
Abstract
Understanding the structure and dynamics of newcomer optoelectronic materials - lead halide perovskites APbX3 [A = Cs, methylammonium (CH3NH3+, MA), formamidinium (CH(NH2)2+, FA); X = Cl, Br, I] - has been a major research thrust. In this work, new insights could be gained by using 207Pb solid-state nuclear magnetic resonance (NMR) spectroscopy at variable temperatures between 100 and 300 K. The existence of scalar couplings 1JPb-Cl of ca. 400 Hz and 1JPb-Br of ca. 2.3 kHz could be confirmed for MAPbX3 and CsPbX3. Diverse and fast structure dynamics, including rotations of A-cations, harmonic and anharmonic vibrations of the lead-halide framework and ionic mobility, affect the resolution of the coupling pattern. 207Pb NMR can therefore be used to detect the structural disorder and phase transitions. Furthermore, by comparing bulk and nanocrystalline CsPbBr3 a greater structural disorder of the PbBr6-octahedra had been confirmed in a nanoscale counterpart, not readily captured by diffraction-based techniques.
Similar content being viewed by others
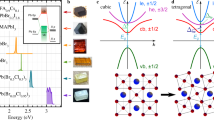
Introduction
Semiconducting lead halide perovskite materials, foremost of APbX3-type [A = Cs, methylammonium (CH3NH3+, MA), formamidinium (CH(NH2)2+, FA); X = Cl, Br, I], have raised tremendous interest over the past years due to their outstanding optoelectronic properties, which find application in solar cells1,2, X-ray3 and gamma detectors4,5,6 and light-emitting devices7,8,9,10,11,12,13,14. These semiconductors exhibit unusually high defect-tolerance, which is the nearly intrinsic semiconducting behaviour in spite of the high abundance of structural imperfections. Such defect-tolerance had been attributed to the specifics of the electronic structure, crystal structure and structural dynamics15,16,17,18,19,20,21. It is therefore fundamental to develop an experimental toolset and a related mind-set for studying the local structure and structural dynamics as well as their relationship to the electronic and physical properties of these semiconductors. Solid-state nuclear magnetic resonance (NMR) is a powerful technique for characterizing solid materials. It is complementary to X-ray diffraction, as it is particularly sensitive to the local environment of nuclei. Chemical composition of APbX3 makes these compounds very well suited for NMR, owing to the range of NMR-active nuclei (1H22,23,24,25, 2H22,23,26,27,28, 13C22,23,24,25,29, 14N22,23,24,25,26,27,29, 15N25,30, 133Cs29,31, 207Pb23,24,25,27,31,32,33, 35Cl, 37Cl, 79Br, 81Br, 127I)34. In this contribution, we focus on 207Pb NMR spectroscopy of APbX3 compounds and report on the existence of scalar lead-halide J-couplings in some of them. 1JPb-Cl of ca. 400 Hz and 1JPb-Br of ca. 2.5 kHz have been measured for MAPbX3 and CsPbX3 compounds. For other compounds within the APbX3 family, scalar couplings are elucidated to be on the order of 2–3 kHz but had not been spectrally resolved. The temperature dependence of the couplings correlates with the known reduction of the structural dynamics and ionic mobility in these perovskites35.
In APbX3 perovskite compounds, corner-sharing lead-halide octahedra form a 3-dimensional (3D) anionic network, charge-stabilized by A-cations filling large 12-fold-coordinated voids in-between the octahedra. Several 3D-polymorphs of these compounds exist, but they differ in the distortion of the lead-halide octahedral lattice (Fig. 1a; see Table S1 for a detailed overview of known structures at various temperatures). This structural data is correlated in the following discussion with our NMR data. The compounds consist of a dynamic inorganic PbX-framework with a high concentration of point defects (higher than 0.4% in MAPbI3 at room temperature36) leading to defect-mediated hopping of the halide anions37. These frameworks are coupled through ion-ion interactions and hydrogen bonds to the A-cation. The rotation and displacement of the A-cations lead to distortions and anharmonic vibrations in the whole perovskite structures38, which has been shown by Whalley et al.39 and Beecher et al.40 for MAPbI3 and by Marronnier et al.41,42 for CsPbI3. All these dynamic processes take place in the picosecond time scale and contribute to the soft structure of the lead halide perovskites. They can be diminished or for some rotations even completely suppressed by reducing the temperature.

(a) Cubic, tetragonal and orthorhombic structures of 3D-perovskites as well as the 1D-structure of orthorhombic CsPbI3. (b) 207Pb NMR spectra of MAPbI3, CsPbI3, MAPbBr3, CsPbBr3, MAPbCl3 and CsPbCl3. The spectra of MAPbI3, CsPbI3, MAPbBr3, and CsPbBr3 were acquired at 16.4 T and the spectra of MAPbCl3 and CsPbCl3 were acquired at 11.7 T at room temperature (RT) using powdered materials. Spinning side bands are marked by asterisks.
NMR studies on lead halide perovskites had already been launched in the 1980s. For MAPbX3, Wasylishen reported in 1985 on the dynamics of the organic cation and phase transitions using 2H and 14N NMR26. Most of the subsequent studies concentrated on 1H, 2H, 13C, 14N, 15N nuclei to characterize and understand the dynamics and mobility of the organic cations (MA or FA)22,23,24,25,26,27,43,44,45. Only a few studies concerned 133Cs NMR29,31,46,47,48,49,50,51. As to the halides, NMR spectroscopy had thus far been hampered by their large quadrupolar constants, leading to massively broadened signals and distorted line shapes52,53. For this reason, halides are more commonly assessed with nuclear quadrupole resonance (NQR) spectroscopy25,44,54,55,56,57,58. Sharma et al.59 (1987) and the dissertation by Ullmann (1998)60 are, to our knowledge, the first reports on 207Pb NMR of lead-halide perovskites. Two decades later, 207Pb NMR studies had been resumed by Rosales et al.33, who studied mixed-halide methylammonium perovskites. The last three years have seen the increasing use of 207Pb NMR for the characterization of APbX3 perovskites and novel lead halide compounds23,24,25,27,44,45,61,62,63,64,65. In these studies, MAPbI3 has been the main focus. It was studied at variable temperatures24,25,27,44, during decomposition61, with dimethylammonium incorporation45 and also with bromine substitution23. 2D NMR and dynamic nuclear polarization (DNP) NMR was measured for micro- and nanocrystalline MAPbX3 at 100 K, which resulted in an enhancement factor of up to 20 for MAPbCl362,65. MAPbCl3 was additionally measured at various temperatures and its utility as an internal thermometer was shown based on the temperature dependence of its 207Pb NMR chemical shift63.
Results and Discussion
Of fundamental importance are the first observations of lead-halide J-coupling in MAPbCl3 (1JPb-Cl) and in CsPbBr3 as well as Cs4PbBr627,32. J-couplings are mediated through bonds by hyperfine interactions between the nuclei and their local electrons. The J-coupling contains information about bond length and angles. It resonates with the notion that the Pb-halide framework in APbX3 compounds exhibits substantial covalency and directionality in the Pb-X bonding, which is needed for efficient through-bond J-coupling66. The existence of these bonds has recently been verified with band structure calculations by Goesten and Hoffmann67.
Lead-halide J-couplings had already been postulated by Dybowski et al. for PbI2 (built from face-sharing Pb-I octahedra), but could not be resolved68. For the 207Pb NMR signal with a full-width at half-maximum (FWHM) of 20 kHz, they calculated a scalar coupling 1JPb-I of 4.9 kHz, which is of a similar magnitude as other scalar couplings involving 207Pb69.
The observation of J-coupling depends on several factors such as nuclear spin and quadrupolar moment of the halide as well as any dynamic changes in the structure or structural defects. The nuclear spin of the halide determines the number of lines present within the coupling pattern, while the large quadrupolar constant of the halides generally broadens the signal and therefore masks the J-coupling70. The next factor that affects the observation of J-coupling is a combined effect of structural inhomogeneities that are primarily due to structural dynamics but also to static structural defects. A distribution of chemical sites, which can be caused, for instance, by vacancies or doping, leads to (inhomogeneous) broadening of the lines. Structure dynamics of the lead halide sublattice falls in the picosecond range35,71, which is too fast for the NMR time scale (µs-to-seconds) and will be averaged out and seen as a quasi-static impact on the observed NMR spectrum. The major type of Pb-halide atomic motion is the tilting of the lead-halide octahedra with respect to each other72,73. This constantly changes the Pb-X-Pb bond angle and therefore affects the orbital overlap thus obscuring the J-coupling. We therefore expect that the J-coupling is a sensitive probe for the structural dynamics or disorder. This disorder was also related to the phonon anharmonicity observed in hybrid perovskites39. It is also important to note that the dynamic disorder of the A-site cation is correlated to that of the Pb-halide framework38. Differences in the Pb-X bond length will result in slightly unequal coupling strengths to the individual halides in the lattice74. A perfect PbX6-octahedron should therefore yield narrower lines compared to a distorted one. This has been shown on 207Pb-19F couplings in amorphous Pb5Ga3F1975. J-couplings to quadrupolar nuclei are known to be self-decoupled, due to their fast relaxation induced by the quadrupole. The fact that in these perovskite materials the couplings are clearly visible, indicates a relatively slow quadrupolar relaxation of the halides76. Once observed, the J-coupling could be, in principle, correlated to the atomic structure. While calculations of J-couplings are sufficiently accurate for light elements (1H, 13C etc.) and are generally possible for solid-state inorganic materials77,78, there is no such theoretical work yet for lead-halide perovskites. At present, a broader experimental survey over diverse lead halide structure is needed to start drawing correlations with the structural motives (bond length/angles, corner/edge/face-sharing connectivity, octahedral or other lead halide building blocks etc.).
All studied APbX3 compounds were synthesized using solution-phase methods, as reported earlier (see Supporting Information for synthesis details)64,79,80. We note that the as-synthesized 3D-polymorph of FAPbI3 (α-phase) rapidly converts into a face-sharing 1D-polymorph (δ-phase) under magic angle spinning (MAS) NMR and therefore we could acquire the spectrum only under static conditions (Fig. S1)64. For CsPbI3, only the 1D-phase (δ-phase) with edge-sharing octahedra is stable at RT81,82. CsPbBr3 NCs were synthesized using colloidal methods with long-chain zwitterionic surfactants as surface capping ligands83. The NCs were precipitated using antisolvents and properly purified before being isolated by centrifugation, dried under vacuum, and measured as a pure solid.
RT MAS solid-state 207Pb NMR spectra of powdered Cs and MA compounds are displayed in Fig. 1b. In agreement with the earlier reports, no scalar couplings were found neither for MAPbI3 (blue left, Fig. 1b) nor for MAPbBr3 (blue middle, Fig. 1b)23,24,33,44,59,61,62. δ-CsPbI3 showed no scalar coupling either (red left, Fig. 1b). However, chloride compounds exhibit pronounced J-couplings of 1JPb-Cl = 400 Hz in CsPbCl3 (red right, Fig. 1b) and 1JPb-Cl = 390 Hz in MAPbCl3 (blue right, Fig. 1b). The latter is similar to that reported by Bernard et al. earlier27. A particularly strong scalar coupling was observed for CsPbBr3; 1JPb-Br = 2.3 kHz (red middle, Fig. 1b). This shows that Pb-Br scalar couplings are possible and that the 207Pb NMR spectra of CsPbBr3 and Cs4PbBr6 acquired and presented in the dissertation of Ullmann appear to exhibit coupling patterns as well although not named and rationalized as such by the author60. The coupling patterns observed for CsPbBr3 as well as for CsPbCl3 and MAPbCl3 (Fig. S2) can be safely attributed to scalar coupling since the splitting between the lines stays unchanged at different magnetic fields but changes with temperature (Fig. 2).

(a) 207Pb NMR spectra of CsPbBr3 acquired at 14.1 T at 100 K (dark blue), 16.4 T at RT (black), 14.1 T at RT (light blue) at 11.7 T and RT (red). The grey lines serve as a guide to the eye by marking the lines of the coupling pattern. The spectra are displayed against a frequency axis centered around 0 Hz. This illustrates most clearly the constant splitting distance (in Hz) between lines, due to a scalar coupling of 1JPb-Br = 2.3 kHz at RT. (b) Coupling trees for six equivalent spins I = 3/2 (top) and the 207Pb NMR spectra of CsPbBr3 (left) and MAPbBr3 (right) acquired at 100 K on a 14.1 T instrument. The acquired spectra are shown in black. The simulated individual lines with intensities obtained from the coupling trees and scalar couplings of 2.5 and 2.4 kHz for CsPbBr3 and MAPbBr3 respectively are displayed in light blue. The sum of the lines is shown in dark blue and is matching the experimental data.
The coupling patterns match expectations for lead coordinated to six equivalent halides in an octahedron. Under 1st order conditions, the signal of a nucleus coupled with n magnetically equivalent nuclei of spin I is a multiplet with 2nI + 1 lines84. Accordingly, for six equivalent halides (n = 6) with spin I = 3/2 (35Cl, 37Cl, 79Br, 81Br) a multiplet with 19 lines is anticipated. The intensity of the individual lines is obtained by constructing a coupling tree for a spin I = 3/2, related to Pascal’s triangle. The coupling pattern of CsPbBr3 and MAPbBr3 at 100 K were simulated by using the experimental scalar couplings of 2.5 and 2.35 kHz, the theoretical intensities from the coupling tree to six equivalent spins I = 3/2 and a Lorentzian line shape with a FWHM of 2.1 and 2.35 kHz. Experimental data and simulations are in excellent agreement (Fig. 2).
Based on the observation that CsPbBr3 and MAPbBr3 possess similar total linewidth in their 207Pb NMR spectra at RT, the hypothesis that an underlying scalar coupling is responsible for the observed overall spectral width is plausible. The absence of visible lines in the multiplet in MAPbBr3 can be explained by the greater broadening of the individual lines. Potential interactions causing such line-broadening in NMR are the dipolar coupling, the chemical shift anisotropy (CSA), site disorder, structural dynamics, and fast relaxation84. Since the sample is spun at the magic angle at 10 kHz or faster, dipolar couplings (80–280 Hz for Pb-X, and 25 Hz for Pb-Cs and Pb-Pb) and CSAs are averaged out85. In a reference experiment, under static conditions, these two interactions also broaden the lines in the 207Pb NMR spectrum of CsPbBr3 to such an extent that they cannot be resolved anymore (Fig. S3). Hence, we conclude that the site-disorder, dynamics or fast relaxation are greater contributors to the line-broadening in MAPbBr3 and MAPbI3 than they are in CsPbBr3, this way obscuring the J-coupling.
Structural disorder, especially of a dynamic nature, is strongly dependent on temperature in the mobile lead halide lattices as is evidenced by the NMR experiments at low temperatures (Figs. 2 and 3). The low-temperature measurements in this study were carried out at 100 K, since at this temperature all lead halide perovskites are reported to be in their lowest-temperature polymorph. No further phase-transitions were reported to occur below 100 K.

207Pb NMR spectra of CsPbBr3 (left), MAPbBr3 (middle) and FAPbBr3 (right). The spectra acquired at RT are shown in red (top) and the ones at 100 K in blue (bottom). The isotropic chemical shifts and the coupling constants are listed in Table 1. A possible coupling in FAPbBr3 is difficult to observe due to the low signal-to-noise ratio.
The isotropic chemical shift of all compounds substantially changes towards higher frequencies upon cooling, which is typical for the 207Pb NMR signal. For CsPbBr3, one can clearly observe an improved resolution of the coupling pattern at 100 K (Fig. 3). The FWHM of the individual lines narrows from 2.7 to 2.1 kHz while the coupling strength increases from 2.3 to 2.5 kHz. The stronger coupling can be explained by the contraction of the unit cell that occurs upon cooling leading to a shortening of the Pb-Br bonds86,87,88. A more prominent improvement of the coupling resolution can be observed for MAPbBr3. After the phase transition to the orthorhombic phase below 150 K the coupling pattern appears (Figs. 3 and S4). In this phase, the motion of the MA-cation is limited to rotation around its C-N axis. This has been shown by Wasylishen et al.26 with 2H and 14N NMR, by Zhu et al.35 where the time-resolved optical Kerr effect at various temperatures was measured and calculated with molecular dynamics simulations by Even et al.89. In its high-temperature phases, the MA cations can be described as an anisotropic molecular liquid while at 77 K it was found to be frozen. The dynamics of the A-cation and the halides are coupled by hydrogen bonds37. A mobile cation with rotational freedom will, therefore, lead to a higher distortion of the PbX6-octahedra. These distortions are so far only visible in synchrotron methods like total scattering or pair distribution function analysis90. In NMR these deformations of the octahedra will result in a smeared out coupling pattern. 207Pb NMR is, therefore, a useful tool to indirectly detect A-cation dynamics and its induced distortion of the inorganic lattice.
For FAPbBr3, no coupling was observed despite cooling, and the total FWHM does not change significantly. This indicates a still highly mobile and disordered surrounding for the 207Pb nuclei. Indeed, it has been shown by single-crystal XRD that the bromides have large displacement factors orthogonal to the Pb-Br bonds even at 100 K91. Additionally, by synchrotron XRD, four distinct positions for Br where detected92. This displacement, therefore, leads to inhomogeneously broadened lines and obscures the coupling pattern. A fast deformation dynamics in hybrid organic inorganic perovskites was also observed by measuring the hot fluorescence emission35. This deformation was attributed to the coupling of the liquid-like motion of the organic cations with the inorganic framework. With 207Pb NMR we could also detect these distortions, present even at 100 K (Fig. 3), making it a powerful method to probe for lattice dynamics at various temperatures. The signal to noise ratio for the low temperature measurement is lower compared to CsPbBr3 and MAPbBr3 measurements, due to the lower number of scans.
The lines of APbBr3 are several times broader than their chlorine analogs (2500 Hz vs. 600 Hz). Both bromine and chlorine possess two isotopes with high natural abundance% 35Cl (76%) and 37Cl (24%) as well as 79Br (51%) and 81Br (49%). The J-couplings between 207Pb and the different isotopes further contribute to the broadening of the individual lines in the coupling pattern, which cannot be quantified at this stage. Another possible contribution to the line broadening could be faster relaxation due to the close vicinity of 207Pb to quadrupoles (halides). All isotopes of chlorine and bromine have a spin of I = 3/2, but the quadrupole moments of the bromine isotopes are four times larger than the ones of the chlorine. This effect should be even more severe for iodine in APbI3 perovskites, with a spin of I = 5/2 and a quadrupole moment more than twice as large as the bromine. Possible Pb-I couplings were calculated by Dybowski et al. in PbI2 to be around 4.9 kHz and by Bernard et al. in MAPbI3 to be between 2 and 3 kHz. So far, none of these couplings could be experimentally resolved.
For MAPbI3, low activation energy for migration of iodine and MA was calculated by Eames et al.36, and confirmed by experimental studies24,93. This ion migration will additionally disturb the PbI6-octahedra. Low-temperature measurements will, therefore, be indispensable for the resolution of Pb-I scalar couplings. At 100 K, a broad tensor was observed for APbI3 (Fig. S5). The isotropic chemical shifts are 990 ppm for CsPbI3 and 1030 ppm for MAPbI3 with FWHM values of around 25 and 20 kHz, respectively. The presence of spinning side bands is an indicator for high anisotropy around the lead nuclei. The spinning side bands overlap complicating the identification of an eventual coupling pattern. Higher spinning speeds would be required to prevent an overlap of the spinning side bands and help resolve the coupling pattern.
Since lead halide couplings were observed for several 3D-perovskite bromides and chlorides, we have looked for the occurrence of 1JPb-X couplings in other octahedrally coordinated lead halides, such as the 0D Cs4PbBr6. Here too, we could observe a well-resolved coupling of 2.0 kHz was detected even at RT (Fig. 4). The FWHM is 1.5 kHz, which is significantly narrower than that of CsPbBr3 at 2.4 kHz. All Pb-Br bonds are identical in Cs4PbBr6 unlike in CsPbBr3, and this leads to a smaller coupling constant distribution that narrows the linewidth94. This clearly shows the effect of lattice distortions on the coupling. For a better insight into this effect, the comparison with 0D hybrid materials with MA and FA would be indispensable. Unfortunately, these materials are so far not known.

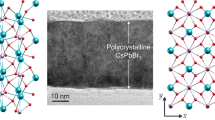
(a) Crystal structure of 0D Cs4PbBr6 with isolated PbBr6-octahedra. (b) Picture of Cs4PbBr6 powder. (c) 207Pb NMR spectrum of Cs4PbBr6 (black) at RT acquired on a 16.4 T spectrometer. The simulated individual lines with intensities obtained from the coupling tree, a FWHM of 1.5 kHz and a scalar coupling of 2.0 kHz are displayed in light blue. The sum of the lines is shown in dark blue and is matching the experimental data. (d) High-Angle Annular Dark-Field Scanning Transmission Electron Microscopy (HAADF-STEM) image of a single CsPbBr3 NC. (e) Colloidal solution of CsPbBr3 NCs under UV excitation. (f) 207Pb NMR spectra of CsPbBr3 bulk (bottom) and NCs (top) acquired at RT on a 11.7 T instrument with identical measurement and processing parameters. The coupling pattern cannot be resolved for the NCs and the total line width increases to 17.6 kHz. This was attributed to higher PbBr6-octahedral disorders over the whole nanocrystal compared to the bulk material.
We have then also probed the effect of dimensionality by comparing bulk material and colloidal nanocrystals (NCs) of CsPbBr3 (Fig. 4). These NCs have recently become an object of intense research due to their outstanding luminescent properties – narrow-band emission with high absolute quantum yields, highly suited for applications in television displays8,21,83. The coupling cannot be resolved at RT, and the total linewidth increases to 17.6 kHz. This is attributed to higher disorder and higher ion-mobility in NCs. In fact, this result correlates well with the model proposing coherent twins and dynamic disorder in these nanocrystals from the analysis of X-ray total scattering techniques and the Debye scattering equation95.
Conclusions
In summary, scalar couplings between 207Pb and halide nuclei (35Cl, 35Cl,79Br, 81Br) have been detected in 207Pb NMR spectra of APbX3 perovskites. The coupling strengths are in the range of 400 Hz for 1JPb-Cl and 2.3 kHz for 1JPb-Br. Only CsPbCl3 and CsPbBr3 exhibit pronounced coupling patterns at RT. The substantial diminishing of structural dynamics in MAPbBr3 at temperatures below 150 K results in the resolution of the J-coupling. For the iodine compounds, a coupling 1JPb-I of ca. 3 kHz can only be postulated based on the overall spectral line width, but it could not be experimentally resolved. Future studies might concentrate on resolving Pb-iodide couplings at lower magnetic fields. 207Pb NMR has shown to be an easily accessible tool to detect permanent and dynamic distortions in the inorganic framework of perovskites. This shows its great potential to better characterize these materials, which is not possible by normal X-ray diffraction. Another important avenue is to probe the relationship between Pb-Br J-couplings and the structural disorder induced by the dimensionality, for instance, in colloidal CsPbBr3 NCs.
Methods
APbX3 (A = Cs, MA, FA; X = Cl, Br, I) compounds were synthesized in the corresponding hydrohalic acid. CsPbX3 was additionally prepared from the solid state by melting together CsX and PbX2 in a 1:1 ratio. CsPbBr3 NCs were prepared by hot injection using long-chain zwitterionic molecules as capping ligands83. See SI for further details. The purity of all compounds was confirmed by powder X-ray diffraction (pXRD). All samples were ground into a fine powder and densely packed into ZrO2 rotors. Solid-state Magic Angle Spinning (MAS) NMR experiments at ambient conditions were performed on three Bruker Avance IIIHD spectrometers (Bruker Biospin, Fällanden, Switzerland). The 11.7 and 16.4 T instruments were equipped with 2.5 mm two-channel and three-channel solid-state probe heads. The spinning frequency was set between 0 and 20 kHz. The 14.1 T magnet was equipped with a 3.2 mm double-channel MAS probe and a MAS spinning rate of 10 kHz was used. Low-temperature experiments were conducted on the 14.1 T Bruker instrument equipped with a 3.2 mm double-channel low-temperature MAS probe using MAS spinning of 10 kHz. 207Pb NMR chemical shifts were referenced to PbMe4. A Hahn echo pulse-sequence was used for all measurements with an echo delay between 0.1 and 0.5 ms96. The rf field of the echo pulses was set to 35.7, 26.3 and 19.8 kHz at 11.7, 14.1 and 16.4 T, respectively, which is strong enough to be refocused the complete spectra of the 3D phases. The 1D phases are too broad (>140 kHz) to be refocused completely.
References
Yang, W. S. et al. High-Performance Photovoltaic Perovskite Layers Fabricated through Intramolecular Exchange. Science 348, 1234–1237 (2015).
Yang, W. S. et al. Iodide Management in Formamidinium-Lead-Halide–Based Perovskite Layers for Efficient Solar Cells. Science 356, 1376–1379 (2017).
Kim, Y. C. et al. Printable Organometallic Perovskite Enables Large-Area, Low-Dose X-Ray Imaging. Nature 550, 87 (2017).
Yakunin, S. et al. Detection of Gamma Photons using Solution-Grown Single Crystals of Hybrid Lead Halide Perovskites. Nat. Photonics 10, 585 (2016).
Wei, H. et al. Dopant Compensation in Alloyed CH3NH3PbBr3−xClx Perovskite Single Crystals for Gamma-Ray Spectroscopy. Nat. Mater. 16, 826 (2017).
He, Y. et al. High Spectral Resolution of Gamma-Rays at Room Temperature by Perovskite CsPbBr3 Single Crystals. Nat. Commun. 9, 1609 (2018).
Sutherland, B. R. & Sargent, E. H. Perovskite Photonic Sources. Nat. Photonics 10, 295 (2016).
Kovalenko, M. V., Protesescu, L. & Bodnarchuk, M. I. Properties and Potential Optoelectronic Applications of Lead Halide Perovskite Nanocrystals. Science 358, 745–750 (2017).
Xing, J. et al. Color-Stable Highly Luminescent Sky-Blue Perovskite Light-Emitting Diodes. Nat. Commun. 9, 3541 (2018).
Cao, Y. et al. Perovskite Light-Emitting Diodes Based on Spontaneously Formed Submicrometre-Scale Structures. Nature 562, 249–253 (2018).
Lin, K. et al. Perovskite Light-Emitting Diodes with External Quantum Efficiency Exceeding 20 Per Cent. Nature 562, 245–248 (2018).
Zhao, X., Ng, J. D. A. & Friend, R. H. Tan Z.-K. Opportunities and Challenges in Perovskite Light-Emitting Devices. ACS Photonics 5, 3866–3875 (2018).
Chiba, T. et al. Anion-Exchange Red Perovskite Quantum Dots with Ammonium Iodine Salts for Highly Efficient Light-Emitting Devices. Nat. Photonics 12, 681–687 (2018).
Shynkarenko, Y. et al. Direct Synthesis of Quaternary Alkylammonium-Capped Perovskite Nanocrystals for Efficient Blue and Green Light-Emitting Diodes. ACS Energy Lett. 4, 2703–2711 (2019).
Zakutayev, A. et al. Defect Tolerant Semiconductors for Solar Energy Conversion. J. Phys. Chem. Lett. 5, 1117–1125 (2014).
Brandt, R. E., Stevanović, V., Ginley, D. S. & Buonassisi, T. Identifying Defect-Tolerant Semiconductors with High Minority-Carrier Lifetimes: Beyond Hybrid Lead Halide Perovskites. MRS Commun. 5, 265–275 (2015).
Walsh, A., Scanlon, D. O., Chen, S., Gong, X. G. & Wei, S.-H. Self-Regulation Mechanism for Charged Point Defects in Hybrid Halide Perovskites. Angew. Chem., Int. Ed. 54, 1791–1794 (2015).
Comin, R. et al. Lattice Dynamics and the Nature of Structural Transitions in Organolead Halide Perovskites. Phys. Rev. B 94, 094301 (2016).
Kang, J. & Wang, L.-W. High Defect Tolerance in Lead Halide Perovskite CsPbBr3. J. Phys. Chem. Lett. 8, 489–493 (2017).
Tan, H. et al. Dipolar Cations Confer Defect Tolerance in Wide-Bandgap Metal Halide Perovskites. Nat. Commun. 9, 3100 (2018).
Huang, H., Bodnarchuk, M. I., Kershaw, S. V., Kovalenko, M. V. & Rogach, A. L. Lead Halide Perovskite Nanocrystals in the Research Spotlight: Stability and Defect Tolerance. ACS Energy Lett. 2, 2071–2083 (2017).
Kubicki, D. J. et al. Cation Dynamics in Mixed-Cation (MA)x(FA)1−xPbI3 Hybrid Perovskites from Solid-State NMR. J. Am. Chem. Soc. 139, 10055–10061 (2017).
Roiland, C. et al. Multinuclear NMR as a Tool for Studying Local Order and Dynamics in CH3NH3PbX3 (X = Cl, Br, I) Hybrid Perovskites. Phys. Chem. Chem. Phys. 18, 27133–27142 (2016).
Senocrate, A. et al. The Nature of Ion Conduction in Methylammonium Lead Iodide: A Multimethod Approach. Angew. Chem., Int. Ed. 56, 7755–7759 (2017).
Senocrate, A., Moudrakovski, I. & Maier, J. Short-Range Ion Dynamics in Methylammonium Lead Iodide by Multinuclear Solid State NMR and 127I NQR. Phys. Chem. Chem. Phys. 20, 20043–20055 (2018).
Wasylishen, R. E., Knop, O. & Macdonald, J. B. Cation Rotation in Methylammonium Lead Halides. Solid State Commun. 56, 581–582 (1985).
Bernard, G. M. et al. Methylammonium Cation Dynamics in Methylammonium Lead Halide Perovskites: A Solid-State NMR Perspective. J. Phys. Chem. A 122, 1560–1573 (2018).
Knop, O., Wasylishen, R. E., White, M. A., Cameron, T. S. & Oort, M. J. M. V. Alkylammonium Lead Halides. Part 2. CH3NH3PbX3 (X = Cl, Br, I) Perovskites: Cuboctahedral Halide Cages with Isotropic Cation Reorientation. Can. J. Chem. 68, 412–422 (1990).
Kubicki, D. J. et al. Phase Segregation in Cs-, Rb- and K-Doped Mixed-Cation (MA)x(FA)1−xPbI3 Hybrid Perovskites from Solid-State NMR. J. Am. Chem. Soc. 139, 14173–14180 (2017).
Milić, J. V. et al. Supramolecular Engineering for Formamidinium-Based Layered 2D Perovskite Solar Cells: Structural Complexity and Dynamics Revealed by Solid-State NMR Spectroscopy. Adv. Energy Mater. 9, 1900284 (2019).
Karmakar, A. et al. Mechanochemical Synthesis of 0D and 3D Cesium Lead Mixed Halide Perovskites. Chem. Commun. 55, 5079–5082 (2019).
Ray, A. et al. Green-Emitting Powders of Zero-Dimensional Cs4PbBr6: Delineating the Intricacies of the Synthesis and the Origin of Photoluminescence. Chem. Mater. 31, 7761–7769 (2019).
Rosales, B. A. et al. Persistent Dopants and Phase Segregation in Organolead Mixed-Halide Perovskites. Chem. Mater. 28, 6848–6859 (2016).
Franssen, W. M. J. & Kentgens, A. P. M. Solid–state NMR of hybrid halide perovskites. Solid State Nucl. Magn. Reson. 100, 36–44 (2019).
Zhu, H. et al. Screening in Crystalline Liquids Protects Energetic Carriers in Hybrid Perovskites. Science 353, 1409–1413 (2016).
Eames, C. et al. Ionic Transport in Hybrid Lead Iodide Perovskite Solar Cells. Nat. Commun. 6, 7497 (2015).
Mosconi, E. & De Angelis, F. Mobile Ions in Organohalide Perovskites: Interplay of Electronic Structure and Dynamics. ACS Energy Lett. 1, 182–188 (2016).
Gallop, N. P. et al. Rotational Cation Dynamics in Metal Halide Perovskites: Effect on Phonons and Material Properties. J. Phys. Chem. Lett. 9, 5987–5997 (2018).
Whalley, L. D., Skelton, J. M., Frost, J. M. & Walsh, A. Phonon Anharmonicity, Lifetimes, and Thermal Transport in CH3NH3PbI3 from Many-Body Perturbation Theory. Phys. Rev. B 94, 220301 (2016).
Beecher, A. N. et al. Direct Observation of Dynamic Symmetry Breaking above Room Temperature in Methylammonium Lead Iodide Perovskite. ACS Energy Lett. 1, 880–887 (2016).
Marronnier, A. et al. Structural Instabilities Related to Highly Anharmonic Phonons in Halide Perovskites. J. Phys. Chem. Lett. 8, 2659–2665 (2017).
Marronnier, A. et al. Anharmonicity and Disorder in the Black Phases of Cesium Lead Iodide Used for Stable Inorganic Perovskite Solar Cells. ACS Nano 12, 3477–3486 (2018).
Baikie, T. et al. A Combined Single Crystal Neutron/X-ray Diffraction and Solid-State Nuclear Magnetic Resonance Study of the Hybrid Perovskites CH3NH3PbX3 (X = I, Br and Cl). J. Mater. Chem. A 3, 9298–9307 (2015).
Franssen, W. M. J., van Es, S. G. D., Dervişoğlu, R., de Wijs, G. A. & Kentgens, A. P. M. Symmetry, Dynamics, and Defects in Methylammonium Lead Halide Perovskites. J. Phys. Chem. Lett. 8, 61–66 (2017).
Franssen, W. M. J., Bruijnaers, B. J., Portengen, V. H. L. & Kentgens, A. P. M. Dimethylammonium Incorporation in Lead Acetate Based MAPbI3 Perovskite Solar Cells. Chem. Phys. Chem. 19, 3107–3115 (2018).
Armstrong, R. L., Lourens, J. A. J. & Stroud, J. D. 133Cs Spin-Lattice Relaxation Study of Phase Transitions in CsPbCl3. Phys. Rev. B 13, 5099–5101 (1976).
Chen, Y. et al. Surface Termination of CsPbBr3 Perovskite Quantum Dots Determined by Solid-State NMR Spectroscopy. J. Am. Chem. Soc. 142, 6117–6127 (2020).
Boziki, A. et al. Atomistic Origins of the Limited Phase Stability of Cs+-Rich FAxCs(1−x)PbI3 Mixtures. Chem. Mater. 32, 2605–2614 (2020).
Kubicki, D. J. et al. Doping and phase segregation in Mn2+- and Co2+-doped lead halide perovskites from 133Cs and 1H NMR relaxation enhancement. J. Mater. Chem. A 7, 2326–2333 (2019).
Prochowicz, D. et al. One-Step Mechanochemical Incorporation of an Insoluble Cesium Additive for High Performance Planar Heterojunction Solar Cells. Nano Energy 49, 523–528 (2018).
Kubicki, D. J. et al. Phase Segregation in Potassium-Doped Lead Halide Perovskites from 39K Solid-State NMR at 21.1 T. J. Am. Chem. Soc. 140, 7232–7238 (2018).
Schurko, R. W. Acquisition of Wideline Solid-State NMR Spectra of Quadrupolar Nuclei (ed. Grant, D. M., Harris, R. K.) 77–90 (John Wiley & Sons Ltd, 2011).
Wasylishen, R. E., Ashbrook S. E., & Wimperis, S. NMR of Quadrupolar Nuclei in Solid Materials (John Wiley & Sons Ltd, 2012).
Tovborg-Jensen, N. NQR Investigation of Phase Transitions in Cesium Plumbochloride. J. Chem. Phys. 50, 559–560 (1969).
Volkov, A. F., Venevtsev, Y. N. & Semin, G. K. Nuclear Quadrupole Resonance (NQR) of 79Br and 81Br in Perovskite and Orthorhombic Forms of CsPbBr3 and CsPbJ3. Phys. Status Solidi B 35, K167–K169 (1969).
Armstrong, R. L. Pure Nuclear Quadrupole Resonance Studies of Structural Phase Transitions. J. Magn. Reson. (1969-1992) 20, 214–231 (1975).
Hidaka, M., Okamoto, Y. & Zikumaru, Y. Structural Phase Transition of CsPbCl3 below Room Temperature. Phys. Status Solidi A 79, 263–269 (1983).
Sharma, S., Weiden, N., Weiss, A. Phase Transitions in CsSnCl3 and CsPbBr3 An NMR and NQR Study. In: Z. Naturforsch. A) (1991).
Sharma, S., Weiden, N., Weiss, A. 207Pb and 205Tl NMR on Perovskite Type Crystals APbX3 (A = Cs, Tl, X = Br, I). In: Z. Naturforsch. A) (1987).
Ullmann, H. Strukturchemische und MAS-NMR-Spektroskopische Untersuchungen mit Quantenchemischen Berechnungen von Binären, Ternären und Quaternären Blei(II)-Halogeniden (Herbert Utz Verlag Wissenschaft, 1998).
Askar, A. M., Bernard, G. M., Wiltshire, B., Shankar, K. & Michaelis, V. K. Multinuclear Magnetic Resonance Tracking of Hydro, Thermal, and Hydrothermal Decomposition of CH3NH3PbI3. J. Phys. Chem. C 121, 1013–1024 (2017).
Rosales, B. A. et al. Lead Halide Perovskites: Challenges and Opportunities in Advanced Synthesis and Spectroscopy. ACS Energy Lett. 2, 906–914 (2017).
Bernard, G. M. et al. Methylammonium Lead Chloride: A Sensitive Sample for an Accurate NMR Thermometer. J. Magn. Reson. 283, 14–21 (2017).
Nazarenko, O. et al. Guanidinium-Formamidinium Lead Iodide: A Layered Perovskite-Related Compound with Red Luminescence at Room Temperature. J. Am. Chem. Soc. 140, 3850–3853 (2018).
Hanrahan, M. P., Men, L., Rosales, B. A., Vela, J. & Rossini, A. J. Sensitivity-Enhanced 207Pb Solid-State NMR Spectroscopy for the Rapid, Non-Destructive Characterization of Organolead Halide Perovskites. Chem. Mater. 30, 7005–7015 (2018).
Hahn, E. L. & Maxwell, D. E. Spin Echo Measurements of Nuclear Spin Coupling in Molecules. Phys. Rev. 88, 1070–1084 (1952).
Goesten, M. G. & Hoffmann, R. Mirrors of Bonding in Metal Halide Perovskites. J. Am. Chem. Soc. 140, 12996–13010 (2018).
Taylor, R. E., Beckmann, P. A., Bai, S. & Dybowski, C. 127I and 207Pb Solid-State NMR Spectroscopy and Nuclear Spin Relaxation in PbI2: A Preliminary Study. J. Phys. Chem. C 118, 9143–9153 (2014).
Kennedy, J. D., McFarlane, W. & Wrackmeyer, B. Indirect Nuclear Spin-Spin Coupling of Lead-207 to Other Magnetic Nuclei. Inorg. Chem. 15, 1299–1302 (1976).
Glatfelter, A. et al. Solid-State 207Pb NMR Studies of Mixed Lead Halides, PbFX (X=Cl, Br, or I). Spectrochim. Acta, Part A 66, 1361–1363 (2007).
Sendner, M. et al. Optical Phonons in Methylammonium Lead Halide Perovskites and Implications for Charge Transport. Mater. Horiz. 3, 613–620 (2016).
Yang, R. X., Skelton, J. M., da Silva, E. L., Frost, J. M. & Walsh, A. Spontaneous Octahedral Tilting in the Cubic Inorganic Cesium Halide Perovskites CsSnX3 and CsPbX3 (X = F, Cl, Br, I). J. Phys. Chem. Lett. 8, 4720–4726 (2017).
Bechtel, J. S. & Van der Ven, A. Octahedral Tilting Instabilities in Inorganic Halide Perovskites. Phys. Rev. Mater. 2, 025401 (2018).
Autschbach, J. & Le Guennic, B. Analyzing and Interpreting NMR Spin–Spin Coupling Constants Using Molecular Orbital Calculations. J. Chem. Educ. 84, 156 (2007).
Martineau, C., et al. Accurate Heteronuclear J-Coupling Measurements in Dilute Spin Systems using the Multiple-Quantum Filtered J-Resolved Experiment. Chem. Commun., 2720–2722 (2007).
Yu, H. et al. Solid-State 63Cu, 65Cu, and 31P NMR Spectroscopy of Photoluminescent Copper(I) Triazole Phosphine Complexes. J. Phys. Chem. A 119, 8279–8293 (2015).
Massiot, D. et al. Detection and Use of small J Couplings in Solid State NMR Experiments. Comptes Rendus Chimie 13, 117–129 (2010).
Kamieńska-Trela, K., Wójcik, J. Applications of spin-spin couplings. In: Nuclear Magnetic Resonance: Volume 41. The Royal Society of Chemistry (2012).
Wells, H. L. Über die Cäsium- und Kalium-Bleihalogenide. Z. Anorg. Allg. Chem. 3, 195–210 (1893).
Nazarenko, O., Yakunin, S., Morad, V., Cherniukh, I. & Kovalenko, M. V. Single Crystals of Caesium Formamidinium Lead Halide Perovskites: Solution Growth and Gamma Dosimetry. NPG Asia Mater. 9, e373 (2017).
Møller, C. K. Crystal Structure and Photoconductivity of Cæsium Plumbohalides. Nature 182, 1436 (1958).
Trots, D. M. & Myagkota, S. V. High-Temperature Structural Evolution of Caesium and Rubidium Triiodoplumbates. J. Phys. Chem. Solids 69, 2520–2526 (2008).
Krieg, F. et al. Colloidal CsPbX3 (X = Cl, Br, I) Nanocrystals 2.0: Zwitterionic Capping Ligands for Improved Durability and Stability. ACS Energy Lett. 3, 641–646 (2018).
Becker, E. D. High Resolution NMR (Second Edition) (Academic Press, 1980).
Lowe, I. J. Free Induction Decays of Rotating Solids. Phys. Rev. Lett. 2, 285–287 (1959).
Rodová, M., Brožek, J., Knížek, K. & Nitsch, K. Phase Transitions in Ternary Cesium Lead Bromide. J. Therm. Anal. Calorim. 71, 667–673 (2003).
Stoumpos, C. C. et al. Crystal Growth of the Perovskite Semiconductor CsPbBr3: A New Material for High-Energy Radiation Detection. Cryst. Growth Des. 13, 2722–2727 (2013).
Wang, K.-H., Li, L.-C., Shellaiah, M. & Wen Sun, K. Structural and Photophysical Properties of Methylammonium Lead Tribromide (MAPbBr3) Single Crystals. Sci. Rep. 7, 13643 (2017).
Even, J., Carignano, M. & Katan, C. Molecular Disorder and Translation/Rotation Coupling in the Plastic Crystal Phase of Hybrid Perovskites. Nanoscale 8, 6222–6236 (2016).
Bernasconi, A. & Malavasi, L. Direct Evidence of Permanent Octahedra Distortion in MAPbBr3 Hybrid Perovskite. ACS Energy Lett. 2, 863–868 (2017).
Schueller, E. C. et al. Crystal Structure Evolution and Notable Thermal Expansion in Hybrid Perovskites Formamidinium Tin Iodide and Formamidinium Lead Bromide. Inorg. Chem. 57, 695–701 (2018).
Protesescu, L. et al. Monodisperse Formamidinium Lead Bromide Nanocrystals with Bright and Stable Green Photoluminescence. J. Am. Chem. Soc. 138, 14202–14205 (2016).
Futscher, M. H. et al. Quantification of Ion Migration in CH3NH3PbI3 Perovskite Solar Cells by Transient Capacitance Measurements. Mater. Horiz. 6, 1497–1503 (2019).
Møller C. K. On the structure of caesium hexahalogeno-plumbates (II) (Munksgaard, 1960).
Bertolotti, F. et al. Coherent Nanotwins and Dynamic Disorder in Cesium Lead Halide Perovskite Nanocrystals. ACS Nano 11, 3819–3831 (2017).
Hahn, E. L. Spin Echoes. Phys. Rev. 80, 580–594 (1950).
Author information
Authors and Affiliations
Contributions
M.K. and R.V. supervised this work. M.A. and L.P. carried out NMR experiments and analyzed the results. O. N., B.B., and F.K. synthesized all samples. M. A. and M. K. wrote the manuscript with the input of all co-authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Aebli, M., Piveteau, L., Nazarenko, O. et al. Lead-Halide Scalar Couplings in 207Pb NMR of APbX3 Perovskites (A = Cs, Methylammonium, Formamidinium; X = Cl, Br, I). Sci Rep 10, 8229 (2020). https://doi.org/10.1038/s41598-020-65071-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-65071-4
This article is cited by
-
The squeezed dark nuclear spin state in lead halide perovskites
Nature Communications (2023)
-
Effect of aliovalent bismuth substitution on structure and optical properties of CsSnBr3
Communications Chemistry (2023)
-
The Landé factors of electrons and holes in lead halide perovskites: universal dependence on the band gap
Nature Communications (2022)
-
Solid-State 207Pb NMR Spectroscopy and Relativistic Quantum Chemical Calculations of Red Pigments: Identification in Cultural Heritage Materials
Applied Magnetic Resonance (2022)
-
NMR spectroscopy probes microstructure, dynamics and doping of metal halide perovskites
Nature Reviews Chemistry (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
