
Abstract
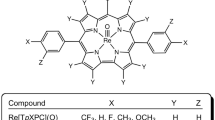
Six-coordinate iridium(III) triarylcorrole derivatives, Ir[TpXPC)]L2, where TpXPC = tris(para-X-phenyl)corrole (X = CF3, H, Me, and OCH3) and L = pyridine (py), trimethylamine (tma), isoquinoline (isoq), 4-dimethylaminopyridine (dmap), and 4-picolinic acid (4pa), have been examined, with a view to identifying axial ligands most conducive to near-infrared phosphorescence. Disappointingly, the phosphorescence quantum yield invariably turned out to be very low, about 0.02 – 0.04% at ambient temperature, with about a two-fold increase at 77 K. Phosphorescence decay times were found to be around ~5 µs at 295 K and ~10 µs at 77 K. Fortunately, two of the Ir[TpCF3PC)]L2 derivatives, which were tested for their ability to sensitize singlet oxygen formation, were found to do so efficiently with quantum yields Φ(1O2) = 0.71 and 0.38 for L = py and 4pa, respectively. Iridium corroles thus may hold promise as photosensitizers in photodynamic therapy (PDT). The possibility of varying the axial ligand and of attaching biotargeting groups at the axial positions makes iridium corroles particularly exciting as PDT drug candidates.
Similar content being viewed by others
Introduction
The 5d transition metal corroles represent an unusual class of size-mismatched metal-ligand assemblies, which combine a large 5d ion and a sterically constrained, macrocyclic corrole ligand1. Although many of the complexes were initially synthesized as part of curiosity-driven exercises, their photophysical properties now promise a wide range of practical applications2,3, as near-IR emitters, as oxygen sensors, and as photosensitizers for photodynamic therapy, dye-sensitized solar cells, and triplet-triplet annihilation upconversion4,5,6,7,8,9. Iridium corroles were among the first 5d metallocorroles to be synthesized10 and reported as exhibiting as near-IR phosphorescence at room temperature4,5. Subsequently, Au11,12,13,14, OsN15, ReO16, and Pt17 corroles were synthesized and found to exhibit significantly stronger phosphorescence6,7,8,9. In this reexamination of six-coordinate Ir corroles (structures depicted below), we attempted to determine whether different axial ligands, including pyridine (py), trimethylamine (tma), isoquinoline (isoq), 4-dimethylaminopyridine (dmap), and 4-picolinic acid (4pa), might be exploited to enhance the phosphorescent behavior.
Results and Discussion
Early spectroscopic and structural studies
Four different meso-tris(para-X-phenyl)corrole ligands, H3[TpXPC] (X = CF3, H, Me, and OMe), and two different axial ligands, pyridine (py) and trimethylamine (tma), were initially investigated. The compounds were all found to exhibit broad, asymmetric Soret bands with a maximum at 416 ± 2 nm and intense Q bands with the maximum varying over 600 ± 5 nm (Fig. 1 and Table 1). Three of the complexes – Ir[TPC]tma2, Ir[TpMePC]tma2, and Ir[TpCF3PC]py2 (Fig. 2, Table 2) – could be structurally characterized, revealing planar metallocorrole macrocycles with Ir-Ncorrole distances of 1.95-1.97 Å and Ir-Naxial distances of 2.17-2.18 Å. Electrochemical studies revealed two reversible oxidation potentials, including a rather low first oxidation potential that ranged from 0.2 to 0.44 V vs. the saturated calomel electrode (Table 1). In general, a reduction potential could not be observed within the available potential window for the solvent (dichloromethane), except for Ir[TpCF3PC]py2, which showed a reduction potential of -1.71 V, a reflection of the highly electron-rich character of the macrocycle in Ir corroles (Table 1). All these properties are qualitatively consistent with those observed for Ir tris(pentafluorophenyl)corrole derivatives10.

Representative UV-vis spectra in dichloromethane: Ir[(TpMePC)]tma2 (top) and Ir[(TpMePC)]py2 (bottom).

X-ray structures (top and side views) of Ir[TPC]tma2 (left), Ir[TpMePC]tma2 (middle), and Ir[TpCF3PC]py2 (right). Bond distances (Å) for Ir[TPC]tma2: Ir-N1 1.9543(16), Ir-N2 1.9728(16), Ir-N3 1.9745(16), Ir-N4 1.9489(17), Ir-N5 2.1795(18), Ir-N6 2.1672(18). Bond distances (Å) for Ir[TpMePC]tma2. Ir-N1 1.953(2), Ir-N2 1.9746(19), Ir-N3 1.978(2), Ir-N4 1.948(2), Ir-N5 2.178(3), Ir-N6 2.176(3). Bond distances (Å) for Ir[TpCF3PC]py2: Ir-N1 1.946(2), Ir-N2 1.9702(18), Ir-N5 2.055(2); note that N5 and N6 are crystallographically equivalent, as are N2 and N3, and N1 and N4.
Near-IR phosphorescence
For the final set of measurements, we chose to focus on the TpCF3PC derivatives but with an expanded set of axial ligands (L) including py, tma, isoquinoline (isoq), 4-dimethylaminopyridine (dmap), and 4-picolinic acid (4pa). All the complexes were found to exhibit weak phosphorescence in the near-infrared part of the spectrum in anoxic solutions (Fig. 3a). The phosphorescence was almost completely quenched in the presence of molecular oxygen. The excitation spectra (Fig. 3b) matched the absorption spectra very well. As expected, the emission spectra were much narrower at 77 K (Fig. 3c), which enabled more precise determination of the triplet state energies from the edge of the emission spectra. Disappointingly, the phosphorescence quantum yields for all the complexes turned out to be very low, which made their precise estimation difficult. Quantum yields of around 0.02 – 0.04% were estimated at ambient temperature with about a two-fold increase at 77 K. The phosphorescence of the bis-pyridine and bis-isoquinoline complexes was found to be stronger than that of bis-tma and bis-dmap complexes. Furthermore, the emission spectra for the pyridine, isoquinoline, and 4-picolinic acid complexes turned out almost identical (Fig. 3a, Table 3). The decay time profiles for these complexes are mono-exponential (Fig. 4). The fits provide very similar values of ~5 µs at 295 K and ~10 µs at 77 K. Interestingly, in the case of the bis-dmap complex, a relatively long-decaying component was observed both at ambient temperature and at 77 K.

Photophysical properties of Ir-TpCF3PC complexes with the axial ligands indicated. (a) Emission spectra in anoxic toluene (unless otherwise mentioned) at 295 K with λex = 595-605 nm (at the maximum of the Q band). The bis-4pa complex was measured in EtOH (anoxic conditions – solid line, air-saturated solvent – dashed line). (b) Excitation spectra at 295 K in the same solvents as in (a). The emission was detected in the maximum of the emission spectra. RG9 long-pass filter was installed in the emission channel to eliminate the monochromator artefacts. (c) Emission spectra in frozen glasses (77 K) with λex = 595-605 nm (at the maximum of the Q-band). The bis-4pa complex was measured in 4:1 v/v ethanol:methanol, while the other complexes were measured in 2:3 v/v toluene:tetrahydrofuran.

Logarithmic plots for the phosphorescence decay of Ir-TpCF3PC complexes in anoxic solutions at 295 K (above) and in frozen glasses at 77 K (below). The inset depicts the phosphorescence decay of the pyridine complex and the fit according to a monoexponential decay model. The solvents are the same as in Fig. 3.
Singlet oxygen sensitization
Many metalloporphyrins and related compounds are known to be powerful sensitizers of singlet oxygen owing to efficient intersystem crossing and long triplet state lifetimes3,4,5,6,7,8,9. The fact that Ir corroles exhibit room temperature phosphorescence indicates that the triplet state is populated to at least some degree. In this study, the Ir-TpCF3PC complexes with py and 4pa axial ligands were evaluated for their 1O2 sensitization capabilities. The assay relied on 9,10-dimethylanthracene as a singlet oxygen acceptor18. Methylene blue, which exhibits a quantum yield for 1O2 formation [Φ(1O2)] of 0.48 and is spectrally compatible with the corroles, was used as the reference7. Fig. 5 shows that Ir(III) corroles efficiently sensitize the formation of singlet oxygen. In fact, Φ(1O2) was found to be 0.71 and 0.38 for L = py and 4pa, respectively. The lower value for 4pa correlates with the lower phosphorescence quantum yield for the same complex (Table 3). These Φ(1O2) values are several fold higher than those reported for Ir(III) tris(4-cyanophenyl)corrole derivatives, which might reflect more efficient radiationless deactivation of the triplet states of the latter (which exhibit phosphorescence quantum yields of 0.01%)5. Efficient sensitization of 1O2 by Ir(III) complexes is of great interest from the standpoint of photodynamic therapy19,20,21 owing to the co-occurrence of two valuable properties: (i) a long excitation wavelength that enables deeper light penetration and (ii) high flexibility in the choice of axial ligands that should facilitate attachment of tumor markers in the axial positions. Covalent attachment via 4-picolinic acid represents a simple, potential synthetic approach for the latter.

Singlet oxygen-induced degradation of 9,10-dimethylanthracene (c = 0.2 mM) as monitored at 358 nm during photosensitization by two Ir[TpCF3PC)]L2 complexes (L = py and 4pa) and methylene blue in ethanol. Excitation of the sensitizers was performed at 575 nm.
Conclusion
Although the photophysical studies of six-coordinate Ir(III) corroles have been previously reported, only a handful of complexes have been examined to date, all exhibiting weak NIR phosphorescence at room temperature. Several additional complexes were accordingly synthesized and examined herein, with a view to identifying axial ligands most conducive to NIR phosphorescence. Unfortunately, regardless of the triarylcorrole and the axial ligands (which varied over pyridine, trimethylamine, isoquinoline, 4-dimethylaminopyridine, and 4-picolinic acid), the phosphorescence quantum yield turned out to be very low, estimated at around 0.02–0.04% at ambient temperature, with about a two-fold increase at 77 K. Phosphorescence decay times were found to be around ~5 µs at 295 K and ~10 µs at 77 K. Fortunately, two of the Ir[TpCF3PC)]L2 derivatives were found to efficiently sensitize singlet oxygen formation, with quantum yields Φ(1O2) = 0.71 and 0.38 for L = py and 4pa, respectively. Iridium corroles thus may hold promise as photosensitizers in photodynamic therapy. The possibility of varying the axial ligand and of attaching tumor-targeting groups at the axial positions makes iridium corroles particularly exciting as drug candidates.
Experimental Section
Materials
Unless otherwise mentioned, all chemicals were obtained from Merck. Silica gel 60 (0.04-0.063 mm particle size, 230-400 mesh) was employed for flash chromatography. Silica gel 60 preparative thin-layer chromatographic plates (20 cm ×20 cm, 0.5 mm thick, Merck) and aluminum oxide 60 preparative thin-layer chromatographic plates (20 cm × 20 cm, 1.5 mm thick, Merck) were used for final purification of all complexes. Free-base corroles were prepared according to previously reported procedures22,23.
Instrumental methods
The methods used were essentially the same as in our earlier work11,13,15,16. UV − visible spectra were recorded on an HP 8453 spectrophotometer. 1H NMR spectra were recorded on a 400 MHz Bruker Avance III HD spectrometer equipped with a 5 mm BB/1H SmartProbe in CDCl3 and referenced to residual CHCl3 7.26 ppm, C6D6 and referenced to residual C6H6 7.16 ppm, C3D6O and referenced to residual C3H6O 2.05 ppm or CD3OD and referenced to residual CH3OH 3.31 ppm. High-resolution electrospray-ionization (HR-ESI) mass spectra were recorded from methanolic solution on an LTQ Orbitrap XL spectrometer.
Cyclic voltammetry was performed at 298 K using an EG&G model 263 A potentiostat with a three-electrode system, comprising a glassy carbon working electrode, a platinum wire counter electrode, and a saturated calomel reference electrode (SCE), in CH2Cl2 (distilled from CaH2) as solvent (as in earlier work11,13,15,16). The reference electrode was separated from the bulk solution by a fritted-glass bridge filled with the electrolyte solution. The electrolyte solution was purged with argon for several minutes, and electrochemical measurements were conducted under an argon blanket. All potentials are referenced to the SCE.
The phosphorescence of the Ir(III) corroles was studied on a Fluorolog 3 fluorescence spectrometer from Horiba (Japan) equipped with an NIR-sensitive photomultiplier R2658 from Hamamatsu (Japan). The spectra were corrected for the sensitivity of the photomultiplier and smoothing processing (adjusting averaging function) was applied to eliminate noise due to low signals. For measurements at room temperature, dye solutions in a sealable quartz cell (Hellma Analytics, Mülheim, Germany) were deoxygenated by bubbling nitrogen (purity 99.9999%, Linde gas, Austria) for 15 min. Measurements at 77 K were conducted in toluene:tetrahydrofuran (4:6 v/v) or ethanol/methanol (4:1 v/v) frozen glass using accessories for deep-temperature measurements from Horiba. Luminescence decay times were measured on the Fluorolog 3 spectrometer equipped with a DeltaHub module (Horiba Scientific) controlling a SpectraLED-460 light source and using DAS-6 Analysis software for data analysis.
Singlet oxygen generation was studied as previously described7. Briefly, a stirred solution containing 9,10-dimethylanthracene (0.2 mM) as a singlet oxygen acceptor and a sensitizer (concentration adjusted to achieve identical absorption for all the sensitizers at λex) was irradiated with light from the xenon lamp of the Fluorolog spectrometer (λex 575 nm). The degradation of the acceptor was assessed via measurement of the UV-vis spectra.
General procedure for the synthesis of Ir[TpXPC]L2 (X = OMe, CH3, H, CF3, L = tma, py, dmap, 4pa, isoq)
The iridium complexes were prepared according to a previously reported procedure10 with slight modifications. Bis(1,5-cyclooctadiene)diiridium(I) dichloride (Merck, 2 eq) and potassium carbonate (10 eq.) were dissolved in a solution of the free-base corrole (~0.1 mmol, 1 eq) in anhydrous tetrahydrofuran (150 mL). After degassing with argon for a few minutes, the solution was brought to reflux under argon. After 1.5 h, a reagent corresponding to the axial ligand (15 eq) was added all at once and the solution was left to reach room temperature (1 h). For L = tma, the reagent used was trimethylamine N-oxide; in the other cases, the unmodifed ligand was used. The reaction mixture was then rotary-evaporated to dryness. Unless otherwise mentioned, the residue was dissolved in a small amount of dichloromethane and subjected to column chromatography (silica, 1:1 CH2Cl2:hexanes) followed by preparative thin-layer chromatography (PTLC, silica, 1:1 CH2Cl2:pentane). Additional details of purification and characterization for each new compound are given below. As in earlier studies of Ir corroles5,10, accurate elemental analyses could not be consistently obtained; proof of composition and purity was accordingly obtained via thin-layer chromatography, clean high-resolution mass spectra that matched theoretical simulations, and, in three cases, single-crystal X-ray structure determinations.
Ir[TPC]tma2
PTLC afforded the product as a dichroic purple-green solid. Yield 37.5 mg (30%). UV-vis (CH2Cl2) λmax (nm) [ϵ x 10-4 (M-1cm-1)]: 384 (3.65, sh), 416 (5.58), 468 (1.59, sh), 596 (2.67). 1H NMR (400 MHz, acetone-d6, δ): 8.88 (d, J = 4.1 Hz, 2H, β-H), 8.62 (d, J = 4.7 Hz, 2H, β-H), 8.39 (d, J = 4.8 Hz, 2H, β-H), 8.24 – 8.19 (m, 4H, 5,15 o-Ph), 8.15 – 8.11 (m, 2H, 10 o-Ph), 8.01 (s, 2H, β-H), 7.78 (dt, J = 14.9, 7.5 Hz, 6H, 5,10,15 m-Ph), 7.69 – 7.63 (m, 3H, 5,10,15 p-Ph), -2.86 (s, 18H, tma-CH3). MS (ESI): M+ = 834.3017 (expt), 834.3019 (calcd for IrC43H41N6).
Ir[TPC]py2
PTLC afforded the product as a dichroic purple-green solid. Yield 83.4 mg (63.6%). UV-vis (CH2Cl2) λmax (nm) [ϵ x 10-4 (M-1cm-1)]: 414 (5.30), 459 (2.66, sh), 562 (0.86, sh), 602 (3.37). 1H NMR (400 MHz, acetone-d6, δ): 8.82 – 8.78 (m, 2H, β-H), 8.62 (dd, J = 4.8, 1.1 Hz, 2H, β-H), 8.38 (d, J = 4.7 Hz, 2H, β-H), 8.28 – 8.22 (m, 4H, 5,15 o-Ph), 8.17 (d, J = 4.1 Hz, 2H, β-H), 8.05 – 8.00 (m, 2H, 10 o-Ph), 7.77 (dd, J = 8.3, 6.8 Hz, 4H, 5,15 m-Ph), 7.70 – 7.59 (m, 5H, 5,15 p-Ph and 10 p,m-Ph), 6.30 (tt, J = 7.6, 1.5 Hz, 2H, p-py), 5.42 – 5.33 (m, 4H, m-py), 1.85 (dt, J = 5.4, 1.5 Hz, 4 H, o-py). MS (ESI): M+ = 874.2403 (expt), 874.2393 (calcd for IrC47H33N6).
Ir[TpMePC]tma2
PTLC with 1:2 CH2Cl2:pentane afforded the product as a bright green solid. Yield 75.7 mg (49.6%). UV-vis (CH2Cl2) λmax (nm) [ϵ x 10-4 (M-1cm-1)]: 384 (3.28, sh), 417 (4.88), 468 (1.54, sh), 598 (2.36). 1H NMR (400 MHz, chloroform-d, δ): 8.88 – 8.82 (m, 2H, β-H), 8.66 (d, J = 4.7 Hz, 2H, β-H), 8.39 (d, J = 4.6 Hz, 2H, β-H), 8.03 (dd, J = 47.5, 7.5 Hz, 8H 5,10,15 o-Ph + β-H, 8H), 7.57 (dd, J = 14.0, 7.6 Hz, 6H, 5,10,15 m-Ph), 1.28 (s, 9H, Ph p-CH3), -2.88 (s, 18H, tma-CH3). MS (ESI): M+ = 876.3488 (expt), 876.3489 (calcd for IrC46H47N6).
Ir[TpMePC]py2
PTLC afforded the product as a dichroic purple-green solid. Yield 36 mg (30.1%). UV-vis (CH2Cl2) λmax (nm) [ϵ × 10-4 (M-1cm-1)]: 417 (6.40), 460 (2.75, sh), 562 (0.98, sh), 603 (3.49). 1H NMR (400 MHz, benzene-d6, δ): 9.14 (dd, J = 4.5, 3.0 Hz, 4H, β-H), 8.97 (d, J = 4.8 Hz, 2H, β-H), 8.80 (d, J = 4.2 Hz, 2H, β-H), 8.46 – 8.41 (m, 4H, 5,15 o-Ph), 8.30 (d, J = 7.9 Hz, 2H, 10 o-Ph), 7.42 (d, J = 7.7 Hz, 4H, 5,15 m-Ph), 7.37 (d, J = 8.0 Hz, 2H, 10 m-Ph), 4.79 (t, J = 7.6 Hz, 2H, p-py), 4.17 – 4.11 (m, 4H, m-py), 2.42 (d, J = 4.2 Hz, 9H, Ph -p-CH3), 2.06 (dt, J = 5.3, 1.5 Hz, 4H, o-py). MS (ESI): M+ = 916.2868 (expt), 916.2863 (calcd for IrC50H39N6).
Ir[TpOMePC]tma2
During initial column chromatography on silica, the eluent was gradually changed from 1:1 CH2Cl2:hexanes to CH2Cl2, and finally to 9:1 CH2Cl2:EtOAc. PTLC with 4:1 EtOAc:CH2Cl2 then afforded the product as a bright green solid. Yield 35.2 mg (29.2%). UV-vis (CH2Cl2) λmax (nm) [ϵ x 10-4 (M-1cm-1)]: 384 (0.74, sh), 417 (1.05), 468 (0.36, sh), 600 (0.48). 1H NMR (400 MHz, chloroform-d, δ): 8.89 (d, J = 3.8 Hz, 2H, β-H), 8.68 (d, J = 4.7 Hz, 2H, β-H), 8.32 (d, J = 4.6 Hz, 2H, β-H), 8.06 (d, J = 8.0 Hz, 4H, 5,15 o-Ph), 7.92 (d, J = 7.9 Hz, 2H, 10 o-Ph), 7.83 (s, 2H, β-H), 7.33 (dd, J = 11.7, 8.2 Hz, 6H, 5,10,15 m-Ph), 4.09 (d, J = 3.6 Hz, 9H, Ph p-OCH3), -2.86 (s, 18H, tma-CH3). MS (ESI): M+ = 924.3336 (expt), 924.3336 (calcd for IrC46H47O3N6).
Ir[TpOMePC]py2
PTLC afforded the product as a bright green solid. Yield 31.2 mg (24.5%). UV-vis (CH2Cl2) λmax (nm) [ϵ x 10-4 (M-1cm-1)]: 416 (3.98), 458 (2.14, sh), 563 (0.77, sh), 605 (2.36). 1H NMR (400 MHz, benzene-d6, δ): 9.19 – 9.15 (m, 4H, 2 β-H), 8.98 (d, J = 4.9 Hz, 2H, β-H), 8.76 (s, 2H, β-H), 8.42 (d, J = 8.0 Hz, 4H, 5,15 o-Ph), 8.08 (d, J = 8.5 Hz, 2H, 10 o-Ph), 7.20 (d, J = 8.8 Hz, 4H, 5,15 m-Ph), 7.11 – 7.07 (m, 2H, 10 m-Ph), 4.81 (ddd, J = 7.7, 6.2, 1.5 Hz, 2H, p-py), 4.20 – 4.16 (m, 4H, m-py), 3.55 (d, J = 2.0 Hz, 9H, Ph p-OCH3), 2.11 – 2.07 (m, 4H, o-py). MS (ESI): M+ = 964.2738 (expt), 964.2710 (calcd for IrC50H39O3N6).
Ir[TpCF3PC]tma2
PTLC with 1:3 CH2Cl2:pentane afforded the product as a dark green solid. Yield 14.7 mg (11.8%). UV-vis (CH2Cl2) λmax (nm) [ϵ x 10-4 (M-1cm-1)]: 418 (2.13), 595 (0.68). 1H NMR (400 MHz, chloroform-d, δ): 8.89 (d, J = 4.2 Hz, 2H, β-H), 8.64 (d, J = 4.8 Hz, 2H, β-H), 8.48 (d, J = 4.8 Hz, 2H, β-H), 8.38 (d, J = 7.8 Hz, 4H, 5,15 o-Ph), 8.29 (d, J = 7.8 Hz, 2H, 10 o-Ph), 8.20 (d, J = 4.2 Hz, 2H, β-H), 8.01 (d, J = 7.9 Hz, 4H, 5,15 m-Ph), 7.98 (d, J = 7.9 Hz, 2H, 10 o-Ph), -2.93 (s, 18H, tma-CH3). MS (ESI): M+ = 1038.2654 (expt), 1038.2641 (calcd for IrC46H38F9N6).
Ir[TpCF3PC]py2
PTLC with 1:2 CH2Cl2:pentane afforded the product as a dark green solid. Yield 16.3 mg (13.7%). UV-vis (CH2Cl2) λmax (nm) [ϵ x 10-4 (M-1cm-1)]: 416 (3.86), 602 (1.48). 1H NMR (400 MHz, benzene-d6, δ): 9.12 (d, J = 4.3 Hz, 2H, β-H), 8.90 (d, J = 4.8 Hz, 2H, β-H), 8.75 (d, J = 4.8 Hz, 2H, β-H), 8.54 (d, J = 4.3 Hz, 2H, β-H), 8.30 (d, J = 8.0 Hz, 4H, 5,15 o-Ph), 8.23 (d, J = 7.9 Hz, 2H, 10 o-Ph), 7.76 (dd, J = 8.1, 6.6 Hz, 6H, 5,10,15 m-Ph), 4.84 (tt, J = 7.7, 1.5 Hz, 2H, p-py), 4.23 – 4.17 (m, 4H, m-py), 1.86 (dt, J = 5.6, 1.5 Hz, 4H, o-py). MS (ESI): M+ = 1078.2032 (expt), 1078.2015 (calcd for IrC50H30F9N6).
Ir[TpCF3PC]dmap2
Initial column chromatography was carried out on neutral alumina with 1:1 CH2Cl2:hexanes as eluent. PTLC on alumina with the same eluent afforded the product as a green solid. Yield 31.7 mg (18.1%). UV-vis (CH2Cl2) λmax (nm) [ϵ x 10-4 (M-1cm-1)]: 418 (1.79), 606 (0.48). 1H NMR (400 MHz, benzene-d6, δ): 9.20 (d, J = 4.2 Hz, 2H, β-H), 8.99 (d, J = 4.8 Hz, 2H, β-H), 8.86 (d, J = 4.8 Hz, 2H, β-H), 8.60 (d, J = 4.2 Hz, 2 H, β-H), 8.45 (d, J = 7.9 Hz, 4 H, 5,15 o-Ph), 8.40 (d, J = 7.9 Hz, 2H, 10 o-Ph), 7.77 (dd, J = 8.3, 2.1 Hz, 6H, 5,10,15 m-Ph), 3.57 – 3.52 (m, 4H, m-py), 1.76 – 1.69 (m, 4 H, o-Ph), 1.07 (s, 6 H, N-CH3), 1.00 (s, 6H, N-CH3). MS (ESI): M+ = 1164.2855 (expt), 1164.2860 (calcd for IrC54H40F9N8).
Ir[TpCF3PC](4pa)2
Initial column chromatography was carried out on neutral alumina with 1:1 CH2Cl2:EtOAc. PTLC on alumina with the same eluent afforded the product as a green solid. Yield 29.6 mg (40.1%). UV-vis (CH2Cl2) λmax (nm) [ϵ x 10-4 (M-1cm-1)]: 414 (1.71), 602 (0.94). 1H NMR (400 MHz, methanol-d4, δ): 8.88 (d, J = 4.2 Hz, 2 H, β-H), 8.61 (d, J = 4.8 Hz, 2 H, β-H), 8.43 – 8.41 (m, 6 H, β-H + 5,15 o-Ph), 8.28 (d, J = 8.0 Hz, 2 H, 10 o-Ph), 8.24 (d, J = 4.3 Hz, 2 H, β-H), 8.08 – 8.03 (m, 4 H, 5,15 m-Ph), 7.99 (d, J = 7.9 Hz, 2 H, 10 m-Ph), 5.61 (d, J = 7.1 Hz, 4 H, m-py), 1.75 (d, J = 7.0 Hz, 4 H, o-py). MS (ESI): M– = 1165.1729 (expt), 1165.1745 (calcd for IrC52H29F9N6O4).
Ir[TpCF3PC]isoq2
Initial column chromatography was carried out on silica with 1:3 CH2Cl2:hexanes as eluent. PTLC on silica with the same eluent afforded the product as a green solid. Yield 12.1 mg (7.9%). UV-vis (CH2Cl2) λmax (nm) [ϵ x 10-4 (M-1cm-1)]: 417 (1.82), 603 (1.02). 1H NMR (400 MHz, chloroform-d, δ): 8.88 (d, J = 4.2 Hz, 2 H, β-H), 8.69 (d, J = 4.8 Hz, 2 H, β-H), 8.46 (d, J = 4.7 Hz, 2 H, β-H), 8.43 (d, J = 7.9 Hz, 4 H, 5,15 o-Ph), 8.29 (s, 2 H, β-H), 8.24 (d, J = 7.9 Hz, 2 H, 10 o-Ph), 8.05 – 7.99 (m, 4 H, 5,15 m-Ph), 7.93 (d, J = 7.9 Hz, 2 H, 10 m-Ph), 7.07 (ddd, J = 8.2, 6.9, 1.2 Hz, 2 H, isoquinoline C7), 6.93 (ddd, J = 8.2, 6.9, 1.1 Hz, 2 H, isoquinoline C6), 6.76 – 6.72 (m, 2H, isoquinoline C8), 6.40 (d, J = 8.4 Hz, 2 H, isoquinoline C5), 5.58 – 5.54 (m, 2 H, isoquinoline C4), 2.36 (s, 2 H, isoquinoline C1), 1.68 (d, J = 6.9 Hz, 2 H, isoquinoline C3). MS (ESI): M– = 1178.2351 (expt), 1178.2329 (calcd for IrC58H34F9N6).
X-ray structure determinations
X-ray data for Ir[TPC]tma2, Ir[TpMePC]tma2, and Ir[TpCF3PC]py2 were collected on beamline 11.3.1 at the Advanced Light Source, Lawrence Berkeley National Laboratory. Samples were mounted on MiTeGen kapton loops and placed in a 100(2)-K (for Ir[TPC]tma2 and Ir[TpCF3PC]py2) or 150(2)-K (for Ir[TpCF3PC]py2) nitrogen cold stream provided by an Oxford Cryostream 800 Plus low-temperature apparatus on the goniometer head of a Bruker D8 diffractometer equipped with a PHOTON100 CMOS detector operating in shutterless mode. Diffraction data were collected using synchrotron radiation monochromated using silicon(111) to a wavelength of 0.7293(1)Å (for Ir[TpMePC]tma2) or 0.7749(1)Å (for Ir[TPC]tma2 and Ir[TpCF3PC]py2). An approximate full-sphere of data was collected using a combination of φ and ω scans with scan speeds of one second per degree for the φ scans and one and three seconds per degree for the ω scans at 2θ = 0° and −45°, respectively. The structures were solved by intrinsic phasing (SHELXT24) and refined by full-matrix least-squares on F2 (SHELXL-201425). All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were geometrically calculated and refined as riding atoms. Additional crystallographic information has been summarized in Table 2.
References
Ghosh, A. Electronic Structure of Corrole Derivatives: Insights from Molecular Structures, Spectroscopy, Electrochemistry, and Quantum Chemical Calculations. Chem. Rev. 117, 3798–3881 (2017).
Teo, R. D., Hwang, J. Y., Termini, J., Gross, Z. & Gray, H. B. Fighting Cancer with Corroles. Chem. Rev. 117, 2711–2729 (2017).
Mahammed, A. & Gross, Z. Corroles as triplet photosensitizers. Coord. Chem. Rev. 379, 121–132 (2019).
Palmer, J. H., Durrell, A. C., Gross, Z., Winkler, J. R. & Gray, H. B. Near-IR Phosphorescence of Iridium(III) Corroles at Ambient Temperature. J. Am. Chem. Soc. 132, 9230–9231 (2010).
Sinha, W., Ravotto, L., Ceroni, P. & Kar, S. NIR-Emissive Iridium(III) Corrole Complexes as Efficient Singlet Oxygen Sensitizers. Dalton Trans. 44, 17767–17773 (2015).
Alemayehu, A. B. et al. Gold Tris(carboxyphenyl)corroles as Multifunctional Materials: Room Temperature Near-IR Phosphorescence and Applications to Photodynamic Therapy and Dye-Sensitized Solar Cells. ACS Appl. Mater. Interfaces 8, 18935–18942 (2016).
Borisov, S. M., Alemayehu, A. & Ghosh, A. Osmium-Nitrido Corroles as NIR Indicators for Oxygen Sensors and Triplet Sensitizers for Organic Upconversion and Singlet Oxygen Generation. J. Mater. Chem. C 4, 5822–5828 (2016).
Lemon, C. M., Powers, D. C., Brothers, P. J. & Nocera, D. G. Gold Corroles as Near-IR Phosphors for Oxygen Sensing. Inorg. Chem. 56, 10991–10997 (2017).
Alemayehu, A. B., McCormick, L. J., Gagnon, K. J., Borisov, S. M. & Ghosh, A. ACS Omega 3, 9360–9368 (2018).
Palmer, J. H. et al. Iridium Corroles. J. Am. Chem. Soc. 130, 7786–7787 (2008).
Alemayehu, A. B. & Ghosh, A. Gold Corroles. J. Porphyrins Phthalocyanines 15, 106–110 (2011).
Rabinovitch, E., Goldberg, I. & Gross, Z. Gold(I) and Gold(III) Corroles. Chem. Eur. J 17, 12294–12301 (2011).
Thomas, K. E., Alemayehu, A. B., Conradie, J., Beavers, C. & Ghosh, A. Synthesis and Molecular Structure of Gold Triarylcorroles. Inorg. Chem. 50, 12844–12851 (2011).
Thomas, K. E. et al. Ligand Noninnocence in Coinage Metal Corroles: A Silver Knife-Edge. Chem. Eur. J 21, 16839–16847 (2015).
Alemayehu, A. B., Gagnon, K. J., Terner, J. & Ghosh, A. Oxidative Metalation as a Route to Size-Mismatched Macrocyclic Complexes: Osmium Corroles. Angew. Chem. Int. Ed 53, 14411–14414 (2014).
Einrem, R. F., Gagnon, K. J., Alemayehu, A. B. & Ghosh, A. Metal-Ligand Misfits: Facile Access to Rhenium-Oxo Corroles by Oxidative Metalation. Chem. Eur. J 22, 517–520 (2016).
Alemayehu, A. B. et al. Platinum Corroles. Chem. Comm 50, 11093–11096 (2014).
Gross, E., Ehrenberg, B. & Johnson, F. M. Singlet Oxygen Generation by Porphyrins and the Kinetics of 9,10‐Dimethylanthracene Photosensitization in Liposomes. Photochem. Photobiol. 57, 808–813 (1993).
Bonnett, R. Chemical Aspects of Photodynamic Therapy. 1-324 (CRC, 2000).
Pandey, R. K., Kessel, D. & Dougherty, T. J. (eds.) Handbook of photodynamic therapy: updates on recent applications of porphyrin-based compounds. 1–564 (World Scientific, 2016).
Nardis, S., Mandoj, F., Stefanelli, M. & Paolesse, R. Metal complexes of corrole. Coord. Chem. Rev. 388, 360–405 (2019).
Gryko, D. T. & Koszarna, B. Refined methods for the synthesis of meso-substituted A3- and trans-A2B-corroles. Org. Biomol. Chem. 1, 350–357 (2003).
Koszarna, B. & Gryko, D. T. Efficient Synthesis of meso-Substituted Corroles in a H2O − MeOH Mixture. J. Org. Chem. 71, 3707–3717 (2006).
Sheldrick, G. M. SHELXT - Integrated Space-Group and Crystal-Structure Determination. Acta Cryst A71, 3–8 (2015).
Sheldrick, G. M. Crystal Structure Refinement with SHELXL. Acta Cryst C71, 3–8 (2015).
Acknowledgements
This work was supported by the Research Council of Norway (grant no. 262229 to AG) and the Arctic Center for Sustainable Energy at UiT – The Arctic University of Norway. The work also used resources of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02-05CH11231.
Author information
Authors and Affiliations
Contributions
I.K.T. carried out all chemical syntheses, L.M.M. did the X-ray structure determinations, and S.M.B. did the photophysical studies. A.G. planned and coordinated the research. All authors contributed to writing the manuscript text.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Thomassen, I.K., McCormick-McPherson, L.J., Borisov, S.M. et al. Iridium Corroles Exhibit Weak Near-Infrared Phosphorescence but Efficiently Sensitize Singlet Oxygen Formation. Sci Rep 10, 7551 (2020). https://doi.org/10.1038/s41598-020-64389-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-64389-3
This article is cited by
-
Phenol- and resorcinol-appended metallocorroles and their derivatization with fluorous tags
Scientific Reports (2022)
-
Synthesis and molecular structure of perhalogenated rhenium-oxo corroles
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
