, 89.79, 28.02 (CH3), 27.43 (CH3); Anal Calcd for C14H14N4O5, C, 52.83, H, 4.43, N, 17.60 found: C, 52.88, H, 4.40, N, 17.67.
(E)-1,3-dimethyl-5-((2-(3,4,5-trimethoxybenzylidene)hydrazinyl)methylene)pyrimidine-2,4,6(1H,3H,5H)-trione (7d). White solid; isolated yield: 94%, mp 197–199 °C; IR (KBr, υ): 3294, 3047, 2931, 1626 cm-1; 1H NMR (301 MHz, DMSO-d6) δ 12.82 – 12.51 (m, 1H, NH), 8.65 (s, 1H, CH=N), 8.49 (s, 1H), 7.07 (s, 2H), 3.86 (s, 6H, 2CH3), 3.74 (s, 3H), 3.20 (s, 6H); 13C NMR (76 MHz, DMSO-d6) δ 164.63, 158.97, 155.50, 150.67, 146.14, 140.03, 126.53, 106.53, 103.88, 94.75, 61.30 (OCH3), 55.46 (OCH3), 27.91 (CH3), 26.53 (CH3); Anal Calcd for C17H20N4O6, C, 54.25, H, 5.36, N, 14.89 found: C, 54.28, H, 5.30, N, 14.88.
(E)-1,3-dimethyl-5-((2-(3-phenoxybenzylidene)hydrazinyl)methylene)pyrimidine-2,4,6(1H,3H,5H)-trione (7e). White solid; isolated yield: 87%, mp 217–219 °C; IR (KBr, υ): 3281, 3056, 2935, 1639 cm-1; 1H NMR (301 MHz, DMSO-d6) δ 12.68 (s, 1H, NH), 8.73 (s, 1H, CH=N), 8.35 (s, 1H, CH=C), 7.52 – 7.41 (m, 4H), 7.32 (s, 1H, H6), 7.21 (t, J = 7.4 Hz, 1H), 7.14 (dt, J = 7.2, 2.3 Hz, 1H), 7.08 (d, J = 7.6 Hz, 2H), 3.18 (s, 6H, 2CH3); 13C NMR (76 MHz, DMSO-d6) δ 157.77, 156.56, 154.46, 153.54, 151.85, 137.88, 135.16, 131.19, 130.66, 126.07, 124.44, 123.56, 121.67, 119.50, 116.88, 90.88, 27.50 (CH3), 26.08 (CH3); Anal Calcd for C20H18N4O4, C, 63.48, H, 4.79, N, 14.81 found: C, 63.41, H, 4.83, N, 14.86.
(E)-5-((2-(4-chlorobenzylidene)hydrazinyl)methylene)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione (7f). White solid; isolated yield: 92%, mp 179–181 °C; IR (KBr, υ): 3290, 3059, 2928, 1631 cm-1; 1H NMR (301 MHz, DMSO-d6) δ 12.73 (d, J = 11.9 Hz, 1H, NH), 8.78 (s, 1H, CH=N), 8.41 (d, J = 11.8 Hz, 1H, CH=C), 7.76 (d, J = 8.5 Hz, 2H, H2&H6), 7.57 (d, J = 8.5 Hz, 2H, H3&H5), 3.21 (s, 3H, CH3), 3.19 (s, 3H, CH3); 13C NMR (76 MHz, DMSO-d6) δ 159.50, 156.03, 153.92, 149.09, 144.03, 141.19, 137.84, 131.96, 129.32, 98.30, 27.89 (CH3), 25.25 (CH3); Anal Calcd for C14H13ClN4O3, C, 52.43, H, 4.09, N, 17.47 found: C, 52.46, H, 4.05, N, 17.52.
(E)-5-((2-(3-bromobenzylidene)hydrazinyl)methylene)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione (7g). White solid; isolated yield: 94%, mp 207–209 °C; IR (KBr, υ): 3293, 3055, 2932, 1637 cm-1; 1H NMR (301 MHz, DMSO-d6) δ 12.75 (s, 1H, NH), 8.73 (s, 1H, CH=N), 8.48 (s, 1H, CH=C), 7.94 (s, 1H, H2), 7.77–7.67 (m, 2H, H4&H6), 7.47 (t, J = 7.9 Hz, 1H, H5), 3.20 (s, 6H, 2CH3); 13C NMR (76 MHz, DMSO-d6) δ 154.44, 151.91, 149.42, 143.87, 138.34, 133.23, 131.75, 129.48, 128.21, 122.74, 121.79, 90.73, 27.07 (CH3), 26.78 (CH3); Anal Calcd for C14H13BrN4O3, C, 46.05, H, 3.59, N, 15.34 found: C, 46.09, H, 3.53, N, 15.30.
(E)-1,3-dimethyl-5-((2-(2-nitrobenzylidene)hydrazinyl)methylene)pyrimidine-2,4,6(1H,3H,5H)-trione (7h). White solid; isolated yield: 92%, mp 178–180 °C; IR (KBr, υ): 3291, 3048, 2928, 1638 cm-1; 1H NMR (301 MHz, DMSO-d6) δ 12.93 (s, 1H, NH), 9.19 (s, 1H, CH=N), 8.43 (d, J = 16.5 Hz, 1H, CH=C), 8.11 (s, 2H), 7.84 (s, 1H, H5), 7.74 (s, 1H, H4), 3.20 (s, 6H, 2CH3); 13C NMR (76 MHz, DMSO-d6) δ 163.30, 162.20, 153.82, 151.88, 148.91, 145.29, 140.42, 137.59, 133.56, 131.36, 128.75, 125.09, 90.60, 28.07 (CH3), 27.45 (CH3); Anal Calcd for C14H13N5O6, C, 48.42, H, 3.77, N, 20.17 found: C, 48.40, H, 3.70, N, 20.20.
(E)-1,3-dimethyl-5-((2-(4-nitrobenzylidene)hydrazinyl)methylene)pyrimidine-2,4,6(1H,3H,5H)-trione (7i). White solid; isolated yield: 86%, mp 171–173 °C; IR (KBr, υ): 3282, 3047, 2934, 1641 cm-1; 1H NMR (301 MHz, DMSO-d6) δ 12.83 (s, 1H, NH), 8.89 (s, 1H, CH=N), 8.47 (s, 1H, CH=C), 8.33 (d, J = 8.6 Hz, 2H, H3&H5), 8.00 (d, J = 8.7 Hz, 2H, H3&H5), 3.21 (s, 6H, 2CH3); 13C NMR (76 MHz, DMSO-d6) δ 162.97, 156.56, 153.92, 149.09, 143.50, 142.14, 138.90, 130.07, 125.85, 96.11, 28.95 (CH3), 27.36 (CH3); Anal Calcd for C14H13N5O5, C, 50.76, H, 3.96, N, 21.14 found: C, 50.79, H, 3.90, N, 21.20.
(E)-5-((2-(2,3-dichlorobenzylidene)hydrazinyl)methylene)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione (7j). White solid; isolated yield: 81%, mp 201–203 °C; IR (KBr, υ): 3296, 3051, 2935, 1635 cm−1; 1H NMR (301 MHz, DMSO-d6) δ 12.93 (d, J = 11.7 Hz, 1H, NH), 9.23 (s, 1H, CH=N), 8.38 (d, J = 11.6 Hz, 1H, CH=C), 7.96 (d, J = 7.9 Hz, 1H, H4), 7.76 (d, J = 8.0 Hz, 1H, H6), 7.45 (t, J = 8.0 Hz, 1H, H5), 3.18 (s, 6H, 2CH3); 13C NMR (76 MHz, DMSO-d6) δ 162.76, 160.22, 155.90, 152.72, 146.60, 143.80, 140.40, 135.48, 128.84, 127.02, 124.72, 124.38, 90.45, 27.78 (CH3), 25.79 (CH3); Anal Calcd for C14H12Cl2N4O3, C, 47.34, H, 3.41, N, 15.77 found: C, 47.30, H, 3.45, N, 15.70.
(E)-5-((2-(2-chloro-5-nitrobenzylidene)hydrazinyl)methylene)-1,3-dimethylpyrimidine 2,4,6(1H,3H,5H)-trione (7k). White solid; isolated yield: 83%, mp 188–190 °C; IR (KBr, υ): 3285, 3052, 2927, 1633 cm-1; 1H NMR (301 MHz, DMSO-d6) δ 13.02 (s, 1H, NH), 9.25 (s, 1H, CH=N), 8.70 (s, 1H, H6), 8.50 (s, 1H, H4), 8.29 (d, J = 11.7 Hz, 1H, CH=C), 7.87 (d, J = 8.9 Hz, 1H), 3.20 (s, 6H, 2CH3); 13C NMR (76 MHz, DMSO-d6) δ 161.84, 157.69, 154.60, 153.61, 148.63, 144.86, 136.33, 133.08, 131.05, 128.49, 126.00, 125.39, 93.02, 25.80 (CH3), 24.88 (CH3). Anal Calcd for C14H12ClN5O5, C, 45.98, H, 3.31, N, 19.15 found: C, 45.96, H, 3.35, N, 19.13.
(E)-5-((2-(5-chloro-2-nitrobenzylidene)hydrazinyl)methylene)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione (7l). White solid; isolated yield: 88%, mp 191–193 °C; IR (KBr, υ): 3286, 3053, 2934, 1630 cm-1; 1H NMR (301 MHz, DMSO-d6) δ 13.01 (s, 1H, NH), 9.17 (s, 1H, CH=N), 8.51 (s, 1H, H6), 8.18–8.13 (m, 2H), 7.84–7.78 (m, 1H, H4), 3.21 (s, 6H, 2CH3); 13C NMR (76 MHz, DMSO-d6) δ 163.82, 161.17, 159.79, 154.89, 153.82, 147.26, 143.33, 140.07, 134.59, 126.90, 123.77, 122.91, 93.08, 26.30 (CH3), 24.57 (CH3); Anal Calcd for C14H12ClN5O5, C, 45.98, H, 3.31, N, 19.15 found: C, 45.96, H, 3.38, N, 19.21.
(E)-1,3-dimethyl-5-((2-(thiophen-2-ylmethylene)hydrazinyl)methylene)pyrimidine2,4,6(1H,3H,5H)-trione (7m). White solid; isolated yield: 81%, mp 164–166 °C; IR (KBr, υ): 3288, 3052, 2930, 1635 cm-1; 1H NMR (301 MHz, DMSO-d6) δ 12.70 (s, 1H, NH), 8.97 (s, 1H, CH=N), 8.30 (s, 1H, CH=C), 7.81 (d, J = 5.0 Hz, 1H, H5), 7.56–7.52 (m, 1H, H3), 7.20 (dd, J = 5.0, 3.7 Hz, 1H, H4), 3.19 (s, 6H, 2CH3); 13C NMR (76 MHz, DMSO-d6) δ 163.30, 162.20, 153.82, 151.88, 148.90, 137.59, 133.56, 131.36, 128.75, 90.60, 28.07 (CH3), 27.45 (CH3); Anal Calcd for C12H12N4O3S, C, 49.31, H, 4.14, N, 19.17 found: C, 49.28, H, 4.17, N, 19.24.
(E)-5-((2-((5-chlorothiophen-2-yl)methylene)hydrazinyl)methylene)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione (7n). White solid; isolated yield: 88%, mp 185–187 °C; IR (KBr, υ): 3293, 3056, 2935, 1638 cm-1; 1H NMR (301 MHz, DMSO-d6) δ 12.93–12.48 (m, 1H, NH), 8.85 (s, 1H, CH=N), 8.37–8.30 (m, 1H, CH=C), 7.47–7.38 (m, 1H, H4), 7.27–7.19 (m, 1H, H3), 3.20 (s, 6H, 2CH3); 13C NMR (76 MHz, DMSO-d6) δ 162.25, 158.07, 153.90, 151.73, 148.03, 136.80, 133.36, 128.75, 124.66, 90.93, 28.12 (CH3), 27.48 (CH3); Anal Calcd for C12H11ClN4O3S, C, 44.11, H, 3.39, N, 17.15 found: C, 44.17, H, 3.42, N, 17.19.
(E)-1,3-dimethyl-5-((2-(naphthalen-1-ylmethylene)hydrazinyl)methylene)pyrimidine-2,4,6(1H,3H,5H)-trione (7o). White solid; isolated yield: 90%, mp 213–115 °C; IR (KBr, υ): 3292, 3057, 2933, 1639 cm−1; 1H NMR (301 MHz, DMSO-d6) δ 12.82 (s, 1H, NH), 9.53 (s, 1H, CH=N), 8.76 (d, J = 8.4 Hz, 1H, H2), 8.58 (s, 1H, CH=C), 8.12–8.02 (m, 3H), 7.73–7.60 (m, 3H), 3.20 (s, 6H, 2CH3); 13C NMR (76 MHz, DMSO-d6) δ 163.43, 155.47, 147.22, 143.86, 135.72, 133.54, 131.66, 130.81, 129.40, 127.03, 124.78, 123.21, 121.65, 117.64, 97.80, 26.21 (CH3), 24.66 (CH3); Anal Calcd for C18H16N4O3, C, 64.28, H, 4.79, N, 16.66 found: C, 64.33, H, 4.72, N, 16.60.
Urease inhibitory activity and kinetic study
All used material and JB urease (EC 3.5.1.5) were purchased from Sigma–Aldrich (USA). Potassium phosphate buffer (PPB) solution with concentration 100 mM and pH = 7.4 was prepared in distilled water. Urease inhibition effects of the synthesized compounds 7a–o was determined using the modified Berthelot spectrophotometric method by a Synergy H1 Hybrid multi-mode microplate reader (BioTek, Winooski, VT, USA) at 625 nm16,17,18,19,20. The enzymatic reactions were performed in PPB solution which reach to the 985 μL by adding urea (850 μL) and the synthesized compound (100 μL, 0–10 mg/mL). Then, urease (15 μL) was added and the concentration of liberated ammonia was measured after 60 min. The corresponding concentration of ammonia was determined by addition of the incubated solution (100 μL) to the mixture of 500 μL of solution I (5.0 g phenol and 25.0 mg sodium nitroprusside in 500 mL water) and 500 μL of solution II (2.5 g sodium hydroxide and 4.2 mL sodium hypochlorite (5% chlorine) in 500 mL water) which was incubated at 37 °C for 30 min. The absorbance was obtained by measuring indophenols. The activity of uninhibited JB urease was considered as the control activity of 100%. The inhibition assays were conducted according to this formula: I (%) = [1 − (T/C)] × 100; where I (%) is the enzyme inhibition, T (test) is the absorbance of the analyzed compounds in the presence of urease solution, and C (control) is the absorbance of the solvent in the presence of urease solution. Data were expressed as mean ± standard error (SD) and run in triplicate. The IC50 values for the all compounds 7a–o were calculated using GraphPad Prism 5 software (GraphPad Software, Inc., San Diego, CA). Thiourea and hydroxyurea were used as the standard inhibitors for urease. For the kinetic study the urea concentrations were changed from 3.12 to 100 mM and concentrations 0, 0.5, and 1 µM of the most potent urease inhibitor was used.
Molecular modeling procedure
In order to find out the interactions mode of designed molecules over urease enzyme, Maestro Molecular Modeling platform (version10.5) by Schrödinger, LLC was performed29. The X-ray crystallographic structure of Jack bean urease (JB urease) (in complex with acetohydroxamic acid, AHA) was downloaded from the Protein Data Bank (PDB ID; 4h9m) (www.rcsb.org). As urease is reported to be functionally active in monomeric state, all the docking studies were performed on single monomer. In addition, prosthetic group and co-factors are not directly involved in urease inhibition, so they totally removed before docking investigation. Water molecules and co-crystallized ligands were removed from the enzymes crystallographic structures. The 2D structures of all synthesized compounds were drawn in Marvin 15.10.12.0 program (http://www.chemaxon.com) and converted into pdb file30. The Protein Preparation Wizard and the LigPrep module were used to prepare protein and ligand structure properly31,32. The missing side chains of the proteins were filled using the Prime tool and missing residues were updated.
The accurate side‑chain, backbone conformational changes or both during ligand binding at the active site of urease enzyme were predicted by IFD method using Glide software (Schrödinger LLC 2018, USA)33. The AHA binding site was used to generate the grid for IFD calculation. The maximum 20 poses with receptor and ligand van der Waals radii of 0.7 and 0.5, respectively considered. Residues within 5 Å of the AHA at the active site were refined followed by side-chain optimization. Structures whose Prime energy is more than 30 kcal/mol are eliminated based on extra precious Glide docking.
The ligand binding energies (ΔG bind) were calculated for each synthesized compound using Molecular mechanics/generalized born surface area (MM‑GBSA) modules (Schrödinger LLC 2018) based on the following equation;
where ΔG Bind is the calculated relative free energy which includes both ligand and receptor strain energy33. EComplex is the MM/GBSA energy of the minimized complex, and ELigand is the MM/ GBSA energy of the ligand after removing it from the complex and allowing it to relax. EReceptor is the MM-GBSA energy of relaxed protein after separating it from the ligand. The MM-GBSA calculation was performed based on the best pose structure obtained from IFD complexes.
Molecular dynamic simulation
Molecular simulations of this study were performed using the Desmond v5.3 using Maestro interface (from Schrödinger 2018‐4 suite)34. The appropriate pose for MD simulation procedure of the compound was achieved by IFD method.
In order to build the system for MD simulation, the protein–ligand complexes were solvated with SPC explicit water molecules and placed in the center of an orthorhombic box of appropriate size in the periodic boundary condition. Sufficient counter‐ions and a 0.15 M solution of NaCl were also utilized to neutralize the system and to simulate the real cellular ionic concentrations, respectively. The MD protocol involved minimization, pre-production, and finally production MD simulation steps. In the minimization procedure, the entire system was allowed to relax for 2500 steps by the steepest descent approach. Then the temperature of the system was raised from 0 to 300 K with a small force constant on the enzyme in order to restrict any drastic changes. MD simulations were performed via NPT (constant number of atoms, constant pressure i.e. 1.01325 bar and constant temperature i.e. 300 K) ensemble. The Nose‐Hoover chain method was used as the default thermostat with 1.0 ps interval and Martyna‐Tobias‐Klein as the default barostat with 2.0 ps interval by applying isotropic coupling style. Long‐range electrostatic forces were calculated based on particle‐mesh‐based ewald approach with the he cut‐off radius for columbic forces set to 9.0 Å. Finally, the system subjected to produce MD simulations for 20 ns for each protein–ligand complex. During the simulation every 1000 ps of the actual frame was stored. The dynamic behavior and structural changes of the systems were analyzed by the calculation of the root mean square deviation (RMSD) and RMSF. Subsequently, the representative frames of the simulation extracted based on the clustering method from the equilibrated trajectory system for investigating of ligand–protein complex interaction.
In silico pharmacokinetic properties of synthesized compounds
QikProp module of Schrodinger35, swissADME36, and pkCSM37 were used to calculate the important physico-chemical properties of the synthesized compounds like drug-likeness, partition coefficient, solubility, cell permeation.
References
Salama, N. R. et al. Life in the human stomach: Persistence strategies of the bacterial pathogen Helicobacter pylori. Nat. Rev. Microbiol. 11, 385–399. https://doi.org/10.1038/nrmicro3016 (2013).
Lacy, B. E. & Rosemore, J. Helicobacter pylori: Ulcers and more: The beginning of an era. J. Nutr. 131, 2789S-2793S. https://doi.org/10.1093/jn/131.10.2789S (2001).
Calvet, X. et al. A meta-analysis of short versus long therapy with a proton pump inhibitor, clarithromycin and either metronidazole or amoxycillin for treating Helicobacter pylori infection. Aliment. Pharmacol. Ther. 14, 603–610. https://doi.org/10.1046/j.1365-2036.2000.00744 (2000).
Rautelin, H. et al. Role of metronidazole resistance in therapy of Helicobacter pylori infections. Antimicrob. Agents Chemother. 36, 163–166. https://doi.org/10.1128/AAC.36.1.163 (1992).
Suerbaum, S. & Michetti, P. Helicobacter pylori infection. N. Engl. J. Med. 347, 1175–1186. https://doi.org/10.1056/NEJMra020542 (2002).
Graham, D. Y. & Miftahussurur, M. Helicobacter pylori urease for diagnosis of Helicobacter pylori infection: A mini review. J. Adv. Res. 13, 51–57. https://doi.org/10.1016/j.jare.2018.01.006 (2018).
Fagoonee, S. & Pellicano, R. Helicobacter pylori: Molecular basis for colonization and survival in gastric environment and resistance to antibiotics. A short review. Infect. Dis. 51, 399–408. https://doi.org/10.1080/23744235.2019.1588472 (2019).
Kafarski, P. & Talma, M. Recent advances in design of new urease inhibitors: A review. J. Adv. Res. 13, 101–112. https://doi.org/10.1080/23744235.2019.1588472 (2018).
de Fátima, Â. et al. Schiff bases and their metal complexes as urease inhibitors—A brief review. J. Adv. Res. 13, 113–126. https://doi.org/10.1016/j.jare.2018.03.007 (2018).
Korona-Glowniak, I. et al. The in vitro activity of essential oils against Helicobacter Pylori growth and urease activity. Molecules 25, 586. https://doi.org/10.3390/molecules25030586 (2020).
Rauf, A. et al. Design and synthesis of new barbituric-and thiobarbituric acid derivatives as potent urease inhibitors: Structure activity relationship and molecular modeling studies. Bioorg. Med. Chem. 23, 6049–6058. https://doi.org/10.1016/j.bmc.2015.05.038 (2015).
Bano, B. et al. Synthesis, in vitro urease inhibitory activity, and molecular docking studies of thiourea and urea derivatives. Bioorg. Chem. 80, 129–144. https://doi.org/10.1016/j.bioorg.2018.06.007 (2018).
Barakat, A. et al. Synthesis, X-ray crystal structures, biological evaluation, and molecular docking studies of a series of barbiturate derivatives. J. Chem. https://doi.org/10.1155/2016/8517243 (2016).
Asadi, M. et al. Synthesis and urease inhibitory activity of some 5-aminomethylene barbituric/thiobarbituric acid derivatives. Lett. Drug Des. Discov. 15, 428–436. https://doi.org/10.2174/1570180814666170727142928 (2018).
Saeed, A. et al. 2-(Hetero (aryl) methylene) hydrazine-1-carbothioamides as potent urease inhibitors. Chem. Biol. Drug Des. 85, 225–230. https://doi.org/10.1111/cbdd.12379 (2015).
Moghimi, S. et al. Synthesis, evaluation, and molecular docking studies of aryl urea-triazole-based derivatives as anti-urease agents. Arch. Pharm. 351, 1800005. https://doi.org/10.1002/ardp.201800005 (2018).
Azizian, H. et al. Large-scale virtual screening for the identification of new Helicobacter pylori urease inhibitor scaffolds. J. Mol. Model. 18, 2917–2927. https://doi.org/10.1007/s00894-011-1310-2 (2012).
Vosooghi, M. et al. Synthesis of novel 5-arylidene (thio) barbituric acid and evaluation of their urease inhibitory activity. J. Iran Chem. Soc. 12, 1487–1491. https://doi.org/10.1007/s13738-015-0617-9 (2015).
Biglar, M. et al. Novel N,N-dimethylbarbituric-pyridinium derivatives as potent urease inhibitors: Synthesis, in vitro, and in silico studies. Bioorg. Chem. 95, 103529. https://doi.org/10.1016/j.bioorg.2019.103529 (2020).
Asgari, M. S. et al. New 1,2,3-triazole–(thio) barbituric acid hybrids as urease inhibitors: Design, synthesis, in vitro urease inhibition, docking study, and molecular dynamic simulation. Arch. Pharm. https://doi.org/10.1002/ardp.202000023 (2020).
Azizian, H. et al. Pantoprazole derivatives: Synthesis, urease inhibition assay and in silico molecular modeling studies. ChemistrySelect 5, 4580–4587. https://doi.org/10.1002/slct.202000578 (2020).
Azizian, H. et al. Arylhydrazone derivatives of naproxen as new analgesic and anti-inflammatory agents: Design, synthesis and molecular docking studies. J. Mol. Graph. Model. 67, 127–136 (2016).
Syakaev, V. V. et al. NMR study of conformation and isomerization of aryl-and heteroarylaldehyde 4-tert-butylphenoxyacetylhydrazones. J. Mol. Struct. 788, 55–62 (2006).
Ünsal-Tan, O., Özden, K., Rauk, A. & Balkan, A. Synthesis and cyclooxygenase inhibitory activities of some N-acylhydrazone derivatives of isoxazolo [4,5-d] pyridazin-4 (5H)-ones. Eur. J. Med. Chem. 45, 2345–2352 (2010).
Hifumi, E. Catalytic features and eradication ability of antibody light-chain UA15-L against Helicobacter pylori. J. Biol. Chem. 283, 899–907. https://doi.org/10.1074/jbc.M705674200 (2008).
Lipinski, C. A., Lombardo, F., Dominy, B. W. & Feeney, P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development q settings. Adv. Drug Deliv. Rev. 46, 3–26 (2001).
van de Waterbeemd, H. & Gifford, E. ADMET in silico modelling: Towards prediction paradise. Nat. Rev. Drug Discov. 2, 192–204. https://doi.org/10.1038/nrd1032 (2003).
Jorgensen, W. L. & Duffy, E. M. Prediction of drug solubility from structure. Adv. Drug Deliv. Rev. 54, 355–366. https://doi.org/10.1016/S0169-409X(02)00008-X (2002).
Schrödinger Suite 2018, Induced Fit Docking protocol; Glide version 5.5, S., LLC, New York, NY, 2009; Prime version 2.1, Schrödinger, LLC, New York, NY (2009).
ChemAxon Marvin was used for drawing, displaying and characterizing Chemical structures, substructures and reactions, Marvin 15.10.12.0 (version number), ChemAxon http://www.chemaxon.com (2015).
Schrödinger Release 2018-4: Schrödinger Release 2018-4 Protein Preparation Wizard; Epik, Schrödinger, LLC, New York, NY (2016); Impact, Schrödinger, LLC, New York, NY (2016); Prime, Schrödinger, LLC, New York, NY (2019).
Schrödinger Release 2018-4: LigPrep, Schrödinger, LLC, New York, NY (2018).
Schrödinger Release 2018-4: Induced Fit Docking protocol; Glide, Schrödinger, LLC, New York, NY, 2018; Prime, Schrödinger, LLC, New York, NY (2018).
Schrödinger Release 2018-4: Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY (2018).
Schrödinger Release 2018-4: QikProp, Schrödinger, LLC, New York, NY (2018).
SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7, 42717 (2017).
Pires, D. E. V., Blundell, T. L. & Ascher, D. B. pkCSM: Predicting small-molecule pharmacokinetic properties using graph-based signatures. J. Med. Chem. 58(9), 4066–4072 (2015).
Author information
Authors and Affiliations
Contributions
K.P., M.S.A., and M.A. did the synthesis of the compounds. M.S.A., M.A. and S.B. characterized the compounds and analyzed the characterization data. H.A., M.N.M., M.A., and M.A. did the biological experiments. M.M.-K., and H.R. performed the docking and compoutational section. M.M.-K., S.B., and B.L. wrote the manuscript and prepared the figures and schemes. M.M. supervised the work, wrote the manuscript and corresponded to the manuscript submision.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pedrood, K., Azizian, H., Montazer, M.N. et al. Arylmethylene hydrazine derivatives containing 1,3-dimethylbarbituric moiety as novel urease inhibitors. Sci Rep 11, 10607 (2021). https://doi.org/10.1038/s41598-021-90104-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-90104-x
This article is cited by
-
[1,2,4]triazolo[3,4-b][1,3,4]thiadiazole derivatives as new therapeutic candidates against urease positive microorganisms: design, synthesis, pharmacological evaluations, and in silico studies
Scientific Reports (2023)
-
New thioxothiazolidinyl-acetamides derivatives as potent urease inhibitors: design, synthesis, in vitro inhibition, and molecular dynamic simulation
Scientific Reports (2023)
-
Different barbiturate derivatives linked to aryl hydrazone moieties as urease inhibitors; design, synthesis, urease inhibitory evaluations, and molecular dynamic simulations
Medicinal Chemistry Research (2023)
-
Application of animation products via multimodal information and semantic analogy
Multimedia Tools and Applications (2023)
-
Design and synthesis of novel nitrothiazolacetamide conjugated to different thioquinazolinone derivatives as anti-urease agents
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
