
Abstract
Human intestinal morphogenesis establishes 3D epithelial microarchitecture and spatially organized crypt–villus characteristics. This unique structure is necessary to maintain intestinal homeostasis by protecting the stem cell niche in the basal crypt from exogenous microbial antigens and their metabolites. Also, intestinal villi and secretory mucus present functionally differentiated epithelial cells with a protective barrier at the intestinal mucosal surface. Thus, re-creating the 3D epithelial structure is critical to building in vitro intestine models. Notably, an organomimetic gut-on-a-chip can induce spontaneous 3D morphogenesis of an intestinal epithelium with enhanced physiological function and biomechanics. Here we provide a reproducible protocol to robustly induce intestinal morphogenesis in a microfluidic gut-on-a-chip as well as in a Transwell-embedded hybrid chip. We describe detailed methods for device fabrication, culture of Caco-2 or intestinal organoid epithelial cells in conventional setups as well as on microfluidic platforms, induction of 3D morphogenesis and characterization of established 3D epithelium using multiple imaging modalities. This protocol enables the regeneration of functional intestinal microarchitecture by controlling basolateral fluid flow within 5 d. Our in vitro morphogenesis method employs physiologically relevant shear stress and mechanical motions, and does not require complex cellular engineering or manipulation, which may be advantageous over other existing techniques. We envision that our proposed protocol may have a broad impact on biomedical research communities, providing a method to regenerate in vitro 3D intestinal epithelial layers for biomedical, clinical and pharmaceutical applications.
Similar content being viewed by others

Introduction
Development of the protocol
It has been experimentally demonstrated that intestinal epithelial Caco-2 cells cultured in a gut-on-a-chip1,2,3,4,5 or in a bilayered microfluidic device6,7 can undergo spontaneous 3D morphogenesis in vitro without a clear understanding of the underlying mechanism. In our recent study, we identified that the removal of morphogen antagonists that are basolaterally secreted from the culture setup is necessary and sufficient to induce 3D epithelial morphogenesis in vitro, verified with both Caco-2 and patient-derived intestinal organoid epithelia4. In this study, we specifically focused on the cellular production and concentration profile of a potent Wnt antagonist, Dickkopf-1 (DKK-1), in both gut-on-a-chip and a modified microfluidic device that contains a Transwell insert, called a ‘hybrid chip’. We confirmed that exogenous additions of Wnt antagonists (e.g., DKK-1, Wnt inhibitory factor 1, secreted frizzled-related protein 1 or Soggy-1) into a gut-on-a-chip result in the inhibition of morphogenesis or the disruption of prestructured 3D epithelial layers, suggesting that the antagonistic pressure during the culture is responsible for intestinal morphogenesis in vitro. Thus, practical approaches for robust morphogenesis on an epithelial interface are to either remove or minimally maintain the level of Wnt antagonists in the basolateral compartment by actively washing away (e.g., in the gut-on-a-chip or hybrid chip platforms) or diffusing the basolateral culture broth (e.g., from the Transwell insert to a large basolateral reservoir in the well).
In this protocol, we provide detailed methods for fabricating a gut-on-a-chip microdevice and a Transwell-insertable hybrid chip (Steps 1–5), culturing intestinal epithelial cells on either a polydimethylsiloxane (PDMS)-based porous membrane (Steps 6A, 7A, 8, 9) or the polyester membrane of a Transwell insert (Steps 6B, 7B, 8, 9), and inducing 3D morphogenesis in vitro (Step 10). We also identify cellular and molecular characteristics that are indicative of tissue-specific histogenesis and lineage-dependent cytodifferentiation by applying multiple imaging modalities (Steps 11–24). We induce morphogenesis using human intestinal epithelial cells such as Caco-2, or intestinal organoids, in both culture formats with technical details including surface modification of a porous membrane, creation of a 2D monolayer and recapitulation of the biochemical and biomechanical microenvironment of the gut in vitro. To induce 3D morphogenesis from a 2D epithelial monolayer, we remove morphogen antagonists in both culture formats by flowing culture medium into the basolateral compartment of the cultures. Finally, we provide representative examples of the utility of the regenerative 3D epithelial layers that can potentially be used for simulating morphogen-dependent epithelial growth, longitudinal host–microbiome co-cultures, pathogen infection, inflammatory injury, epithelial barrier dysfunctions and probiotic-based therapeutic effect.
Applications of the method
Our protocol can potentially provide a broad impact to a wide spectrum of scientists in both basic (e.g., intestinal mucosal biology, stem cell biology and developmental biology) and applied research (e.g., preclinical drug testing, disease modeling, tissue engineering and gastroenterology). Since our protocol is reproducible and robust to induce 3D morphogenesis of an intestinal epithelium in vitro, we envision that our technical strategy can be disseminated to audiences who study the dynamics of cellular signaling during intestinal development, regeneration or homeostasis. Also, our protocol can be useful for audiences who interrogate mechanisms of disease pathology and pathogenesis under various infectious agents such as norovirus8, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), Clostridioides difficile, Salmonella enterica Typhimurium9 or Vibrio cholerae. The use of the gut-on-a-chip microphysiological system can potentially allow a longitudinal co-culture10 and subsequent assessments of host defense, immune response and pathogen-associated damage repair in the gastrointestinal (GI) tract11. Other GI diseases that involve villous atrophy, crypt shortening, mucosal damage or impaired epithelial barrier germane to leaky gut syndrome, celiac disease, Crohn’s disease, ulcerative colitis, pouchitis or irritable bowel syndrome can be emulated when a 3D intestinal epithelial layer is prepared using patient’s biopsy- or stem-cell-derived intestinal organoids12,13. To better mimic a higher complexity of a disease milieu, readers can contemplate adding disease-relevant cell types such as tissue-specific immune cells derived from a patient’s peripheral blood mononuclear cells (PBMCs) in the model that contains 3D intestinal villus–crypt microarchitecture3,5.
Since the 3D epithelial microstructure can be fixed and visualized without a sectioning process, audiences who work on spatial transcriptomics and high- or super-resolution imaging may potentially be interested in our technique to map the spatiotemporal dynamics of genes and proteins on the epithelial niche in response to microbial or immune stimulations. In addition, longitudinal host–microbiome crosstalk that orchestrates intestinal homeostasis can be established in a 3D intestinal mucosal layer10,14, especially in the gut-on-a-chip platform, by co-culturing various microbial species, microbial communities or fecal microbiota. This approach will be particularly compelling to audiences who study mucosal immunology, gastroenterology, human microbiome, culturomics with an effort to culture previously uncultured gut microbiota in laboratories, and clinical microbiology. If our in vitro morphogenesis protocol can be adapted to a scalable culture format such as multiwell porous inserts in a 24-, 96- or 384-well plate that can continuously replenish the basolateral compartment, the protocol can be also disseminated to audiences who develop a high-throughput screening or a validation platform in the pharmaceutical, biomedical or food industries. As a proof-of-principle, we recently demonstrated the feasibility of a multiplex high-throughput morphogenesis system scalable to a 24-well plate format15. In addition, multiple organ-on-a-chip products have been commercialized16,17,18. Hence, the validation of our in vitro morphogenesis method can be accelerated and potentially adopted in many research laboratories, industries or government and regulatory agencies to understand cellular reprogramming of in vitro intestinal morphogenesis at the transcriptome level19, to test absorption and transport of pharmaceutical or biotherapeutic candidates using the 3D gut surrogates or to assess reproducibility of the intestinal morphogenic process using customized or commercialized organ-on-a-chip models.
Comparison with other methods
A limited number of human-relevant experimental models have been utilized to study intestinal epithelial morphogenesis, mainly because of a lack of implementable protocol to induce in vitro 3D morphogenesis. Indeed, a majority of the current knowledge on intestinal morphogenesis is based on animal studies (e.g., zebrafish20, mouse21 or chicken22). However, they are labor and cost intensive, may be ethically questionable and, most importantly, do not accurately dictate the developmental process in humans. These models are also substantially limited for testing in a multiplex scalable way. Thus, our protocol to regenerate a 3D tissue architecture in vitro is advantageous over in vivo animal models as well as other conventional static 2D cell culture models. As previously mentioned, utilizing a 3D epithelial structure allows us to examine the spatial localization of differentiated cells in a crypt–villus axis in response to various mucosal or immune stimulations. The 3D epithelial layer can provide a space to investigate how microbial cells compete to make a spatial niche and ecologically evolve in response to host factors (e.g., inner versus outer mucus layers, secretion of IgA and antimicrobial peptides). Furthermore, 3D epithelial morphology may allow us to understand how the gut microbiota structures their communities and synergistically produces microbial metabolites (e.g., short-chain fatty acids) that shape the cellular organization and stem cell niche in the basal crypt. These features can only be demonstrated when 3D epithelial layers are established in vitro.
There are a couple of in vitro approaches besides our method to create 3D intestinal epithelial structures. The intestinal organoid culture is the most advanced tissue engineering technique based on an intestinal stem cell culture under defined morphogen conditioning23,24,25. However, it is often challenging to use the 3D organoid models for running transport assays or host–microbiome co-cultures because the intestinal lumen is enclosed inside an organoid body, and thus, introduction of luminal components such as microbial cells or exogenous antigens is limited. Using a microinjector can improve the accessibility to the organoid lumen26,27, but this method is invasive and labor intensive and requires expertise to execute. Furthermore, the conventional organoid culture is maintained under a static condition in a hydrogel scaffold, which does not accurately reflect active in vivo biomechanics.
Other approaches that several research groups have adopted leverage a prestructured 3D hydrogel scaffold to mimic the intestinal epithelial architecture by culturing dissociated human intestinal cells on the gel surface28,29,30. Hydrogel scaffolds were fabricated using 3D printed, micromilled or lithographically made molds. This method has shown a self-organized alignment of dissociated epithelial cells with a physiologically relevant morphogen gradient in vitro, establishment of a high-aspect ratio epithelial structure and stromal–epithelial crosstalk by including stromal cells in the scaffold. However, the nature of prestructured scaffolds may preclude demonstrating a spontaneous morphogenesis process per se. These models also do not offer dynamic luminal or interstitial flow, lacking fluid shear stress that intestinal cells need to undergo morphogenesis and gain physiological functions. Another recent study used a hydrogel scaffold in a microfluidic platform and patterned intestinal epithelial structure using a laser etching technology31. Mouse intestinal organoids formed an intestinal tubular structure following the etched pattern, and a luminal fluid flow can be recapitulated using a microfluidic module. However, this model also neither demonstrates a spontaneous morphogenesis process nor includes intestinal mechanobiological motions. The same group’s 3D printing technology enabled an establishment of a mini-intestinal tube with a spontaneous morphogenesis process32. Although this study sophisticatedly fabricated different segments of the gut within a tube, the model also lacks luminal fluid flow and mechanical deformations. Furthermore, the manipulability of the model may be limited, especially to perturb experimental conditions or intercellular interactions once the bioprinting process is completed. On the contrary, our proposed protocol offers spontaneous intestinal morphogenesis, physiologically relevant shear stress, biomechanics emulating intestinal peristalsis, accessibility of apical and basolateral compartments independently, and modularity for re-creating a complex biological microenvironment. Hence, our protocol for in vitro 3D morphogenesis may provide a complementary approach to overcome the challenges of the existing methods.
Limitations
Our protocol focuses entirely on the 3D epithelial morphogenesis, having only the epithelial cells in cultures without other types of surrounding cells such as mesenchymal, endothelial and immune cells. As stated earlier, the core of our protocol is to induce epithelial morphogenesis by removing morphogen inhibitors secreted in the basolateral side in the introduced medium4. While the strong modularity of our gut-on-a-chip and a hybrid chip allows us to re-create undulated 3D epithelial layers, additional biological complexity such as epithelial–mesenchymal interactions33,34, extracellular matrix (ECM) deposition for 3D regeneration35 and crypt–villus characteristics that convey a stem cell niche in the basal crypt36 remains to be further considered in our model. Stromal cells in the mesenchyme (e.g., fibroblasts) play a critical role in producing ECM proteins and regulating intestinal morphogenesis in vivo35,37,38. Inclusion of mesenchymal cells in our model may enhance the morphogenesis process and cell attachment efficiency. Endothelial layers (i.e., capillary vasculatures or lymphatic vessels) play seminal roles in regulating molecular transport39 and immune cell recruitment40 in the intestinal microenvironment. Also, when tissue models aim to demonstrate multiorgan interactions, the vasculature component that can connect between the tissue models is a prerequisite. Therefore, including endothelial cells may be required for simulating more accurate physiological features with organ-level resolutions. In the case of modeling intestinal diseases, patient-derived immune cells are also essential for demonstrating innate immune responses, antigen presentation, innate-adaptive immune crosstalk and tissue-specific immunity.
The use of a hybrid chip is more straightforward than that of a gut-on-a-chip because the device setup is simpler and the use of a Transwell insert allows scalable cultures of intestinal epithelium. However, the commercially available Transwell insert with a polyester membrane is not elastic to emulate intestinal peristalsis-like movements. Furthermore, the apical compartment of the Transwell insert placed in a hybrid chip remains static, lacking shear stress on the apical side. Obviously, a static nature in the apical compartment is seldom capable of long-term bacterial co-cultures in a hybrid chip. Although we can robustly induce 3D morphogenesis in a Transwell insert when using a hybrid chip, a shortage of physiologically relevant biomechanics and apical fluid flow may restrict the implementability of the hybrid chip platform for potential applications.
In both gut-on-a-chip and hybrid chip cultures, the full-size re-creation of the human crypt–villus axis has not been fully established. Since the morphogenesis is initiated from an epithelial monolayer, the 3D microarchitecture does not necessarily provide morphological similarity with an in vivo crypt. Although we characterized the population of proliferative cells near the basal crypt domain in the microengineered 3D epithelium2, the crypt and villus regions are not clearly compartmentalized. Although a taller upper channel of a gut-on-a-chip induces an increased height of a microengineered epithelium4, the maximum height is still limited to ~300–400 µm. The actual depth of human intestinal crypts in vivo in the small and large intestines is ~135 µm and ~400 µm in depth, respectively, and the height of a small intestinal villus is ~600 µm41.
From an imaging standpoint, in situ super-resolution imaging of the 3D microarchitecture may be limited in a gut-on-a-chip because the required working distance from an objective to the epithelial layer is on the order of a couple of millimeters. To overcome this issue, a long-distance objective may be required. Furthermore, because of the high elasticity of PDMS, it is challenging to make a thin section for imaging specimen preparation. In addition, since the layer-by-layer microfabrication of a gut-on-a-chip involves a permanent bonding between each layer, it is extremely challenging to open or remove the upper layer to examine the surface structure of the epithelial layers, for example, by using scanning electron microscopy (SEM).
Hydrophobicity of PDMS has been a limiting factor in microfluidics-based studies dealing with hydrophobic small molecules because PDMS can nonspecifically adsorb such hydrophobic molecules. Other polymeric materials may be considered to substitute PDMS. Alternatively, surface modification of PDMS (e.g., coating with lipophilic material42 or poly(ethylene glycol)43) may be contemplated to minimize the adsorption of hydrophobic molecules.
Finally, our approach has not been fully characterized for providing a high-throughput screening or a ‘one-fit-to-all’-type user-friendly experimental platform. The current protocol requires one syringe pump per each microdevice, which takes up space in a CO2 incubator, preventing large-scale experiments. This limitation may be substantially improved by innovating the scalability of the culture format (e.g., 24-, 96- or 384-well porous inserts that allow the continuous replenishment and removal of basolateral medium).
Experimental design
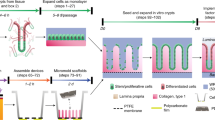
To induce 3D morphogenesis of human intestinal epithelium in vitro, we use a microfluidic gut-on-a-chip device that contains two parallel microchannels and an elastic porous membrane in the middle to create a lumen–capillary interface. We also demonstrate the use of a single channel microfluidic device, a hybrid chip, that can offer continuous basolateral flow below a polarized epithelial layer grown on a Transwell insert. In both of the platforms, morphogenesis of various human intestinal epithelial cells can be demonstrated by applying a directional manipulation of flow to remove morphogen antagonists from the basolateral compartment. The overall experimental procedure (Fig. 1) is composed of five parts: (i) microfabrication of a gut-on-a-chip or a Transwell-insertable hybrid chip (Steps 1–5; Box 1), (ii) preparation of intestinal epithelial cells (either Caco-2 cells or human intestinal organoids; Boxes 2–5), (iii) culture of intestinal epithelial cells on a gut-on-a-chip or a hybrid chip (Steps 6–9), (iv) induction of in vitro 3D morphogenesis (Step 10) and (v) characterization of 3D epithelial microarchitectures (Steps 11–24). Finally, appropriate control groups (discussed further below) are designed to verify the validity of in vitro morphogenesis by comparing the epithelial morphogenesis with spatial, temporal, conditional or procedural controls.

We use two different culture platforms: a gut-on-a-chip either with a straight channel or a nonlinear convoluted channel, or a hybrid chip that includes a Transwell (TW) insert in a microfluidic device, fabricated as described in Box 1 and Steps 1–5. ‘Device fabrication’ displays the major steps for making either a gut-on-a-chip or a hybrid chip. ‘Culture of human intestinal epithelium’ explains the cell sources used in this protocol (Caco-2 or human intestinal organoids) and the culture process. ‘In vitro morphogenesis’ shows the overall steps of Caco-2 or organoid-derived epithelial cell culture on a gut-on-a-chip or on the Transwell insert of a hybrid chip followed by the induction of 3D morphogenesis and characterization of the formed epithelial structure. Procedure step numbers or box numbers are indicated below each arrow. Applications provides examples that the established intestinal epithelial layers can be utilized for, such as characterization of cytodifferentiation, study of intestinal physiology, establishment of a host–microbiome ecosystem and disease modeling. An immunofluorescence image in ‘Cytodifferentiation’ shows nuclei, F-actin and MUC2 expressed in a 3D Caco-2 epithelial layer produced on a gut-on-a-chip. The MUC2 signal is found in both the goblet-like cells and the secreted mucus on the mucosal surface. A fluorescence image in ‘Intestinal physiology’ displays the production of mucus by staining sialic acid and N-acetylglucosaminyl residues using fluorescent wheat germ agglutinin. Two overlaid images in ‘Host–microbe co-culture’ show the representative host-microbiome co-culture in a gut-on-a-chip. The left image shows the co-culture of green fluorescence protein (GFP)-expressing Escherichia coli with a microengineered 3D Caco-2 epithelium. The right image visualizes the localization of GFP E. coli co-cultured with 3D Caco-2 epithelium followed by the immunofluorescence staining with F-actin (red) and nuclei (blue). Disease modeling illustrates a demonstration of healthy versus leaky gut in a gut inflammation-on-a-chip under physiological challenges by a bacterial antigen (e.g., lipopolysaccharide, LPS) and immune cells (e.g., PBMCs; green). Caco-2 cells were cultured to establish the 3D epithelial layers. Scale bars, 50 µm. Images in bottom row: ‘Cytodifferentiation’ adapted with permission from ref. 2, Oxford University Press; ‘Intestinal physiology’ reproduced with permission from ref. 5, NAS; ‘Host–microbe co-culture’ adapted with permission from ref. 3, NAS; ‘Disease modeling’ adapted with permission from ref. 5, NAS.
Microfabrication of a gut-on-a-chip or a Transwell-insertable hybrid chip (Box 1 and Steps 1–5)
Both the gut-on-a-chip and the hybrid chip are fabricated using PDMS replicas demolded from silicon molds patterned with SU-8 by soft lithography1,44. The design of microchannels in each chip was determined by considering fluid dynamics such as shear stress and hydrodynamic pressure1,4,12. The original design of the gut-on-a-chip (Extended Data Fig. 1a) that contains two parallel straight-lined microchannels apposed to each other has evolved to a convoluted gut-on-a-chip (Extended Data Fig. 1b) that includes a pair of curved microchannels to induce increased fluid residence time, nonlinear flow pattern and the multiaxial deformation of cultured cells (Fig. 2a–f)12. The convoluted gut-on-a-chip may be chosen when more complex intestinal biomechanics need to be reconstituted. We have verified that the convoluted gut-on-a-chip also robustly induces 3D morphogenesis at a similar extent of epithelial growth in a similar time frame regardless of the cultured cell types compared with the original gut-on-a-chip12. For the purpose of inducing 3D morphogenesis, thus, the linear and the convoluted gut-on-a-chip designs are interchangeable. The PDMS replica cured on the silicon molds with SU-8 patterns provides a negative feature once they are demolded (Fig. 2a). To fabricate a gut-on-a-chip, a prepared upper PDMS layer is sequentially bonded to a porous PDMS membrane, then subsequently aligned to the lower PDMS layer by performing irreversible bonding using a corona treater (Fig. 2b–f). To fabricate a hybrid chip, a cured PDMS replica is bonded to a glass slide to establish a single-channel microfluidic device that can hold a Transwell insert (Fig. 2h and Extended Data Fig. 2). The bonding process is performed by treating the surface of the PDMS replica and the glass with oxygen plasma or corona treatment. After sterilization of a microfabricated device connected to silicone tubing, the device setup is ready to use for performing 3D morphogenesis of intestinal epithelium (Fig. 2g).

a, A schematic to prepare a PDMS part from a SU-8-patterned silicon mold. Uncured PDMS solution is poured on a silicon mold (left), cured at 60 °C (middle) and demolded (right). The demolded PDMS is cut into several pieces and cleaned for further use. b, A photograph of a silicon mold for preparing an upper PDMS layer. c, A photograph of a silicon mold for fabricating PDMS porous membranes. d, A series of photos of upper and lower PDMS parts as well as an assembled gut-on-a-chip device. e, A schematic diagram of the alignment of the upper, membrane and lower PDMS parts. Each layer is irreversibly bonded by either plasma or corona treatment. f, A schematic of a fabricated gut-on-a-chip device that has superimposed convoluted microchannels and vacuum chambers. g, Setup of a gut-on-a-chip for microfluidic cell culture. The fabricated gut-on-a-chip assembled with silicone tubing and syringes is placed on a cover slip. The chip setup is placed on a lid of a 150 mm Petri dish for handling. Binder clips are used to close the silicone tubing. h, A visual snapshot of the fabrication of a hybrid chip and the use of a hybrid chip for 3D morphogenesis. A Transwell insert that is independently prepared to culture a 2D monolayer of intestinal epithelial cells is inserted into a hybrid chip to induce intestinal 3D morphogenesis. Culture medium is perfused through the microchannel underneath the cell layer established on the Transwell insert. Scale bars, 1 cm. h reproduced with permission from ref. 4, Elsevier.
Preparation of intestinal epithelial cells (Boxes 2–5 and Step 8)
In this protocol, Caco-2 cell line and intestinal organoids are used as epithelial sources (Fig. 3a). Both types of cells are independently cultured (Boxes 2 and 5) and used for seeding in an ECM-coated microchannel of a gut-on-a-chip or on a Transwell insert. Caco-2 cells (passage number between 10 and 50) routinely cultured in a T flask are harvested when the cells are confluent (>95% coverage in a flask) to prepare a dissociated cell suspension by trypsinization (Box 2). Human intestinal organoids derived from intestinal biopsies or surgical resections are cultured in a dome of Matrigel scaffold plated in a 24-well plate to support the structural microenvironment. Culture medium that contains essential morphogens (e.g., Wnt, R-spondin and Noggin) and growth factors, prepared as described in Box 3, is replenished every other day until the organoids grow up to ~500 µm in diameter. Fully grown organoids are harvested and dissociated into single cells for a seeding into a gut-on-a-chip or on a Transwell insert (Box 5). As we previously reported, diverse intestinal organoid lines can be established and used depending on the disease type12,13 (e.g., ulcerative colitis, Crohn’s disease, colorectal cancer or normal donors), the site of lesion (e.g., diseased versus nondiseased region) and the location in the GI tract (e.g., duodenum, jejunum, ileum, cecum, colon or rectum). We provide an optimized protocol in Box 5 for culturing colonic organoids (colonoids) that typically require a higher concentration of morphogens than small intestinal organoids.

a, A workflow to induce intestinal morphogenesis in a gut-on-a-chip. Both Caco-2 human intestinal epithelium and intestinal organoids are used in this protocol to demonstrate 3D morphogenesis. The dissociated epithelial cells are seeded in a prepared gut-on-a-chip device (Chip prep). Once the cells are seeded (Seeding) and attached (Attachment) on a PDMS porous membrane on day 0 (D0), apical (AP) flow is initiated and maintained for the first 2 d (Flow, AP, D0–D2). When an intact 2D monolayer is formed, basolateral (BL) flow is also initiated along with a cyclic stretching motion (Stretching, Flow, AP and BL). Intestinal 3D morphogenesis spontaneously occurs after 5 d of microfluidic culture (Morphogenesis, D5). Phase-contrast images show representative morphologies of Caco-2 cells at each experimental step or timepoint (Bar, 100 µm). Four schematics illustrate the corresponding cascade of intestinal morphogenesis (right top). Dashed arrows in the schematics indicate the direction of fluid flow. b, An SEM image shows the surface topology of the established 3D Caco-2 epithelium (left). An inset that highlights a zoomed-in area (a white dashed box) shows the microvilli regenerated on the 3D Caco-2 layer (right). c, Immunofluorescence confocal visualization of the tight junction protein (ZO-1, red) and the continuous brush-border membrane labeled for F-actin (green) and nuclei (blue) in a horizontal en face view of an established Caco-2 3D epithelium in a gut-on-a-chip. Arrows directed to the schematic image in the middle indicate the location of the focal plane of each confocal view. d, A time course of morphological changes of an organoid epithelium cultured in a gut-on-a-chip acquired by a phase-contrast microscope on days 3, 7, 9, 11 and 13. The insets (right top) show a high magnification of the provided images. e, A DIC micrograph of an organoid 3D epithelium established in a gut-on-a-chip taken on day 7. f, Overlaid immunofluorescence images showing the markers of stem cells (LGR5; magenta), goblet cells (MUC2; green), F-actin (gray) and nuclei (cyan) on the organoid epithelial layers grown in gut-on-a-chips for 3 (left) and 13 d (middle), respectively. See also Extended Data Fig. 3, which highlights LGR5 signal without MUC2 signal. A fluorescent image shows the epithelial microstructure of a 3D organoid epithelium established in a gut-on-a-chip visualized by staining the plasma membrane using a CellMask dye at day 13 of the culture (right). Scale bars 50 µm, unless otherwise indicated. b reproduced with permission from ref. 2, Oxford University Press; c adapted with permission from ref. 2, Oxford University Press; e and f adapted with permission from ref. 12 under a Creative Commons licence CC BY 4.0.
Culture of intestinal epithelial cells on a gut-on-a-chip or on the Transwell insert of a hybrid chip (Steps 6–9)
In the gut-on-a-chip, it is necessary to modify the hydrophobic surface of the PDMS porous membrane for a successful ECM coating. In this protocol, we apply two different methods to modify the PDMS membrane’s hydrophobicity. For culturing Caco-2 cells, surface activation via UV/ozone treatment alone is enough to reduce hydrophobicity of the PDMS surface, coat the ECM and attach Caco-2 cells on the PDMS membrane. However, the microfluidic culture of organoid epithelium requires chemical-based surface functionalization to achieve efficient deposition of ECM proteins by applying polyethylenimine (PEI) and glutaraldehyde, sequentially, to the PDMS microchannels13. After the surface modification, ECM proteins are deposited to coat the functionalized PDMS surface followed by the introduction of dissociated organoid epithelium. After cell attachment, microfluidic cell culture begins by perfusing culture medium only to the upper microchannel until the cells form an intact monolayer while the lower microchannel maintains static conditions. This optimized approach for surface activation and ECM coating enables the attachment of organoid epithelium to induce 3D morphogenesis on the PDMS surface13.
Transwell cultures also require ECM coating prior to cell seeding; however, Transwell cultures do not require complex pretreatment steps for activating the surface of a porous insert. For growing Caco-2 cells on a Transwell insert, an ECM coating on the porous insert accelerates the attachment of dissociated Caco-2 cells (<1 h) and the formation of tight junction barrier1 (<1–2 d). For culturing organoid cells on a Transwell insert, dissociated organoid cells are seeded on the ECM-coated insert, attached on the membrane surface (<3 h) and maintained until the organoid cells form an intact monolayer with barrier integrity. The Transwell culture is performed in a 24-well plate, and the hybrid chip is not used at this point.
Induction of in vitro 3D morphogenesis (Step 10)
The in vitro 3D morphogenesis can be initiated by applying a fluid flow on the basolateral side of the established epithelial layer. In the gut-on-a-chip, epithelial morphogenesis begins when the culture medium is perfused into both the upper and the lower microchannels (Fig. 3a). As previously described4, it is critical to introduce fluid flow in the lower (basolateral) compartment to continuously remove the directionally secreted morphogen inhibitors. To supply sufficient nutrients and serum to the cells bound on the porous membrane as well as to create a luminal shear stress, we typically apply dual flow in a gut-on-a-chip. In the hybrid chip, a Transwell insert that contains an epithelial monolayer is inserted into a hybrid chip. Then, the culture medium is applied through the microchannel, beneath the basolateral side of the porous Transwell insert. Intestinal morphogenesis occurs in 3–5 d after the basolateral flow is initiated in both culture platforms.
Characterization of 3D epithelial microarchitectures (Steps 11–24)
The morphological characteristics of microengineered 3D epithelial layers can be analyzed by applying various imaging modalities including phase-contrast microscopy, differential interference contrast (DIC) microscopy, SEM or immunofluorescence confocal microscopy (Figs. 3 and 4). Phase-contrast or DIC imaging can be readily performed at any time during the culture to monitor the shape and the protrusion of 3D epithelial layers. Thanks to the optical transparency of PDMS and the polyester membrane, both the gut-on-a-chip and hybrid chip platforms offer real-time in situ imaging without sectioning or disassembly of the device. When immunofluorescence imaging is performed (Figs. 1, 3c,f and 4b,c), cells are generally fixed with 4% (wt/vol) paraformaldehyde (PFA), then permeabilized and blocked with Triton X-100 and 2% (wt/vol) bovine serum albumin (BSA), sequentially. Depending on the cell type, different fixatives, permeabilizing reagents and blocking reagents may be used. Primary antibodies targeting lineage-dependent cells or regional markers are used for highlighting the cells fixed in situ in the chips, followed by treatment with secondary antibodies as well as counterstaining dyes that target either nucleus (e.g., 4′,6-diamidino-2-phenylindole, DAPI) or F-actin (e.g., fluorescence-labeled phalloidin). Fluorescence-based real-time imaging can be also performed in situ to detect mucus production (Fig. 1, ‘Cytodifferentiation’ and ‘Intestinal physiology’), stochastic colonization of microbial cells (Fig. 1, ‘Host–microbe co-culture’), recruitment of immune cells (Fig. 1, ‘Disease modeling’) or the contours of the 3D epithelial morphology (Figs. 3c,f and 4b,c). When the gut-on-a-chip is modified to separate the upper layer from the lower microchannel layer as described in ref. 2, 3D epithelial morphology as well as the microvilli on the apical brush border can be visualized by SEM (Fig. 3b). Expression of differentiation markers may be assessed by performing quantitative PCR5 or single-cell RNA sequencing19. In this case, epithelial cells of a 3D layer grown in either a gut-on-a-chip or a hybrid chip are harvested by trypsinization, then used for molecular or genetic analyses.

a, A workflow to induce intestinal morphogenesis in a hybrid chip. Both Caco-2 and intestinal organoids are used in this protocol to demonstrate 3D morphogenesis in a hybrid chip platform. The dissociated epithelial cells are seeded in a prepared Transwell insert (TW prep; see the schematic below). Once the cells are seeded (Seeding) and attached on a polyester membrane in a Transwell insert, all the cells are cultured under static conditions (TW culture). After 7 d, an individual Transwell insert that contains a 2D monolayer of epithelial cells is incorporated into a hybrid chip to introduce basolateral flow (Flow, BL) that eventually leads to a generation of a 3D epithelial layer (Morphogenesis). Phase-contrast micrographs show the morphological profile of human organoid epithelial cells derived from the ascending colon of a normal donor (C103 line) at each experimental step or timepoint. Schematics in the upper layer illustrate the experimental configuration at each step. b, The hybrid chip (left schematic) can lead to the 3D morphogenesis of organoid epithelial cells, where the top-down confocal microscopic views taken at different Z-positions (upper, middle and lower; see the right schematic with corresponding dashed lines) show distinct morphological characteristics. F-actin (cyan), nuclei (gray). c, Fluorescence confocal micrographs (3D angled views) of the organoid-derived epithelial cells cultured in a static Transwell (TW; an inset inside a white dashed box) versus a hybrid chip (the largest full shot) comparing the 2D versus 3D morphologies, respectively. A pair of 2D vertical cross-cut views (insets on the right top; ‘XZ’) also display 2D versus 3D characteristics. Scale bars, 100 µm. c reproduced with permission from ref. 4, Elsevier.
Preparation of the control groups
Control groups can be prepared by culturing the same cells (either Caco-2 or intestinal organoid epithelium) into a 2D monolayer in conventional static culture conditions. It is noted that it may be challenging to maintain static conditions in a gut-on-a-chip microdevice for a long time because the limited volume capacity of the microchannels (i.e., ~4 µL in the top channel of the original gut-on-a-chip design) may cause nutrient depletion. Thus, epithelial morphologies before and after applying basolateral flow can be also compared.
Materials
Biological materials
Caution
The cell lines used in your study should be regularly checked to ensure that they are authentic and are not infected with mycoplasma.
-
Caco-2 human colon cancer cells (Harvard Digestive Diseases Center, RRID: CVCL_1096)
-
Human intestinal organoids
Caution
Human intestinal organoids are derived from clinical samples such as biopsies or resected tissue pieces, as described in Box 4. The collection of human clinical samples should be regulated by the human subject research guidelines approved by the relevant institutional review board. In addition, biopsies or surgical resections may contain mucosal bacteria that require excessive washing with antibiotic-containing solutions. Make sure to add Primocin (final concentration 100 µg/mL) in all solutions used to establish germ-free organoid lines.
-
Cultrex R-spondin1-producing 293T cells (Trevigen, cat. no. 3710-001-K; RRID: CVCL_RU08)
-
Noggin-producing 293T cells (Digestive Diseases Center, Baylor College of Medicine)
Reagents
Microfabrication
-
Photomasks (CAD/Art Services, custom-made)
Critical
Use computer-aided design (CAD) software such as AutoCAD to design photomasks. See Extended Data Fig. 1a,b,d for the designs of the gut-on-a-chip and Extended Data Fig. 2 for the design of the hybrid chip. When designing photomasks, consider having enough space between patterned features; place a maximum of four device features per mask with a 3 inch wafer. For instance, one 3 inch silicon wafer may hold up to four patterns of either an upper or a lower layer of the gut-on-a-chip (Fig. 2a,b) or two features of the hybrid chip. Request a negative polarity printing (i.e., black background with clear features) at a resolution of 20,000 dots per inch (dpi).
-
Photoresist SU-8 2100 (Microchem, cat. no. Y111075 0500L 1GL)
Caution
SU-8 is combustible and can cause irritation on skin. Take safety precautions when handling the material.
-
Silicon wafers (100 mm SSP Si Wafer Mechanical Grade; University Wafer Product, ID 1196)
-
Sulfuric acid (Sigma-Aldrich, cat. no. 258105)
Caution
Sulfuric acid is corrosive and can cause severe skin burns and eye damage. Take safety precautions when handling the material.
-
Hydrogen peroxide (Fisher Chemical, cat. no. H312)
Caution
Hydrogen peroxide is a strong oxidizer and can cause severe skin burns and eye damage. Take safety precautions when handling the material.
-
SU-8 photoresist developer (polypylene glycol monomethyl ether acetate; Microchem, cat. no. Y020100 4000L 1PE)
Caution
SU-8 developer is flammable and may damage fertility or the unborn child. Take safety precautions when handling the material.
-
Isopropyl alcohol (IPA; Acros Organics, cat. no. 149320025)
Caution
IPA is flammable and may cause irritation. Take safety precautions when handling the material.
-
Acetone (Acros Organics, cat. no. 177170025)
Caution
Acetone is flammable and causes eye and respiratory tract irritation. Take safety precautions when handling the material.
-
Trichloro(1H,1H,2H,2H-perfluorooctyl)silane (Sigma-Aldrich, cat. no. 448931)
Caution
Take safety precautions when handling the material because it is combustible and causes severe skin burns and eye damage.
-
Silicon mold prepatterned with an array of etched pillars for the porous membrane of the gut-on-a-chip (Applied Novel Devices, custom-made)
Critical
Please see Extended Data Fig. 1d for the dimension of patterns. The diameter of pillars is 10 µm, center-to-center distance between pillars is 25 µm and the height of pillars is 20 µm. The suggested size of a silicon mold is ~5 cm × 5 cm (Fig. 2c). However, if the silicon mold fits into a 3″ Petri dish, any size that once cut up can cover the area of a device is feasible for making a porous membrane.
-
Sylgard 184 silicone elastomer kit, PDMS and curing agent (Dow Corning, cat. no. 4019862)
-
Ethyl alcohol (EtOH), 200 proof (Fisher Scientific, cat. no. BP2818500)
Caution
Take safety precautions when handling the material because it is flammable and causes serious eye irritation and damage to organs.
-
70% (vol/vol) EtOH (LabChem, cat. no. LC22210)
General cell culture
-
Sterile deionized water (DIW)
-
Trypan blue, 0.4% (wt/vol) (VWR International, cat. no. VWRVK940-100ML)
Caco-2 and 293T cell culture
-
Dulbecco’s modified Eagle medium (DMEM; Gibco, cat. no. 10564029)
-
Fetal bovine serum (FBS), heat-inactivated (Gibco, cat. no. 10438034)
-
100× penicillin–streptomycin (Gibco, cat. no. 10378016)
-
Zeocin (Thermo Fisher Scientific, cat. no. R25005)
-
Puromycin dihydrochloride (Gibco, cat. no. A1113803)
-
0.25% trypsin/1 mM ethylenediaminetetraacetic acid (EDTA) (Thermo Fisher Scientific, cat. no. 25200072)
-
Phosphate-buffered saline (PBS), pH 7.4, Ca2+- Mg2+-free (Thermo Fisher Scientific, cat. no. 14190250)
Organoid culture
-
0.5 M EDTA (VWR International LLC, cat. no. 97062-836)
-
Matrigel, growth factor reduced (83% protein that gels; Corning, cat. no. 356234)
-
Advanced DMEM/F12 (Gibco, cat. no. 12634028)
-
Primocin, 50 mg/mL (Invivogen, cat. no. ant-pm-1)
-
GlutaMAX, 100× (Gibco, cat. no. 35050061)
-
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 1 M (HEPES; Gibco, cat. no. 15630080)
-
N-2 MAX Media Supplement, 100× (R&D Systems, cat. no. AR009)
-
B-27 Supplement, 50× (Gibco, cat. no. 17504044)
-
Nicotinamide (Sigma Aldrich, cat. no. N0636)
-
N-acetylcysteine (MP Biomedicals, cat. no. 0219460305)
-
Wnt Surrogate-Fc Fusion protein (U-Protein Express, cat. no. N001-0.5 mg)
-
Mouse epidermal growth factor (EGF) recombinant protein (PeproTech, cat. no. 315-09)
-
SB202190 (Sigma Aldrich, cat. no. S7067)
-
A-8301 (Sigma Aldrich, cat. no. SML0788)
-
[Leu15]-Gastrin I human (Sigma Aldrich, cat. no. G9145)
-
Y-27632, dihydrochloride (STEMCELL Technologies, cat. no. 72302)
-
Dimethyl sulfoxide (DMSO), sterile-filtered (Sigma Aldrich, cat. no. D2650)
Caution
Take safety precautions when handling the material because it is combustible liquid.
-
Cell Recovery solution (Corning, cat. no. 354253)
-
TrypLE Express (Gibco, cat. no. 12605028)
On-chip culture
-
Rat-tail collagen Type I, 3 mg/mL (Gibco, cat. no. A1048301)
-
Steriflip-HV, 0.45 µm, polyvinylidene fluoride, radio-sterilized (MilliporeSigma, cat. no. Se1M003M00)
-
PEI solution, average molecular weight ~2,000 g/mol, 50 weight % in H2O (Sigma, cat. no. 408700)
-
Glutaraldehyde, 50% (wt/vol) aqueous solution, electron microscopy grade (Electron Microscopy Sciences, cat. no. 16320)
Caution
Take safety precautions when handling the material because it causes eye and skin burns.
Immunofluorescence imaging
-
4% (wt/vol) PFA solution, diluted in PBS (Alfa Aesar, cat. no. J19943-K2)
Caution
Take safety precautions when handling the material because it may cause allergenic skin reaction and serious eye damage.
-
Triton X-100 (Thermo Fisher Scientific, cat. no. 85111)
-
BSA (MP Biomedicals, cat. no. 08810683)
-
Phalloidin CruzFluor 647 conjugate (Santa Cruz Biotechnology, cat. no. sc-363797)
-
Rabbit anti-ZO-1 polyclonal antibody (Thermo Fisher Scientific, cat. no. 61-7300; RRID: AB_2533938)
-
Rabbit anti-LGR5 polyclonal antibody (Abcam, cat. no. ab75732; RRID: AB_1310281)
-
Mouse anti-MUC2 monoclonal antibody (Santa Cruz Biotechnology, cat. no. sc-515032; RRID: AB_2815005)
-
Goat anti-mouse IgG DyLight 488 (Abcam, cat. no. ab96871; RRID: AB_10680543)
-
Goat anti-rabbit IgG Alexa Fluor 555 (Abcam, cat. no. ab150078; RRID: AB_2722519)
-
DAPI solution, 1 mg/mL (Thermo Fisher Scientific, cat. no. 62248)
-
CellMask Deep Red Plasma membrane stain (Thermo Fisher Scientific, cat. no. c10046)
Equipment
-
Glass dishes (PYREX, cat. no. 3140190)
-
Disposable plastic cups (Dixie, 12 Oz, cat. no. CP12DX)
-
Fluoropolymer coated polyester film (3M Scotchpak Release Liners, product no. 9744)
-
Scalpel blades, #11 (Fine Science Tools, cat. no. 1001100)
-
Scalpel handles, #3 (Defender, cat. no. 10604)
-
Razor blades (Fisher Scientific, cat. no. 12-640)
-
Swiss wafer style tweezers, 120 mm length (SPI SUPPLIES, Item No. S2WFG-XD)
-
Tweezers, stainless steel (SPI SUPPLIES, Item No. W2ANCB-XD)
-
150 mm Petri dishes (Thermo Scientific, cat. no. 168381)
-
Biopsy puncher, diameter 2 mm (GE Healthcare, cat. no. WB100029)
-
Biopsy puncher, diameter 8 mm (Med Vet International, cat. no. BP8MM)
-
One-side frosted glass slides (Corning, cat. no. 2948-75×25)
-
Glass slides, 75 × 50 mm (Corning, cat. no. 2947-75×50)
-
Coverslips, No. 1, 60 × 48 mm (Epredia, cat. no. 48×60-1-002G)
-
Silicone tubing, 0.8 mm inner diameter, 2.4 mm outer diameter (Saint Gobain Performance Plastics, cat. no. ABW00001)
-
Y-connectors, 1.57 mm inner diameter (Eldon James, cat. no. Y0-1NN)
-
Blunt-end needles, 18 gauge, 1 inch long (Shintop, cat. no. JJ-ZT001-100-LVS)
-
5 min epoxy glue (Devcon, cat. no. 14250)
-
Binder clips, width 12.7 mm (Universal, cat. no. UNV10199)
-
1 mL syringes, slip tip (BD Biosciences, cat. no. 309659)
-
Vacuum desiccator No. 5, plate diameter 23 cm (Bel-Art, cat. no. F42025-0000)
-
Scissors, 20 cm (Office Depot, cat. no. 458612)
-
Scotch Magic Invisible tape, 1.9 cm × 22 m (3M, cat. no. 452913)
-
Lead balls, #7.5, diameter 2.39 mm (RotoMetals, part no. EagleShot7new)
-
Tin containers for lead balls, 150 mL (Fizz, model no. 8541994739)
-
15 mL conical tubes (Corning, cat. no. 430790)
-
50 mL conical tubes (Corning, cat. no. 430829)
-
10 mL serological pipettes (CELLTREAT, cat. no. 229210B)
-
Pipette aid (Drummond Scientific, cat. no. 4-000-101)
-
P20 micropipette (Eppendorf, cat. no. 3124000040)
-
P100 micropipette (Eppendorf, cat. no. 3124000075)
-
P1000 micropipette (Eppendorf, cat. no.3124000121)
-
P1000 pipette tips (CELLTREAT, cat. no. 229018)
-
P100 pipette tips (CELLTREAT, cat. no. 229021)
-
Vacuum filter unit, 500 mL, 0.2 µm cutoff (Fisher Scientific, cat. no. FB12566504)
-
Vacuum suction tubing (ADI MEDICAL, cat. no. D4808)
-
Pasteur glass pipettes (Fisher Scientific, cat. no. 13-678-20D)
-
Syringe filter, cutoff size 0.2 µm (VWR International, cat. no. CA28145-477)
-
Hemocytometer (LW Scientific, cat. no. CTL-HEMM-GLDR)
-
Cell strainers, 100 µm (CELLTREAT, cat. no. 229485)
-
T flasks, surface area 75 cm2 (Fablab, cat. no. FL7341)
-
500 mL glass bottles (PYREX, cat. no. 1395500)
-
24-well culture plates (Corning, cat. no. 3524)
-
Transwell insert, polyester membrane, 0.4 µm pore (Corning, cat. no. 3470)
-
Luer lock syringes, 3 mL sterile (BD Biosciences, cat. no. 309657)
-
Spin coater (Laurell, model no. WS-400B-6NPP/AS2)
-
Mask aligner for UV exposure (Karl Suss America, model no. MA6/BA6)
Caution
Take safety precautions when handling the equipment. Avoid being exposed to UV light.
-
Hot plates (Thermo Scientific, cat. no. HP88857100)
Caution
Take safety precautions with high temperature.
-
Biosafety cabinet (NuAire, model no. NU-437-400)
-
Dry oven (Fisher Scientific, cat. no. 15-103-0519)
-
Plasma cleaner (Femto Science, cat. no. COVANCE-1MPR)
-
UV/ozone generator (Jelight Company, model no. 342)
-
Corona treater (Electro-Technic Products, model no. BD-20AC)
Caution
Take safety precautions when handling the equipment. Avoid being exposed to corona plasma beam.
-
CO2 incubator, 37 °C, humidified, 5% CO2 (Eppendorf, model no. C170i)
-
Syringe pump (Braintree Scientific, model no. BS-8000 120V)
-
Centrifuge (Eppendorf, model no. 5910R)
-
Water bath (VWR, cat. no. 10128-126)
-
Inverted phase-contrast microscope (DMi1, Leica microsystems)
-
Phase-contrast microscope (Axiovert 40CFL, Zeiss)
-
TCS SPE confocal microscope (DMi8, Leica microsystems)
-
Confocal microscope (Leica SP5 X MP DMi-6000)
-
FlexCell FX-5000 Tension system (Flexcell International Corporation)
-
Scanning electron microscope (Vega III SEM, Tescan USA)
-
Water purification system (MiliporeSigma, Direct-Q Water Purification System, cat. no. ZRQS0P3WW)
Software
-
LAS X imaging software (Leica microsystems; provided with the microscopes)
-
MetaMorph (Molecular Devices, https://www.moleculardevices.com/products/cellular-imaging-systems/acquisition-and-analysis-software/metamorph-microscopy#gref)
-
IMARIS (IMARIS x64; Bitplane Scientific Software, https://imaris.oxinst.com/packages)
-
AutoQuant (AutoQuant X, Version X3.0.1; Media Cybernetics, https://www.mediacy.com/autoquantx3)
Reagent setup
Caco-2 culture medium
Add 100 mL of heat-inactivated FBS (final concentration 20% (vol/vol)) and 5 mL of 100× penicillin–streptomycin to 395 mL of DMEM in a biosafety cabinet. Store the medium at 4 °C for up to 6 months.
R-spondin culture medium
Add 150 µL of 100 mg/mL Zeocin (final concentration 300 µg/mL) and 5 mL of heat-inactivated FBS (final concentration 10%, (vol/vol)) to 45 mL of DMEM. Store the medium at 4 °C for up to 6 months.
Noggin culture medium
Add 50 µL of 10 mg/mL puromycin (final concentration 10 µg/mL) and 5 mL of heat-inactivated FBS (final concentration 10% (vol/vol)) to 45 mL of DMEM. Store the medium at 4 °C for up to 6 months.
Antibiotic-free 293T cell medium
Add 5 mL of heat-inactivated FBS (final concentration 10% (vol/vol)) to 45 mL of DMEM. Store the medium at 4 °C for up to 6 months.
Medium for collecting R-spondin- and Noggin-conditioned medium (Step 9, Box 3)
Add 40 mL of heat-inactivated FBS (final concentration 8% (vol/vol)) and 5 mL of GlutaMAX to 455 mL of Advanced DMEM/F12. Store the medium at 4 °C for up to 6 months.
Organoid basal medium
Add 5 mL of GlutaMAX, 5 mL of 1 M HEPES (final concentration 10 mM) and 5 mL of 100× penicillin–streptomycin to 485 mL of Advanced DMEM/F12. Store the medium at 4 °C for up to 6 months.
Organoid culture medium
In a sterile 500 mL bottle, add 337.7 mL of organoid basal medium, 500 µL of 100 µg/mL human recombinant Wnt3a (final concentration 100 ng/mL), 100 mL of R-spondin-conditioned medium (Box 3), 50 mL of Noggin-conditioned medium (Box 3), 1 mL of 50 mg/mL Primocin (final concentration 100 µg/mL), 2.5 mL of N-2, 5 mL of B-27, 2.5 mL of 1 M nicotinamide (dissolved in DIW; final concentration 10 mM), 500 µL of 500 mM N-acetylcysteine (dissolved in sterile DIW; final concentration 1 mM), 250 µL of 100 µg/mL mouse recombinant EGF (dissolved in PBS; final concentration 50 ng/mL), 33.15 µL of SB202190 (25 mg/mL, dissolved in DMSO; final concentration 10 µM), 10.5 µL of A-8301 (5 mg/mL dissolved in DMSO; final concentration 500 nM) and 10.4 µL of gastrin (0.5 mg/mL dissolved in PBS; final concentration 10 nM). Mix the contents by gently inverting the bottle two to three times, and filter the medium using a vacuum filter (cutoff size 0.2 µm). Store aliquots (40 mL per tube) of the organoid culture medium at −20 °C for up to 3 months. The recipe for the organoid culture medium was established on the basis of previously published studies23,25,45.
Organoid differentiation medium
Add 465.7 mL of organoid basal medium, 25 mL of Noggin-conditioned medium (Box 3), 1 mL of Primocin (50 mg/mL; final concentration 100 µg/mL), 2.5 mL of N-2, 5 mL of B-27, 500 µL of N-acetylcysteine (500 mM dissolved in sterile DIW; final concentration 1 mM), 250 µL of mouse recombinant EGF (100 µg/mL dissolved in PBS; final concentration 50 ng/mL), 10.5 µL of A-8301 (5 mg/mL dissolved in DMSO; final concentration 500 nM) and 10.4 µL of gastrin (0.5 mg/mL dissolved in PBS; final concentration 10 nM) to a sterile 500 mL bottle. Mix the contents by inverting the bottle two to three times, then filter the mixture with a vacuum filter (cutoff size 0.2 µm). Store aliquots (40 mL per tube) of the organoid differentiation medium at −20 °C for up to 3 months.
PEI solution
Add 1 mL of 50% (wt/vol) PEI solution to 49 mL of PBS (final concentration 1%(wt/vol)) in a 50 mL conical tube. Mix the solution by inverting the tube two or three times. Filter the solution using a syringe filter (cutoff size 0.2 µm). Store the tube at 4 °C for up to 6 months.
Glutaraldehyde solution
Add 100 µL of glutaraldehyde stock solution (50% (wt/vol) to 49.9 mL of PBS in a 50 mL conical tube to produce working solution (final concentration 0.1% (wt/vol)). Mix the solution by inverting the tube two or three times. Filter the solution using a 0.2 µm syringe filter. Store the tube at 4 °C for up to 6 months.
Blocking solution (2% (wt/vol) BSA solution)
Dissolve 1 g of BSA in 50 mL of PBS. Filter the solution using a 0.2 µm syringe filter. Store aliquots (10 mL per tube) at 4 °C for up to 6 months.
Permeabilizing solution
Add 150 µL of Triton X-100 to 50 mL of blocking solution (final concentration of Triton X-100 0.3% (vol/vol)). Filter the mixture using a 0.2 µm syringe filter. Store aliquots (10 mL per tube) at 4 °C for up to 6 months.
Primary antibody solution
Dilute primary antibodies in blocking solution to the following final concentrations: anti-ZO-1 (final concentration 20 µg/mL), anti-MUC2 (final concentration 10 µg/mL) and anti-LGR-5 (1:50 dilution) for in situ on-chip immunofluorescence staining.
Secondary antibody solution
Dilute secondary antibody solutions in blocking solution, using a 1:50 dilution factor for the goat anti-mouse IgG DyLight 488 and 1:200 dilution factor for the goat anti-rabbit IgG Alexa Fluor 555.
Counterstaining solutions
Dilute the Phalloidin CruzFluor 647 conjugate and the DAPI solutions 1:500 in PBS.
Procedure
Microfabrication
Timing 2–4 d
-
1
Clean the silicon molds prepared as described in Box 1 (Fig. 2a (‘Silicon mold’) and 2b) with 100% EtOH. Remove residual EtOH with an air gun and completely dry the molds in a 60 °C dry oven for at least 2 h.
-
2
Mix PDMS base elastomer and curing agent with 15:1 mass ratio in a disposable cup and stir the mixture vigorously for at least 60 s. Degas the mixture in a vacuum desiccator at room temperature (RT, 20–25 °C) for 20 min.
Critical step
The most conventional mixing ratio of PDMS base elastomer and curing agent is 10:1 mass ratio. However, we use 15:1 ratio to increase the elasticity of the device.
-
3
Place each silicon mold into a 100 mm Petri dish. Pour the degassed PDMS solution onto the silicon molds and degas the poured PDMS in a vacuum desiccator for 20 min. The desired thicknesses of cured PDMS layers of the upper and the lower layers are ~7 mm and ~1 mm, respectively. The desired thickness of a PDMS layer for making a hybrid chip is ~7 mm. The thickness can be estimated by looking at the Petri dish from the side. Remove floating bubbles using an air gun, then put the silicon molds containing uncured PDMS in a 60 °C dry oven (Fig. 2a ‘PDMS casting’). Make sure the shelf in the oven is leveled. Incubate for at least 4 h to completely cure the PDMS solution.
-
4
Demold the cured PDMS parts from the silicon molds by cutting the edge with a scalpel and lifting up the PDMS layers using tweezers (Fig. 2a ‘Demolded PDMS’). Cut the PDMS pattern into a desired size with sufficient margins (at least 0.5 cm apart from the patterns) using a scalpel or a razor blade (Fig. 2d).
-
5
To fabricate a gut-on-a-chip, follow option A. To make a Transwell-insertable hybrid chip, follow option B.
-
(A)
Microfabrication of a gut-on-a-chip
-
(i)
Prepare 1-cm-thick PDMS slabs by gently pouring uncured degassed PDMS solution (15:1 mass ratio, base polymer:curing agent) into a Petri dish, curing in a 60 °C dry oven overnight and cutting using a scalpel to the size that covers the patterned area of the membrane wafer.
Critical step
The desired thickness of a PDMS slab can be determined by looking at the Petri dish from the side while pouring the degassed PDMS mix.
-
(ii)
Prepare a 3 kg weight by filling a tin container with lead balls.
Critical step
Conventional 3 kg weights can be also used. However, we use tin containers filled with lead balls to provide a contact surface that is as flat as possible and maximize the contact surface area.
-
(iii)
To produce a porous PDMS membrane, pour ~1 mL of uncured PDMS onto a prepatterned silicon mold that contains an array of micropillars (diameter 10 µm; Fig. 2c and Extended Data Fig. 1d; see ‘Reagents’), then cover with a fluoropolymer-coated polyester film (3M Scotchpak Release Liners) without bubbles. Next, place a 1-cm-thick PDMS slab (from Step 5A(i)) on top of the fluoropolymer-coated polyester film, lay frosted glasses on the flat PDMS slab (frosted side down to the PDMS slab) and put the 3 kg weight from Step 5A(ii) on top. Incubate this setup at RT for 2 h or until the film does not freely move on the mold. Move the setup to a 60 °C dry oven and cure PDMS for >4 h.
-
(iv)
To prepare an upper layer, punch holes (outer diameter 2 mm) at the connecting ports of the inlet and outlet of microchannels as well as the vacuum reservoirs of the PDMS replica from Step 4. Clean the punched holes by flushing them with 100% EtOH and remove residual EtOH using an air gun. Clean the surface of the PDMS parts with Scotch Magic tape and completely dry the parts in a 60 °C dry oven for 4 h or more.
-
(v)
To prepare a lower layer, clean the surface of a lower PDMS layer (from Step 4) with Scotch Magic tape and place it on a No. 1 coverslip.
Caution
Handling a lower PDMS layer is challenging because the layer is very thin (~1 mm). Make sure to avoid bubble trapping or wrinkling when a lower PDMS layer is placed on a coverslip.
Critical step
The fabricated lower PDMS layer does not need to be bonded with a coverslip. No surface activation step is needed.
-
(vi)
Demold the PDMS membrane layer attached to the fluoropolymer-coated polyester film from the silicon membrane mold in Step 5A(iii), and keep it in a clean Petri dish to avoid adsorption of lint until use.
-
(vii)
Keep all the prepared PDMS parts in a lint-free condition (e.g., keep the parts in a container with a lid).
-
(viii)
Place PDMS parts of an upper layer and a porous membrane in a Petri dish, with the surface to be treated facing upward, and expose to oxygen plasma (atmospheric gas, target pressure at 1 × 10−5 torr, power at 125 W) for 1.5 min in the plasma cleaner.
-
(ix)
Take out the plasma-treated PDMS layers immediately once the process is done and place the patterned surface of the upper PDMS layer onto the PDMS surface of the membrane. Press the PDMS surface of the upper layer gently and examine whether any air bubbles are trapped between the two layers. If an air bubble is found, keep pressing the upper PDMS part for 3–5 s.
-
(x)
Incubate the PDMS setup in an 80 °C oven for irreversible bonding for >4 h.
-
(xi)
After bonding, detach the assembly of the upper PDMS layer bonded with the porous membrane from the polyester film by facing the membrane surface up and remove the membrane from the area covering the vacuum chambers and the ports that connect to the lower microchannels (Fig. 2e) using fine-tip forceps, under an optical microscope or a stereoscope.
Critical step
This step is needed to make completely through-hole ports to connect tubing to the lower layer and to make empty vacuum chambers.
-
(xii)
Activate the surface of the membrane-bound upper PDMS part and the lower PDMS layer placed on a coverslip in Step 5A(v) by treating with corona plasma for at least 1 min each.
-
(xiii)
Place the membrane-bound upper PDMS layer and the lower layer together, and under the microscope, align the microchannels and tap the PDMS surface using tweezers to enhance bonding.
-
(xiv)
Incubate the whole setup for bonding in an 80 °C dry oven for >12 h to make a complete gut-on-a-chip device (Fig. 2f).
-
(xv)
Assemble tubing for inlets and outlets of the upper and the lower microchannels of a gut-on-a-chip (Fig. 2g). To make an inlet connecting part, join 4-cm-long silicone tubing to all the branches of a Y-connector. To one end, connect an 18 G blunt-end needle which will serve as a connecting port of a Luer-lock syringe for medium supply. To another end, connect a bent stainless-steel connector that is made by separating the shaft part of a 18 G blunt-end needle from the needle hub and bending it to 90–100°. Insert the cut metal part into the silicone tubing. The remaining branch will serve as a bypass tubing. Prepare two inlet parts per chip so they can be connected to both upper and lower microchannels.
Critical step
The purpose of the bypass tubing is to manipulate pressure when handling syringes. During a cell culture in a gut-on-a-chip, open the bypass tubing while closing the outlet every time when removing and reassembling syringes to avoid generating unnecessary pressure on the cells in the channel. This procedure is explained in detail in the latter steps.
-
(xvi)
To make an outlet part, connect a bent metal connector to 4-cm-long silicone tubing. Prepare two sets per chip.
-
(xvii)
Assemble the prepared inlet and outlet parts on the gut-on-a-chip device by inserting stainless steel connectors that link to tubing into the holes in the gut-on-a-chip.
-
(xviii)
Connect two 1 mL syringes containing 70% EtOH to the inlet ports and flow EtOH through the microchannels. Inspect whether any leakage occurs. If there is a leakage, discard the device.
-
(xix)
To test whether the membrane integrated into a gut-on-a-chip is porous, close the outlet of the lower microchannel and bypass tubing of both upper and lower microchannels using binder clips and push the syringe connected to the lower microchannel, making the fluid flow from the lower to the upper microchannel. Inspect whether the fluid comes out to the outlet of the upper microchannel without any leakage or pressure. Repeat the same test by closing the upper microchannel and pushing the syringe connected to the upper microchannel.
-
(xx)
Remove syringes from the hub of the device and dry the device in a 60 °C dry oven overnight.
-
(i)
-
(B)
Microfabrication of a hybrid chip
-
(i)
Punch a hole using a biopsy puncher (outer diameter 8 mm) in the center of the channel of the PDMS replica from Step 4, and punch holes to the marked connecting ports using a biopsy puncher (outer diameter 2 mm).
-
(ii)
Wash the PDMS part with 100% EtOH and dry completely in a 60 °C dry oven for at least 4 h (Fig. 2h ‘Microchannel layer’).
-
(iii)
Clean the PDMS part and a glass slide using Scotch Magic tape to remove lint.
-
(iv)
Place the PDMS part and the glass slide in a Petri dish, facing the surface to be treated upward, and expose to oxygen plasma (atmospheric gas, target pressure at 1 × 10-5 torr, power at 125 W) for 1.5 min in a plasma cleaner.
-
(v)
Take out the plasma-treated PDMS part and the glass slide immediately once the plasma treatment process is done, and place the PDMS part on the surface-activated glass slide (Fig. 2h). Tap the PDMS part gently and examine whether any air bubble is trapped between the two layers. If an air bubble is found, press the PDMS part gently for 3–5 s.
-
(vi)
Incubate the setup in an 80 °C dry oven for bonding for >4 h to produce a complete hybrid chip device (Fig. 2h).
-
(vii)
Prepare a set of inlet and outlet tubing, as described in Step 5A(xv) and (xvi), and attach to the ports.
Pause point
Keep the fabricated devices at RT in a covered container. The fabricated devices can be stored indefinitely under this condition.
Critical step
The estimated success rate of microfabrication is >90% that does not cause any leakages due to the misalignment. However, the success rate may vary depending on the level of proficiency in the fabrication process and training. See Table 1 for troubleshooting.
-
(i)
-
(A)
Surface activation and ECM coating for Caco-2 culture
Timing 2 h to 1 d
-
6
For a gut-on-a-chip experiment, follow option A. For a hybrid chip experiment, follow option B because a Transwell insert does not need surface activation. If the device will be used for Caco-2 cell culture, follow this step, but if you intend instead to use it for organoid culture, please skip to Step 7.
-
(A)
Surface activation and ECM coating in a gut-on-a-chip
-
(i)
Incubate a gut-on-a-chip device in a 60 °C dry oven for 30 min to remove any residual moisture.
-
(ii)
Put the gut-on-a-chip device setup in a UV/ozone generator. Expose to UV and ozone for 40 min.
Critical step
Keep the distance of the device from the UV lamp ~3 cm or less. Also, avoid any tangling of tubing so the surface of microchannels is efficiently activated during the treatment.
-
(iii)
Prepare an ECM mixture by adding 50 µL of 3 mg/mL rat-tail collagen I (final concentration 30 µg/mL) and 50 µL of Matrigel (100× dilution) to 5 mL of ice-cold DMEM. Keep this solution at 4 °C or on ice until the UV/ozone treatment is over.
Critical step
It is recommended that the ECM mixture be prepared right before the UV/ozone treatment is done.
-
(iv)
Bring the device to a biosafety cabinet immediately after the UV/ozone treatment, and expose the chip to UV light while the chip is cooled down to RT for 5 min.
-
(v)
Prepare two 1 mL syringes containing 300 µL of the cold ECM mixture from Step 6A(iii).
-
(vi)
Close the bypass tubing in both upper and lower microchannels using binder clips. Keep the outlet tubing of both the upper and the lower microchannels open.
-
(vii)
Attach the ECM mixture containing syringes from Step 6A(v) to the inlets of the upper and the lower microchannels of the device.
-
(viii)
Introduce the ECM mixture into the microchannels. Keep both outlets open without clamping.
Critical step
The ECM mixture can be introduced only to the upper microchannel if the lower microchannel does not need to be coated. This can be determined empirically. Caco-2 cells have relatively strong binding on the ECM-coated PDMS surface, compared with organoid-derived cells. If the cell attachment is repeatedly successful without coating the lower microchannel, only the upper microchannel may be coated with ECM. In this case, leave the lower microchannel filled with air by clamping the outlet of the lower microchannel with a binder clip.
-
(ix)
Incubate the device at 37 °C in a humidified 5% CO2 incubator for 1 h.
-
(x)
During the incubation time, prepare 3 mL syringes containing prewarmed Caco-2 culture medium. Degas the warm Caco-2 culture medium using a Steriflip to minimize air bubble generation in the medium. Attach the Steriflip to a 50 mL conical tube that contains the warm Caco-2 culture medium. Flip the tube over and attach a vacuum suction tubing to the connecting port on the Steriflip. Wait until the medium is filtered through the membrane of the Steriflip and wait for an additional 1 min for degassing. Tap the Steriflip tube to generate bubbles of dissolved gas while running vacuum suction. Remove the Steriflip unit.
-
(xi)
After 1 h of ECM coating, take out the device from the CO2 incubator and place it inside the biosafety cabinet.
-
(xii)
Close the outlet tubing that is connected to the upper and lower microchannels using binder clips.
-
(xiii)
Open the bypass tubing connected to the lower microchannel.
-
(xiv)
Remove the 1 mL syringe connected to the lower microchannel and aspirate residual ECM solution in the blunt-end needle hub. Close the outlet tubing connected to the lower microchannel with a binder clip, while keeping the bypass tubing open.
-
(xv)
Attach a 3 mL Luer-lock syringe that contains Caco-2 culture medium from Step 6A(x) to the blunt-end needle connected to the lower microchannel of the gut-on-a-chip. Perfuse the medium (~50 µL) to the bypass tubing. Next, close the bypass tubing connected to the lower microchannel using a binder clip once the tubing is completely filled with the introduced medium.
-
(xvi)
Open the outlet tubing linked to the lower microchannel. Gently flow the Caco-2 culture medium through the lower microchannel. After perfusion, clamp the outlet tubing attached to the lower microchannel with a binder clip.
Critical step
Remove air bubbles that can be trapped inside the microchannel.
-
(xvii)
Repeat Steps 6A(xiii)–(xvi) to replace the 1 mL syringe used for ECM introduction to the upper microchannel with a 3 mL Luer-lock syringe containing Caco-2 culture medium.
-
(xviii)
Open both outlet tubing by removing binder clips once two 3 mL syringes are firmly connected to the gut-on-a-chip device.
Pause point
Optionally, when there is not enough time to continue the process, this Procedure can be paused here overnight. In this case, flow the Caco-2 culture medium through the microchannels at 30 µL/h in the 37 °C humidified CO2 incubator overnight. However, this process should be cautiously performed because the coated ECM proteins can be perturbed or washed off, which can lead to a poor attachment of dissociated epithelial cells.
-
(i)
-
(B)
ECM coating of a Transwell insert
-
(i)
Prepare an ECM mixture following the recipe in Step 6A(iii) (30 µg/mL of collagen I and 100× diluted Matrigel).
-
(ii)
Add 100 µL of the ECM mixture to the apical side of a Transwell insert.
-
(iii)
Incubate the Transwell insert at 37 °C in a humidified CO2 incubator for 1 h.
-
(iv)
Remove the ECM mixture using a micropipette.
-
(v)
Add 100 µL of Caco-2 culture medium to the Transwell insert and add 500 µL of the same medium in the basolateral compartment. Keep this setup at 37 °C in the humidified CO2 incubator until cell seeding.
-
(i)
-
(A)
Surface activation and ECM coating for organoid culture
Timing 1 d
-
7
For a gut-on-a-chip experiment, follow option A. For a hybrid chip experiment, follow option B because the Transwell insert does not need surface activation. If the device will be used for organoid cultures, follow this step, but this step can be skipped if you have carried out Step 6 already and are using the device for Caco-2 cell culture.
-
(A)
Surface activation and ECM coating in a gut-on-a-chip
-
(i)
Follow Step 6A(i)–(ii) for the activation of PDMS surface.
-
(ii)
Close the bypass tubing and the outlet of the lower microchannel using binder clips.
-
(iii)
Remove the outlet tubing set linked with a bent stainless-steel connector from the upper microchannel. Keep the bypass tubing of the upper microchannel open.
-
(iv)
Introduce 30 µL of 1% PEI solution through the outlet hole of the upper microchannel using a P100 micropipette.
-
(v)
Reconnect the outlet tubing in the upper microchannel and close both bypass and outlet tubing of the upper microchannel using binder clips.
-
(vi)
Repeat Step 7A(ii)–(v) for the lower microchannel.
-
(vii)
Incubate the device at RT for 10 min.
-
(viii)
Repeat Steps 7A(ii)–(vi) but using 0.1% glutaraldehyde solution instead of 1% PEI.
-
(ix)
Incubate the device setup at RT for 20 min.
-
(x)
Wash both microchannels by flowing 1 mL of sterile DIW using 1 mL syringes.
-
(xi)
Dry the chip setup in a 60 °C dry oven overnight.
-
(xii)
Repeat Step 6A(iii) and (v) to prepare syringes containing ECM mixture.
-
(xiii)
After cooling down the dried gut-on-a-chip setup in a biosafety cabinet for 10 min, attach the ECM mixture containing syringes to both microchannels by following Step 6A(vi)–(vii), and introduce the mixture by manually pushing the plungers.
-
(xiv)
Incubate the device setup at 37 °C in a humidified CO2 incubator for 1 h.
-
(xv)
Replenish the ECM solution only in the upper channel by pushing the syringe plunger of the upper channel gently, and incubate the device in the incubator for another 1 h.
-
(xvi)
Prepare two 1 mL syringes containing cold organoid culture medium that is depleted of A8301 but supplemented with Y-27632. Connect the syringes to the chip and gently flow the medium through the channels by following Step 6A(xv)–(xviii). Flow the channels one by one (i.e., when the upper channel is handled, make sure the lower channel tubing is closed, and vice versa).
Critical step
This step should be cautiously performed. Flow the organoid culture medium slowly, and make sure to handle one microchannel at a time. The coated ECM proteins can be easily perturbed or washed off, which can lead to poor attachment of dissociated epithelial cells.
-
(i)
-
(B)
ECM coating of a Transwell insert
-
(i)
Follow the methods described in Step 6B(i)–(iv).
-
(ii)
Add 100 µL of organoid culture medium, depleted of A8301 but supplemented with Y-27632 to the membrane insert, and add 500 µL of the same medium in the basolateral compartment.
-
(i)
-
(A)
Cell preparation for seeding
Timing 30 min
-
8
Harvest the fully cultured cells (Boxes 2 and 5) for seeding in either a gut-on-a-chip or on a Transwell insert. Follow option A or B for seeding the Caco-2 cells or the human organoids epithelial cells, respectively.
-
(A)
Preparation of Caco-2 cells for seeding
-
(i)
Wash a T75 flask containing 95% confluent Caco-2 cells from Box 2 by rinsing the cells with 5 mL of warm PBS twice after aspiration of the culture medium.
-
(ii)
Remove PBS, add 1 mL of 0.25% trypsin/1 mM EDTA and incubate the flask in a humidified CO2 incubator at 37 °C for 10 min.
-
(iii)
Add 9 mL of warm Caco-2 culture medium and collect the cell suspension by pipetting in a 15 mL sterile conical tube.
-
(iv)
Centrifuge the tube at 300g at 4 °C for 3 min and remove supernatant to obtain a cell pellet.
-
(v)
Resuspend the cell pellet in 1 mL of Caco-2 culture medium and count the cell number using a hemocytometer under a phase-contrast microscope.
-
(vi)
Adjust the density of the dissociated Caco-2 cells to 1 × 107 cells/mL with Caco-2 culture medium.
-
(i)
-
(B)
Preparation of human organoid cells for seeding
-
(i)
Remove organoid culture medium by vacuum suction and add 500 µL of ice-cold Cell Recovery solution in each well that contains fully grown organoids in a 24-well plate from Box 5.
Critical step
Typically, a well containing fully grown organoids (~300 organoids) gives enough dissociated cells for seeding one gut-on-a-chip or one Transwell insert.
-
(ii)
Using a P1000 micropipette, mechanically break Matrigel domes down for 5 s. Then, collect organoid suspension in a 15 mL conical tube.
-
(iii)
Centrifuge at 100g at 4 °C for 5 min and remove supernatant, while leaving up to 500 µL of the cell layer if there is no clear cell pellet layer with a cloudy Matrigel gradient.
-
(iv)
Add 1 mL of warm TrypLE Express solution containing 10 µM Y-27632 (final concentration) and resuspend the cell pellet by pipetting with a P1000 micropipette.
-
(v)
Incubate the cell suspension in a 37 °C water bath for 5 min.
-
(vi)
Add 5 mL of warm PBS and pipette aggressively using the P1000 micropipette until the cell suspension becomes turbid without noticeable cell clumps.
-
(vii)
Filter the cell suspension through a cell strainer (cutoff 100 µm) to remove Matrigel debris and large cell clumps.
-
(viii)
Centrifuge the tube at 300g at 4 °C for 3 min and resuspend in organoid culture medium depleted of A8301 but containing 10 µM Y-27632 (final concentration). It is recommended that 20 µL of organoid culture medium be used for resuspension of a cell pellet harvested from one well (i.e., use 120 µL of organoid culture medium when six wells were harvested).
-
(ix)
Adjust the density of viable cells to 1 × 107 cells/mL with organoid culture medium depleted of A8301 but containing 10 µM Y-27632 (final concentration). Perform a viability estimation with a hemocytometer by mixing 10 µL of the cell suspension and 10 µL of Trypan blue and observing the cells under a microscope.
-
(i)
-
(A)
Formation of a 2D cell monolayer
Timing 2–3 d
-
9
Seed the harvested cells from Step 8 in an ECM-coated gut-on-a-chip or on the Transwell insert of a hybrid chip. Then, culture the cells to form a confluent 2D monolayer prior to inducing 3D morphogenesis. For the gut-on-a-chip experiment, follow option A. For the hybrid chip experiment, follow option B.
-
(A)
Formation of a 2D cell monolayer in a gut-on-a-chip
-
(i)
Remove the outlet tubing connected to the upper microchannel from the device, open the bypass tubing connected to the inlet of the upper microchannel and close both inlet and outlet of the lower microchannel using binder clips.
-
(ii)
Introduce 20 µL of the cell suspension from Step 8 to the open hole of the upper microchannel using a P100 or P20 micropipette.
-
(iii)
Close the bypass tubing of the upper microchannel. Then carefully reconnect the outlet tubing to the open port of the outlet of the upper microchannel while keeping the tubing open. Close the outlet tubing reconnected to the upper channel slowly using a binder clip.
-
(iv)
Confirm under a microscope if the cell suspension is uniformly dispersed across the upper microchannel.
Caution
Carry the device horizontally after a cell seeding by balancing the level. Avoid any physical shock that may perturb the cell distribution in the microchannel.
-
(v)
Incubate the device in a humidified CO2 incubator at 37 °C. Caco-2 cells are attached in 1 h, while human organoid cells take ~3 h for attachment.
-
(vi)
When there is not enough time to continue the procedure or cell attachment does not look good under the microscope, extend the attachment time overnight.
-
(vii)
Carefully introduce fresh medium and remove unbound cells using a syringe plunger. Handle each microchannel independently to avoid pressure induced damage to the cell monolayer. Alternatively, the device can be hooked up to a syringe pump that can be used to flow fresh medium.
-
(viii)
Inspect under the microscope if unbound cells are washed out.
-
(ix)
Hook up the syringe linked to the upper microchannel of the gut-on-a-chip to a syringe pump placed inside the humidified CO2 incubator. Start perfusion of culture medium at 30 µL/h. In the case of culturing intestinal organoid cells in a gut-on-a-chip, change the medium into organoid culture medium including A8301 but without Y-27632 at this point.
Caution
Make sure the diameter of the syringe is applied to the setting of the syringe pump before running a microfluidic culture.
-
(x)
To maintain the intestinal epithelial cell culture on-chip, keep perfusing the culture medium to the upper microchannel until the cells form a confluent 2D monolayer, which you can assess via a phase contrast microscope. Replenish the medium filled in the lower microchannel every day by manually flowing fresh medium while the continuous flow is applied to the upper microchannel. Caco-2 and organoid epithelial cells form epithelial monolayers in ~24–36 h (Fig. 3a) and 2–3 d (Fig. 3d), respectively.
-
(i)
-
(B)
Formation of a 2D cell monolayer on a Transwell insert
-
(i)
Adjust the cell density to 2 × 106 cells/mL with a corresponding medium.
-
(ii)
Add 100 µL of the cell suspension to the insert and 500 µL of the culture medium to the basolateral well of the Transwell, and incubate the Transwell-containing plate in a 37 °C CO2 incubator for 16 h.
-
(iii)
Inspect cell attachment under a microscope.
-
(iv)
Remove culture medium from both the apical and basolateral compartments and add the same amount of fresh medium (i.e., 100 µL to the apical and 500 µL to the basolateral) after cell attachment. When intestinal organoids are cultured on a Transwell insert, use organoid culture medium including A8301 but without Y-27632.
-
(v)
To maintain the intestinal epithelial cell culture on the Transwell insert, change the culture medium every other day in both apical and basolateral compartments until the cells form a confluent 2D monolayer. An epithelial monolayer of Caco-2 or organoid epithelial cells is formed in ~2 days and 5 days (Fig. 4a), respectively.
-
(i)
-
(A)
Induction of 3D morphogenesis in vitro
Timing 3–5 d
-
10
Once confluent 2D monolayers are formed, 3D morphogenesis can be induced. For a gut-on-a-chip experiment, follow option A. For a hybrid chip experiment, follow option B.
-
(A)
Induction of in vitro 3D morphogenesis in a gut-on-a-chip
-
(i)
Flow the corresponding culture medium (for Caco-2 or organoid cells) into the lower microchannel to induce intestinal morphogenesis in a gut-on-a-chip (Fig. 3a ‘Flow, AP’). Flow medium into both upper and lower microchannels using a syringe pump at 50 µL/h volumetric flow rate to establish 0.02 dyne/cm2 of shear stress in a 500-µm-height microchannel (Extended Data Fig. 1c)10.
Critical step
The volumetric flow rate can vary depending on the geometric configuration of the microchannels in a gut-on-a-chip.
-
(ii)
Connect a Flexcell connector to the vacuum chamber ports in the gut-on-a-chip when the dual microfluidic flow is initiated. Start applying stretching motions to the cells growing in the gut-on-a-chip using a Flexcell module with 10% of cell strain and 0.15 Hz of frequency.
-
(iii)
Maintain this culture setup for at least 2–3 d. Typically, morphogenesis occurs after this point (Fig. 3a ‘Stretching, flow, AP and BL’).
-
(iv)
If human intestinal organoids are cultured in a gut-on-a-chip, change the organoid culture medium into organoid differentiation medium after 3D morphogenesis is observed under the microscope to induce cell differentiation.
-
(i)
-
(B)
Induction of in vitro 3D morphogenesis on a Transwell insert
-
(i)
When intestinal morphogenesis is to be induced on a Transwell insert, transfer the Transwell insert that contains a cell monolayer into a hybrid chip. The insert can be physically inserted into the punched hole of the device (Fig. 1 ‘Culture platforms’ and 4a).
-
(ii)
Apply epoxy glue around the Transwell insert at the interface with the PDMS part to avoid any leakage if needed. Cure the epoxy glue at RT in a biosafety cabinet for 5 min.
-
(iii)
Connect a syringe containing an appropriate culture medium (for Caco-2 or organoid culture) to the hybrid chip and start to flow at 30 µL/h.
-
(iv)
Change the medium in the apical compartment of the Transwell insert every other day using a micropipette. Morphogenesis occurs after 2–3 d in the hybrid chip (Fig. 4a).
-
(i)
-
(A)
Characterization of a 3D epithelial layer
Timing 3 d
-
11
Place the device on a microscope to image the established 3D epithelium. For live-cell imaging, a phase-contrast, DIC or bright-field microscope can be applied.
-
12
Perform fluorescence live-cell imaging to monitor the contour of 3D epithelial microarchitecture grown on either a gut-on-a-chip or a hybrid chip using a membrane dye (e.g., CellMask; Fig. 3f ‘Plasma membrane’). Dilute the CellMask solution at 1:500 in a corresponding culture medium. Transfer the diluted solution to two 1 mL syringes in case of the gut-on-a-chip and one syringe in case of the hybrid chip. Detach the syringes that were used for culture and attach the CellMask-containing syringes to the chips. Slowly fill the microchannels and flow the CellMask-containing medium at 30 µL/h in a CO2 incubator at 37°C for 10 min. For the apical compartment of the hybrid chip, remove the culture medium and add 100 µL of the CellMask-containing medium. Change the CellMask syringes back to the culture-medium-containing syringes to wash the CellMask solution out briefly. Perform fluorescence imaging using a confocal microscope. Culture may be resumed after the imaging when the cells are needed for other experiments.
-
13
To perform immunofluorescence staining and imaging, fix the cells by introducing 30 µL of 4% PFA solution to both microchannels in case of the gut-on-a-chip at RT for 15 min. At this point, a micropipette can be used to introduce solutions instead of using syringes, following the same method described in Step 7A(ii)–(v). For a hybrid chip, separate the Transwell insert from the hybrid chip and transfer the insert to a 24-well plate for easier handling. Add 100 µL of 4% PFA to the apical compartment of a Transwell insert. Incubate the Transwell insert at RT for 15 min.
-
14
Wash the cells by flowing 100 µL of PBS through the microchannels in case of the gut-on-a-chip. With the Transwell, remove PFA by pipetting and add 100 µL of PBS to the apical side of the insert. Repeat three times for the Transwell insert.
-
15
Permeabilize the cells by introducing 30 µL of the permeabilization solution (see ‘Reagent setup’) to both the upper and lower microchannels of the gut-on-a-chip. Or, add 100 µL of the permeabilization solution to the apical compartment of the Transwell insert after removing PBS. Incubate the devices at RT for 30 min.
-
16
Repeat Step 14.
-
17
Block the cells to prevent nonspecific binding by introducing 30 µL of the blocking solution (see ‘Reagent setup’) to both the upper and lower microchannels of the gut-on-a-chip. Or, add 100 µL of the blocking solution to the apical side of the Transwell insert after removing PBS. For the gut-on-a-chip, leave the pipette tips inserted in the gut-on-a-chip to prevent evaporation from the inlet and outlet ports. Incubate at RT for 1 h or at 4 °C for overnight.
-
18
Prepare a primary antibody solution (see ‘Reagent setup’). Introduce 30 µL of the primary antibody solution to both the upper and lower microchannels of the gut-on-a-chip. Incubate at RT for 3 h and at 4 °C for overnight, sequentially. Leave the pipette tips inserted in the gut-on-a-chip to prevent evaporation from the inlet and outlet ports. In the case of the Transwell, add 100 µL of the primary antibody solution to the apical side of an insert and incubate at RT for 3 h or at 4 °C for overnight.
-
19
Repeat Step 14.
-
20
Prepare a secondary antibody solution (see ‘Reagent setup’) and follow the method described in Step 18.
Caution
From this step onward, the device setup must be covered by aluminum foil to avoid photobleaching.
-
21
Repeat Step 14.
-
22
Prepare F-actin and nuclei counterstaining solutions, individually, or in a mix (see ‘Reagent setup’). Introduce 30 µL of the F-actin and/or nuclei staining solution to both the upper and the lower microchannels of the gut-on-a-chip. Add 100 µL of the counterstaining solution mix to the Transwell insert. Incubate at RT under light protection for 30 min.
-
23
Repeat Step 14.
-
24
Perform fluorescence imaging using a confocal microscope. The goal of this imaging step is to achieve structural, functional or locational information by visualizing the shape or the localization of the target cells. To observe the structure of the 3D epithelial layers, a 25× long-distance objective is typically used. Adjust parameters (e.g., laser power, gain, pinhole, scanning speed) to obtain optimal fluorescence signals.
Troubleshooting
Troubleshooting advice can be found in Table 1.
Timing
-
Steps 1–5, microfabrication: 2–4 d
-
Step 6, surface activation and ECM coating for Caco-2 culture: 2 h to 1 d
-
Step 7, surface activation and ECM coating for organoid culture: 1 d
-
Step 8, cell preparation for seeding: 30 min
-
Step 9, formation of a 2D cell monolayer: 2–3 d
-
Step 10, induction of 3D morphogenesis in vitro: 3–5 d
-
Steps 11–24, characterization of a 3D epithelial layer: 3 d
Anticipated results
Our protocol produces reproducible results enabling spontaneous intestinal morphogenesis and 3D epithelial structure in a gut-on-a-chip (Fig. 3) or a Transwell-embedded hybrid chip (Fig. 4). Within 3 d of introducing basolateral fluid flow, a condensation of epithelial cells is sporadically found across the microchannel (Fig. 3a,d), followed by the continuous protrusion of 3D epithelial cell layers toward the lumen reaching 120–150 µm in height (Figs. 3 and 4)1,2, up to a couple of hundred micrometers4. This morphological change can be monitored using a phase-contrast, DIC or any bright-field microscopy. It is also plausible to apply real-time imaging modalities by using live-cell staining dyes that target produced mucus on the apical membrane (Fig. 1 ‘Intestinal physiology’) or the plasma membrane of a living epithelial layer (Fig. 3f, right).
It has been noted that intestinal epithelial cells cultured in a gut-on-a-chip have a taller epithelial height compared with the epithelial protrusion established on a Transwell insert in a hybrid chip. For instance, when the same organoid cells are cultured on the Transwell insert of a hybrid chip and morphogenesis is induced, the epithelial height is observed to be ~70 µm (Fig. 4c), compared with ~150 µm of epithelial height established in a gut-on-a-chip (Fig. 3f)12. We postulate that the static conditions in the apical compartment may limit the 3D growth of the cell layer. Also, a lack of physical deformation that can induce cellular proliferation in a hybrid chip system may be another factor that makes a substantial difference in height compared to the 3D epithelial morphology in a gut-on-a-chip. Thus, the Transwell-based intestinal morphogenesis may be greatly advanced if the culture system could support continuous flow in both the apical and basolateral compartments as well as including the mechanical dynamics of normal growth conditions.
One of the advanced features of the gut-on-a-chip platform is the feasibility of in situ visualization using various imaging modalities. For instance, we can adapt conventional laser-scanning confocal microscopy to verify that the fully grown intestinal epithelium shows differentiated and spatially organized cell phenotypes (Fig. 3f, see refs. 2,12). An intestinal organoid-derived epithelial layer can be differentiated by applying organoid differentiation medium as detailed in the Procedure23,46. Importantly, different cell types are anticipated to be localized in physiologically relevant locations2. In our previous study2 that used Caco-2 cells, we showed that absorptive (sucrase-isomaltase), goblet (mucin (MUC) 2, MUC3 and MUC17) and enteroendocrine markers (chromogranin A) may be detected in the villus area, whereas the Paneth cell marker (lysozyme) is strongly expressed in the basal crypt region. We also visually verified that regional differentiation markers such as keratin 20 (KRT20), villin, carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) and cytochrome P450 3A4 (CYP3A4) were considerably expressed in the villus region, suggesting cytodifferentiation of the 3D epithelial layers in the gut-on-a-chip. In this protocol, we demonstrate the localization of representative markers for goblet cells (MUC2), stem cells (leucine-rich repeat-containing G-protein-coupled receptor 5, LGR5) and the formation of tight junctions (zonula occludens-1, ZO-1) in the 3D epithelial layer in vitro (Figs. 1 and 3c,f). The expression, and localization along the crypt–villus axis, of other lineage-associated intestinal epithelial markers can also be visualized in a microengineered 3D intestinal epithelium.
Furthermore, visualization technologies including real-time imaging allow us to monitor the growth of living gut bacteria (Fig. 1) that occupy a spatial niche at an intestinal crevicular space. This visualization of a 3D epithelial microarchitecture re-created in a gut-on-a-chip will be extremely useful to investigate host–microbiome interactions during various lesion-specific pathological interactions when we apply this technology to build patient-specific models of GI diseases such as intestinal inflammation, Crohn’s disease or colorectal cancer (Fig. 1). Combining immunofluorescence imaging with targeted RNA visualization (e.g., fluorescence in situ hybridization47,48) may also provide an innovative approach to visually map the host transcriptomes, proteomes and microbiomes in a gut-on-a-chip at various pathophysiological microenvironments.
The formed 3D epithelial layers using both a gut-on-a-chip and a hybrid chip have regenerative potential, confirmed in various studies1,2,4,6,12,15,19. For instance, when the basolateral flow is ceased, established 3D epithelial microstructure is destroyed, which can regenerate once the basolateral medium flow is resumed4. When the proliferative cells were marked with EdU pulse (a proliferation marker that incorporates into newly synthesized DNA), EdU-positive cells were predominantly found near the basal crypt area, which could migrate upward to the microengineered villous tip in a couple of hours, demonstrating the active regeneration of Caco-2 epithelium in the gut-on-a-chip2. The 3D epithelial structure can also be destroyed by adding a barrier-disrupting chemical such as dextran sodium sulfate (DSS), in which a DSS-free culture medium could restore the 3D epithelial structure in a gut-on-a-chip5. This evidence supports the robustness of our protocol, which could be used in longitudinal studies of intestinal epithelial biology, intestinal homeostasis and wound healing mechanisms. The regenerative potential of the microengineered 3D epithelial layers can be further exploited to study human intestinal diseases that involve villous atrophy or crypt shortening in celiac disease or inflammatory bowel disease12.
The in vitro intestinal microenvironment may be manipulated by co-culturing other types of cells. Lymphatic or vascular endothelial cells can be cultured on the opposite side of the porous membrane, and gut bacteria can be cultured on top of the formed 3D intestinal epithelium. Methods to manipulate intestinal microenvironment in a gut-on-a-chip, including co-culture of gut bacteria (Fig. 1), were discussed in earlier publications44,49. The reconstituted microenvironment is anticipated to be used for various applications, including drug testing, fundamental mechanistic studies and disease modeling, as mentioned earlier. Desired assays can be performed by either harvesting supernatant and cells or imaging the cells using live-cell dyes or fixative for further immunofluorescence imaging.
Taking these findings together, we provide a robust and reliable protocol to regenerate 3D intestinal epithelial layers in vitro using a gut-on-a-chip or a hybrid chip device with a Transwell insert. The formed 3D epithelial structure will enable reconstitution of a more physiological intestinal microenvironment in vitro. Applications of the formed intestinal architecture in various biomedical studies may bring about translational research outcomes.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Data presented in the below listed figure panels are available from the original references as listed in the figure legends. All the other data were generated for this article. Any raw data are available for research purposes from the corresponding author upon request. Intestinal organoid lines used in this paper are available from the corresponding author upon request, under a material transfer agreement between institutions.
References
Kim, H. J., Huh, D., Hamilton, G. & Ingber, D. E. Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow. Lab Chip 12, 2165–2174 (2012).
Kim, H. J. & Ingber, D. E. Gut-on-a-Chip microenvironment induces human intestinal cells to undergo villus differentiation. Integr. Biol. 5, 1130–1140 (2013).
Kim, H. J., Li, H., Collins, J. J. & Ingber, D. E. Contributions of microbiome and mechanical deformation to intestinal bacterial overgrowth and inflammation in a human gut-on-a-chip. Proc. Natl Acad. Sci. USA 113, E7–15 (2016).
Shin, W., Hinojosa, C. D., Ingber, D. E. & Kim, H. J. Human intestinal morphogenesis controlled by transepithelial morphogen gradient and flow-dependent physical cues in a microengineered gut-on-a-chip. iScience 15, 391–406 (2019).
Shin, W. & Kim, H. J. Intestinal barrier dysfunction orchestrates the onset of inflammatory host-microbiome cross-talk in a human gut inflammation-on-a-chip. Proc. Natl Acad. Sci. USA 115, E10539–E10547 (2018).
Maurer, M. et al. A three-dimensional immunocompetent intestine-on-chip model as in vitro platform for functional and microbial interaction studies. Biomaterials 220, 119396 (2019).
Tan, H.-Y. et al. A multi-chamber microfluidic intestinal barrier model using Caco-2 cells for drug transport studies. PloS ONE 13, e0197101 (2018).
Ettayebi, K. et al. Replication of human noroviruses in stem cell-derived human enteroids. Science 353, 1387–1393 (2016).
Gazzaniga, F. S. et al. Harnessing colon chip technology to identify commensal bacteria that promote host tolerance to infection. Front. Cell Infect. Microbiol. 11, 638014 (2021).
Shin, W. et al. A robust longitudinal co-culture of obligate anaerobic gut microbiome with human intestinal epithelium in an anoxic–oxic interface-on-a-chip. Front. Bioeng. Biotechnol. 7, 13 (2019).
Bein, A. et al. Microfluidic organ-on-a-chip models of human intestine. Cell Mol. Gastroenterol. Hepatol. 5, 659–668 (2018).
Shin, Y. C. et al. Three-dimensional regeneration of patient-derived intestinal organoid epithelium in a physiodynamic mucosal interface-on-a-chip. Micromachines 11, 633 (2020).
Shin, W. et al. Robust formation of an epithelial layer of human intestinal organoids in a polydimethylsiloxane-based gut-on-a-chip microdevice. Front. Med. Technol. 2, 2 (2020).
Sontheimer-Phelps, A. et al. Human colon-on-a-chip enables continuous in vitro analysis of colon mucus layer accumulation and physiology. Cell Mol. Gastroenterol. Hepatol. 9, 507–526 (2020).
Chong, H. B., Youn, J., Shin, W., Kim, H. J. & Kim, D. S. Multiplex recreation of human intestinal morphogenesis on a multi-well insert platform by basolateral convective flow. Lab Chip 21, 3316–3327 (2021).
Zhang, B. & Radisic, M. Organ-on-a-chip devices advance to market. Lab Chip 17, 2395–2420 (2017).
Ribas, J., Pawlikowska, J. & Rouwkema, J. Microphysiological systems: analysis of the current status, challenges and commercial future. Microphysiol. Syst. 2, 10 (2018).
Allwardt, V. et al. Translational roadmap for the organs-on-a-chip industry toward broad adoption. Bioengineering 7, 112 (2020).
Shin, W., Su, Z., Yi, S. S. & Kim, H. J. Single-cell transcriptomics elucidates in vitro reprogramming of human intestinal epithelium cultured in a physiodynamic gut-on-a-chip. Preprint at bioRxiv https://doi.org/10.1101/2021.09.01.458444 (2021).
Wallace, K. N., Akhter, S., Smith, E. M., Lorent, K. & Pack, M. Intestinal growth and differentiation in zebrafish. Mech. Dev. 122, 157–173 (2005).
Kim, B. M., Mao, J., Taketo, M. M. & Shivdasani, R. A. Phases of canonical Wnt signaling during the development of mouse intestinal epithelium. Gastroenterology 133, 529–538 (2007).
Theodosiou, N. A. & Tabin, C. J. Wnt signaling during development of the gastrointestinal tract. Dev. Biol. 259, 258–271 (2003).
Sato, T. et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 141, 1762–1772 (2011).
Sato, T. & Clevers, H. Growing self-organizing mini-guts from a single intestinal stem cell: mechanism and applications. Science 340, 1190–1194 (2013).
Shin, W. et al. Spatiotemporal gradient and instability of Wnt induce heterogeneous growth and differentiation of human intestinal organoids. iScience 23, 101372 (2020).
Bartfeld, S. & Clevers, H. Organoids as model for infectious diseases: culture of human and murine stomach organoids and microinjection of Helicobacter pylori. J. Vis. Exp. 105, e53359 (2015).
Forbester, J. L. et al. Interaction of Salmonella enterica serovar Typhimurium with intestinal organoids derived from human induced pluripotent stem cells. Infect. Immun. 83, 2926–2934 (2015).
Hinman, S. S., Wang, Y., Kim, R. & Allbritton, N. L. In vitro generation of self-renewing human intestinal epithelia over planar and shaped collagen hydrogels. Nat. Protoc. 16, 352–382 (2021).
Creff, J. et al. Fabrication of 3D scaffolds reproducing intestinal epithelium topography by high-resolution 3D stereolithography. Biomaterials 221, 119404 (2019).
Verhulsel, M. et al. Developing an advanced gut on chip model enabling the study of epithelial cell/fibroblast interactions. Lab Chip 21, 365–377 (2021).
Nikolaev, M. et al. Homeostatic mini-intestines through scaffold-guided organoid morphogenesis. Nature 585, 574–578 (2020).
Brassard, J. A., Nikolaev, M., Hubscher, T., Hofer, M. & Lutolf, M. P. Recapitulating macro-scale tissue self-organization through organoid bioprinting. Nat. Mater. 20, 22–29 (2021).
Valenta, T. et al. Wnt ligands secreted by subepithelial mesenchymal cells are essential for the survival of intestinal stem cells and gut homeostasis. Cell Rep. 15, 911–918 (2016).
Regan, M. C., Flavin, B. M., Fitzpatrick, J. M. & O’Connell, P. R. Stricture formation in Crohn’s disease: the role of intestinal fibroblasts. Ann. Surg. 231, 46–50 (2000).
Roulis, M. & Flavell, R. A. Fibroblasts and myofibroblasts of the intestinal lamina propria in physiology and disease. Differentiation 92, 116–131 (2016).
Wang, Y. et al. Formation of human colonic crypt array by application of chemical gradients across a shaped epithelial monolayer. Cell Mol. Gastroenterol. Hepatol. 5, 113–130 (2018).
Goke, M., Kanai, M. & Podolsky, D. K. Intestinal fibroblasts regulate intestinal epithelial cell proliferation via hepatocyte growth factor. Am. J. Physiol. 274, G809–818 (1998).
Powell, D. W., Pinchuk, I. V., Saada, J. I., Chen, X. & Mifflin, R. C. Mesenchymal cells of the intestinal lamina propria. Annu. Rev. Physiol. 73, 213–237 (2011).
Pappenheimer, J. R. & Michel, C. C. Role of villus microcirculation in intestinal absorption of glucose: coupling of epithelial with endothelial transport. J. Physiol. 553, 561–574 (2003).
Agace, W. W. T-cell recruitment to the intestinal mucosa. Trends Immunol. 29, 514–522 (2008).
Dutton, J. S., Hinman, S. S., Kim, R., Wang, Y. & Allbritton, N. L. Primary cell-derived intestinal models: recapitulating physiology. Trends Biotechnol. 37, 744–760 (2019).
van Meer, B. J. et al. Small molecule absorption by PDMS in the context of drug response bioassays. Biochem. Biophys. Res. Commun. 482, 323–328 (2017).
Gokaltun, A., Kang, Y. B. A., Yarmush, M. L., Usta, O. B. & Asatekin, A. Simple surface modification of poly(dimethylsiloxane) via surface segregating smart polymers for biomicrofluidics. Sci. Rep. 9, 7377 (2019).
Huh, D. et al. Microfabrication of human organs-on-chips. Nat. Protoc. 8, 2135–2157 (2013).
Stewart, C. J., Estes, M. K. & Ramani, S. Establishing human intestinal enteroid/organoid lines from preterm infant and adult tissue. Methods Mol. Biol. 2121, 185–198 (2020).
Kasendra, M. et al. Development of a primary human Small Intestine-on-a-Chip using biopsy-derived organoids. Sci. Rep. 8, 2871 (2018).
Ambrosini, Y. M. et al. Recapitulation of the accessible interface of biopsy-derived canine intestinal organoids to study epithelial–luminal interactions. PLoS ONE 15, e0231423 (2020).
Silahtaroglu, A. N. et al. Detection of microRNAs in frozen tissue sections by fluorescence in situ hybridization using locked nucleic acid probes and tyramide signal amplification. Nat. Protoc. 2, 2520–2528 (2007).
Shin, W. & Kim, H. J. Pathomimetic modeling of human intestinal diseases and underlying host-gut microbiome interactions in a gut-on-a-chip. Methods Cell Biol. 146, 135–148 (2018).
Acknowledgements
This work was supported in part by the National Cancer Institute of the National Institutes of Health (NIH/NCI) F99/K00 Predoctoral to Postdoctoral Transition Award (K00CA245801 to W.S.), Asan Foundation Biomedical Science Scholarship (W.S.), Mogam Science Scholarship (W.S.), Technology Impact Award of the Cancer Research Institute (UTA18-000889 to H.J.K.), the NIH/NCI (R21CA236690 to H.J.K.) and the Leona M. & Harry B. Helmsley Charitable Trust (1912-03604 to H.J.K.).
Author information
Authors and Affiliations
Contributions
W.S. and H.J.K. designed and performed the experiments, analyzed the data, generated the figures and wrote and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Protocols thanks Stephanie Descroix and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Key references using this protocol
Shin, W. et al. iScience 15, 391–406 (2019): https://doi.org/10.1016/j.isci.2019.04.037
Kim, H. J. & Ingber, D. E. Integr. Biol. 5, 1130–1140 (2013): https://doi.org/10.1039/C3IB40126J
Kim, H. J. et al. Lab Chip 12, 2165–2174 (2012): https://doi.org/10.1039/C2LC40074J
Extended data
Extended Data Fig. 1 Dimensions of the linear and the convoluted gut-on-a-chip designs.
a,b, Dimensions of the linear gut-on-a-chip (a) and the convoluted gut-on-a-chip (b). The design of the upper and the lower microchannels of the linear gut-on-a-chip is identical. In contrast, the design of the upper and the lower microchannel of the nonlinear convoluted gut-on-a-chip has a slightly different geometry. c, Height of the upper and the lower microchannels as well as the porous membrane of the gut-on-a-chip device. d, Dimensions and top-down view of the porous membrane. The individual circles indicate the pores of a membrane. This pattern can be used to make the membrane silicon mold that contains an array of micropillars (5 cm by 5 cm in x–y dimension of the pore array) with 20 µm height.
Extended Data Fig. 2 Dimensions of the hybrid chip.
Dimensions of the hybrid chip. A biopsy puncher will be used to make a Transwell-insertable module (see Step 5B(i)).
Extended Data Fig. 3 Expression of a stem cell marker LGR5.
The fluorescence image visualizes the expression of LGR5 on the organoid epithelium cultured in a gut-on-a-chip at days 3 and 13. Nuclei (cyan) and F-actin (gray) are also shown. This figure is comparable to Fig. 3f, where the co-localization of LGR5 with MUC2 was independently visualized by removing the MUC2 signal. Adapted with permission from ref. 12 under Creative Commons licence CC BY 4.0.
Supplementary information
Rights and permissions
About this article

Cite this article
Shin, W., Kim, H.J. 3D in vitro morphogenesis of human intestinal epithelium in a gut-on-a-chip or a hybrid chip with a cell culture insert. Nat Protoc 17, 910–939 (2022). https://doi.org/10.1038/s41596-021-00674-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41596-021-00674-3
This article is cited by
-
Microfluidic high-throughput 3D cell culture
Nature Reviews Bioengineering (2024)
-
Hybrid chips to enable a sustainable internet of things technology: opportunities and challenges
Discover Materials (2024)
-
Revolutionizing the female reproductive system research using microfluidic chip platform
Journal of Nanobiotechnology (2023)
-
Modelling host–microbiome interactions in organ-on-a-chip platforms
Nature Reviews Bioengineering (2023)
-
Generating human blastoids modeling blastocyst-stage embryos and implantation
Nature Protocols (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
