
Abstract
As part of our efforts to develop rhenium-oxo corroles as photosensitizers for oxygen sensing and photodynamic therapy, we investigated the potential β-perhalogenation of five ReO meso-tris(para-X-phenyl)corroles, Re[TpXPC](O) (X = CF3, H, F, CH3, and OCH3), with elemental chlorine and bromine. With Cl2, β-octachlorinated products Re[Cl8TpXPC](O) were rapidly obtained for X = CF3, H, and CH3, but X = OCH3 resulted in overchlorination on the meso-aryl groups. Full β-octabromination proved slower relative to Cu and Ir corroles, but the desired Re[Br8TpXPC](O) products were finally obtained for X = H and F after a week at room temperature. For X = CH3 and OCH3, these conditions led to undecabrominated products Re[Br11TpXPC](O). Compared to the β-unsubstituted starting materials, the β-octahalogenated products were found to exhibit sharp 1H NMR signals at room temperature, indicating that the aryl groups are locked in place by the β-halogens, and substantially redshifted Soret and Q bands. Single-crystal X-ray structures of Re[Cl8TpCF3PC](O), Re[Cl8TpCH3PC](O), and Re[Br8TpFPC](O) revealed mild saddling for one Cl8 structure and the Br8 structure. These structural variations, however, appear too insignificant to explain the slowness of the β-octabromination protocols, which seems best attributed to the deactivating influence of the high-valent Re center.
Similar content being viewed by others
Introduction
The remarkable β-octachlorination and β-octabromination of metallotetraarylporphyrins was first reported by the Traylor, Dolphin and their research groups in the 1980s1,2. During the 1990s, iron and manganese complexes of β-octahalogenoporphyrins were intensively investigated as rugged, synthetic models of cytochrome P4503,4,5,6. β-Octahalogeno-meso-tetraarylporphyrin derivatives also provided textbook examples of saddling, a nonplanar distortion in which the pyrrole rings are alternately tilted up and down relative to the mean porphyrin plane7,8,9,10,11. The compounds became the subject of a battery of spectroscopic, electrochemical, and structural studies, which yielded a rich body of insights on substituent effects in porphyrin derivatives10,12,13,14,15.
With the advent of simple, one-pot syntheses16,17,18,19 of corroles20,21, β-octabromination was also found to work for certain corrole derivatives22,23. In our laboratory, we prepared some of the first β-octabromo-meso-triarylcorroles, initially the copper complexes24,25 and subsequently also the free bases26,27,28. Remarkably, a number of crystal structures of β-octabrominated metallocorroles revealed planar corrole rings, underscoring the rigidity of the corrole ring system relative to porphyrins23,29,30,31. Today we know that saddling in corroles is largely limited to copper32,33,34,35,36,37 corroles (and in part to silver38 corroles but not gold39,40,41,42 corroles), where it is thought to be a manifestation of ligand noninnocence, i.e., a distortion mode that facilitates CuII(dx2−y2)-corrole⋅2− antiferromagnetic coupling43,44,45,46.
The present study is part of our ongoing efforts to functionalize and derivatize 5d metallocorroles38,39,40,41,42,47,48,49,50,51,52,53,54. These complexes provide unusual examples of a large transition metal ion coordinated to a sterically constrained macrocyclic ligand. Despite the steric mismatch inherent in their structures, a good fraction of these complexes exhibit remarkable thermal, chemical, and photochemical stability. Many also exhibit near-IR phosphorescence and also efficiently sensitize singlet oxygen formation, which has led to applications in oxygen sensing, photodynamic therapy, and dye-sensitized solar cells55,56,57,58,59,60,61,62,63. The chemical reactivity of these complexes, by and large, remains poorly explored, with only a handful of reports on the subject. β-Octachlorination has been reported for an OsN corrole64, while gold corroles have been polyiodinated, with 4–5 iodines attached to the β-positions65,66. A couple of examples of metal-centered reactivity have also been documented; thus, MoO67 and ReO50 corroles have been transformed to the MX2 (X = Cl, Ph) derivatives, so-called Viking helmet corroles68, while OsN corroles have been found to act as unusual π-acceptor metallaligands toward Pt(II)64. Herein we document our efforts to halogenate rhenium(V)-oxo triarylcorroles with elemental chlorine and bromine (Fig. 1).

ReO corroles synthesized in this work.
The main contributions of this work may be described as threefold. First and foremost are the products themselves, which should serve as starting materials in a variety of cross-coupling reactions, affording, for example, ReO undecaarylcorroles via the Suzuki–Miyaura reaction. The products thus obtained are likely to further extend the growing range of applications of ReO corroles60,61. Second, three of the β-octahalogenated products have yielded single-crystal X-ray structures, shedding light on potential distortion modes available to these sterically congested species. Third, although the “messy” reaction conditions did not allow us to devise kinetic studies, qualitative observations indicate major differences in the times required for β-octabromination as a function of the coordinated metal, which appear to be ascribable to the electronic effects of the coordinated metal, as described below.
Results and discussion
Synthetic method development
Optimizing the conditions for β-octachlorination proved relatively straightforward69. In the final, optimized protocol, a saturated, greenish-yellow solution of chlorine (Cl2) in chloroform was added dropwise, over a period of 20 min, to a benzene solution of β-unsubstituted ReO corroles, Re[TpXPC](O), maintained at 0 °C in an ice bath. The ice bath was removed after 1 h and the reaction was allowed to continue at room temperature for 24 h. After work-up, HR-ESI mass spectrometry and 1H NMR spectroscopy showed that all eight β-hydrogens had been fully replaced by chlorine atoms for X = CF3, H and CH3. For X = OCH3, the most electron-donating substituent, however, overchlorination was observed, with Cl8, Cl9 and Cl10 products appearing in a ratio of approximately 15:100:80 (see Figure S11 in the Supplementary Material).
Finding the optimum conditions for β-octabromination, in contrast, involved a fair amount of trial and error. Initial experiments with up to 100 equiv liquid bromine (Br2) in chloroform led after 4 h to a mixture of Br4, Br5, Br6, and Br7 products, with only traces of Br8. Increasing the reaction time to 16 h also led to the same complex mixtures. Increasing the reaction time to 48 h, however, led to the selective formation of Br6 and Br7 derivatives as the major products, with the Br8 appearing as a minor product. We surmised that increasing the concentration of elemental bromine and lengthening the reaction time even further might result in full β-octabromination. Accordingly, we increased the amount of elemental bromine threefold and extended the reaction time to 7 days. As before, we began with 100 equiv of Br2 added dropwise over a 20-min period. On the 2nd day, we added another 100 equiv of Br2 dropwise over 20 min. We did the same on the 3rd day and let the reaction run for an additional 4 days, i.e., a total of 7 days. After work up, HR-MS and 1H NMR showed that octabrominated complexes Re[Br8TpXPC](O) had cleanly formed for X = H and F. For X = CH3 and OCH3, on the other hand, over-bromination had occurred, resulting in the undecabrominated complexes Re[Br11TpXPC](O).
The long times needed for β-octabromination of ReO corroles, and presumably also for OsN corroles, may be contrasted with the rapid reactions observed for Cu24 and Ir23 corroles. The difference is most simply ascribed to the higher oxidation state of the central metal in the case of the ReO and OsN complexes, which presumably deactivates the corrole toward electrophilic substitution. Such a rationale is in line with the redox potentials of the metallocorroles; the oxidation potentials of ReO50 and OsN52 corroles are substantially higher than those of analogous Cu24,38 and Ir23 corroles.
1H NMR and UV–Vis spectroscopy
Both types of spectra clearly reflect the effect of β-octahalogenation. The most obvious change in the 1H NMR spectra is associated with the disappearance of the β-proton signals between 8.5 and 10 ppm (Fig. 2). Another highly characteristic change is that unlike the room-temperature 1H NMR spectra of starting complexes50, the spectra of the β-perhalogenated products are already sharp at room temperature. The broad 1H NMR spectra of Re[TpXPC](O) at room temperature reflect partially restricted rotation of the meso-aryl groups and only around − 20 °C or so do the aryl ortho and meta signals split into distinguishable o,o´and m,m´signals. In β-perhalogenated products, the aryls are effectively locked in place even at room temperature.

1H NMR spectra of Re[Cl8TPC](O) (top) and Re[Br8TPC](O) (bottom) in CDCl3 at 298 K.
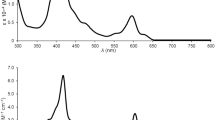
Like a number of other classes of 5d metallocorroles40,51,52,54, ReO triarylcorroles exhibit sharp, intense Soret bands and characteristic, double-humped Q bands50. The qualitative shapes of these features persist relatively unaltered upon β-octahalogenation. β-Octahalogenation does, however, engender significant redshifts for each of these features. Thus, for β-octachlorination, the Soret and Q bands redshift by around 9 and 13–16 nm, respectively, while for β-octabomination, the corresponding shifts are 17–19 and 21–22 nm, respectively (Table 1 and Fig. 3).

UV–Vis spectra in dichloromethane for (a) Re[Cl8TpXPC](O) (X = CF3, H and CH3), (b) Re[Br8TpXPC](O) (X = F and H) and (c) Re[Br11TpXPC](O) (X = CH3 and OCH3). (d) Comparative UV–Vis spectra for Re[TPC](O) (black), Re[Cl8TPC](O) (blue), and Re[Br8TPC](O) (red).
X-ray crystallography and molecular structure
The molecular structures of β-perhalogenated ReO corroles were expected to be of unusual interest as a window into potential deformation pathways of the corrole macrocycle in response to extreme peripheral crowding70. In general, β-octahalogenation does not result in significant nonplanar deformations for metallocorroles, reflecting the rigidity of the direct pyrrole–pyrrole linkage23,28,29,30,31. Coinage metal corroles, especially Cu32,33,34,35,36,37 corroles but also Ag38 corroles, constitute the major exceptions to this generalization. These metallocorroles are intrinsically saddled, as a result of a specific metal(d)-corrole(π) orbital interaction, which results in a noninnocent, partial MII-corrole⋅2− character of the complexes. Importantly, the degree of saddling in Cu corroles, while substantial even in β-unsubstituted triarylcorrole derivatives, can be greatly enhanced by β-octasubstitution. The same orbital interaction, however, is energetically unfavorable for Au corroles40. Accordingly, even undecasubstituted Au corroles are fairly rigorously planar29,30,38,40,41,42. So, as a matter of fact, are six-coordinate Ir corroles, including Ir β-octabrominated derivatives23,56,62. Diboron corroles provide another example of a dramatic structural influence of β-octasubstitution. Thus, while simple corroles yield strongly domed complexes with cisoid FBOBF groups71, β-octabromo-meso-triarylcorroles yield unbridged bis-BF2 complexes, with the BF2 groups on opposite sides of corrole macrocycle72.
Three of the products obtained here, including one octabrominated and two octachlorinated products, yielded single-crystal X-ray structures (Table 2). The Re–O and Re–N bond distances, as well as the displacement of the Re atom above the mean N4 plane, all turned out to be essentially identical to those observed for β-unsubstituted ReO corroles (Fig. 4)50. Interestingly, modest variations were observed for the conformations of the corrole macrocycles. Aligning the mean N4 planes of β-H8, β-Cl8, and β-Br8 structures showed that the corrole macrocycles in these systems might be described as slightly domed, planar, and slightly, if somewhat irregularly, saddled, respectively (Fig. 5 and Table 3). The β-Br8 crystal structure reported here thus represents a rare example of a saddled corrole, aside from the coinage metal corroles.

Thermal ellipsoid plots (50%) for (a) Re[Cl8TpCF3PC](O), (b) Re[Cl8TpCH3PC](O), and (c) Re[Br8TpFPC](O). Selected distances (Å) for (a) Re[Cl8TpCF3PC](O): Re1-N1 1.997(5), Re1-N2 2.015(4), Re1-N3 2.025(4), Re1-N4 1.995(4), and Re1-O1 1.670(4); Re2-N101 1.989(4), Re2-N102 2.014(4), Re2-N103 2.019(4), Re2-N104 1.980(4), Re2-O2 1.672. Selected distances (Å) for Re[Cl8TpCH3PC](O): Re1-N1 1.995(3), Re1-N2 2.019(3), Re1-N3 2.017(3), Re1-N4 1.992(3), and Re1-O1 1.677(3). Selected distances (Å) for Re[Br8TpFPC](O): Re1-N1 1.996(2), Re1-N2 2.019(2), Re1-N3 2.011(2), Re1-N4 1.997(2), and Re1-O1 1.673(2).

Mercury overlay of the nitrogen atoms of Re[TPC](O) (black), molecule 1 of Re[Cl8TpCF3PC](O) (blue) and Re[Br8TpFPC](O). (a) View from above C1–C19 toward C10. (b) View along C5–C15.
Might the above structural differences play a role in explaining the slow rates of β-octabromination of ReO triarylcorroles relative to Cu and Ir triarylcorroles? Given that the above differences are rather minor (Table 3), we believe that the answer is essentially ‘no’; as stated above, the high oxidation state of the Re center provides the most plausible explanation for the slowness of the octabromination.
Conclusion
In summary, we have optimized reaction conditions leading to β-perhalogenation of ReO triarylcorroles with elemental chlorine and bromine. β-Perhalogenation is accompanied by highly characteristic changes in the 1H NMR and UV–Vis spectra of the compounds. Three of the β-octahalogenated products, including two of the octachlorinated complexes and one octabrominated complex, yielded single-crystal X-ray structures. On the whole, the structures were remarkably similar to those of β-unsubstituted ReO corroles. Minor variations were observed in regard to macrocycle conformation. Thus, whereas β-unsubstituted ReO corroles are generally slightly domed, one octachlorinated complex and the octabrominated complex were found to exhibit slightly saddled macrocycles. These structural differences, however, appear to be too minor to explain the comparative slowness of β-octabromination of ReO triarylcorroles, relative to their Cu and Ir counterparts. The slowness is more plausibly attributed to the high oxidation state of the Re center, which leads to a higher oxidation potential for the corrole macrocycle and, in turn, a lower susceptibility to electrophilic attack. It is hoped that the β-perhalogenated complexes reported herein will act as substrates in Suzuki–Miyaura and other palladium-catalyzed transformations, thereby affording additional avenues for the elaboration of ReO corroles.
Experimental section
Materials
Rhenium-oxo meso-triarylcorroles, Re[TpXPC](O), were synthesized as previously reported50. Chlorine gas (Cl2), liquid bromine (Br2), benzene, and chloroform were purchased from Sigma-Aldrich. Silica gel 60 (0.04–0.063 mm particle size, 230–400 mesh, Merck) was used for flash chromatography and silica gel 60 preparative thin-layer chromatography (PTLC) plates (20 cm × 20 cm, 0.5 mm thick; Merck) were used for final purification of all complexes.
Instrumental methods
UV–visible spectra were recorded on an HP 8453 spectrophotometer. 1H NMR spectra (298 K, CDCl3) were recorded on a 400 MHz Bruker Avance III HD spectrometer equipped with a 5-mm BB/1H SmartProbe and referenced to residual CHCl3 at 7.26 ppm. High-resolution electrospray-ionization (HR-ESI) mass spectra were recorded from methanolic solution on an LTQ Orbitrap XL spectrometer.
General procedure for the synthesis of Re[Cl8TpXPC](O)
To a 10-mL benzene solution of Re[TpXPC](O) (X = CF3: 15 mg, 0.016 mmol; X = H: 25 mg, 0.034 mmol; X = CH3: 20 mg, 0.026 mmol) chilled to 0 °C in an ice-bath, was added a greenish-yellow, saturated solution of chlorine (Cl2, 10 mL) in chloroform over a period of 20 min. After an hour at 0 °C, the ice-bath was removed and the reaction was allowed to continue under stirring at room temperature for a total of 24 h. The reaction mixture was then quenched by washing twice with 20% aqueous sodium metabisulfite solution (20 mL × 2). The organic phase was thoroughly washed with distilled water, dried with sodium sulfate, and rotary-evaporated to dryness. The resulting crude product was dissolved in a minimum amount of dichloromethane and loaded onto a silica gel column prepared with 3:1 hexane/dichloromethane and eluted with the same solvent system. The resulting greenish-red product was collected, evaporated to dryness, and further purified via preparative thin-layer chromatography with the same solvent system. Yields and analytical details of new compounds are given below. X-ray-quality crystals of Re[Cl8TpCF3PC](O) and Re[Cl8TpCH3PC](O) were obtained by slow diffusion of methanol vapor into concentrated dichloromethane solutions of the complexes.
Re[Cl8TpCF3PC](O)
Yield 11.5 mg (59.22%). UV–Vis (CH2Cl2) λmax [nm, ε × 10−4 (M−1 cm−1)]: 349 (2.17), 447 (11.04), 565 (1.58), 599 (2.24). 1H NMR (400 MHz, 25 °C) δ: 8.29 (d, 3H, 3JHH = 7.92 Hz, 5,10,15-o1-Ph); 8.09 (d, 2H, 3JHH = 7.96 Hz, 5,15-o2-Ph); 8.05 (d, 1H, 3JHH = 7.60 Hz, 10-o2-Ph); 7.98 (d, 4H, 3JHH = 6.40 Hz, 5,15-m1& m2-Ph); 7.92 (d, 1H, 3JHH = 9.80 Hz, 10-m1-Ph); 7.84 (d, 1H, 3JHH = 8.00 Hz, 10-m2-Ph). Elemental analysis calcd for C40H12ON4F9Cl8Re: C 39.86, H 1.00, N 4.65; found: C 40.27, H 1.21. N 4.24. MS (ESI): M+ = 1203.7915 (expt), 1203.7880 (calcd for C40H12ON4F9Cl8Re).
Re[Cl8TPC](O)
Yield 23 mg (66.71%). UV–Vis (CH2Cl2) λmax [nm, ε × 10−4 (M−1 cm−1)]: 349 (2.04), 448 (9.93), 567 (1.41), 599 (2.11). 1H NMR (400 MHz, 25 °C): δ 8.12 (d, 3H, 3JHH = 7.48 Hz, 5,10,15-o1-Ph); 7.85–7.73 (m, 8H, Ph); 7.70–7.60 (m, 4H, Ph). Elemental analysis calcd for C37H15ON4Cl8Re: C 44.38, H 1.51, N 5.60; found; C 44.07, H 1.37, N 5.18. MS (ESI): M+ = 999.8272 (expt), 999.8258 (calcd for C37H15ON4Cl8Re).
Re[Cl8TpCH3PC](O)
Yield 15.3 mg (56.39%). UV–Vis (CH2Cl2) λmax [nm, ε × 10−4 (M−1 cm−1)]: 350 (2.54), 449 (11.00), 568 (1.70), 601 (2.47). 1H NMR (400 MHz, 25 °C): δ 7.97 (d, 3H, 3JHH = 8.24 Hz, 5,10,15-o1-Ph); 7.64 (d, 2H, 3JHH = 7.96 Hz, 5,15-o2-Ph); 7.60 (d, 2H, 3JHH = 7.68 Hz, 5,15-m1-Ph); 7.55 (d, 1H, 3JHH = 8.92 Hz, 10-o2-Ph); 7.49 (d, 3H, 3JHH = 7.72 Hz, 10-m1& 5,15-m2-Ph); 7.43 (d, 1H, 3JHH = 7.72 Hz, 10-m2-Ph); 2.72 (s, 6H, 5,15-p-CH3); 2.69 (s, 3H, 10-p-CH3). MS (ESI): Elemental analysis calcd for C40H21ON4Cl8Re: C 46.04, H 2.03, N 5.37; found: C 46.11, H 1.67, N 5.60. MS (ESI): M+ = 1041.8746 (expt), 1041.8728 (calcd for C40H21ON4Cl8Re).
General procedure for the synthesis of Re[Br8TpXPC](O) and Re[Br11TpXPC](O)
To a chloroform solution of Re[TpXPC](O) (10 mL; X = F: 8 mg, 0.0102 mmol; X = H: 16 mg, 0.022 mmol; X = CH3: 10 mg, 0.013 mmol, and X = OCH3: 15 mg, 0.018 mmol) was added a solution of liquid bromine (a total of 300 equiv) solution in chloroform in three 10-mL portions (each containing 100 equiv Br2 and added over 20 min) at 24-h intervals over 3 days. The reaction was then allowed to proceed under stirring at room temperature for a total of 7 days. The resulting mixture was quenched by washing with 20% aqueous sodium metabisulfite (25 mL × 3). The organic phase was then washed with distilled water (50 mL), dried over sodium sulfate, and rotary evaporated to dryness. The crude reaction mixture was loaded onto a silica gel column prepared with 4:1 hexane/dichloromethane and eluted with the same solvent system. The greenish-red product was rotary evaporated to dryness and further purified via preparative thin-layer chromatography with the same eluent. Detailed analytical and respective yield of the compounds synthesized are given below. X-ray-quality crystals of Re[Br8TpFPC](O) were obtained by slow diffusion of methanol vapor into a concentrated benzene solution of the complex.
Re[Br8TpFPC](O)
Yield 9 mg (62.09%). UV–Vis (CH2Cl2) λmax [nm, ε × 10−4 (M−1 cm−1)]: 357 (2.08), 456 (9.77), 574 (1.43), 607 (2.12). 1H NMR (400 MHz, 25 °C): δ 8.11 (m, 1H, 10-o1-Ph); 8.05 (m, 2H, 5,15-o1-Ph); 7.78 (m, 2H, 3JHH = 7.68 Hz, 5,15-o2-Ph); 7.65 (m, 1H, 10-o2-Ph); 7.55–7.36 (m, 6H, 5,10,15-m1 & m2-Ph). Elemental analysis calcd for C37H12F3ON4Br8Re: C 31.50, H 0.86, N 3.97; found: C 31.78, H 1.24, N 3.68. MS (ESI): M+ = 1411.3885 (expt), 1411.3889(calcd for C37H12F3ON4Br8Re).
Re[Br8TPC](O)
Yield 20 mg (66.76%). UV–Vis (CH2Cl2) λmax [nm, ε × 10−4 (M−1 cm−1)]: 357 (2.28), 456 (10.45), 575 (1.59), 607 (2.40). 1H NMR (400 MHz, 25 °C): δ 8.14 (d, 1H, 3JHH = 7.48 Hz, 10-o1-Ph); 8.08 (d, 2H, 3JHH = 7.40 Hz, 5,15-o1-Ph); 7.86–7.62 (m, 12H, Ph). Elemental analysis calcd for C37H15ON4Br8Re: C 32.75, H 1.11, N 4.13; found: C 32.49, H 0.95, N 3.88. MS (ESI): M+ = 1357.4166 (expt), 1357.4174(calcd for C37H15ON4Br8Re).
Re[Br11TpCH3PC](O)
Yield 12 mg (56.36%). UV–Vis (CH2Cl2) λmax [nm, ε × 10−4 (M−1 cm−1)]: 359 (2.07), 458 (9.70), 574 (1.44), 608 (2.10). 1H NMR (400 MHz, 25 °C): δ 8.35 (s, 1H, Ph); 8.28 (s, 1H, Ph); 8.02 (s, 1H, Ph); 7.97 (d, 1H, 3JHH = 7.68 Hz, Ph); 7.92 (d, 2H, 3JHH = 8.44 Hz, Ph); 7.68 (d, 3H, 3JHH = 8.08 Hz, Ph); 7.59 (d, 1H, 3JHH = 7.84 Hz, Ph); 7.53 (s, 1H, Ph); 2.78 (s, 6H, 5,15-pCH3); 2.72 (s, 3H, 10-pCH3). MS (ESI): Elemental analysis calcd for C40H18ON4Br11Re: C 29.37, H 1.11, N 3.43; found: C 29.59, H 1.41, N 3.33. M+ = 1635.1998 (expt), 1635.1936(calcd for C40H18ON4Br11Re).
Re[Br11TpOCH3PC](O)
Yield 19 mg (59.81%). UV–Vis (CH2Cl2) λmax [nm, ε × 10−4 (M−1 cm−1)]: 356 (1.82), 459 (8.59), 574 (1.15), 608 (1.72). 1H NMR (400 MHz, 25 °C): δ 8.34 (s, 1H, Ph); 8.28 (s, 1H, Ph); 8.01 (s, 1H, Ph); 7.71 (d, 2H, 3JHH = 8.72 Hz, Ph); 7.57 (d, 2H, 3JHH = 8.36 Hz, Ph); 7.35 (d, 3H, 3JHH = 9.32 Hz, Ph); 7.19 (d, 1H, 3JHH = 8.20 Hz, Ph); 4.23 (s, 6H, 5,15-pOCH3); 4.18 (s, 3H, 10-pOCH3). Elemental analysis calcd for C40H18O4N4Br11Re: C 28.53, H 1.08, N 3.33; found: C 28.17, H 1.35, N 2.98. MS (ESI): M+ = 1683.1775 (expt), 1683.1784(calcd for C40H18O4N4Br11Re).
X-ray structure determinations
All X-ray diffraction data were collected on beamline 12.2.1 at the Advanced Light Source of Lawrence Berkeley National Laboratory, Berkeley, California. The samples were mounted on MiTeGen kapton loops and placed in a 100(2) K nitrogen cold stream provided by an Oxford Cryostream 700 Plus low temperature apparatus on the goniometer head of a Bruker D8 diffractometer equipped with PHOTONII CPAD detector. Diffraction data were collected using synchrotron radiation monochromated with silicon(111) to a wavelength of 0.7288(1) Å. In each case, an approximate full-sphere of data was collected using 1° ϕ and ω scans. Absorption corrections were applied using SADABS73. The structure was solved by intrinsic phasing (SHELXT)74 and refined by full-matrix least squares on F2 (SHELXL-2014)75 using the ShelXle GUI76. Appropriate scattering factors were applied using the XDISP77 program within the WinGX suite78. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were geometrically calculated and refined as riding atoms.
Accession codes
The crystal structures described in this paper have been deposited at the Cambridge Crystallographic Data Centre and been assigned the following deposition numbers CCDC 1532043-1532045.
References
Traylor, T. G. & Tsuchiya, S. Perhalogenated tetraphenylhemins: stable catalysts of high turnover catalytic hydroxylations. Inorg. Chem. 26, 1338–1339 (1987).
Dolphin, D., Traylor, T. G. & Xie, L. Y. Polyhaloporphyrins: unusual ligands for metals and metal-catalyzed oxidations. Acc. Chem. Res. 30, 251–259 (1997).
Grinstaff, M. W., Hill, M. G., Labinger, J. A. & Gray, H. B. Mechanism of catalytic oxygenation of alkanes by halogenated iron porphyrins. Science 264, 1311–1313 (1994).
Lyons, J. E., Ellis, P. E. & Myers, H. K. Halogenated metalloporphyrin complexes as catalysts for selective reactions of acyclic alkanes with molecular oxygen. J. Catal. 155, 59–73 (1995).
Groves, J. T., Shalyaev, K. V., Bonchio, M. & Carofiglio, T. Rapid catalytic oxygenation of hydrocarbons with perhalogenated ruthenium porphyrin complexes. Stud. Surf. Sci. Catal. 110, 865–872 (1997).
Costas, M. Selective C–H oxidation catalyzed by metalloporphyrins. Coord. Chem. Rev. 255, 2912–2932 (2011).
Mandon, D. et al. β-Halogenated-pyrrole porphyrins. Molecular structures of 2,3,7,8,12,13,17,18-octabromo-5,10,15,20-tetramesitylporphyrin, nickel(II) 2,3,7,8,12,13,17,18-octabromo-5,10,15,20-tetramesitylporphyrin, and nickel(II) 2,3,7,8,12,13,17,18-octabromo-5,10,15,20-tetrakis(pentafluorophenyl)porphyrin. Inorg. Chem. 31, 2044–2049 (1992).
Grinstaff, M. W. et al. Structures, electronic properties, and oxidation–reduction reactivity of halogenated iron porphyrins. Inorg. Chem. 34, 4896–4902 (1995).
Birnbaum, E. R. et al. 19F NMR spectra and structures of halogenated porphyrins. Inorg. Chem. 34, 3625–3632 (1995).
Thomassen, I. K., Vazquez-Lima, H., Gagnon, K. J. & Ghosh, A. Octaiodoporphyrin. Inorg. Chem. 54, 11493–11497 (2015).
Ochsenbein, P. et al. Conformational effects on the redox potentials of tetraarylporphyrins halogenated at the β-pyrrole positions. Angew. Chem. Int. Ed. 33, 348–350 (1994).
Bhyrappa, P. & Krishnan, V. Octabromotetraphenylporphyrin and its metal derivatives: Electronic structure and electrochemical properties. Inorg. Chem. 30, 239–245 (1991).
Ghosh, A. et al. Electrochemistry of nickel and copper β-octahalogeno-meso-tetraarylporphyrins. Evidence for important role played by saddling-induced metal(dx2-y2)−porphyrin(“a2u”) orbital interactions. J. Phys. Chem. B 105, 8120–8124 (2001).
Kadish, K. M. & Van Caemelbecke, E. Electrochemistry of metalloporphyrins in nonaqueous media. In Encyclopedia of Electrochemistry Bioelectrochemistry Vol. 9 (ed. Wilson, G. S.) 175–228 (Wiley-VCH, Weinheim, 2002).
Shao, J., Steene, E., Hoffman, B. M. & Ghosh, A. EPR, ENDOR, and DFT studies on (β-Octahalo-meso-tetraarylporphyrin)copper complexes: characterization of the metal(dx2-y2)-porphyrin(a2u) orbital interaction. Eur. J. Inorg. Chem. 2005, 1609–1615 (2005).
Gross, Z., Galili, N. & Saltsman, I. The first direct synthesis of corroles from pyrrole. Angew. Chem. Int. Ed. 38, 1427–1429 (1999).
Paolesse, R. et al. 5,10,15-Triphenylcorrole: a product from a modified Rothemund reaction. Chem. Commun. 14, 1307–1308 (1999).
Ghosh, A. A perspective of pyrrole–aldehyde condensations as versatile self-assembly processes. Angew. Chem. Int. Ed. 43, 1918–1931 (2004).
Orłowski, R., Gryko, D. & Gryko, D. T. Synthesis of corroles and their heteroanalogs. Chem. Rev. 117, 3102–3137 (2017).
Ghosh, A. Electronic structure of corrole derivatives: insights from molecular structures, spectroscopy, electrochemistry, and quantum chemical calculations. Chem. Rev. 117, 3798–3881 (2017).
Nardis, S., Mandoj, F., Stefanelli, M. & Paolesse, R. Metal complexes of corrole. Coord. Chem. Rev. 388, 360–405 (2019).
Golubkov, G. et al. High-valent manganese corroles and the first perhalogenated metallocorrole catalyst. Angew. Chem. Int. Ed. 40, 2132–2134 (2001).
Palmer, J. H., Durrell, A. C., Gross, Z., Winkler, J. R. & Gray, H. B. Iridium corroles. J. Am. Chem. Soc. 130, 7786–7787 (2008).
Wasbotten, I. H., Wondimagegn, T. & Ghosh, A. Electronic absorption, resonance raman, and electrochemical studies of planar and saddled copper(III) meso-triarylcorroles. Highly substituent-sensitive soret bands as a distinctive feature of high-valent transition metal corroles. J. Am. Chem. Soc. 124, 8104–8116 (2002).
Alemayehu, A. B., Hansen, L. K. & Ghosh, A. Nonplanar, noninnocent, and chiral: a strongly saddled metallocorrole. Inorg. Chem. 49, 7608–7610 (2010).
Capar, C., Thomas, K. E. & Ghosh, A. Reductive demetalation of copper corroles: first simple route to free-base β-octabromocorroles. J. Porphyrins Phthalocyanines 12, 964–967 (2008).
Capar, C., Hansen, L.-K., Conradie, J. & Ghosh, A. β-octabromo-meso-tris(pentafluorophenyl)corrole: reductive demetalation-based synthesis of a heretofore inaccessible, perhalogenated free-base corrole. J. Porphyrins Phthalocyanines 14, 509–512 (2010).
Capar, J. et al. Improved syntheses of β-octabromo-meso-triarylcorrole derivatives. J. Inorg. Biochem. 153, 162–166 (2015).
Rabinovitch, E., Goldberg, I. & Gross, Z. Gold(I) and gold(III) corroles. Chem. Eur. J. 17, 12294–12301 (2011).
Thomas, K. E., Gagnon, K. J., McCormick, L. J. & Ghosh, A. Molecular structure of gold 2,3,7,8,12,13,17,18-octabromo-5,10,15-tris(4′′-pentafluorosulfanylphenyl)corrole: Potential insights into the insolubility of gold octabromocorroles. J. Porphyrins Phthalocyanines 22, 596–601 (2018).
Norehim, H.-K. et al. Ligand noninnocence in FeNO corroles: insights from β-octabromocorrole complexes. Dalton Trans. 45, 681–689 (2018).
Alemayehu, A. B., Gonzalez, E., Hansen, L. K. & Ghosh, A. Copper corroles are inherently saddled. Inorg. Chem. 48, 7794–7799 (2009).
Thomas, K. E., Conradie, J., Hansen, L. K. & Ghosh, A. A metallocorrole with orthogonal pyrrole rings. Eur. J. Inorg. Chem. 2011, 1865–1870 (2011).
Berg, S., Thomas, K. E., Beavers, C. M. & Ghosh, A. Undecaphenylcorroles. Inorg. Chem. 51, 9911–9916 (2012).
Thomas, K. E. et al. Halterman corroles and their use as a probe of the conformational dynamics of the inherently chiral copper corrole chromophore. Inorg. Chem. 57, 4270–4276 (2018).
Thomassen, I. K., McCormick, L. J. & Ghosh, A. Synthesis and molecular structure of a copper octaiodocorrole. ACS Omega 3, 5106–5110 (2018).
Thomas, K. E., Settineri, N. S., Teat, S. J., Steene, E. & Ghosh, A. Molecular structure of copper and μ-oxodiiron octafluorocorrole derivatives: insights into ligand noninnocence. ACS Omega 5, 10176–10182 (2020).
Thomas, K. E. et al. Ligand noninnocence in coinage metal corroles: a silver knife-edge. Chem. Eur. J. 21, 16839–16847 (2015).
Alemayehu, A. B. & Ghosh, A. Gold corroles. J. Porphyrins Phthalocyanines 15, 106–110 (2011).
Thomas, K. E., Alemayehu, A. B., Conradie, J., Beavers, C. & Ghosh, A. Synthesis and molecular structure of gold triarylcorroles. Inorg. Chem. 50, 12844–12851 (2011).
Thomas, K. E., Beavers, C. M. & Ghosh, A. Molecular structure of a gold β-octakis(trifluoromethyl)-meso-triarylcorrole: an 85° difference in saddling dihedral relative to copper. Mol. Phys. 110, 2439–2444 (2012).
Capar, J. et al. Demetalation of Copper undecaarylcorroles: molecular structures of a free-base undecaarylisocorrole and a gold undecaarylcorrole. J. Inorg. Biochem. 162, 146–153 (2016).
Brückner, C., Briñas, R. P. & Bauer, J. A. K. X-ray structure and variable temperature nmr spectra of [meso-Triarylcorrolato]copper(III). Inorg. Chem. 42, 4495–4497 (2003).
Steene, E., Dey, A. & Ghosh, A. β-octafluorocorroles. J. Am. Chem. Soc. 125, 16300–16309 (2003).
Bröring, M., Brégier, F., Tejero, E. C., Hell, C. & Holthausen, M. C. Revisiting the electronic ground state of copper corroles. Angew. Chem. Int. Ed. 46, 445–448 (2007).
Lim, H. et al. X-ray absorption spectroscopy as a probe of ligand noninnocence in metallocorroles: the case of copper corroles. Inorg. Chem. 58, 6722–6730 (2019).
Buckley, H. L. & Arnold, J. Recent developments in out-of-plane metallocorrole chemistry across the periodic table. Dalton Trans. 44, 30–36 (2015).
Ziegler, J. A., Buckley, H. L. & Arnold, J. Synthesis and reactivity of tantalum corrole complexes. Dalton Trans. 46, 780–785 (2017).
Alemayehu, A. B., Vazquez-Lima, H., Gagnon, K. J. & Ghosh, A. Tungsten biscorroles: new chiral sandwich compounds. Chem. Eur. J. 22, 6914–6920 (2016).
Einrem, R. F., Gagnon, K. J., Alemayehu, A. B. & Ghosh, A. Metal-ligand misfits: facile access to rhenium-oxo corroles by oxidative metalation. Chem. Eur. J. 22, 517–520 (2016).
Alemayehu, A. B., Teat, S. J., Borisov, S. M. & Ghosh, A. Rhenium-imido corroles. Inorg. Chem. 59, 6382–6389 (2020).
Alemayehu, A. B., Gagnon, K. J., Terner, J. & Ghosh, A. Oxidative metalation as a route to size-mismatched macrocyclic complexes: osmium corroles. Angew. Chem. Int. Ed. 53, 14411–14414 (2014).
Alemayehu, A. B. et al. Platinum corroles. Chem. Commun. 50, 11093–11096 (2014).
Alemayehu, A. B., McCormick, L. J., Gagnon, K. J., Borisov, S. M. & Ghosh, A. Stable platinum(IV) corroles: synthesis, molecular structure, and room-temperature near-IR phosphorescence. ACS Omega 3, 9360–9368 (2018).
Palmer, J. H., Durrell, A. C., Gross, Z., Winkler, J. R. & Gray, H. B. Near-IR phosphorescence of iridium(III) corroles at ambient temperature. J. Am. Chem. Soc. 132, 9230–9231 (2010).
Sinha, W., Ravotto, L., Ceroni, P. & Kar, S. NIR-emissive iridium(III) corrole complexes as efficient singlet oxygen sensitizers. Dalton Trans. 44, 17767–17773 (2015).
Borisov, S. M., Alemayehu, A. & Ghosh, A. Osmium-nitrido corroles as NIR indicators for oxygen sensors and triplet sensitizers for organic upconversion and singlet oxygen generation. J. Mater. Chem. C 4, 5822–5828 (2016).
Lemon, C. M., Powers, D. C., Brothers, P. J. & Nocera, D. G. Gold corroles as near-IR phosphors for oxygen sensing. Inorg. Chem. 56, 10991–10997 (2017).
Alemayehu, A. B. et al. Gold tris(carboxyphenyl)corroles as multifunctional materials: room temperature near-ir phosphorescence and applications to photodynamic therapy and dye-sensitized solar cells. ACS Appl. Mater. Interfaces 8, 18935–18942 (2016).
Borisov, S. M., Einrem, R. F., Alemayehu, A. B. & Ghosh, A. Ambient-temperature near-IR phosphorescence and potential applications of rhenium-oxo corroles. Photochem. Photobiol. Sci. 18, 1166–1170 (2019).
Einrem, R. F., Alemayehu, A. B., Borisov, S. M., Ghosh, A. & Gederaas, O. A. Amphiphilic rhenium-oxo corroles as a new class of sensitizers for photodynamic therapy. ACS Omega 5, 10596–10601 (2020).
Thomassen, I. K., McCormick-McPherson, L. J., Borisov, S. M. & Ghosh, A. Iridium corroles exhibit weak near-infrared phosphorescence but efficiently sensitize singlet oxygen formation. Sci. Rep. 10, 7551 (2020).
Teo, R. D., Hwang, J. Y., Termini, J., Gross, Z. & Gray, H. B. Fighting cancer with corroles. Chem. Rev. 117, 2711–2729 (2017).
Reinholdt, A., Alemayehu, A. B., Gagnon, K. J., Bendix, J. & Ghosh, A. Electrophilic activation of osmium-nitrido corroles. The OsN triple bond as a π-acceptor metalla-ligand in a heterobimetallic OsVIN-PtII complex. Inorg. Chem. 59, 5276–5280 (2020).
Soll, M. et al. One-pot conversion of fluorophores to phosphorophores. Org. Lett. 18, 5840–5843 (2016).
Sudhakar, K. et al. Effect of selective CF3 substitution on the physical and chemical properties of gold corroles. Angew. Chem. Int. Ed. 56, 9837–9841 (2017).
Johansen, I. et al. Substituent effects on metallocorrole spectra: insights from chromium-oxo and molybdenum-oxo triarylcorroles. J. Porphyrins Phthalocyanines 15, 1335–1344 (2011).
Schweyen, P. et al. Viking helmet corroles: activating inert oxidometal corroles. Chem. Eur. J. 23, 13897–13900 (2017).
Mahammed, A., Botoshanskya, M. & Gross, Z. Chlorinated corroles. Dalton Trans. 41, 10938–10940 (2012).
Thomas, K. E., Alemayehu, A. B., Conradie, J., Beavers, C. M. & Ghosh, A. The structural chemistry of metallocorroles: combined X-ray crystallography and quantum chemistry studies afford unique insights. Acc. Chem. Res. 45, 1203–1214 (2012).
Albrett, A. M. et al. Corrole as a binucleating ligand: preparation, molecular structure and density functional theory study of diboron corroles. J. Am. Chem. Soc. 130, 2888–2889 (2008).
Albrett, A. M. et al. Mono- and diboron corroles: factors controlling stoichiometry and hydrolytic reactivity. Inorg. Chem. 53, 5486–5493 (2014).
Krause, L., Herbst-Irmer, R., Sheldrick, G. M. & Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Cryst. 48, 3–10 (2015).
Sheldrick, G. M. SHELXT—integrated space-group and crystal-structure determination. Acta Cryst. A71, 3–8 (2015).
Sheldrick, G. M. Crystal structure refinement with SHELXL. Acta Cryst. C71, 3–8 (2015).
Hübschle, C. B., Sheldrick, G. M. & Dittrich, B. ShelXle: a Qt graphical user interface for SHELXL. J. Appl. Cryst. 44, 1281–1284 (2011).
Kissel, L. & Pratt, R. H. Corrections to tabulated anomalous-scattering factors. Acta Cryst. A46, 170–175 (1990).
Farrugia, L. J. WinGX and ORTEP for windows: an update. J. Appl. Cryst. 45, 849–854 (2012).
Acknowledgements
We acknowledge Grant No. 262229 (to AG) from the Research Council of Norway. This research used resources of the Advanced Light Source, a U.S. DOE Office of Science User Facility under Contract No. DE-AC02-05CH11231.
Author information
Authors and Affiliations
Contributions
A.B.A. and R.F.E. carried out the synthetic work; L.J.M.M. and N.S.S. carried out the X-ray structure determinations. A.G. planned and supervised the project. The manuscript was largely composed by A.B.A. and A.G.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Alemayehu, A.B., Einrem, R.F., McCormick-McPherson, L.J. et al. Synthesis and molecular structure of perhalogenated rhenium-oxo corroles. Sci Rep 10, 19727 (2020). https://doi.org/10.1038/s41598-020-76308-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-76308-7
This article is cited by
-
Phenol- and resorcinol-appended metallocorroles and their derivatization with fluorous tags
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
