
Abstract
Boron tribromide-mediated demethylation of rhenium-oxo and gold meso-tris(4-methoxyphenyl)corrole and meso-tris(3,5-dimethoxyphenylcorrole), M[TpOMePC] and M[T(3,5-OMe)PC] (M = ReO, Au), have yielded the corresponding phenol- and resorcinol-appended metallocorroles, M[TpOHPC] and M[T(3,5-OH)PC], in good yields. The latter compounds proved insoluble in dichloromethane and chloroform but soluble in THF. The M[T(3,5-OH)PC] derivatives also proved moderately soluble in 0.05 M aqueous KOH. Unlike oxidation-prone aminophenyl-substituted corroles, the phenol- and resorcinol-appended metallocorroles could be readily handled in air without special precautions. The phenolic metallocorroles could be readily alkylated with 4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-heptadecafluoroundecyl iodide (“FtI”) to afford the fluorous-tagged metallocorroles M[TpOFtPC] and M[T(3,5-OFt)PC] in > 90% yields. The simplicity of the synthetic protocols promise a wide range of phenolic and fluorous-tagged porphyrin analogues with potential applications to diverse fields such as sensors, catalysis, and photodynamic therapy, among others.
Similar content being viewed by others
Introduction
Corroles, which were mere curiosities just 25 years ago1,2,3,4,5, are now a major class of macrocyclic ligands with applications rivaling those of porphyrins6,7. Besides key photophysical properties8,9,10,11,12,13,14,15, biomedical applications such as photodynamic therapy (PDT)16,17,18,19 require water-soluble and amphiphilic ligands for effective biodelivery20,21,22,23,24,25,26,27,28. An attractive approach to effective biodelivery in PDT involves nanodroplets of locally-perfluorinated (fluorous29,30,31,32,33,34,35) porphyrin analogues dissolved in a fluorocarbon solvent with high oxygen-carrying capacity. Finally, new strategies for functionalization are a key first step for novel bio- and nanoconjugation of porphyrin analogues36,37,38,39,40,41,42. Against this backdrop, we present here simple synthetic routes to amphiphilic phenol- and resorcinol-appended metallocorroles43 (analogous to other similarly functionalized porphyrin analogues44,45,46,47) and their elaboration to highly fluorophilic fluorous-tagged derivatives (Fig. 1).

Schematic illustration of the synthesis of phenol- and resorcinol-appended metallocorroles and their derivatization with a fluorous tag.
Results and discussion
Synthesis of phenol- and resorcinol-appended metallocorroles
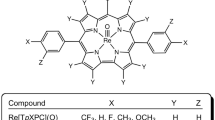
Rhenium-oxo48,49,50,51,52,53 and gold54,55,56,57,58,59,60,61,62,63 tris(4-methoxyphenyl)corrole, M[TpOMePC] (M = ReO, Au), and tris(3,5-dimethoxyphenyl)corrole, M[T(3,5-OMe)PC] (M = ReO, Au), which rank among the most readily accessible 5d metallocorroles64,65,66,67,68,69, were used as starting materials. The choice of the two metals was dictated by the fact that they yield rugged, electronically innocent complexes that have been shown to act as triplet photosensitizers in oxygen sensing and in vitro photodynamic therapy experiments. The complexes underwent smooth demethylation70,71,72,73 with boron tribromide in dichloromethane at – 78 °C, affording phenol- and resorcinol-appended metallocorroles M[TpOHPC] and M[T(3,5-OH)PC] in 55 to > 90% yields, with the higher yields observed for M = ReO. The products were purified via silica-gel column chromatography, followed by recrystallization, and characterized by UV–vis spectroscopy, 1H NMR spectroscopy, and high-resolution electrospray ionization mass spectrometry. 1H NMR spectra of the new compounds indicated complete disappearance of the methoxy protons at around 4 ppm and the appearance of two new singlets between 8.46 and 8.80 ascribable to hydroxy protons (Figs. 2, 3). HRMS proved consistent with the expected structural assignments and also indicated the absence of partially demethylated products and also of higher-mass byproducts.

1H NMR spectra of M[TpOHPC] in THF-d8: M = ReO (above) and Au (below).

1H NMR spectra of M[T(3,5-OH)PC] in THF-d8: M = ReO (above) and Au (below).
The phenolic metallocorroles proved insoluble in dichloromethane and chloroform so UV–vis (Table 1 and Fig. 4) and 1H NMR spectra were acquired in THF and THF-d8, respectively. As far as UV–vis spectra are concerned, the phenolic metallocorroles were found to exhibit very similar peak positions relative to their methoxy precursors. In contrast, modest differences in peak positions were observed between the 4-methoxy/hydroxy and 3,5-dimethoxy/dihydroxy derivatives. Interestingly, the resorcinol-appended complexes M[T(3,5-OH)PC] (M = ReO, Au) were found to exhibit moderate solubility and modest spectral redshifts in 0.05 M aqueous KOH, consistent with (partial) deprotonation of the phenolic OH groups74,75,76,77,78.

UV–vis spectra of new compounds prepared in this study. Phenolic and fluorous-tagged metallocorroles were analyzed in THF and dichloromethane (with a drop of hexafluorobenzene), respectively. Sample concentrations were in the 10–20 μM range.
Synthesis of fluorous-tagged metallocorroles
The phenol- and resorcinol-appended metallocorroles could be readily derivatized with the fluorous-tagging reagent 4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-heptadecafluoroundecyl iodide (“FtI”) and K2CO3 in refluxing acetone over 24 h, whereupon the fluorous-tagged products M[TpOFtPC] and M[T(3,5-OFt)PC] (M = ReO, Au) were obtained in > 90% yields. Upon removal of the solvent from the reaction mixture, the residues were dissolved in a small quantity of hexafluorobenzene and purified via column chromatography on a silica gel column with dichloromethane as eluent (i.e., the mobile phase was effectively dichloromethane with a small quantity of hexafluorobenzene). The products were found to be freely soluble in hexafluorobenzene but sparingly so in nonfluorinated solvents including dichloromethane and chloroform. Evidence for exhaustive fluorous tagging came from both 1H and 19F NMR spectroscopy (in CDCl3 with a drop of hexafluorobenzene) and HRMS (Fig. 5). 1H NMR spectral analyses showed the complete disappearance of the OH singlets between 8.46 and 8.80 ppm and the appearance of new alkyl proton signals between 2.08 and 4.39 ppm and with an intensity (relative to corrole protons) that exactly matched the expected structure. Clean 19F NMR spectra further confirmed this conclusion. Electrospray ionization HRMS also did not reveal any evidence of incompletely fluorous-tagged products. For UV–vis spectroscopy, the fluorous-tagged metallocorroles were dissolved in a minimum volume of hexafluorobenzene followed by dilution with dichloromethane to the required volume. The spectra, unsurprisingly, proved similar to those of simple 5d metallocorroles, with Soret maxima at ~ 420 nm for M = Au and at ~ 440 nm for M = ReO and the usual double-humped Q bands.

1H and 19F NMR spectra of M[TpOFtPC] and M[T(3,5-OFt)PC] (M = ReO, Au).
Concluding remarks
In summary, we have described simple routes to amphiphilic phenol- and resorcinol-appended rhenium-oxo and gold corroles, which could be readily derivatized to highly fluorophilic fluorous-tagged derivatives. Although straightforward in retrospect, the successful synthesis of phenolic metallocorroles was a priori somewhat uncertain; corroles are more electron-rich than analogous porphyrins and it was unclear whether appending electron-rich phenol and resorcinol moieties would lead to fragile, air-sensitive products. Thus, compared with aminophenyl-substituted porphyrins79,80,81,82, aminophenyl-substituted corroles are far more susceptible to aerial oxidation and accordingly far trickier to handle83,84,85,86. These fears proved unfounded for our compounds, which could be readily manipulated in air without special precautions.
The simple access to fluorous-tagged metallocorroles promises a wide range of related products in the near future. These include (a) fluorous-tagged complexes with other metals, including electroactive metals such as manganese and iron87,88,89, (b) light fluorous-tagged complexes with one or two fluorous tags based on so-called A2B triarylcorroles90,91, and (c) environmentally friendlier (i.e., more biodegradable) complexes with shorter fluorinated chains92,93,94,95.
The above products and their logical successors promise a plethora of applications, in fields as diverse as sensors89,96 photodynamic and related therapies97,98, and catalysis99,100,101, among others.
Experimental section
Materials
All chemicals were purchased from Sigma Aldrich (Merck). Silica gel 60 (0.04–0.063 mm particle size, 230–400 mesh) was employed for flash chromatography. Metallocorrole starting materials were prepared as previously described48,56,60.
Re[T(3,5-OCH3)PC](O)
This previously unreported complex was prepared from H3[T(3,5-OMe)PC], dirhenium decacarbonyl (Re2(CO)10, 2 equiv), and K2CO3 (100 mg) using the standard method for Re insertion48. Yield 90.12 mg (70.23%). UV–vis (CH2Cl2) λmax (nm) and ε × 10–4 (M−1 cm−1): 441 (11.40), 554 (1.77), 586(2.28). 1H NMR (400 MHz, 25 °C, CDCl3): δ 9.60 (d, 2H, 3JHH = 4.4 Hz, β-H); 9.41 (d, 2H, 3JHH = 4.4 Hz, β-H); 9.40 (d, 2H, 3JHH = 4.9 Hz, β-H); 9.20 (d, 2H, 3JHH = 4.9 Hz, β-H); 7.77 (s, 2H, 5,15(3,5-o1Ph)); 7.71 (s, 1H, 10(3,5-o1Ph)a); 7.27 (s, 2H, 5,15(3,5-o2Ph)); 7.08 (s, 1H, 10(3,5-o2Ph)); 6.93 (m, 3H, 5,10,15-pPh); 4.07 (brs, 6H, 5,15(3,5-OCH3)a; 4.04 (s, 3H, 10(3,5-OCH3)a); 3.95 (brs, 6H, 5,15(3,5-OCH3)b); 3.90 (s, 3H, 10(3,5-OCH3)b). HRMS (ESI): [M−] = 906.2126 (expt), 906.2133 (calcd for C37H23N4O7Re, major isotopomer).
Instrumental methods
The instrumentation used was essentially the same as in our earlier work50,51,52,53. UV–visible spectra were recorded on an HP 8453 spectrophotometer. 1H NMR spectra were recorded on a 400 MHz Bruker Avance III HD spectrometer equipped with a 5 mm BB/1H SmartProbe in either CDCl3 (referenced to residual CHCl3 at 7.26 ppm) or THF-d8 (referenced to residual C4H8O at 3.58 and 1.73 ppm. 19F NMR spectra were acquired on the same spectrometer and referenced to hexaflurobenzene (C6F6, – 164.9 ppm). High-resolution electrospray-ionization mass spectra were recorded on methanolic solutions on an Orbitrap Exploris 120 (Thermo Fisher Scientific) spectrometer.
General synthetic procedure for metallotris(4-hydroxyphenyl)corrole complexes, M[TpOHPC] (M = ReO, Au)
To a solution of boron tribromide (1.51 mmol) in dry dichloromethane (10 mL) cooled to − 78 °C was added M[TpOMePC] (M = ReO, Au; 0.121 mmol), also dissolved in dry dichloromethane (10 mL), over a period of 20 min. The mixture was stirred for 2 h at − 78 °C and then for an additional 12 h at 25 °C. The solution was then cooled to 0 °C in an ice bath and methanol was added to quench any remaining boron tribromide. The reaction mixture was rotary-evaporated to dryness and the solid residue obtained was chromatographed on a silica gel column with 95:5 v/v dichloromethane/methanol as eluent. The final product was recrystallized from 5:1 v/v chloroform/methanol. Yields and spectroscopic data are given below.
Synthesis of metallotris(3,5-dihydroxyphenyl)corrole complexes, M[T(3,5-OH)PC] (M = ReO, Au)
To a solution of boron tribromide (3.02 mmol) in dry dichloromethane (10 mL) cooled to − 78 °C was added M[T(3,5-OMe)PC] (M = ReO, Au; 0.121 mmol), also dissolved in dry dichloromethane (10 mL), over a period of 20 min. The mixture was stirred for 2 h at − 78 °C and then for an additional 12 h at 25 °C. The solution was then cooled to 0 °C in an ice bath and methanol was added to quench any remaining boron tribromide. The reaction mixture was rotary-evaporated to dryness and the solid residue obtained was chromatographed on a silica gel column with 9:1 v/v dichloromethane/methanol as eluent. The final product was recrystallized from 3:1 v/v chloroform/methanol. Yields and spectroscopic data are given below.
General synthesis of M[TpOFtPC] (M = ReO, Au)
A 250-mL round-bottom flask equipped with a stir-bar and a reflux condenser was charged with M[TpOHPC] (0.026 mmol), 4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-heptadecafluoroundecyl iodide (49 mg, 3.2 equiv, 0.083 mmol), and potassium carbonate (100 mg dissolved in 50 mL acetone). The reaction mixture was then refluxed for 24 h, followed by removal of the solvent under reduced pressure. The crude product was dissolved in a minimum amount of hexafluorobenzene and loaded onto a silica gel column and eluted with dichloromethane, affording the desired fluorous-tagged metallocorroles.
General synthesis of M[T(3,5-OFt)PC] (M = ReO, Au)
A 250-mL round-bottom flask equipped with a stir-bar and reflux condenser was charged with M[T(3,5-OH)PC] (0.026 mmol), 4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-heptadecafluoroundecyl iodide (98 mg, 6.4 equiv, 0.167 mmol), and potassium carbonate (200 mg dissolved in 50 mL acetone). The reaction mixture was then refluxed for 24 h, followed by removal of the solvent under reduced pressure. The crude product was dissolved in a minimum amount of hexafluorobenzene and loaded onto a silica gel column and eluted with dichloromethane, affording the desired fluorous-tagged metallocorroles.
Re[TpOHPC](O)
Yield 88.5 mg (87.2%). UV–vis (THF) λmax (nm) and ε × 10–4 (M−1 cm−1): 441 (14.35), 556(1.95), 591(2.98). 1H NMR (400 MHz, 25 °C, THF-d8): δ 9.64 (d, 2H, 3JHH = 4.4 Hz, β-H); 9.34 (d, 2H, 3JHH = 4.8 Hz, β-H); 9.32 (d, 2H, 3JHH = 4.4 Hz, β-H); 9.14 (d, 2H, 3JHH = 4.8 Hz, β-H); 8.80 (s, 2H, 5,15-pOHPh); 8.76 (s, 1H, 10-pOHPh); 8.39 (br s, 2H, 5,15-o1Ph); 8.32 (dd, 1H, 3JHH = 8.0, 2.32 Hz, 10-o1Ph); 7.90 (br s, 2H, 5,15-o2Ph); 7.74 (dd, 1H, 3JHH = 8.2, 2.32 Hz, 10-o2Ph); 7.27 (m, 5H, 10-m1Ph & 5,15-mPh); 7.13 (dd, 1H, 3JHH = 8.3, 2.72 Hz, 10-m2Ph). HRMS (ESI): [M+] = 774.1274 (expt), 774.1273 (calcd for C37H23N4O4Re, major isotopomer).
Au[TpOHPC]
Yield 70.7 mg (70.2%). UV–vis (THF) λmax (nm) and ε × 10–4 (M−1 cm−1): 420 (11.70), 559 (1.88), 580 (2.95). 1H NMR (400 MHz, 25 °C, THF-d8): δ 9.16 (m, 2H, β-H); 9.03 (d, 2H, 3JHH = 4.8 Hz, β-H); 8.82 (d, 2H, 3JHH = 4.4 Hz, β-H); 8.80 (d, 2H, 3JHH = 4.8 Hz, β-H); 8.72(s, 2H, 5,15-pOHPh); 8.68 (s, 1H, 10-pOHPh); 8.07 (d, 4H, 3JHH = 8.3 Hz, 5,15-oPh); 7.95 (d, 2H, 3JHH = 8.4 Hz, 10-oPh); 7.21 (d, 4H, 3JHH = 8.6 Hz, 5,15-mPh); 7.16 (d, 2H, 3JHH = 8.4 Hz, 10-mPh). HRMS (ESI) [M−] = 767.1367 (expt), 767.1363 (calcd for C37H23N4O3Au, major isotopomer).
Re[T(3,5-OH)PC](O)
Yield 99.6 mg (90.1%). UV–vis (THF) λmax (nm) and ε × 10–4 (M−1 cm−1): 440 (10.96), 556 (1.65), 587(2.24). 1H NMR (400 MHz, 25 °C, THF-d8): δ 9.64 (d, 2H, 3JHH = 4.4 Hz, β-H); 9.44 (d, 2H, 3JHH = 5.0 Hz, β-H); 9.42 (d, 2H, 3JHH = 4.4 Hz, β-H); 9.24 (d, 2H, 3JHH = 4.8 Hz, β-H); 8.67 (br s, 2H, 5,15(3,5-OHPh)a); 6.63 [s, 1H, 10(3,5-OH)a]; 8.47 [br s, 2H, 5,15(3,5-OHPh)b]; 8.42 [s, 1H, 10(3,5-OHPh)b]; 7.50 (br s, 2H, 5,15-o1Ph); 7.44 (s, 1H, 10-o1Ph); 6.92 (br s, 2H, 5,15-o2Ph); 6.77 (s, 1H, 10-o2Ph); 6.66 (m, 3H, 5,10,15-pPh). HRMS (ESI): [M−] = 821.1046 (expt), 821.1053 (calcd for C37H23N4O7Re, major isotopomer).
Au[T(3,5-OH)PC]
Yield 60.4 mg (55.0%). UV–vis (THF) λmax (nm) and ε × 10–4 (M−1 cm−1): 419(9.65), 576(2.64). 1H NMR (400 MHz, 25 °C, THF-d8): δ 9.22 (d, 2H, 3JHH = 4.4 Hz, β-H); 9.19 (d, 2H, 3JHH = 4.9 Hz, β-H); 8.98 (d, 2H, 3JHH = 4.5 Hz, β-H); 8.94 (d, 2H, 3JHH = 5.0 Hz, β-H); 8.50(s, 4H, 5,15(3,5-OHPh); 8.47 (s, 2H, 10(3,5-OHPh); 7.18 (d, 4H, 4JHH = 2.2 Hz, 5,15-oPh); 7.09 (d, 2H, 4JHH = 2.2 Hz, 10-oPh); 6.61 (m, 3H, 3JHH = 8.6 Hz, 5,10,15-pPh). HRMS (ESI): [M−] = 815.1206 (expt), 815.1210 (calcd for C37H23N4O6Au, major isotopomer).
Re[TpOFtPC](O)
Yield 53.8 mg (96.1%). UV–vis ((CH2Cl2/C6F6) λmax (nm) and ε × 10–4 (M−1 cm−1): 442(8.78), 556(1.14), 590(1.64). 1H NMR (400 MHz, 25 °C, CDCl3): δ 9.58 (d, 2H, 3JHH = 4.4 Hz, β-H); 9.31 (d, 4H, 3JHH = 5 Hz, β-H); 9.11 (d, 2H, 3JHH = 5 Hz, β-H); 8.50 (br s, 2H, 5,15-o1Ph); 8.43 (d, 1H, 10-o1Ph); 8.00 (br s, 2H, 5,15-o2Ph); 7.82 (d, 1H, 10-o2Ph); 7.47–7.29 (m, 5H, 10-m1Ph & 5,15-mPh); 7.24 (d, 1H, 3JHH = 8.4 Hz, 10-m2Ph); 4.35 (m, 6H, 5,10,15–OCH2–); 2.59 (m, 6H, 5,10,15-CH2-CF2-); 2.32 (m, 6H, 5,10,15–CH2–CH2O–); 19F NMR (C6F6): δ − 83.81, m 9F, CF3-); − 117.29, m, 6F, –CF2–; − 124.68, m, 6F, –CF2–; − 124.92, m 12F, –CF2–; − 125.71, m 6F, –CF2–; − 126.38, m 6F, –CF2–; − 129.22, m, 6F, –CF2–. HRMS (ESI): [M+] = 2154.1637 (expt), 2154.1636 (calcd for C70H38F51N4O4Re, major isotopomer).
Re[T(3,5-OFt)PC](O)
Yield 88.1 mg (94.6%). UV–vis (CH2Cl2/C6F6) λmax (nm) and ε × 10–4 (M−1 cm−1): 440(9.17), 555(1.39), 585(1.89). 1H NMR (400 MHz, 25 °C, CDCl3): δ 9.59 (d, 2H, 3JHH = 4.5 Hz, β-H); 9.38 (d, 4H, 3JHH = 4.6 Hz, β-H); 9.18 (d, 2H, 3JHH = 5.0 Hz, β-H); 7.77 (s, 2H, 5,15-o1Ph); 7.72 (s, 1H, 10-o1Ph); 7.25 (s, 2H, 5,15-o2Ph); 7.08 (s, 1H, 10-o2Ph); 6.92 (s, 3H, 5,10,15-pPh) 4.39–4.09 (m, 12H, 5,10,15–OCH2–); 2.55–2.08 (m, 24H, 5,10,15–CH2–CH2–CF2–); 19F NMR (C6F6): δ − 83.98, m 18F, CF3–); − 117.40, m, 12F, –CF2–; − 124.78, m, 12F, –CF2–; − 125.07, m 24F, –CF2–; − 125.85, m 12F, –CF2–; − 126.52, m 12F, –CF2–; − 129.26, m, 12F, –CF2–. MS (ESI): [M+] = 3582.14 (expt), 3582.18 (calcd for C103H53F102N4O7Re, major isotopomer). Elemental analysis found C 34.58, H 1.47, N 1.52; calcd C 34.53, H 1.49, N 1.56.
Au[TpOFtPC]
Yield 53.0 mg (95.0%). UV–vis (CH2Cl2/C6F6) λmax (nm) and ε × 10–4 (M−1 cm−1): 420 (9.35), 560(1.42), 580(2.20). 1H NMR (400 MHz, 25 °C, CDCl3): δ 9.19 (d, 2H, 3JHH = 4.4 Hz, β-H); 9.03 (d, 2H, 3JHH = 4.9 Hz, β-H); 8.86 (d, 2H, 3JHH = 4.4 Hz, β-H); 8.80 (d, 2H, 3JHH = 4.8 Hz, β-H); 8.21 (d, 4H, 5,15-oPh); 8.10 (d, 2H, 10-oPh); 7.34 (d, 4H, 3JHH = 8.7 Hz, 5,15-mPh); 7.29 (d, 2H, 3JHH = 8.7 Hz, 10-mPh); 4.34 (m, 6H, 5,10,15–OCH2–); 2.50 (m, 6H, 5,10,15–CH2–CF2–); 2.30 (m, 6H, 5,10,15–CH2–CH2O–); 19F NMR (C6F6): δ − 83.87, m 9F, CF3–); − 117.31, m, 6F, –CF2–; − 124.72, m, 6F, –CF2–; − 124.98, m 12F, –CF2–; − 125.78, m 6F, –CF2–; − 126.37, m 6F, –CF2–; − 129.17, m, 6F, –CF2–. HRMS (ESI): [M+] = 2148.1795 (expt), 2148.1790 (calcd for C70H38F51N4O3Au, major isotopomer).
Au[T(3,5-OFt)PC]
Yield 86.5 mg (93.1%). UV–vis (CH2Cl2/C6F6) λmax (nm) and ε × 10–4 (M−1 cm−1): 419(10.48), 562(2.13), 572(2.35). 1H NMR (400 MHz, 25 °C, CDCl3): δ 9.22 (d, 2H, 3JHH = 4.4 Hz, β-H); 9.11 (d, 2H, 3JHH = 4.9 Hz, β-H); 8.95 (d, 2H, 3JHH = 4.4 Hz, β-H); 8.88 (d, 2H, 3JHH = 5.0 Hz, β-H); 7.47 (d, 4H, 4JHH = 2.4 Hz, 5,15-oPh); 7.38 (d, 2H, 4JHH = 2.3 Hz, 10-oPh); 6.87 (m, 3H, 5,10,15-pPh); 4.24 (m, 12H, 5,10,15-OCH2–); 2.38 (m, 12H, 5,10,15-CH2–CF2–); 2.20 (m, 12H, 5,10,15-CH2–CH2O–); 19F NMR (C6F6): δ − 83.98, m 18F, CF3–); − 117.56, m, 12F, –CF2–; − 124.83, m, 12F, –CF2–; − 125.08, m 24F, –CF2–; − 125.88, m 12F, –CF2–; − 126.54, m 12F, –CF2–; − 129.30, m, 12F, –CF2–. MS (ESI): [M+] = 3577.2 (expt), 3577.2 (calcd for C103H53F102N4O6Au, major isotopomer) (Supplementary Information S1).
Data availability
All data generated or analyzed in this study are included in this published article and its supplementary information.
References
Johnson, A. W., Kay, I. T. Corroles. I. Synthesis. J. Chem. Soc. 1620–1629 (1965).
Vogel, E. et al. Metallocorroles with formally tetravalent iron. Angew. Chem. Int. Ed. 33, 731–735 (1994).
Gross, Z., Galili, N. & Saltsman, I. The first direct synthesis of corroles from Pyrrole. Angew. Chem. Int. Ed. 38, 1427–1429 (1999).
Paolesse, R. et. al. 5,10,15-Triphenylcorrole: a product from a modified Rothemund reaction. Chem. Comm. 1307–1308 (1999).
Erben, C., Will, S., Kadish, K. M. Metallocorroles: Molecular structure, spectroscopy and electronic states. in The Porphyrin Handbook (eds. Kadish, K. M., Smith, K. M. & Guilard, R.) Vol. 2, Ch. 12, 303–349 (Academic, San Diego, 2002).
Ghosh, A. Electronic structure of corrole derivatives: Insights from molecular structures, spectroscopy, electrochemistry, and quantum chemical calculations. Chem. Rev. 117, 3798–3881 (2017).
Natale, C. d., Gros, C. P. & Paolesse, R. Corroles at work: A small macrocycle for great applications. Chem. Soc. Rev. 51, 1277–1335 (2022).
Mahammed, A. & Gross, Z. Corroles as triplet photosensitizers. Coord. Chem. Rev. 379, 121–132 (2019).
Lemon, C. M. Corrole photochemistry. Pure Appl. Chem. 92, 1901–1919 (2019).
Palmer, J. H., Durrell, A. C., Gross, Z., Winkler, J. R. & Gray, H. B. Near-IR phosphorescence of Iridium(III) corroles at ambient temperature. J. Am. Chem. Soc. 132, 9230–9231 (2010).
Borisov, S. M., Alemayehu, A. & Ghosh, A. Osmium-nitrido corroles as NIR indicators for oxygen sensors and triplet sensitizers for organic upconversion and singlet oxygen generation. J. Mater. Chem. C 4, 5822–5828 (2016).
Lemon, C. M., Powers, D. C., Brothers, P. J. & Nocera, D. G. Gold corroles as near-IR phosphors for oxygen sensing. Inorg. Chem. 56, 10991–10997 (2017).
Alemayehu, A. B., McCormick, L. J., Gagnon, K. J., Borisov, S. M. & Ghosh, A. Stable Platinum(IV) corroles: Synthesis, molecular structure, and room-temperature near-IR phosphorescence. ACS Omega 3, 9360–9368 (2018).
Borisov, S. M., Einrem, R. F., Alemayehu, A. B. & Ghosh, A. Ambient-temperature near-IR phosphorescence and potential applications of rhenium-oxo corroles. Photochem. Photobiol. Sci. 18, 1166–1170 (2019).
Thomassen, I. K., McCormick-McPherson, L. J., Borisov, S. M. & Ghosh, A. Iridium corroles exhibit weak near-infrared phosphorescence but efficiently sensitize singlet oxygen formation. Sci. Rep. 10, 7551 (2020).
Bonnett, R. Chemical Aspects of Photodynamic Therapy. 1–324 (CRC, 2000).
Teo, R. D., Hwang, J. Y., Termini, J., Gross, Z. & Gray, H. B. Fighting cancer with corroles. Chem. Rev. 117, 2711–2729 (2017).
Pandey, R. K., Kessel, D. & Dougherty, T. J. (eds.) Handbook of photodynamic therapy : updates on recent applications of porphyrin-based compounds. 1–564 (World Scientific, 2016).
Lopes, S.M., Pineiro, M. & Pinho e Melo, T. M. Corroles and hexaphyrins: synthesis and application in cancer photodynamic therapy. Molecules 25, 3450 (2020).
Pasternack, R. F. et al. Aggregation of meso-substituted water-soluble porphyrins. J. Am. Chem. Soc. 94, 4511–4517 (1972).
Hambright, P. Chemistry of Water Soluble Porphyrins. in The Porphyrin Handbook (eds. Kadish, K. M., Smith, K. M. & Guilard, R.). Vol. 3, Ch. 18, 129–210 (Academic, San Diego, 2000).
Sibrian-Vazquez, M., Jensen, T. J., Hammer, R. P. & Vicente, M. G. H. Peptide-mediated cell transport of water soluble porphyrin conjugates. J. Med. Chem. 49, 1364–1372 (2006).
Dąbrowski, J. M. et al. Synthesis, photophysical studies and anticancer activity of a new halogenated water-soluble porphyrin. Photochem. Photobiol. 83, 897–903 (2007).
Singh, S. et al. Glycosylated porphyrins, phthalocyanines, and other porphyrinoids for diagnostics and therapeutics. Chem. Rev. 115, 10261–10306 (2015).
Luciano, M. & Brückner, C. Modifications of porphyrins and hydroporphyrins for their solubilization in aqueous media. Molecules 22, 980 (2017).
Alemayehu, A. B. et al. Gold Tris(carboxyphenyl)corroles as multifunctional materials: Room temperature near-IR phosphorescence and applications to photodynamic therapy and dye-sensitized solar cells. ACS Appl. Mater. Interfaces 8, 18935–18942 (2016).
Einrem, R. F., Alemayehu, A. B., Borisov, S. M., Ghosh, A. & Gederaas, O. A. Amphiphilic rhenium-oxo corroles as a new class of sensitizers for photodynamic therapy. ACS Omega 5, 10596–10601 (2020).
Thomassen, I. K., Rasmussen, D., Einrem, R. F. & Ghosh, A. Simple, axial ligand-mediated route to water-soluble iridium corroles. ACS Omega 6, 16683–16687 (2021).
Horváth, I. T. & Rábai, J. Facile catalyst separation without water: Fluorous biphase hydroformylation of olefins. Science 266, 72–75 (1994).
Fish, R. H. Fluorous biphasic catalysis: A new paradigm for the separation of homogeneous catalysts from their reaction substrates and products. Chem. Eur. J. 5, 1677–1680 (1999).
Barthel-Rosa, L. P. & Gladysz, J. A. Chemistry in fluorous media: A user’s guide to practical considerations in the application of fluorous catalysts and reagents. Coord. Chem. Rev. 190, 587–605 (1999).
Cametti, M., Crousse, B., Metrangolo, P., Milani, R. & Resnati, G. The fluorous effect in biomolecular applications. Chem. Soc. Rev. 41, 31–42 (2012).
Horvath, I. T. & Anastas, P. T. Innovations and green chemistry. Chem. Rev. 107, 2169–2173 (2007).
Day, R. A., Estabrook, D. A., Logan, J. K. & Sletten, E. M. Fluorous photosensitizers enhance photodynamic therapy with perfluorocarbon nanoemulsions. Chem. Comm. 53, 13043–13046 (2017).
Miller, M. A. & Sletten, E. M. A general approach to biocompatible branched fluorous tags for increased solubility in perfluorocarbon solvents. Org. Lett. 20, 6850–6854 (2018).
Mahammed, A., Gray, H. B., Weaver, J. J., Sorasaenee, K. & Gross, Z. Amphiphilic corroles bind tightly to human serum albumin. Bioconjug. Chem. 15, 738–746 (2004).
Xue, X., Lindstrom, A. & Li, Y. Porphyrin-based nanomedicines for cancer treatment. Bioconjug. Chem. 30, 1585–1603 (2019).
Gomez, S., Tsung, A. & Hu, Z. Current targets and bioconjugation strategies in photodynamic diagnosis and therapy of cancer. Molecules 25, 4964 (2020).
Pathak, P., Zarandi, M. A., Zhou, X. & Jayawickramarajah, J. Synthesis and applications of porphyrin-biomacromolecule conjugates. Front. Chem. 9, 764137 (2021).
Montaseri, H., Kruger, C. A. & Abrahamse, H. Recent advances in porphyrin-based inorganic nanoparticles for cancer treatment. Int. J. Mol. Sci. 21, 3358 (2020).
Wang, A., Ye, J., Humphrey, M. G. & Zhang, C. Graphene and carbon-nanotube nanohybrids covalently functionalized by porphyrins and phthalocyanines for optoelectronic properties. Adv. Mater. 30, 1705704 (2018).
Magna, G. et al. Recent advances in chemical sensors using porphyrin-carbon nanostructure hybrid materials. Nanomaterials 11, 997 (2021).
Tortora, L. et al. Supramolecular sensing mechanism of corrole thin films. Sens. Actuators B Chem. 187, 72–77 (2013).
Berenbaum, M. C. et al. meso-Tetra(hydroxyphenyl)porphyrins, a new class of potent tumour photosensitisers with favourable selectivity. Br. J. Cancer 54, 717–725 (1986).
Bonnett, R., White, R. D., Winfield, U. J. & Berenbaum, M. C. Hydroporphyrins of the meso-tetra(hydroxyphenyl)porphyrin series as tumour photosensitizers. Biochem. J. 261, 277–280 (1989).
James, D. A., Arnold, D. P. & Parsons, P. G. Potency and selective toxicity of tetra(hydroxyphenyl)- and tetrakis(dihydroxyphenyl)porphyrins in human melanoma cells, with and without exposure to red light. Photochem. Photobiol. 59, 441–447 (1994).
Bhyrappa, P., Wilson, S. R. & Suslick, K. S. Hydrogen-bonded porphyrinic solids: Supramolecular networks of octahydroxy porphyrins. J. Am. Chem. Soc. 119, 8492–8502 (1997).
Einrem, R. F., Gagnon, K. J., Alemayehu, A. B. & Ghosh, A. Metal-ligand misfits: Facile access to rhenium-oxo corroles by oxidative metalation. Chem. Eur. J. 22, 517–520 (2016).
Einrem, R. F. et al. Synthesis and molecular structure of 99Tc Corroles. Chem. Eur. J. 22, 18747–18751 (2016).
Alemayehu, A. B., Teat, S. J., Borisov, S. M. & Ghosh, A. Rhenium-imido corroles. Inorg. Chem. 59, 6382–6389 (2020).
Alemayehu, A. B., Einrem, R. F., McCormick-McPherson, L. J., Settineri, N. S. & Ghosh, A. Synthesis and molecular structure of perhalogenated rhenium-oxo corroles. Sci. Rep. 10, 19727 (2020).
Alemayehu, A. B., McCormick-McPherson, L. J., Conradie, J. & Ghosh, A. Rhenium corrole dimers: electrochemical insights into the nature of the metal-metal quadruple bond. Inorg. Chem. 60, 8315–8321 (2021).
Einrem, R. F. et al. Regioselective formylation of rhenium-oxo and gold corroles: Substituent effects on optical spectra and redox potentials. RSC Adv. 11, 34086–34094 (2021).
Alemayehu, A. B. & Ghosh, A. Gold corroles. J. Porphyr. Phthalocyanines 15, 106–110 (2011).
Rabinovitch, E., Goldberg, I. & Gross, Z. Gold(I) and Gold(III) corroles. Chem. Eur. J. 17, 12294–12301 (2011).
Thomas, K. E., Alemayehu, A. B., Conradie, J., Beavers, C. & Ghosh, A. Synthesis and molecular structure of gold triarylcorroles. Inorg. Chem. 50, 12844–12851 (2011).
Thomas, K. E., Beavers, C. M. & Ghosh, A. Molecular structure of a gold β-Octakis(trifluoromethyl)-meso-triarylcorrole: An 85° difference in saddling dihedral relative to copper. Mol. Phys. 110, 2439–2444 (2012).
Thomas, K. E. et al. Ligand noninnocence in coinage metal corroles: A silver knife-edge. Chem. - Eur. J. 21, 16839–16847 (2015).
Capar, J. et al. Demetalation of copper undecaarylcorroles: molecular structures of a free-base undecaarylisocorrole and a gold undecaarylcorrole. J. Inorg. Biochem. 162, 146–153 (2016).
Norheim, H. K., Schneider, C., Gagnon, K. J. & Ghosh, A. One-Pot Synthesis of a bis-Pocket Corrole through a 14-fold Bromination Reaction. ChemistryOpen 6, 221–225 (2017).
Thomas, K. E., Gagnon, K. J., McCormick, L. J. & Ghosh, A. Molecular structure of gold 2,3,7,8,12,13,17,18-octabromo-5,10,15-tris(4′-pentafluorosulfanylphenyl)corrole: Potential insights into the insolubility of gold octabromocorroles. J. Porphyr. Phthalocyanines 22, 596–601 (2018).
Thomas, K. E. et al. Gold dipyrrin-bisphenolates: A combined experimental and DFT study of metal–ligand interactions. RSC Adv. 10, 533–540 (2020).
Sahu, K. et al. NIR-emissive, singlet-oxygen–sensitizing gold tetra (thiocyano) corroles. Dalton Trans. 51, 13236–13245 (2022).
Thomas, K. E., Alemayehu, A. B., Conradie, J., Beavers, C. M. & Ghosh, A. The structural chemistry of metallocorroles: Combined X-ray crystallography and quantum chemistry studies afford unique insights. Acc. Chem. Res. 45, 1203–1214 (2012).
Buckley, H. L. & Arnold, J. Recent developments in out-of-plane metallocorrole chemistry across the periodic table. Dalton Trans. 44, 30–36 (2015).
Alemayehu, A. B., Thomas, K. E., Einrem, R. F. & Ghosh, A. The story of 5d metallocorroles: From metal-ligand misfits to new building blocks for cancer phototherapeutics. Acc. Chem. Res. 54, 3095–3107 (2021).
Alemayehu, A. B., Gagnon, K. J., Terner, J. & Ghosh, A. Oxidative metalation as a route to size-mismatched macrocyclic complexes: Osmium corroles. Angew. Chem. Int. Ed. 53, 14411–14414 (2014).
Palmer, J. H., Durrell, A. C., Gross, Z., Winkler, J. R. & Gray, H. B. Iridium corroles. J. Am. Chem. Soc. 130, 7786–7787 (2008).
Alemayehu, A. B. et al. Platinum corroles. Chem. Comm. 50, 11093–11096 (2014).
McOmie, J. F. W., Watts, M. L. & West, D. E. Demethylation of aryl methyl ethers by boron tribromide. Tetrahedron 24, 2289–2292 (1968).
Vickery, E.H., Pahler, L.F. & Eisenbraun, E.J. Selective O-demethylation of catechol ethers. Comparison of boron tribromide and iodotrimethylsilane. J. Org. Chem. 44, 4444–4446 (1979).
Karunakaran, S. C. et al. In vitro demonstration of apoptosis mediated photodynamic activity and NIR nucleus imaging through a novel porphyrin. ACS Chem. Biol. 8, 127–132 (2013).
Marydasan, B. et al. In vitro and in vivo demonstration of human-ovarian-cancer necrosis through a water-soluble and near-infrared-absorbing chlorin. J. Med. Chem. 61, 5009–5019 (2018).
Manna, B. K., Bera, S. C. & Rohatgi-Mukherjee, K. K. Effect of solvent and pH on the spectral characteristics of meso-tetrakis(p-hydroxyphenyl)porphyrin in dimethylformamide and dimethylformamide + water mixed solvents. Spectrochim. Acta A Mol. Biomol. Spectrosc. 51, 1051–1060 (1995).
Guo, H. et al. UV-vis spectrophotometric titrations and vibrational spectroscopic characterization of meso-(p-Hydroxyphenyl)porphyrins. J. Phys. Chem. B 108, 10185–10191 (2004).
Guo, H. et al. Sequential Deprotonation of meso-(p-Hydroxyphenyl)porphyrins in DMF: From Hyperporphyrins to Sodium Porphyrin Complexes. J. Phys. Chem. B 110, 587–594 (2006).
Wasbotten, I. H., Conradie, J. & Ghosh, A. Electronic absorption and resonance Raman signatures of hyperporphyrins and nonplanar porphyrins. J. Phys. Chem. B 107, 3613–3623 (2003).
Wamser, C. C. & Ghosh, A. The hyperporphyrin concept: A contemporary perspective. JACS Au 2, 1543–1560 (2022).
Bettelheim, A., White, B. A., Raybuck, S. A. & Murray, R. W. Electrochemical polymerization of amino-, pyrrole-, and hydroxy-substituted tetraphenylporphyrins. Inorg. Chem. 26, 1009–1017 (1987).
Walter, M. G. & Wamser, C. C. Synthesis and characterization of electropolymerized nanostructured aminophenylporphyrin films. J. Phys. Chem. C 114, 7563–7574 (2010).
Rudine, A. B., DelFatti, B. D. & Wamser, C. C. Spectroscopy of protonated tetraphenylporphyrins with amino/carbomethoxy substituents: Hyperporphyrin effects and evidence for a monoprotonated porphyrin. J. Org. Chem. 78, 6040–6049 (2013).
Conradie, J., Wamser, C. C. & Ghosh, A. Understanding hyperporphyrin spectra: TDDFT calculations on diprotonated tetrakis(p-aminophenyl)porphyrin. J. Phys. Chem A 125, 9953–9961 (2021).
Friedman, A., Landau, L., Gonen, S., Gross, Z. & Elbaz, L. Efficient bio-inspired oxygen reduction electrocatalysis with electropolymerized cobalt corroles. ACS Catal. 8, 5024–5031 (2018).
Zhao, Y. et al. A corrole-based covalent organic framework featuring desymmetrized topology. Angew. Chem. Int. Ed. 59, 4354–4359 (2020).
Collman, J. P. & Decréau, R. A. 5, 10, 15-Tris (o-aminophenyl) corrole (TAPC) as a versatile synthon for the preparation of corrole-based hemoprotein analogs. Org. Lett. 7(6), 975–978 (2005).
Thomassen, I. K. & Ghosh, A. Protonation-Induced Hyperporphyrin Spectra of meso-Aminophenylcorroles. ACS Omega 5, 9023–9030 (2020).
Aggarwal, A., Singh, S., Samson, J. & Drain, C. M. Adaptive organic nanoparticles of a teflon-coated iron (III) porphyrin catalytically activate dioxygen for cyclohexene oxidation. Macromol. Rapid Commun. 33, 1220–1226 (2012).
Vincent, J. M., Contel, M., Pozzi, G. & Fish, R. H. How the Horváth paradigm, Fluorous biphasic catalysis, affected oxidation chemistry: Successes, challenges, and a sustainable future. Coord. Chem. Rev. 380, 584–599 (2019).
Update, An. Aggarwal, A., Bhupathiraju, N. D. K., Farley, C. & Singh, S. Applications of Fluorous Porphyrinoids. Photochem. Photobiol. 97, 1241–1265 (2021).
Gryko, D.T. & Jadach, K. A Simple and versatile one-pot synthesis of meso-substituted trans-A2B-Corroles. J. Org. Chem. 66, 4267–4275.
Alemayehu, A., Conradie, M. M. & Ghosh, A. Electronic absorption spectra of copper corroles: Unexpected substituent effects in trans-meso-A2B-triarylcorrole complexes. J. Porphyr. Phthalocyanines. 16, 695–704 (2012).
Reid, C. S., Zhang, Y. & Li, C. J. Fluorous tagging: an enabling isolation technique for indium-mediated allylation reactions in water. Org. Biomol. Chem. 5, 3589–3591 (2007).
Zhang, W. Green chemistry aspects of fluorous techniques—opportunities and challenges for small-scale organic synthesis. Green Chem. 11, 911–920 (2009).
Lo, A. S. & Horváth, I. T. Fluorous ethers. Green Chem. 17, 4701–4714 (2015).
Law, C. K. E. & Horváth, I. T. Synthesis and applications of fluorous phosphines. Org. Chem. Front. 3, 1048–1062 (2016).
Paolesse, R., Nardis, S., Monti, D., Stefanelli, M. & di Natale, C. Porphyrinoids for chemical sensor applications. Chem. Rev. 117, 2517–2583 (2017).
Qidwai, A. et al. Role of nanocarriers in photodynamic therapy. Photodiagnosis Photodyn. Ther. 30, 101782 (2020).
Day, R. A. & Sletten, E. M. Perfluorocarbon nanomaterials for photodynamic therapy. Curr. Opin. Colloid Interface Sci. 54, 101454 (2021).
Kobayashi, Y. et al. Unprecedented Asymmetric Epoxidation of Isolated Carbon–Carbon Double Bonds by a Chiral Fluorous Fe (III) Salen Complex: Exploiting Fluorophilic Effect for Catalyst Design. Eur. J. Org. Chem. 2401–2408 (2019).
Ishihara, K. et al. A recyclable and highly stereoselective multi-fluorous proline catalyst for asymmetric aldol reactions. Tetrahedron Lett. 61, 151657 (2020).
Mika, L. T. & Horváth, I. T. Fluorous Catalysis. in Green Techniques for Organic Synthesis and Medicinal Chemistry (eds. Zhang, W. & Cue, B. W., Jr.) 137–184 (Wiley, New York, 2012).
Acknowledgements
This work was supported by Research Council of Norway (grant no. 324139 to AG).
Funding
Open access funding provided by UiT The Arctic University of Norway (incl University Hospital of North Norway).
Author information
Authors and Affiliations
Contributions
A.B.A. conducted all the experimental work and both authors planned and coordinated the research. Both authors contributed to writing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Alemayehu, A.B., Ghosh, A. Phenol- and resorcinol-appended metallocorroles and their derivatization with fluorous tags. Sci Rep 12, 19256 (2022). https://doi.org/10.1038/s41598-022-23889-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-23889-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
