
Abstract
Classification of tumors into subtypes can inform personalized approaches to treatment including the choice of targeted therapies. The two most common lung cancer histological subtypes, lung adenocarcinoma and lung squamous cell carcinoma, have been previously divided into transcriptional subtypes using microarray data, and corresponding signatures were subsequently used to classify RNA-seq data. Cross-platform unsupervised classification facilitates the identification of robust transcriptional subtypes by combining vast amounts of publicly available microarray and RNA-seq data. However, cross-platform classification is challenging because of intrinsic differences in data generated using the two gene expression profiling technologies. In this report, we show that robust gene expression subtypes can be identified in integrated data representing over 3500 normal and tumor lung samples profiled using two widely used platforms, Affymetrix HG-U133 Plus 2.0 Array and Illumina HiSeq RNA sequencing. We tested and analyzed consensus clustering for 384 combinations of data processing methods. The agreement between subtypes identified in single-platform and cross-platform normalized data was then evaluated using a variety of statistics. Results show that unsupervised learning can be achieved with combined microarray and RNA-seq data using selected preprocessing, cross-platform normalization, and unsupervised feature selection methods. Our analysis confirmed three lung adenocarcinoma transcriptional subtypes, but only two consistent subtypes in squamous cell carcinoma, as opposed to four subtypes previously identified. Further analysis showed that tumor subtypes were associated with distinct patterns of genomic alterations in genes coding for therapeutic targets. Importantly, by integrating quantitative proteomics data, we were able to identify tumor subtype biomarkers that effectively classify samples on the basis of both gene and protein expression. This study provides the basis for further integrative data analysis across gene and protein expression profiling platforms.
Similar content being viewed by others

Introduction
Lung cancer is the leading cause of cancer mortality (1.8 million deaths per year globally) and although multiple treatment options are available, the five-year survival rate remains low and there is an unmet need for better therapies1,2,3. Lung cancer is a heterogeneous disease and the classification of tumors using histological and molecular features can inform personalized approaches to treatment, in particular the choice of targeted therapies4. The increasing use of biomarkers and targeted therapies against receptor tyrosine kinases, angiogenic factors and inhibitory immune checkpoint proteins has indeed resulted in improved patient outcomes5,6,7,8. Lung cancer is generally divided into small cell lung cancer and non-small cell lung cancer (NSCLC) which comprises lung adenocarcinoma (LUAD), squamous cell carcinoma (LUSC) and large cell lung cancer9.
The two most common NSCLC histological subtypes (LUAD and LUSC) have been classified into molecular subtypes associated with clinically relevant characteristics including prognosis and survival, oncogenic drivers, and response to targeted therapies. Transcriptional subtypes (three in LUAD and four in LUSC) were initially identified by clustering gene expression microarray data from three LUAD (total 231 patients) and five LUSC (total 382 patients) discovery cohorts10,11. Gene expression signatures were further applied to RNA-seq data by The Cancer Genome Atlas (TCGA) and successfully classified tumors into corresponding subtypes12,13. The natural extension of these analyses, namely subtype discovery in combined data from the two platforms, presents challenges because of intrinsic differences in data generated using different gene expression profiling technologies, although previous studies showed that dedicated normalization methods enabled cross-platform pattern discovery and classification14,15, and comparative differential expression analyses also showed good agreement between the two gene expression profiling platforms16.
In this study, we explored cross-platform subtype discovery in LUAD and LUSC using public gene expression data from over 3500 normal and tumor lung samples. We tested 384 combinations of preprocessing, cross-platform normalization, and unsupervised feature selection methods. The results were evaluated based on the agreement between subtypes identified in single-platform and cross-platform normalized data using various statistics including clustering comparison measures. We show that unsupervised learning can be achieved with combined microarray and RNA-seq data. We further show that tumor subtype biomarkers can be identified in integrated gene expression and quantitative proteomics data.
Results
Classification of lung cancer subtypes
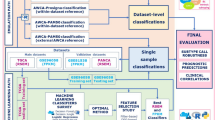
Our main objective was to identify robust expression subtypes in combined microarray and RNA-seq data for the two most common lung cancer histological subtypes (LUAD and LUSC). We collected a total of 2079 lung microarrays (500 normal, 1134 LUAD and 445 LUSC) and 1673 lung RNA-seq samples (532 normal, 640 LUAD and 501 LUSC). Although cross-platform analysis of a large number of samples can facilitate expression subtype analyses, clustering may nevertheless be sensitive to the presence of experimental (platform and batch) effects, outliers (e.g. low quality or misdiagnosed samples) as well as to data processing procedures including normalization and feature selection17,18,19,20,21,22,23,24,25. We therefore evaluated various single and cross-platform normalization and unsupervised feature selection methods to identify an optimal combination of data processing methods for cross-platform classification of lung cancer subtypes as detailed in Fig. 1.

Overview of the workflow for selecting the best combination of data processing methods for cross-platform classification of lung cancer into tumor subtypes. In brief, microarray data were pre-processed using two methods, and RNA-seq data were pre-processed using six methods. Data from the two platform were combined and cross-platform normalization was performed using four methods. After filtering data by removing samples with low confidence regarding main class labels (LUAD, LUSC and normal lung), single-platform and cross-platform normalized data were submitted to unsupervised feature selection using eight methods, and then to consensus clustering. Clustering results were compared between single-platform and cross-platform normalized data using various statistics. Clustering results from the top-ranking combination of data processing methods were selected for a final round of supervised classification into tumor subtypes. Microarray pre-processing methods: mas5, microarray suite 5.0; rma, robust multi-array average. RNA-seq pre-processing methods: cpm, counts per million; fpkm, fragments per kilobase per million; normTransform, shifted logarithm transformation; rpkm, reads per kilobase per million; voom, variance modeling at the observational level; vst, variance stabilizing transformation. Cross-platform normalization methods: ComBat, empirical Bayes batch effect correction; FSQN, feature-specific quantile normalization; quantile, quantile normalization; TDM, training distribution matching. Unsupervised feature selection methods: disr, diversity-induced self-representation; lscore, Laplacian score; mad, median absolute deviation; mcfs, multi-cluster feature selection; specu, unsupervised spectral feature selection; spufs, structure preserving unsupervised feature selection; svde, singular value decomposition entropy; udfs, unsupervised discriminative features selection. Clustering statistics: entropy, platform entropy; ITCC, information theoretic clustering comparison; min(O/E), minimum observed to expected ratio; NbClust: optimal number of clusters; purity: maximum agreement between single and cross-platform data; randomness: platform randomness within clusters.
After preprocessing single-platform data and performing cross-platform normalization, we proceeded to a first round of iterative ensemble classification (Supplementary Fig. S1) to clean data by removing a small number of samples with low confidence (< 75% of votes) regarding the main class labels (1.1–3.5% of microarrays and 0.9–1.3% of RNA-seq samples, depending on the data preprocessing method). Consensus clustering26 analyses were subsequently performed on expression data from a total of 384 data processing combinations, and resulting clusters were evaluated using different statistics. Cluster purity27 and clustering comparison measures28 were used to evaluate agreement between clusters identified using single-platform and combined data, while entropy27, a measure of association29 and randomness30,31 were used to evaluate the tendency of data to cluster by platform rather than by subtype. Lastly, we used min (O/E) , the minimum ratio of observed (size of the smallest cluster) relative to expected (number of samples divided by number of clusters), to identify data containing spurious clusters.
First, the analysis of platform entropy revealed that two cross-platform normalization methods, feature-specific quantile normalization (FSQN)15 and ComBat21,32, performed well as evidenced by their high entropy. In contrast, the two other methods tested, training distribution matching (TDM)14 and quantile normalization33, performed rather poorly showing entropy below 0.1 (Supplementary Fig. S2). For TDM, in addition to using normalized values as input, we also used raw RNA-seq counts as recommended by the authors of the TDM software34, which yielded similar results with entropy close to zero (data not shown). Based on these results, TDM and quantile normalization were excluded from further analyses, leaving 192 data processing combinations for each class.
Next, the following selected filters were applied: purity > 0.8 to retain only results with good agreement between single-platform and combined data, and min(O/E) > 0.1 to eliminate results containing spurious clusters. These filters had little effect on the proportion of single-platform processing or unsupervised feature selection methods in the remaining combinations; however, the vast majority of clustering results remaining after filtering were associated with three LUAD subtypes and two LUSC subtypes (Supplementary Fig. S3). To confirm this observation, we also analyzed the frequency of the best number of clusters for each dataset evaluated using the R package NbClust35 combined with the above filters. This analysis supported the same result showing three LUAD subtypes and two LUSC subtypes (Supplementary Fig. S4).
Remaining data processing combinations retained in both LUAD and LUSC (78 combinations) were then ranked using various statistics27,28,29,30,31. The analysis of absolute correlation between these measures revealed four groups (Supplementary Fig. S5), each of which was weighted equally for the final ranking: (1) single vs. cross-platform clustering agreement: purity, adjusted mutual information, adjusted Rand index, normalized information distance, normalized variation information; (2) measures related to platform entropy and platform-cluster association: entropy, Cramér’s V; (3) platform randomness within clusters: number of runs divided by sample size, rank version of von Neumann's ratio and (4) min(O/E) for minimizing the presence of spurious clusters. The top-ranking data processing combination was: microarray suite 5.0 (mas5)36 for microarray data, edgeR reads per kilobase per million (rpkm)37 for RNA-seq, FSQN15 for cross-platform normalization, and median absolute deviation (mad) for unsupervised feature selection (Supplementary Table S1).
Using clusters resulting from the top-ranking data processing combination as class labels, all lung samples (normal, LUAD and LUSC) were submitted to a final round of supervised classification, which in our experience improves the classification of some of the samples that are more difficult to classify using unsupervised methods alone. The heatmap in Fig. 2 represents the results of this final classification. Clustering of cross-platform normalized data showed consistent expression patterns with good separation between subtypes and even distribution of samples from the two expression profiling platforms for all tumor subtypes.

Heatmap and hierarchical clustering of lung cancer and normal lung microarray and RNA-seq data. LNOR, normal (healthy) lung; LUAD-(1–3), lung adenocarcinoma subtypes 1–3; LUSC-(1–2), lung squamous cell carcinoma subtypes 1–2. This figure was produced using R version 4.0.4 (https://www.r-project.org/).
Characteristics of lung cancer subtypes
To take advantage of our robust cross-platform tumor subtype classification, we proceeded to a reanalysis of LUAD and LUSC genomic alterations and patient outcomes and compared findings to those from the original studies12,13. Since the publication of these studies by TCGA, numerous additional patients have been enrolled and all data have been reanalyzed and harmonized to a newer version of the human genome (GRCH38)38 by the Genomics Data Commons (GDC)39.
Lung adenocarcinoma and LUSC expression subtypes from our analysis were compared to those identified in TCGA studies using annotations from TCGAbiolinks40. Lung adenocarcinoma subtypes 1–3 correspond to the proximal-proliferative, proximal-inflammatory and terminal respiratory unit subtypes, respectively, whereas LUSC-1 regroups the basal, primitive and secretory subtypes and LUSC-2 corresponds to the classical subtype. Analysis of available survival data (1,734 patients) revealed significant (p < 1e−10) differences between subtypes (Supplementary Fig. S6). Overall, LUAD-1 patients had the worst overall prognosis, whereas LUAD-3 patients had the best prognosis (both overall and relapse-free survival). For LUSC, subtype 1 had a better relapse-free survival than subtype 2. Follow-up times were relatively short for TCGA data as noted before41 but were in general longer for microarray data42,43,44,45,46,47 which strengthened the analysis for the combined survival data.
Focal copy number amplifications were assessed in each tumor subtypes using masked copy number segments from GDC analyzed with GISTIC 2.048. Table 1 lists amplified focal copy number regions that overlap with genes coding for targets of clinical-stage or approved lung cancer therapeutic targets2,5,8,49 in any of the five lung cancer subtypes. Lung adenocarcinoma subtypes 1–2 had the highest number of focal amplifications containing potential oncogenes (ERBB2, FGFR1, KRAS, MET), whereas LUAD-3 contained none. Interestingly, LUAD subtypes 1–2 also contained KDR (a.k.a VEGFR) amplification, which was not reported in the original TCGA LUAD study13. Epithelia growth factor receptor (EGFR) was most frequently amplified in four subtypes (LUAD-1, LUAD-2, LUSC-1, LUSC-2). We further evaluated the percentage of samples carrying mutations using averages from MuTect50, VarScan 251, Somatic Sniper52 and MuSE53 in lung cancer therapeutic targets (Table 2). Percentages of samples with mutations within each tumor subtype were highly consistent between the different variant-calling software. Again, LUAD subtypes carried the largest load of somatic mutations in potential oncogenes, and the most frequent mutated gene was KRAS. However, the LUAD-3 subtype had the most frequent mutations in EGFR. Lung squamous cell carcinoma had much less mutations as compared with LUAD, however some genes including e.g. KDR and ROS1 were frequently mutated in LUSC.
Because of the importance of protein assays in the clinic54, we further sought to integrate quantitative proteomics data with gene expression data, taking advantage of the recently released data for LUAD by the Clinical Proteomic Tumor Analysis Consortium (CPTAC)55. Our goal was to select a small set of biomarkers for subtype classification, capable of classifying LUAD tumors using either gene expression or quantitative proteomics data. To illustrate the robustness of the biomarkers, we combined gene expression data with proteomics data without any special cross-platform normalization other than scaling the mean and variance using microarray data as reference. Labels for CPTAC samples (205 classified samples with both RNA-seq and proteomics data) were assigned using classification of RNA-seq samples as described above. The top-10 features were selected for each one-against-one (OAO) class comparison using the log2 fold change (log2FC) and the overlap of locally adaptive kernel densities56. Figure 3 shows that biomarkers selected across platforms are able to accurately separate samples by clustering for all OAO class comparisons.

Biomarkers selected across three platforms for one-against-one classification of LUAD subtypes and normal lung. (A) Normal lung vs. LUAD-1; (B) normal lung vs. LUAD-2; (C) normal lung vs. LUAD-3; (D) LUAD-1 vs. LUAD-2; (E) LUAD-1 vs. LUAD-3; (F) LUAD-2 vs. LUAD-3. HGNC gene symbols are used to identify all biomarkers. This figure was produced using R version 4.0.4 (https://www.r-project.org/).
Discussion
Lung adenocarcinoma and LUSC have previously been classified into transcriptional subtypes associated with important characteristics such as response to targeted therapies10,11. Transcriptional subtypes can further be integrated with other data such as somatic mutations and DNA methylation into multi-omics subtypes12,13. Previous studies have also shown that classification of NSCLC into histological subtypes can be achieved using relatively simple methods, using both microarray and RNA-seq data, for example a nearest class centroid approach using differentially expressed genes and Pearson correlation as a similarity measure57, or a two gene (KRT5 and AGR2) expression ratio which classified LUAD and LUSC samples with relatively high accuracy58. However, to our knowledge, systematic evaluation of data processing methods for unsupervised classification of LUAD and LUSC transcriptional subtypes across gene expression profiling platforms has not been performed previously. In this study, we used an unsupervised approach combining 384 data processing methods to analyze public gene expression data (2,079 microarrays and 1673 RNA-seq samples). This analysis provided insights into the optimal combination of data processing methods for cross-platform clustering, and enabled the identification of robust LUAD and LUSC expression subtypes in combined microarray and RNA-seq data.
Combinations of data processing methods were evaluated using single and cross-platform consensus clustering, and various statistics including cross-platform purity, clustering comparison measures, platform entropy, as well as randomness and min(O/E). Whereas cluster purity is generally used to evaluate the ability of a clustering method to recover known classes27, here it was used to evaluate the maximum agreement between single and cross-platform data. Measures of randomness enabled the evaluation of the tendency of samples to regroup by platform within clusters. In addition, the use of min(O/E) allowed effective filtering of small, spurious clusters. For unsupervised feature selection methods, the simplest and fastest method, namely median absolute deviation (mad), performed well and ranked above more complex and computationally intensive methods. The approach of binning genes by expression level also helped to avoid enrichment of features sets with low-expressed genes.
For cross-platform normalization, FSQN15 performed better than other methods tested in this study. This method normalizes RNA-seq data in a feature-specific manner, using microarray data as a reference (quantiles for each gene). This method performs best with larger numbers of samples, as was the case in our study. As discussed in Franks et al.15, its superior performance can be attributed to the fact that FSQN preserves “distribution information about the center and spread of each individual gene”. Interestingly, ComBat21, a method originally developed to adjust microarray data for batch effects, also performed relatively well for removing platform effects. The latter method estimates model parameters by pooling information across genes and experimental conditions. The resulting empirical Bayes estimates are used to adjust the data for unwanted sources of experimental variation. The other two methods tested for cross-platform normalization, namely quantile normalization33 and TDM14, did not perform well as evidenced by low entropy, meaning that unsupervised learning with data normalized using these methods would identify primarily platform-specific clusters. Quantile normalization ranks features using expression levels, and assigns to each feature the average value of other features with the same rank in other samples. This method can be used with a single matrix, or with a target and a reference matrix. Training distribution matching uses a similar approach, whereas target distributions are adjusted to match certain properties of a reference distribution (interquartile range, spread of the tails, extreme values) and expression data in a target sample is mapped into a range from the minimum to the maximum of the reference data. Altogether, this analysis showed that feature-specific methods such as FSQN perform better for cross-platform normalization, especially for unsupervised learning which is more sensitive than supervised approaches to experimental biases including platform effects.
After the filtering and ranking of data processing methods, we found that RNA-seq data normalized using effective gene length (edgeR rpkm37 and DeSeq2 fpkm59) performed well as input for classification after cross-platform normalization. Although RNA-seq counts normalized using gene or transcript length are generally used to compare gene expression within samples, here we show that such units are very compatible with both supervised and unsupervised classification approaches, and integrate better with microarray data for cross-platform normalization. This can be explained by the fact that methods used to summarize microarray data (mas5 or RMA) are averages across probes, and there is no direct link between gene length and expression level. RNA-seq data normalized using effective gene length are thus more similar to, and integrate better with microarray data for cross-platform normalization and machine learning tasks such as feature selection and classification.
The three LUAD subtypes identified in our analysis were highly concordant with those identified in a previous study10. However, single and cross-platform clustering data provided strong evidence for only two LUSC expression subtypes, in line with results from a previous study60, but opposed to another study that identified four subtypes11. This may be explained by the fact that the study by Wilkerson et al.11 used microarray data only, and a relatively small number of samples, although subtypes were validated across several datasets. The most limiting factor for the number of clusters was cross-platform purity, whereas only two subtypes were consistent between single-platform and cross-platform unsupervised learning in LUSC. The three subtypes previously identified within LUSC-1 (basal, primitive and secretory) may have some utility in terms of prognostics/diagnostics, but our results show that they are grouped into a single class by our data-driven, unsupervised cross-platform tumor subtype identification methodology. This is a strength in our approach, that only the most robust clusters, identified across platforms, are retained as candidate tumor subtypes. Robust classification schemes are more likely to successfully transfer to real-world applications by minimizing reliance on features that are variable due to batch or platform effects.
Each tumor subtype was analyzed separately for focal amplifications and somatic mutations in genes coding for targets of clinical-stage or approved lung cancer therapeutics, and patterns presented here show that patients within each subtype may benefit from a particular subset of targeted therapeutics. In addition, to demonstrate the robustness of transcriptional subtypes identified in this study, we showed that biomarkers accurately separating the different tumor subtypes and normal tissues can be selected and validated with mass spectrometry-based quantitative proteomics data, even though the number of proteins quantified is limited as compared with gene expression data, and generally biased towards highly expressed genes. With further validation, the identified proteins may be used as biomarkers to classify tumors using classical protein-based methods such as immunohistochemistry.
Cross-platform classification of microarray and RNA-seq data is challenging because of intrinsic differences and biases in data distribution between the two platforms. A careful selection of data processing and machine learning methods enabled cross-platform classification of lung tumor expression subtypes. Our study confirmed three LUAD expression subtypes, but only two subtypes in LUSC as opposed to four subtypes previously identified. Such classification provides insights into clinical management and drug development for LUAD and LUSC, in particular with respect to identifying subtype-specific targets for antibody-based therapeutics. This study provides the basis for further integrative analysis of microarray, RNA-seq and quantitative proteomics data, and for the classification of tumors into expression subtypes.
Methods
Data acquisition and preprocessing
Raw data corresponding to a total of 2,079 samples profiled using microarrays (500 normal, 1134 LUAD and 445 LUSC) were obtained from GEO61 and 1673 lung samples profiled using RNA-seq (532 normal, 640 LUAD and 501 LUSC) were obtained from the Sequence Read Archive (SRA)62 (dbGap accession number phs000424.v8.p2, fresh frozen and PAXgene-preserved samples only) and GDC39 (projects TCGA and CPTAC-3). All data were processed using GDC reference files (GRCh38.d1.vd1, GENCODE 22): a custom chip definition (21,552 genes) file was created for microarray data using the methods of Dai et al.63, and GTEx samples were re-aligned to GRCh38.d1.vd1 using GDC mRNA analysis pipeline (STAR two-pass)64. Microarray data were combined into one expression set and processed using R library affy65. RNA-seq data were combined into a single count matrix and processed using edgeR37, DESeq266 and limma (voom)67 (Table 3). Reduced ranges of coding exons were used for fragments per kilobase per million (fpkm) calculations. Batch (series) effect were corrected using ComBat21,32. A subset of 17,095 protein-coding genes represented on both platforms was selected for further analyses. For cross-platform analysis, we first scaled the RNA-seq data to have a similar distribution (mean and variance) to that of microarray data and then merged and normalized the data from the two platforms using R libraries FSQN15, TDM14, sva32 and preprocessCore68 (Table 4).
Unsupervised feature selection and clustering
Normalized data (single-platform and cross-platform) were submitted to unsupervised feature selection with eight different methods using R libraries stats69, Rdimtools70 and an in-house package for singular value decomposition entropy implemented using Rcpp71 (Table 5). For each dataset, a total of 210 features were equally selected from eight bins delineated using gene expression means, to avoid enrichment of low-expression features which are more noisy. This number of features was deemed optimal as evaluated by cross-platform purity for a range of features between 28 and 212 (Supplementary Fig. S7). Data were then submitted to consensus clustering26 as well as evaluation of the optimal number of clusters (min = 2, max = 6) using NbClust35. Agreement between clusters identified using single-platform and combined data were evaluated using R package aricode72 as well as a custom R function to evaluate purity (maximum agreement between single and cross-platform data). The tendency of cross-platform data to cluster by platform rather than by cancer subtype was evaluated using NMF package73 (function entropy) and DescTools package74 (functions CramerV29, RunsTest75 and BartelsRankTest31). Spurious clusters were evaluated using min(O/E), the size of the smallest cluster over sample size divided by number of clusters.
Supervised classification
Samples were submitted to five rounds of a Monte Carlo iterative ensemble classification algorithm, modified from56 to include rounds of repeated random sampling (Supplementary Fig. S1). At each round, a total of 100 iterations were performed, in which 100 samples per class were randomly sampled with replacement, and classifiers were constructed for each \(\left(\genfrac{}{}{0pt}{}{w}{2}\right)\) pair of classes OAO, for three supervised feature selection methods, three classification methods and six increasing number of features. All remaining samples were classified, by generating votes only where labels generated by OAO classifiers were maximal (i.e. number of classes minus one). Then, labels assigned with high confidence (> 90% of votes) by the ensemble of experts (5400 votes from 100 iterations, three feature selection methods, three classification methods, and six increasing number of features) were fed back into the data and used for subsequent feature selection and training of the classifiers. This procedure was repeated until the number of predictions was stabilized over a number of iterations (convergence was considered achieved when the number of classified samples reached a plateau). At this point, all samples with moderate to high confidence (> 75% of votes) were assigned class labels and retained for further analysis. For supervised classification purposes, filter-based feature selection was performed by selecting the top \({\left({2}^{k}\right)}_{k=5}^{10}\) features ranked using three different statistics: q-values derived from linear models for microarray (Limma) moderated t-test76,77, the overlapping coefficient of locally adaptive kernel density estimates78,79, and the weights of support vectors (WSV)80. Locally adaptive kernel densities and overlapping coefficients were computed using an in-house R package implemented using Rcpp71. The WSV were computed using the e1071 R package81. Classification was achieved using three algorithms implemented in the RWeka package82: k-nearest neighbors83, random forests84 and support vectors machines85.
Characterization of tumor subtypes and biomarker selection
Survival data were obtained from the TCGA pan-cancer clinical resource41 for RNA-seq data, and from GEOmetadb86 for microarray data. Survival analysis was performed using the R package survival87 using default parameters. Masked copy number segments were obtained from GDC and processed using GISTIC 2.048. The search for focal amplifications was restricted to peaks covering less than 3 Mb as recommended in Krijgsman et al.88. Somatic calls from MuTect50, VarScan 251, Somatic Sniper52 and MuSE53 were obtained from the GDC, and lung tumor RNA-seq data were analyzed using VarDict89. These data were analyzed using the R packages maftools90 and VariantAnnotation91.
To select biomarkers for LUAD, we used microarray data processed using mas536 and RNA-seq data normalized using DESEq292 and log2 protein expression data from CPTAC55. Proteomics and RNA-seq data were scaled (mean and variance) using microarray data as reference. Features were selected to maximize the log2FC and to minimize the overlap of locally adaptive kernel densities56 with a total of 10 features for each OAO class comparison.
Data availability
Raw data analysed in this study are available to download from GEO61, SRA62 and GDC39 repositories. Some restrictions may apply to the availability of these data, which were used under license for the current study. Processed data analyzed as part of the current study are available from the corresponding authors on request.
References
Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424 (2018).
Herbst, R. S., Morgensztern, D. & Boshoff, C. The biology and management of non-small cell lung cancer. Nature 553, 446–454 (2018).
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 70, 7–30 (2020).
Travis, W. D. et al. The 2015 World Health Organization classification of lung tumors: Impact of genetic, clinical and radiologic advances since the 2004 classification. J. Thorac. Oncol. 10, 1243–1260 (2015).
Hirsch, F. R. et al. Lung cancer: Current therapies and new targeted treatments. Lancet 389, 299–311 (2017).
Bernicker, E. H., Allen, T. C. & Cagle, P. T. Update on emerging biomarkers in lung cancer. J. Thorac. Dis. 11, S81–S88 (2019).
Sankar, K., Gadgeel, S. M. & Qin, A. Molecular therapeutic targets in non-small cell lung cancer. Expert Rev. Anticancer Ther. 20, 1–15 (2020).
Reck, M. & Rabe, K. F. Precision diagnosis and treatment for advanced non-small-cell lung cancer. N. Engl. J. Med. 377, 849–861 (2017).
Herbst, R. S., Heymach, J. V. & Lippman, S. M. Lung cancer. N. Engl. J. Med. 359, 1367–1380 (2008).
Hayes, D. N. et al. Gene expression profiling reveals reproducible human lung adenocarcinoma subtypes in multiple independent patient cohorts. J. Clin. Oncol. 24, 5079–5090 (2006).
Wilkerson, M. D. et al. Lung squamous cell carcinoma mRNA expression subtypes are reproducible, clinically important, and correspond to normal cell types. Clin. Cancer Res. 16, 4864–4875 (2010).
Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525 (2012).
Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550 (2014).
Thompson, J. A., Tan, J. & Greene, C. S. Cross-platform normalization of microarray and RNA-seq data for machine learning applications. PeerJ 4, e1621 (2016).
Franks, J. M., Cai, G. & Whitfield, M. L. Feature specific quantile normalization enables cross-platform classification of molecular subtypes using gene expression data. Bioinformatics 34, 1868–1874 (2018).
Marioni, J. C., Mason, C. E., Mane, S. M., Stephens, M. & Gilad, Y. RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 18, 1509–1517 (2008).
Quackenbush, J. Microarray data normalization and transformation. Nat. Genet. 32(Suppl), 496–501 (2002).
Li, P., Piao, Y., Shon, H. S. & Ryu, K. H. Comparing the normalization methods for the differential analysis of Illumina high-throughput RNA-Seq data. BMC Bioinform. 16, 347 (2015).
Butnor, K. J. Avoiding underdiagnosis, overdiagnosis, and misdiagnosis of lung carcinoma. Arch. Pathol. Lab. Med. 132, 1118–1132 (2008).
Oyelade, J. et al. Clustering algorithms: Their application to gene expression data. Bioinform. Biol. Insights 10, 237–253 (2016).
Johnson, W. E., Li, C. & Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127 (2007).
Leek, J. T. et al. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 11, 733–739 (2010).
Alelyani, S., Tang, J. & Liu, H. Feature selection for clustering: A review. In Data Clustering Algorithms and Applications (eds Aggarwal, C. C. & Reddy, C. K.) 29 (CRC Press, Berlin, 2013).
Hancer, E., Xue, B. & Zhang, M. A survey on feature selection approaches for clustering. Artif. Intell. Rev. https://doi.org/10.1007/s10462-019-09800-w (2020).
Conesa, A. et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 17, 13 (2016).
Wilkerson, M. D. & Hayes, D. N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 26, 1572–1573 (2010).
Kim, H. & Park, H. Sparse non-negative matrix factorizations via alternating non-negativity-constrained least squares for microarray data analysis. Bioinformatics 23, 1495–1502 (2007).
Vinh, N. X., Epps, J. & Bailey, J. Information theoretic measures for clusterings comparison: Variants, properties, normalization and correction for chance. J. Mach. Learn. Res. 11, 2837–2854 (2010).
Cramér, H. Mathematical Methods of Statistics Vol. 43 (Princeton University Press, 1999).
Wald, A. & Wolfowitz, J. On a test whether two samples are from the same population. Ann. Math. Stat. 11, 147–162 (1940).
Bartels, R. The rank version of von Neumann’s ratio test for randomness. J. Am. Stat. Assoc. 77, 40–46 (1982).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883 (2012).
Bolstad, B. M., Irizarry, R. A., Astrand, M. & Speed, T. P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19, 185–193 (2003).
Thompson, J. A. & Greene, C. S. TDM: R Package for Normalizing RNA-seq Data to Make Them Comparable to Microarray Data. (Accessed 25 June 2020); https://github.com/greenelab/TDM (2016).
Charrad, M., Ghazzali, N., Boiteau, V., Niknafs, A. & Charrad, M. M. Package ‘NbClust’. J. Stat. Softw. 61, 1–36 (2014).
Hubbell, E., Liu, W. M. & Mei, R. Robust estimators for expression analysis. Bioinformatics 18, 1585–1592 (2002).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Schneider, V. A. et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 27, 849–864 (2017).
Grossman, R. L. et al. Toward a shared vision for cancer genomic data. N. Engl. J. Med. 375, 1109–1112 (2016).
Colaprico, A. et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 44, e71 (2016).
Liu, J. et al. An Integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 173, 400–416 (2018).
Kuner, R. et al. Global gene expression analysis reveals specific patterns of cell junctions in non-small cell lung cancer subtypes. Lung Cancer 63, 32–38 (2009).
Hou, J. et al. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS ONE 5, e10312 (2010).
Okayama, H. et al. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Can. Res. 72, 100–111 (2012).
Botling, J. et al. Biomarker discovery in non-small cell lung cancer: Integrating gene expression profiling, meta-analysis, and tissue microarray validation. Clin. Cancer Res. 19, 194–204 (2013).
Rousseaux, S. et al. Ectopic activation of germline and placental genes identifies aggressive metastasis-prone lung cancers. Sci. Transl. Med. 5, 186 (2013).
Der, S. D. et al. Validation of a histology-independent prognostic gene signature for early-stage, non-small-cell lung cancer including stage IA patients. J. Thorac. Oncol. 9, 59–64 (2014).
Mermel, C. H. et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41 (2011).
National Cancer Institute. Drugs Approved for Lung Cancer (Accessed 10 September 2020); https://www.cancer.gov/about-cancer/treatment/drugs/lung (2018).
Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219 (2013).
Koboldt, D. C. et al. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576 (2012).
Larson, D. E. et al. SomaticSniper: Identification of somatic point mutations in whole genome sequencing data. Bioinformatics 28, 311–317 (2012).
Fan, Y. et al. MuSE: accounting for tumor heterogeneity using a sample-specific error model improves sensitivity and specificity in mutation calling from sequencing data. Genome Biol. 17, 178 (2016).
Powers, A. D. & Palecek, S. P. Protein analytical assays for diagnosing, monitoring, and choosing treatment for cancer patients. J. Healthcare Eng. 3, 503–534 (2012).
Gillette, M. A. et al. Proteogenomic characterization reveals therapeutic vulnerabilities in lung adenocarcinoma. Cell 182, 200–225 (2020).
Fauteux, F. et al. Computational selection of antibody-drug conjugate targets for breast cancer. Oncotarget 7, 2555–2571 (2016).
Girard, L. et al. An expression signature as an aid to the histologic classification of non-small cell lung cancer. Clin. Cancer Res. 22, 4880–4889 (2016).
Li, X. et al. A qualitative transcriptional signature for the histological reclassification of lung squamous cell carcinomas and adenocarcinomas. BMC Genomics 20, 881 (2019).
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628 (2008).
Inamura, K. et al. Two subclasses of lung squamous cell carcinoma with different gene expression profiles and prognosis identified by hierarchical clustering and non-negative matrix factorization. Oncogene 24, 7105–7113 (2005).
Edgar, R., Domrachev, M. & Lash, A. E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210 (2002).
Leinonen, R., Sugawara, H. & Shumway, M. The sequence read archive. Nucleic Acids Res. 39, D19–D21 (2011).
Dai, M. et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 33, e175 (2005).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Gautier, L., Cope, L., Bolstad, B. M. & Irizarry, R. A. affy-analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20, 307–315 (2004).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Bolstad, B. M. PreprocessCore: A Collection of Pre-processing Functions. R package version 1.52.1 (2020).
R Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, Vienna, Austria, 2020).
You, K. Rdimtools: An R package for Dimension Reduction and Intrinsic Dimension Estimation. Preprint at http://arXiv.org/2005.11107 (2020).
Eddelbuettel, D. et al. Rcpp: Seamless R and C++ integration. J. Stat. Softw. 40, 1–18 (2011).
Chiquet, J., Rigaill, G. & Sundqvist, M. Aricode: Efficient Computations of Standard Clustering Comparison Measures. R Package Version 1.0.0 (2020).
Gaujoux, R. & Seoighe, C. A flexible R package for nonnegative matrix factorization. BMC Bioinform. 11, 367 (2010).
Signorell, A. et al. DescTools: Tools for Descriptive Statistics. R Package Version 0.99.40 (2020).
Wackerly, D., Mendenhall, W. & Scheaffer, R. L. Mathematical Statistics with Applications (Cengage Learning, 2014).
Storey, J. D. The positive false discovery rate: A Bayesian interpretation and the q-value. Ann. Stat. 31, 2013–2035 (2003).
Smyth, G. K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3, 3 (2004).
Abramson, I. S. On bandwidth variation in Kernel estimates-a square root law. Ann. Stat. 10, 1217–1223 (1982).
Schmid, F. & Schmidt, A. Nonparametric estimation of the coefficient of overlapping—Theory and empirical application. Comput. Stat. Data Anal. 50, 1583–1596 (2006).
Guyon, I., Weston, J., Barnhill, S. & Vapnik, V. Gene selection for cancer classification using support vector machines. Mach. Learn. 46, 389–422 (2002).
Meyer, D., Dimitriadou, E., Hornik, K., Weingessel, A. & Leisch, F. e1071: Misc functions of the Department of Statistics, Probability Theory Group. R Package Version 1.7-4 (2020).
Hornik, K., Buchta, C. & Zeileis, A. Open-source machine learning: R meets Weka. Comput. Stat. 24, 225–232 (2009).
Aha, D. W., Kibler, D. & Albert, M. K. Instance-based learning algorithms. Mach. Learn. 6, 37–66 (1991).
Breiman, L. Random forests. Mach. Learn. 45, 5–32 (2001).
Platt, J. C. Fast training of support vector machines using sequential minimal optimization. In Advances in Kernel Methods Vol. 3 (eds Schoelkopf, C. B. & Smola, A.) 185–208 (MIT Press, 1998).
Zhu, Y., Davis, S., Stephens, R., Meltzer, P. S. & Chen, Y. GEOmetadb: Powerful alternative search engine for the gene expression Omnibus. Bioinformatics 24, 2798–2800 (2008).
Therneau, T. M. & Grambsch, P. M. Modeling Survival Data: Extending the Cox Model (Springer, 2013).
Krijgsman, O., Carvalho, B., Meijer, G. A., Steenbergen, R. D. & Ylstra, B. Focal chromosomal copy number aberrations in cancer—Needles in a genome haystack. Biochem. Biophys. Acta 1843, 2698–2704 (2014).
Lai, Z. et al. VarDict: A novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 44, e108 (2016).
Mayakonda, A., Lin, D. C., Assenov, Y., Plass, C. & Koeffler, H. P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 28, 1747–1756 (2018).
Obenchain, V. et al. VariantAnnotation: A bioconductor package for exploration and annotation of genetic variants. Bioinformatics 30, 2076–2078 (2014).
Love, M. I., Anders, S. & Huber, W. Analyzing RNA-seq data with DESeq2. In R package Reference Manual (2017).
Irizarry, R. A. et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264 (2003).
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Genome Biol. 11, R106 (2010).
Law, C. W., Chen, Y., Shi, W. & Smyth, G. K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29 (2014).
Rousseeuw, P. J. & Croux, C. Alternatives to the median absolute deviation. J. Am. Stat. Assoc. 88, 1273–1283 (1993).
Varshavsky, R., Gottlieb, A., Linial, M. & Horn, D. Novel unsupervised feature filtering of biological data. Bioinformatics 22, e507–e513 (2006).
Liu, Y., Liu, K., Zhang, C., Wang, J. & Wang, X. Unsupervised feature selection via diversity-induced self-representation. Neurocomputing 219, 350–363 (2017).
He, X., Cai, D. & Niyogi, P. Laplacian score for feature selection. In Advances in Neural Information Processing Systems 507–514.
Cai, D., Zhang, C. & He, X. Unsupervised feature selection for multi-cluster data. In Proc. 16th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining 333–342.
Zhao, Z. & Liu, H. Spectral feature selection for supervised and unsupervised learning. In Proc. 24th International Conference on Machine Learning 1151–1157.
Lu, Q., Li, X. & Dong, Y. Structure preserving unsupervised feature selection. Neurocomputing 301, 36–45 (2018).
Yang, Y., Shen, H. T., Ma, Z., Huang, Z. & Zhou, X. L2, 1-norm regularized discriminative feature selection for unsupervised. In Twenty-Second International Joint Conference on Artificial Intelligence.
Acknowledgements
The Genotype Tissue-Expression Project (dbGaP accession number phs000424.v8.p2) was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The results published here are in part based upon data generated by The Cancer Genome Atlas (http://cancergenome.nih.gov) managed by the NCI and NHGRI. Some data used in this publication were generated by the Clinical Proteomic Tumor Analysis Consortium (NCI/NIH).
Author information
Authors and Affiliations
Contributions
F.F. performed data analysis and drafted the manuscript. A.S. contributed to data analysis. Y.P. contributed to statistical analysis. S.M. contributed to interpretation of results. F.F. and J.J.H. designed the study and interpreted results. All authors have edited and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fauteux, F., Surendra, A., McComb, S. et al. Identification of transcriptional subtypes in lung adenocarcinoma and squamous cell carcinoma through integrative analysis of microarray and RNA sequencing data. Sci Rep 11, 8709 (2021). https://doi.org/10.1038/s41598-021-88209-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-88209-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
