
Abstract
Macroautophagy and microautophagy are highly conserved eukaryotic cellular processes that degrade cytoplasmic material in lysosomes. Both pathways involve characteristic membrane dynamics regulated by autophagy-related proteins and other molecules, some of which are shared between the two pathways. Over the past few years, the application of new technologies, such as cryo-electron microscopy, coevolution-based structural prediction and in vitro reconstitution, has revealed the functions of individual autophagy gene products, especially in autophagy induction, membrane reorganization and cargo recognition. Concomitantly, mutations in autophagy genes have been linked to human disorders, particularly neurodegenerative diseases, emphasizing the potential pathogenic implications of autophagy defects. Accumulating genome data have also illuminated the evolution of autophagy genes within eukaryotes as well as their transition from possible ancestral elements in prokaryotes.
Similar content being viewed by others

Introduction
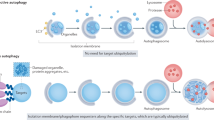
Autophagy (‘self-eating’) is a collection of processes by which cellular components such as proteins and organelles are delivered to the lysosome or vacuole for degradation. Two autophagy pathways are well conserved in eukaryotes: macroautophagy and microautophagy (Box 1). In macroautophagy, a cup-shaped membrane structure (known as the phagophore or isolation membrane) emerges near the endoplasmic reticulum (ER), elongates, bends and finally closes via membrane fission to form a double-membraned structure called the autophagosome, capturing part of the cytoplasm inside itself (Fig. 1). The autophagosome then fuses with lysosomes (or the vacuole in yeasts and plants), where its contents are degraded. In microautophagy, the endosomal or lysosomal membrane invaginates inwards to capture part of the cytoplasm directly (Fig. 2).

a, (1) At initiation of macroautophagy, the ULK complex assembles near the ER membrane upon starvation and recruits ATG9 vesicles via its interaction with the ATG13–ATG101 subcomplex. (2) Alternatively, cargo adaptors such as p62, NDP52 and TAX1BP1 induce assembly of the ULK complex via interaction with FIP200, whereas ATG9 vesicles are recruited by OPTN. At the membrane-elongation step, the ULK complex recruits the class III phosphatidylinositol 3–kinase complex I (PI3KC3–C1) that produces PI(3)P, which further recruits its effector proteins, DFCP1 to omegasomes and WIPI2 and WIPI4 to phagophores. WIPI4 directs ATG2 to the phagophore membrane, which transfers phospholipids from the ER in concert with ATG9, VMP1 and TMEM41B (see b). WIPI2 recruits the ATG12–ATG5–ATG16L1 complex to promote LC3 lipidation on the phagophore membrane. Autophagosomes are closed by the action of the ESCRT machinery. Subsequently, PLEKHM1, EPG5 and RAB7 tether autophagosomes with lysosomes, and the two SNARE complexes, STX17–SNAP29–VAMP7/8 and YKT6–SNAP29–STX7, trigger fusion. After prolonged starvation, lysosomal membrane proteins on autolysosomes are recycled via autophagic lysosome reformation (ALR), whereas autophagosomal membrane proteins are recycled via autophagosomal components recycling (ACR). b, The lipid transfer protein ATG2 tethers the ER and phagophore membranes and transfers phospholipids from the ER to the phagophore. ATG9 on the phagophore membrane and VMP1 and TMEM41B on the ER membrane scramble phospholipids. c, In selective macroautophagy, cargos are recognized in a ubiquitin (Ub)-dependent manner (through Ub-binding adaptors) or a Ub-independent manner. Cargo adaptors/receptors bind to ATG8 on the autophagic membrane. d, In macro-fluidophagy, the phagophore membrane adheres to fluid-like condensates via membrane wetting.

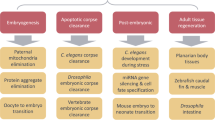
a, Microautophagy involves the invagination of endosomal membranes (left) or lysosomal membranes (right) to incorporate cytoplasmic material. The resulting intraluminal vesicles are degraded inside lysosomes or the vacuole. b, Cargos are recognized by (1) ATG8, (2) Nbr1, (3) HSC70 or (4) other proteins. Microautophagy can also uptake cytoplasmic materials non-selectively. c, Several types of microautophagy in mammals, yeasts, flies and plants are summarized. The numbers in the ‘Recognition’ column correspond to those in panel b. The ‘ATGs’ column indicates dependency on autophagy-related (ATG) proteins. Asterisks indicate dependence on only ATG8. Sp, Schizosaccharomyces pombe. Ub, ubiquitin.
The macroautophagy pathway was first observed in mammalian cells in the 1960s, and its discovery in Saccharomyces cerevisiae in the early 1990s fuelled rapid progress in the field, including the isolation of autophagy-deficient mutants, the identification of autophagy-related (ATG) genes and the characterization of related functional protein complexes1. Similarly, much of the progress in microautophagy research occurred in S. cerevisiae and other yeast species2.
The most fundamental physiological function common to both macro- and microautophagy is thought to be the supply of nutrients in times of need, such as during starvation or development2,3,4,5,6. In addition, autophagy maintains cellular homeostasis by selectively degrading proteins, organelles and foreign entities2,3,4; this process is particularly important for long-living cells such as neurons. Occasionally, autophagy also delivers vacuolar hydrolases to fulfill biosynthetic roles in yeasts7,8. Multiple ATG genes have also been implicated in the function of non-autophagic pathways, including endocytosis, secretion and extracellular vesicle biogenesis9. Owing to these versatile functions, ATG genes have attracted attention in various research fields, and the scope of research continues to grow.
In recent years, much progress has been made in understanding the functions of individual ATG genes as well as their roles in disease and physiology. The molecular functions of key players have been revealed, and the importance of cargo-driven mechanisms and phase separation have been highlighted. These findings were made possible by the application of new technologies such as cryo-electron microscopy, coevolution-based structural prediction and in vitro reconstitution. In addition to the molecular machinery, another focus of autophagy research is its role in pathophysiology. Genetic analyses of human diseases (for example, neurodegenerative disorders and autoimmune diseases) have identified several ATG genes as causative genes or risk factors in the past decade4,10. Last, recent genome data have elucidated the evolution of autophagy genes within eukaryotes and possibly from prokaryotes.
In this Review, we summarize recent progress in the molecular and cellular biology of genes involved in macro- and microautophagy, the pathological relevance of these genes and key evolutionary aspects. We do not cover the third type of autophagy, chaperone-mediated autophagy, which does not involve membrane dynamics and is regulated by an entirely different set of proteins11.

Genes regulating macroautophagy
Initiation of macroautophagy
The ULK complex — which is composed of the scaffold protein FIP200 (also known as RB1CC1), ATG13, ATG101 and the serine/threonine kinases ULK1 or ULK2 — is the central regulator in the initiation of macroautophagy (Table 1, Fig. 1a). Its activity is suppressed primarily by mechanistic target of rapamycin complex 1 (mTORC1). Upon starvation, mTORC1 is inactivated, leading to the activation and assembly of the ULK complex in the vicinity of the ER membrane, which recruits downstream ATG proteins to initiate autophagosome formation (Fig. 1a, Initiation). In yeasts, the homologous Atg1 complex forms similar assembled structures known as pre-autophagosomal structures (PASs). The PAS is a higher-order assembly of the Atg1 complexes, in which Atg13 tethers Atg1 (homologous to ULK1/2) and the Atg17–Atg29–Atg31 subcomplex (Atg17 is homologous to part of FIP200)12,13. The PAS is a fluid-like condensate resulting from liquid–liquid phase separation, which is driven by the multivalent interactions between Atg1, Atg13 and Atg17 (ref. 13), and provides an environment that intermolecularly auto-activates the Atg1 kinase14. Thus, ULK/Atg1 complex assembly is a key step in initiating macroautophagy.
In addition to starvation-induced assembly, ULK/Atg1 complex assembly is also driven by autophagy cargos (Fig. 1a, Initiation). The soluble autophagy cargo adaptor SQSTM1 (also called p62) forms fluid-like condensates with ubiquitinated proteins and interacts with FIP200 to recruit the ULK complex15. (In this Review, ‘adaptor’ refers to a soluble protein that mediates binding between a cargo and the autophagy machinery, whereas ‘receptor’ refers to a cargo-resident protein that binds to the autophagy machinery.) During Parkin-dependent mitophagy (see below) and selective degradation of Salmonella, NDP52 (also called CALCOCO2) — another soluble autophagy adaptor — localizes to ubiquitinated mitochondria and the cytosol-invading bacteria and recruits the ULK complex via interaction with FIP200 (refs. 16,17). TAX1BP1 also recruits FIP200 to SQSTM1 condensates18. Ubiquitin-independent selective macroautophagy also involves the cargo-driven assembly of the ULK complex: the ER-phagy receptor CCPG1 interacts with the Claw domain of FIP200 to recruit the ULK complex19,20.
Upon assembly, the ULK complex recruits ATG9 vesicles (Fig. 1a, ‘Initiation’), which are thought to be the seeds for autophagosome formation21. ATG9 vesicle recruitment is achieved via interactions between the HORMA domains of the ATG13–ATG101 subcomplex and the most C-terminal region of ATG9A22 (Table 1). During Parkin-dependent mitophagy, ATG9 vesicles are also recruited through binding with the autophagy adaptor OPTN, which is present on ubiquitinated mitochondria23. Similarly, in yeast, Atg9 vesicles are recruited via two pathways: one through interaction with the HORMA domain of Atg13 during starvation-induced macroautophagy, and the other through the cargo adaptor Atg11 during selective macroautophagy24.
Membrane elongation by lipid transfer
The ULK complex also recruits the class III phosphatidylinositol 3-kinase complex I (PI3KC3–C1), which produces PI(3)P in autophagic membranes (Fig. 1a, ‘Membrane elongation’). The PI3KC3–C1 is composed of five subunits: VPS34, VPS15, BECN1 (Vps30 in yeast), ATG14 and NRBF2 (Atg38 in yeast) (Table 1). PI3KC3–C1 binds to membranes via ATG14, BECN1 and VPS34, after which VPS34 generates PI(3)P25,26.
The PI(3)P effectors include the β-propellers that bind polyphosphoinositides (PROPPIN) family proteins (WIPI proteins in mammals, and Atg18, Atg21 and Hsv2 in yeast), which further recruit ATG2. ATG2 forms a rod-shaped structure that attaches to the ER membrane with its N-terminal tip and the autophagic membrane with its C-terminal tip27,28 (Fig. 1b). In vitro studies have shown that phospholipids are transferred through a hydrophobic cavity in ATG2, suggesting that this process occurs between the ER and phagophore in vivo29,30,31. However, the mechanism by which the transfer activity is regulated and what drives it remain to be elucidated.
There are four PROPPIN family proteins (WIPI1–4) in mammals. ATG2 is directed to PI(3)P-rich autophagic membranes, probably through the interaction with WIPI3 or WIPI4 (refs. 27,32). Of the four homologues, WIPI2 functions dominantly, as depletion of only WIPI2 profoundly suppresses autophagy33, upstream of WIPI3 and WIPI4 (ref. 34). WIPI2 binds to ATG16L1, a component of the ATG12–ATG5–ATG16L1 complex (Table 1), which has an E3-like activity to promote lipidation of ATG8 proteins (LC3 and GABARAP family proteins in mammals, which are collectively called ATG8 hereafter). ATG8 can further recruit ATG2, as ATG2 has a LC3-interacting region (LIR) (see below)35. Thus, WIPI2 also contributes to ATG2 recruitment indirectly.
After ATG2 transfers lipids to the outer leaflet of the autophagic membrane, ATG9 scrambles phospholipids in the membrane (Fig. 1b). ATG9 forms a trimer and translocates phospholipids between the outer and inner leaflets36,37,38. There are also two phospholipid scramblases required for autophagosome formation in the ER membrane: VMP1 and TMEM41B38,39,40 (Fig. 1b). Both proteins are multi-spanning membrane proteins with a conserved DedA domain predicted to have two characteristic re-entrant loops41,42 (Box 1). Because ATG2A interacts with ATG9 as well as VMP1 and TMEM41B38, VMP1/TMEM41B–ATG2–ATG9 may be considered a lipid transfer unit. These scramblases can equilibrate the imbalance of phospholipid density on each leaflet caused by lipid transfer (Fig. 1b). However, if local lipid synthesis in the outer leaflet of the ER membrane produces pressure for directional lipid transfer, lipid scrambling may weaken that activity. Moreover, VMP1 and TMEM41B are important for the formation of not only autophagosomes but also lipid droplets, lipoproteins and the double-membrane structures required for the replication of several RNA viruses (including SARS-CoV-2)43. Therefore, their scrambling activity may be crucial for a fundamental function of the ER, not only in providing lipids to ATG2. Yeast has a TMEM41B-like protein, Tvp38, but it is not required for autophagosome formation41. The mechanism by which yeast can form autophagosomes without DedA family proteins has yet to be elucidated.
Two ubiquitin-like conjugation systems, the ATG8 and ATG12 systems, also have key roles in autophagosome formation. In concert with the E1-like enzyme ATG7 and the E2-like enzymes ATG3 and ATG10, ATG8 and ATG12 are conjugated to phosphatidylethanolamine (PE) and ATG5, respectively. The ATG12–ATG5–ATG16(L1) complex has an E3-like activity for ATG8–PE conjugation. Although ATG conjugation systems are essential for autophagosome formation in yeasts, seemingly normal autophagosomes can be generated in mammalian cells even when these systems are disrupted44,45,46. They seem to be more important for fusion with lysosomes or the degradation of the autophagosomal inner membrane47,48 and, therefore, are still crucial for autophagic flux.
Cargo recognition
Because autophagosomes engulf part of the cytoplasm (approximately 0.5–1 μm in diameter), the majority of soluble cargos are believed to be incorporated non-selectively. However, autophagosomes can also selectively recognize various cargos, including damaged organelles, intracellular bacteria and certain proteins49. Besides its crucial role in membrane dynamics, ATG8 has a central function in cargo recognition in selective macroautophagy (Fig. 1c). ATG8 directly interacts with the LIR motifs (or the Atg8-interacting motif in yeast) in cargos or autophagy adaptors, which are classified into ubiquitin-dependent and -independent types (Table 2). Ubiquitin-recognizing soluble cargo adaptors include SQSTM1, NBR1, NDP52, OPTN, TAX1BP1 and TOLLIP49. The ubiquitin-independent category includes organelle-bound receptors for ER-phagy (for example, CCPG1, TEX264, FAM134B, SEC62, RTN3L and ATL3) and mitophagy (for example, BNIP3, BNIP3L/NIX, FUNDC1, FKBP8 and BCL2L13) as well as LIR-containing soluble proteins such as CALCOCO1 (ref. 49). Besides cargo recognition, LIRs are used for the recruitment of some core autophagy factors, including FIP200, ULK1 and ATG13 (ref. 50), as well as the tethering factors PLEKHM1 and EPG5 (refs. 51,52). As discussed above, selective cargos can also recruit the ULK complex, constituting another layer of cargo recognition (Fig. 1a, ‘Initiation’).
In addition to organelles and individual proteins, fluid-like condensates formed by liquid–liquid phase separation are degraded by macroautophagy entirely or in a piecemeal manner (termed fluidophagy)53 (Fig. 1d). As shown in the degradation of SQSTM1 condensates, the adherence of fluid-like condensates to ATG8-positive autophagic membranes is promoted by a membrane-wetting effect53.
Closure of autophagosomes
Autophagosome closure is mediated by membrane scission of the phagophore membrane into the outer and inner autophagosomal membranes (Fig. 1a). This is topologically identical to membrane scission of the intraluminal vesicle formation in multivesicular bodies and virus budding at the plasma membrane. Indeed, like these two processes, the endosomal sorting complex required for transport (ESCRT) complex has a pivotal role in autophagosome closure54,55,56 (Table 2). How the ESCRT complex localizes to the open rim of the phagophore remains unknown in mammals. However, in yeast, Rab5-dependent interactions between Atg17 and Snf7, an ESCRT-III component, may account for the ESCRT recruitment to phagophores55.
Autolysosome formation and recycling
After closure, autophagosomes fuse with lysosomes to become autolysosomes. In mammals, fusion is mediated by the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins STX17 and YKT6, which are recruited to autophagosomes when they are closed57,58 (Table 2), thereby preventing premature fusion between unclosed autophagosomes and lysosomes. STX17 and YKT6 form SNARE bundles with cytosolic SNAP29 and lysosomal VAMP7/VAMP8 and STX7, respectively (Fig. 1a, ‘Lysosome fusion’). The relationship and functional differences between these two autophagosomal SNAREs (STX17 and YKT6) are not well understood; thus far, they seem to function redundantly. The role of YKT6 in autophagosome fusion is conserved in yeast59,60, but STX17 is absent in yeast. Tethering between autophagosomes and lysosomes is mediated by multiple tethering factors, including HOPS, PLEKHM1 and EPG5 (ref. 61). PLEKHM1 (ref. 51) and EPG5 (ref. 52) have a LIR motif and therefore interact with autophagosomal ATG8.
After fusing with lysosomes, the inner autophagosomal membrane is degraded. In yeast, autophagosomes fuse with the large vacuole, releasing autophagic bodies surrounded by the autophagosomal inner membranes. The vacuolar phospholipase Atg15 is responsible for degrading the membranes of autophagic bodies (derived from the autophagosomal inner membrane)62. Most organisms, including mammals, do not have Atg15 homologues but instead possess multiple lysosomal phospholipases, which might be redundantly involved in inner membrane degradation. One major remaining question is how degradation is limited to only the inner membrane when both the outer and inner membranes, which are derived from the same phagophore membrane, are exposed to lysosomal enzymes.
After prolonged starvation, autolysosomes deform to generate protolysosomes that then mature into functional lysosomes, in a process called autophagic lysosome reformation (ALR)63 (Fig. 1a, ‘Recycling’). This process recycles lysosomal membrane proteins and is triggered by the reactivation of mTORC1 in response to increased amino acid levels owing to prolonged macroautophagy. Upon induction of ALR, the PI(4)P 5-kinase PIP5K1B produces PI(4,5)P2 on the membrane of autolysosomes, generating PI(4,5)P2-rich microdomains that are further organized by clathrin and adaptor protein 2 (AP2)64 (Table 2). Lysosomal membrane proteins are captured in these microdomains, which subsequently undergo tubulation driven by the kinesin heavy chain KIF5B65. The mechanism by which protolysosomes mature into lysosomes remains to be elucidated.
An additional autolysosome recycling mechanism called autophagosomal components recycling (ACR) was recently reported66. This mechanism recycles autophagosome-derived membrane-anchoring proteins such as ATG9 and STX17 via the budding of autolysosomal membranes depending on the SNX4–SNX5–SNX17 recycler complex and the dynein–dynactin complex (Table 2) (Fig. 1a, ‘Recycling’). Thus, autophagosomal and lysosomal membrane proteins are recycled by ACR and ALR, respectively. ACR occurs earlier than ALR.
Non-autophagic functions
Although ATG proteins were originally identified in yeast as factors required for autophagy, it is now apparent that most of them also participate in non-autophagic processes67 (Table 1). We discuss only a few of these functions here owing to space limitations. Many of the non-autophagic functions of ATG proteins are related to membrane dynamics. BECN1 is a component of not only PI3KC3–C1 but also PI3KC3–C2 (containing UVRAG instead of ATG14), which is involved in the endocytic pathway. The tumour-suppressing activity of BECN1 is at least partially attributable to its non-autophagic function in PI3KC3–C2 (ref. 68). Another well recognized non-autophagic process is the conjugation of ATG8 to single-membrane compartments in the endocytic pathway rather than double-membrane autophagosomes, which is known as conjugation of ATG8 to endolysosomal single membranes (CASM) or single-membrane ATG8 conjugation (SMAC), including LC3-associated phagocytosis (LAP) and LC3-associated endocytosis (LANDO)69,70,71. LAP is one of the phagocytotic pathways and degrades its contents by fusion with lysosomes. LAP does not require the ULK complex but does require the ATG conjugation systems. Notably, the WD40 repeat domain of ATG16L1, which is absent in yeast Atg16 (Table 1), is essential for LAP through binding to V-ATPase, but not for canonical autophagy72,73,74,75,76. The PI3KC3–C2 component UVRAG and RUBCN are also required. A recent study reported that during LAP (or CASM), ATG8 is conjugated not only to PE, but also to phosphatidylserine (PS)77. It is unknown whether ATG8–PS has a unique function distinct from that of ATG8–PE. Secretory autophagy (autophagy-based unconventional secretion) is another type of non-autophagic process. In secretory autophagy, closed autophagosomes do not fuse with lysosomes, but instead fuse with the plasma membrane, secreting cytosolic proteins lacking conventional leader sequences. ATG proteins are required to form autophagosomes in this pathway, and inhibition of autophagosome–lysosome fusion leads to upregulation of secretory autophagy78. In addition, autophagy-related genes are involved in viral replication and transmission. Many viruses exploit autophagosome-like vesicles for replication and exocytosis79. A genome-wide CRISPR screen identified TMEM41B and VMP1 as host factors for flaviviruses and coronaviruses, including SARS-CoV-2, where they are thought to participate in the formation of specialized replication organelles43. Other examples of non-autophagic membrane-related processes that require ATG genes include ER-to-Golgi trafficking (ULK1 and ULK2)80, protection against plasma membrane permeabilization (ATG9A)81, and TFEB activation during lysosomal damage or lysosomal transient receptor potential mucolipin channel 1 (TRPML1) activation (ATG conjugation systems)82,83,84. In apicomplexan parasites such as Plasmodium and Toxoplasma, ATG8 (and the ATG conjugation systems) are essential for the biogenesis of apicoplasts85,86, non-photosynthetic plastids specific to this lineage that support key metabolic functions.
ATG genes can also regulate various non-membrane-related processes, such as cell cycle progression and cell death. ATG7 directly interacts with p53, and ATG7-deficient cells show impaired cell cycle arrest and increased apoptosis (both are p53-mediated) upon nutrient starvation87. ATG12 promotes mitochondrial apoptosis by binding to and inactivating Bcl-2 family proteins88. GABARAPs are required for interferon-γ (IFNγ)-mediated antimicrobial responses through binding to ADP-ribosylation factor 1 (ref. 89).
Genes regulating microautophagy
Membrane dynamics of microautophagy
Microautophagy was first described in mammalian cells as a process involving the invagination of lysosomal membranes that incorporate cytosolic material into lysosomes, followed by membrane fission and degradation90,91 (Fig. 2a). Because observing microautophagy in small lysosomes is difficult by light microscopy, the underlying molecular mechanisms have been primarily revealed in yeasts and plants, where the vacuole is sufficiently large for optical observation. Like macroautophagy, microautophagy can be both non-selective and selective; the process non-selectively enwraps cytosolic material but also selectively recognizes organelles, such as peroxisomes (micropexophagy), mitochondria (micromitophagy), lipid droplets (microlipophagy), a subdomain of the ER (microER-phagy), a portion of the nucleus (micronucleophagy), and photodamaged chloroplasts (microchlorophagy)2,92,93. Microautophagy requires the ESCRT machinery at the membrane fission step2. However, its dependency on ATG proteins is complicated and may differ among cargo types and inducing conditions. While ATG proteins seem to be dispensable in general microautophagy and microER-phagy, at least some of the ATG proteins are required for microlipophagy94,95,96, micropexophagy97,98, micromitophagy99 and micronucleophagy100,101 in yeast as well as microchlorophagy102 in plants (Fig. 2b). In mammals, micromitophagy and microlipophagy seem to be independent of ATG proteins103,104, whereas micronucleophagy mediated by cGAS requires the ATG8 conjugation system for cargo recognition105. Thus, microautophagy can be roughly divided into two types: ATG-independent and ATG-dependent (Fig. 2c). In the latter type, ATG proteins can be involved in the formation of additional membrane structures, the remodelling of vacuolar morphology, and/or the recognition of selective cargos (see below)2,92,93.
In mammalian cells, multivesicular body formation of endosomes is considered to be a type of microautophagy referred to as endosomal microautophagy106. Endosomal microautophagy occurs constitutively and is also induced during early periods of amino acid starvation, leading to the degradation of cytosolic proteins, particularly selective macroautophagy adaptors, such as SQSTM1, NDP52, NBR1, TAX1BP1 and NCOA4 (an adaptor for ferritin)5 (Fig. 2b). The multivesicular body pathway in yeast is also known to be induced by starvation and contributes to early proteome remodelling during starvation6. Starvation-induced endosomal microautophagy in mammals requires ESCRT-III (CHMP4B) and VPS4 but not ESCRT-0, -I or -II (ref. 5). The necessity of ATG proteins in this pathway is also complicated. The ATG8 conjugation system is required for the degradation of SQSTM1 and NDP52 and partially required for NBR1 and TAX1BP1 but not for NCOA4, whereas FIP200 and VPS34 are not required for any of them5. Therefore, ATG proteins should be important for cargo recognition rather than membrane dynamics in this case (Fig. 2c).
Endosomal intraluminal vesicles formed by microautophagy are directed to lysosomes for degradation, but they are also secreted to the extracellular space in mammals. Several RNA-binding proteins, including HNRNPK and SAFB, are incorporated into endosomal intraluminal vesicles by the LC3-dependent endosomal microautophagy-like pathway, referred to as LC3-dependent extracellular vesicle loading and secretion (LDELS)107. This process differs from canonical endosomal microautophagy in that it is independent of ESCRTs; however, it is dependent on ceramide produced by neutral sphingomyelinase 2 (nSMase2, also known as SMPD3), which is an alternative pathway of endosomal membrane invagination (Table 2).
Cargo recognition
For cargo recognition, ATG-dependent microautophagy in yeast utilizes Atg8 and/or Atg11, which interact(s) with organelle-bound selective macroautophagy receptors, including Atg30 in micropexophagy108 and Atg39 in micronucleophagy101 (part (1) of Fig. 2b,c). Additionally, in mammals, the ER-phagy receptor SEC62 mediates microER-phagy in an ATG8 binding-dependent manner109.
By contrast, cargo recognition in ATG-independent microautophagy is not well understood. Specific subdomain formation may be important. For example, in microER-phagy in yeast, the ER membrane forms multilamellar whorls consisting of ribosome-free ER membrane to be subjected to microautophagy110. In Schizosaccharomyces pombe, microautophagy is used for a biosynthesis pathway for vacuolar enzymes, termed the Nbr1-mediated vacuolar targeting (NVT) pathway (part (2) of Fig. 2b,c), which is functionally similar to the cytoplasm-to-vacuole targeting (Cvt) pathway in S. cerevisiae but uses a different route8,111. This pathway is independent of ATG proteins but requires Nbr1 to recognize its cargos, such as aminopeptidases (Ape2, Ape4 and Lap2) and α-mannosidase (Ams1). In the NVT pathway, recruitment of Nbr1 to the endosomal membranes is mediated by ubiquitin8, in sharp contrast to the process of macroautophagy, in which mammalian NBR1 (or its yeast homologue Atg19) is recruited by ATG8.
In mammalian cells, fluid-like ferritin–NCOA4 condensates are subjected to macroautophagy and endosomal microautophagy, both of which require TAX1BP1 for incorporation112,113. Because TAX1BP1 interacts with NCOA4, TAX1BP1 can bridge autophagosomal ATG8 and ferritin–NCOA4 condensates in macroautophagy. However, the mechanism by which TAX1BP1 mediates the incorporation of ferritin–NCOA4 condensates to endosomes has yet to be elucidated, because it is largely independent of ATG8 (ref. 5).
During endosomal microautophagy in mammals, cargo recognition is achieved, in part, by the cytosolic chaperone HSC70/HSPA8 (part (3) of Fig. 2b,c)106. HSC70 recognizes the KFERQ-like motif contained in selective cargos and incorporates them into endosomes (the KFERQ-like motif was originally identified as a signal for chaperone-mediated autophagy11) (Fig. 2b). In this process, HSC70 binds to PS on the endosomal membrane via its cationic domain and induces inward membrane deformation106,114. This KFERQ-dependent endosomal microautophagy is also conserved in Drosophila, despite its lack of chaperone-mediated autophagy115. Notably, this pathway requires Atg1 and Atg13, but not Atg5, Atg7 or Atg12.
In LDELS, ATG8 is required for recognizing the LIR sequence of RNA-binding proteins such as HNRNPK and SAFB107. Membrane invagination occurs even without ATG8 lipidation, but resultant intraluminal vesicles do not contain selective cargos. How ATG8 translocates to endosomes is unknown. A mechanism similar to LAP may be used. Another issue warranting investigation is why only a subset of microautophagy cargos depend on ATG8 to be recognized in both ESCRT-dependent endosomal microautophagy and LDELS.
Autophagy gene mutations and polymorphisms in human diseases
Given the crucial roles of autophagy in various physiological processes, including stress responses and intracellular clearance, it has been postulated that autophagy is involved in the pathogenesis of human diseases. However, it is difficult to determine which diseases are associated with changes in autophagy owing to a lack of methods with which to measure autophagic activity in humans. Nevertheless, recent genetic studies have identified a number of mutations in autophagy-related genes associated with human diseases, suggesting that autophagy alteration contributes to the development of these diseases. Moreover, studies using acute systemic Atg7 knockout and Fip200 knockout mice116,117 and brain-rescued systemic Atg5 knockout mice118 suggest that organs highly susceptible to autophagy deficiency include the nervous system, immune system, liver and intestine. Consistent with these findings, these tissues are often affected in autophagy-gene-related diseases (Tables 3,4). In this section, mutations and polymorphisms of genes involved in general and selective autophagy are discussed. However, it is important to consider that, as emphasized above, most of these autophagy genes also have non-autophagic functions (Table 1). Therefore, the identification of mutations in autophagy genes does not directly implicate a defect in canonical autophagy in the disease phenotype. The involvement of non-autophagic function should always be considered in the interpretation of these mutations.
Mendelian disorders caused by autophagy gene mutations
Autophagy-related diseases include Mendelian disorders caused by mutations in autophagy genes (Table 3). The most frequently affected tissue seems to be the nervous system. Homozygous mutations in ATG5 and ATG7 were found to be associated with human neurological diseases119,120. Autophagy is suppressed in these diseases, but only partially, because small amounts of either the ATG12–ATG5 conjugate or LC3-II (the lipidated form) can be detected. Patients with these diseases arising from mutations in ATG5 and ATG7 show some overlapping phenotypes, including ataxia and developmental delay. Patients with ATG7 mutations also show abnormal cerebellum and corpus callosum structure and facial dysmorphism (it is unknown whether patients with ATG5 mutations have these abnormalities). SQSTM1 accumulates inpatient-derived cells, confirming reduced autophagic flux119,120.
Mutations in the PROPPIN family of proteins also cause neurodegenerative diseases, but their phenotypes are somewhat different. A homozygous mutation in WIPI2 was found in patients with a complex developmental disorder known as intellectual developmental disorder with short stature and variable skeletal anomalies (IDDSSA)121,122,123,124. The detected Val249Met mutation reduces WIPI2–ATG16L1 binding and autophagic flux123. Homozygous nonsense mutations in WDR45B/WIPI3 cause neurodevelopmental disorder with spastic quadriplegia and brain abnormalities with or without seizures (also called El-Hattab–Alkuraya syndrome)125,126,127,128. The WDR45/WIPI4 gene is found on the X chromosome, and its heterozygous mutations in women and hemizygous mutations in men cause β-propeller protein-associated neurodegeneration (BPAN; originally called static encephalopathy of childhood with neurodegeneration in adulthood)129,130,131. This is a biphasic disease that demonstrates infant-onset, non-progressive psychomotor retardation, epilepsy and autism as well as adolescent-onset dystonia, Parkinsonism and dementia. Iron accumulation in the globus pallidus and substantia nigra is one of the hallmarks of this disease, but its relationship with ferritinophagy is unclear because iron accumulation has not been reported in other diseases related to autophagy gene mutations. Given that the deletion of either WIPI3 or WIPI4 suppresses autophagy only mildly compared with deletion of WIPI2, ATG5 or ATG7 (N.M., unpublished results), it is plausible that defects in yet-unknown non-autophagic functions of WIPI3 and WIPI4 may account for the severe phenotype observed in these diseases.
Mutations in genes related to selective autophagy also cause disease (Table 3). The mitophagy-related genes PARK2/PRKN (encoding Parkin) and PARK6/PINK1 are mutated in juvenile-onset familial Parkinson disease132. Parkin, a ubiquitin ligase, ubiquitinates various proteins in depolarized mitochondria in a PINK1-dependent manner, recruiting autophagy adaptors such as NDP52 (ref. 16) and OPTN (Fig. 1a, ‘Initiation’)23. Although Parkin- and PINK1-dependent mitophagy is clearly observed in cell culture, its physiological relevance was initially unclear because Prkn or Pink1 knockout mice show almost normal basal mitophagy levels without an obvious phenotype under normal conditions133,134. However, recent studies revealed that aged Prkn knockout mice develop locomotor impairments associated with dopaminergic neuronal loss135. Intestinal infection could also promote neurodegeneration in Prkn knockout mice136. Furthermore, Parkin- and PINK1-dependent mitophagy is physiologically important to suppress the release of mitochondrial DNA into the cytosol and subsequent inflammation under stress conditions in vivo137,138. By contrast, another report suggests that PINK1-dependent mitophagy in endothelial cells could be pro-inflammatory via the release of mitochondrial formyl peptides139. Thus, the pathophysiological role of Parkin- and PINK1-dependent mitophagy may depend on cell type or context. Mutations in autophagy adaptors such as SQSTM1 (ref. 140 and the ER-phagy receptor FAM134B141 are also found in childhood-onset neurodegeneration and hereditary sensory and autonomic neuropathy type II, respectively, suggesting that defects in selective autophagy may cause these diseases (Table 3).
Although the diseases listed above are recessive, some exhibit dominant inheritance, which includes amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD) (these two are often associated) and Paget disease of bone. Autosomal dominant mutations in the selective autophagy-related genes SQSTM1, OPTN and TBK1 are found in association with these diseases (Table 4). TBK1 phosphorylates and regulates OPTN, but in addition to this canonical role, TBK1 can also directly recruit the PI3KC3–C1 complex in OPTN-dependent mitophagy142. In general, many ALS/FTD-related gene mutations, for example, those in SOD1 and TARDBP (encoding TDP-43), are thought to be gain-of-function mutations and show dominant inheritance143,144. However, the ALS/FTD-related mutations of OPTN and TBK1 are likely to be loss-of-function mutations144. Both autosomal recessive and dominant inheritance patterns have been reported in ALS with OPTN mutations145. Thus, haploinsufficiency of OPTN and TBK1 could contribute to the ALS/FTD spectrum. Although some of the ALS/FTD-associated mutations affect the adaptor function of SQSTM1, OPTN and TBK1 (refs. 146,147), it remains unknown whether this is the general mechanism. The pathogenic effects of these mutations might be mediated by their non-autophagic roles; for example, the TBK1–OPTN axis is also important for the innate immune and RIPK1-dependent cell death pathways144. However, a gain-of-function hypothesis cannot be entirely excluded. Of particular interest is that, like other ALS-related proteins148, autophagy adaptors such as SQSTM1 (refs. 149,150,151) and OPTN152 form liquid-like biomolecular condensates. Mutations of these genes may exhibit some gain of toxicity.
Although most Mendelian disorders associated with autophagy gene mutations are related to the nervous system, there are some diseases involving other tissues and organs (Table 3). An example is Paget disease of bone, which is characterized by one or multiple focal regions with increased bone remodelling. Of its causative genes, SQSTM1 is the major one; however, it remains elusive whether its autophagy adaptor function is involved in the pathogenesis of this disease153,154.
Genetic risk factors for human diseases
The second category includes diseases whose susceptibility is associated with polymorphism of autophagy-related genes (Table 4). The core autophagy gene first shown to be associated with human disease by a genome-wide association study (GWAS) is ATG16L1; the single nucleotide polymorphism resulting in the Thr300Ala (T300A) substitution is a risk factor for Crohn’s disease155,156; Thr300 is located immediately upstream of the WD40 repeat domain (Table 1). Atg16L1T300A knock-in mice exhibit abnormalities in Paneth cells in the intestine157 and gut microbiota158. The WD40 repeat domain in ATG16L1 is essential for LAP but not for canonical autophagy72; however, the effect of the T300A substitution on autophagy and LAP is relatively small or undetectable157,159,160. Nevertheless, it is possible that autophagy is associated with Crohn’s disease because the disorder has also been linked to other autophagy genes (ULK1, ATG9A, NDP52 and ATG4C)161,162,163,164. However, the fact that ATG16L2, which is considered to be unnecessary for autophagy165,166, is also associated with Crohn’s disease167 suggests that a yet unknown non-autophagic function shared by ATG16L1 and ATG16L2 may be involved.
Autophagy genes such as ATG5, ATG7 and MAP1LC3B have also been identified as susceptibility genes in autoimmune diseases, including systemic lupus erythematosus (SLE)168 (Table 4). This may reflect the role of autophagy in mitochondrial quality control to suppress the release of SLE-inducible damage-associated molecular patterns from mitochondria169,170. In addition to canonical autophagy, these genes are also required for LAP. Because LAP is important for interferon production in response to the incorporation of DNA-containing immune complexes171, the role of autophagy genes in LAP may be related to their genetic association with autoimmune diseases. Association of ATG16L2 with SLE was also identified, but the role of ATG16L2 in LAP is unclear.
Whole-exome sequencing and subsequent missense variant searches in patients with non-alcoholic fatty liver disease revealed an enrichment of the Phe426Leu and Val471Ala variants of ATG7 (ref. 172); both are loss-of-function mutations. However, these findings are inconsistent with results obtained in mice showing that a loss of autophagy instead suppresses liver steatosis173. Thus, partially reduced autophagic activity in humans may have an impact on the liver different from that caused by the complete loss of autophagy observed in autophagy gene knockout mice.
The relationship between autophagy and cancer has attracted much attention. However, although there are numerous reports suggesting the association of specific tumours with autophagy gene single nucleotide polymorphisms, recurrent or driver mutations of core autophagy genes in human cancers are rather rare174. Thus, autophagy may be still functional in most cancers and could even be important. In fact, mouse studies have suggested that, while the deletion of autophagy genes might promote tumorigenesis, it also affects tumour growth either through cell-autonomous or -nonautonomous mechanisms175. Nevertheless, mutations in core ATG genes might be associated with familial cancers. For example, a linkage study identified an association of a germline nonsense mutation of ATG7 (c.2000C>T p.Arg659*) with familial cholangiocarcinoma176. In cancer cells, somatic deletion of ATG7 occurs in the complementary allele, leading to complete inhibition of autophagy. This case suggests that autophagy suppression could also be tumorigenic in humans.
Conclusions and perspective
A quarter century has passed since the first autophagy gene ATG1, named APG1 initially, was cloned in yeast in 1997 (ref. 177). During this period, our understanding of the molecular biology underlying autophagy has grown exponentially. In particular, recent structural biological approaches have provided crucial evidence to explain the unique membrane dynamics of autophagy at the molecular level. However, despite the increasing clarity of the functions of individual autophagy gene products, several key cell biological questions remain unanswered. For example, what is the mechanism of unidirectional transport of lipids from the ER to autophagosomes? How is the size of autophagosomes regulated? How is the timing of autophagosome–lysosome fusion regulated? To address these questions, new approaches, including biophysics, theoretical modelling and molecular dynamics simulation, will be useful.
Although we have aimed to summarize the latest knowledge about microautophagy, our efforts may seem incomplete because its mechanisms are still less understood than those of macroautophagy. It is intriguing that some ATG proteins (for example, ATG8) and selective autophagy adaptors (for example, NBR1) are used by both macroautophagy and microautophagy. Whether these molecules exert similar functions in both pathways needs to be elucidated in future studies. In addition, some cargos (for example, ferritin) are selectively degraded by both pathways, but the regulation mechanisms of sorting remain unknown. Further research will reveal a more complete picture of autophagy.
As we mentioned above, one of the apparent bottlenecks in autophagy research is the lack of methods with which to monitor autophagy in humans. It would be ideal if we could estimate autophagic activity by measuring some metabolites (that is, biomarkers) in the blood or urine that are secreted via autophagy-dependent pathways. Alternative techniques may be noninvasive imaging such as fluorescence molecular tomography178 and positron emission tomography179.
Finally, although many diseases have been found to be linked to autophagy gene mutations or associated with polymorphisms of autophagy genes, the phenotypes of these diseases are diverse. Thus, it is still difficult to offer a unified explanation of their pathogenesis. This may be due to complementation by homologues, differences in tissue expression and involvement of non-autophagic functions. More investigations will be required to reveal the exact mechanisms by which autophagy gene defects cause a wide range of human diseases.
References
Nakatogawa, H. Mechanisms governing autophagosome biogenesis. Nat. Rev. Mol. Cell Biol. 21, 439–458 (2020).
Schuck, S. Microautophagy — distinct molecular mechanisms handle cargoes of many sizes. J. Cell Sci. 133, jcs246322 (2020).
Mizushima, N. & Komatsu, M. Autophagy: renovation of cells and tissues. Cell 147, 728–741 (2011).
Levine, B. & Kroemer, G. Biological functions of autophagy genes: a disease perspective. Cell 176, 11–42 (2019).
Mejlvang, J. et al. Starvation induces rapid degradation of selective autophagy receptors by endosomal microautophagy. J. Cell Biol. 217, 3640–3655 (2018).
Müller, M. et al. The coordinated action of the MVB pathway and autophagy ensures cell survival during starvation. eLife 4, e07736 (2015).
Lynch-Day, M. A. & Klionsky, D. J. The Cvt pathway as a model for selective autophagy. FEBS Lett. 584, 1359–1366 (2010).
Liu, X. M. et al. ESCRTs cooperate with a selective autophagy receptor to mediate vacuolar targeting of soluble cargos. Mol. Cell 59, 1035–1042 (2015).
Leidal, A. M. & Debnath, J. Emerging roles for the autophagy machinery in extracellular vesicle biogenesis and secretion. FASEB Bioadv. 3, 377–386 (2021).
Mizushima, N. & Levine, B. Autophagy in human diseases. N. Engl. J. Med. 383, 1564–1576 (2020).
Kaushik, S. & Cuervo, A. M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 19, 365–381 (2018).
Fujioka, Y. et al. Structural basis of starvation-induced assembly of the autophagy initiation complex. Nat. Struct. Mol. Biol. 21, 513–521 (2014).
Yamamoto, H. et al. The intrinsically disordered protein Atg13 mediates supramolecular assembly of autophagy initiation complexes. Dev. Cell 38, 86–99 (2016).
Fujioka, Y. et al. Phase separation organizes the site of autophagosome formation. Nature 578, 301–305 (2020). This paper reports liquid–liquid phase separation of the Atg1 complex that initiates autophagosome formation in yeast.
Turco, E. et al. FIP200 claw domain binding to p62 promotes autophagosome formation at ubiquitin condensates. Mol. Cell 74, 330–346.e311 (2019).
Vargas, J. N. S. et al. Spatiotemporal control of ULK1 activation by NDP52 and TBK1 during selective autophagy. Mol. Cell 74, 347–362.e346 (2019).
Ravenhill, B. J. et al. The cargo receptor NDP52 initiates selective autophagy by recruiting the ULK complex to cytosol-invading bacteria. Mol. Cell 74, 320–329.e326 (2019). The 2019 papers by Turco et al., Vargas et al. and Ravenhill et al. report the recruitment of the ULK complex by selective autophagy adaptors.
Turco, E. et al. Reconstitution defines the roles of p62, NBR1 and TAX1BP1 in ubiquitin condensate formation and autophagy initiation. Nat. Commun. 12, 5212 (2021).
Smith, M. D. et al. CCPG1 is a non-canonical autophagy cargo receptor essential for ER-phagy and pancreatic ER proteostasis. Dev. Cell 44, 217–232 (2018).
Zhou, Z. et al. Phosphorylation regulates the binding of autophagy receptors to FIP200 Claw domain for selective autophagy initiation. Nat. Commun. 12, 1570 (2021).
Sawa-Makarska, J. et al. Reconstitution of autophagosome nucleation defines Atg9 vesicles as seeds for membrane formation. Science 369, eaaz7714 (2020).
Ren, X. et al. Structural basis for ATG9A recruitment to the ULK1 complex in mitophagy initiation. Preprint at bioRxiv https://doi.org/10.1101/2022.07.12.499634 (2022).
Yamano, K. et al. Critical role of mitochondrial ubiquitination and the OPTN–ATG9A axis in mitophagy. J. Cell Biol. 219, e201912144 (2020).
Coudevylle, N. et al. Mechanism of Atg9 recruitment by Atg11 in the cytoplasm-to-vacuole targeting pathway. J. Biol. Chem. 298, 101573 (2022).
Baskaran, S. et al. Architecture and dynamics of the autophagic phosphatidylinositol 3-kinase complex. eLife 3, e05115 (2014).
Hurley, J. H. & Young, L. N. Mechanisms of autophagy initiation. Annu. Rev. Biochem. 86, 225–244 (2017).
Zheng, J. X. et al. Architecture of the ATG2B-WDR45 complex and an aromatic Y/HF motif crucial for complex formation. Autophagy 13, 1870–1883 (2017).
Chowdhury, S. et al. Insights into autophagosome biogenesis from structural and biochemical analyses of the ATG2A–WIPI4 complex. Proc. Natl. Acad. Sci. USA 115, E9792–E9801 (2018).
Maeda, S., Otomo, C. & Otomo, T. The autophagic membrane tether ATG2A transfers lipids between membranes. eLife 8, e45777 (2019).
Osawa, T. et al. Atg2 mediates direct lipid transfer between membranes for autophagosome formation. Nat. Struct. Mol. Biol. 26, 281–288 (2019).
Valverde, D. P. et al. ATG2 transports lipids to promote autophagosome biogenesis. J. Cell Biol. 218, 1787–1798 (2019). The 2019 papers by Maeda et al., Osawa et al. and Valverde et al. report that mammalian ATG2A and yeast Atg2 have lipid transfer activity.
Ren, J. et al. Multi-site-mediated entwining of the linear WIR-motif around WIPI β-propellers for autophagy. Nat. Commun. 11, 2702 (2020).
Dooley, H. C. et al. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol. Cell 55, 238–252 (2014).
Bakula, D. et al. WIPI3 and WIPI4 beta-propellers are scaffolds for LKB1–AMPK–TSC signalling circuits in the control of autophagy. Nat. Commun. 8, 15637 (2017).
Bozic, M. et al. A conserved ATG2–GABARAP family interaction is critical for phagophore formation. EMBO Rep. 21, e201948412 (2020).
Maeda, S. et al. Structure, lipid scrambling activity and role in autophagosome formation of ATG9A. Nat. Struct. Mol. Biol. 27, 1194–1201 (2020).
Matoba, K. et al. Atg9 is a lipid scramblase that mediates autophagosomal membrane expansion. Nat. Struct. Mol. Biol. 27, 1185–1193 (2020). The 2020 papers by Maeda et al. and Matoba et al. demonstrate that mammalian ATG9A and yeast Atg9 have lipid scramblase activity.
Ghanbarpour, A., Valverde, D. P., Melia, T. J. & Reinisch, K. M. A model for a partnership of lipid transfer proteins and scramblases in membrane expansion and organelle biogenesis. Proc. Natl. Acad. Sci. USA 118, e2101562118 (2021).
Li, Y. E. et al. TMEM41B and VMP1 are scramblases and regulate the distribution of cholesterol and phosphatidylserine. J. Cell Biol. 220, e202103105 (2021).
Huang, D. et al. TMEM41B acts as an ER scramblase required for lipoprotein biogenesis and lipid homeostasis. Cell Metab. 33, 1655–1670.e1658 (2021). The 2021 papers by Ghanbarpour et al., Li et al. and Huang et al. demonstrate that the ER membrane proteins TMEM41B and VMP1 have lipid scramblase activity.
Okawa, F. et al. Evolution and insights into the structure and function of the DedA superfamily containing TMEM41B and VMP1. J. Cell Sci. 134, jcs255877 (2021).
Mesdaghi, S., Murphy, D. L., Sanchez Rodriguez, F., Burgos-Marmol, J. J. & Rigden, D. J. In silico prediction of structure and function for a large family of transmembrane proteins that includes human Tmem41b. F1000Res 9, 1395 (2020).
Hama, Y., Morishita, H. & Mizushima, N. Regulation of ER-derived membrane dynamics by the DedA domain-containing proteins VMP1 and TMEM41B. EMBO Rep. 23, e53894 (2022).
Fujita, N. et al. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol. Biol. Cell 19, 4651–4659 (2008).
Sou, Y. S. et al. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol. Biol. Cell 19, 4762–4775 (2008).
Uemura, T. et al. A cluster of thin tubular structures mediates transformation of the ER to autophagic isolation membrane. Mol. Cell. Biol. 34, 1695–1706 (2014).
Tsuboyama, K. et al. The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science 354, 1036–1041 (2016).
Nguyen, T. N. et al. Atg8 family LC3/GABARAP proteins are crucial for autophagosome–lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J. Cell Biol. 215, 857–874 (2016). The 2016 papers by Tsuboyama et al. and Nguyen et al. report that the mammalian ATG8 conjugation system is important for degradation of the autophagosomal inner membrane or autophagosome–lysosome fusion but not essential for autophagosome formation.
Lamark, T. & Johansen, T. Mechanisms of selective autophagy. Annu. Rev. Cell Dev. Biol. 37, 143–169 (2021).
Alemu, E. A. et al. ATG8 family proteins act as scaffolds for assembly of the ULK complex: sequence requirements for LC3-interacting region (LIR) motifs. J. Biol. Chem. 287, 39275–39290 (2012).
McEwan, D. G. et al. PLEKHM1 regulates autophagosome–lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol. Cell 57, 39–54 (2015).
Wang, Z. et al. The Vici syndrome protein EPG5 is a Rab7 effector that determines the fusion specificity of autophagosomes with late endosomes/lysosomes. Mol. Cell 63, 781–795 (2016).
Agudo-Canalejo, J. et al. Wetting regulates autophagy of phase-separated compartments and the cytosol. Nature 591, 142–146 (2021). This paper reports macro-fluidophagy of the fluid-like SQSTM1 condensates that wet to autophagosomal membranes.
Takahashi, Y. et al. An autophagy assay reveals the ESCRT-III component CHMP2A as a regulator of phagophore closure. Nat. Commun. 9, 2855 (2018).
Zhou, F. et al. Rab5-dependent autophagosome closure by ESCRT. J. Cell Biol. 218, 1908–1927 (2019).
Zhen, Y. et al. ESCRT-mediated phagophore sealing during mitophagy. Autophagy 16, 826–841 (2020).
Itakura, E., Kishi-Itakura, C. & Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151, 1256–1269 (2012).
Matsui, T. et al. Autophagosomal YKT6 is required for fusion with lysosomes independently of syntaxin 17. J. Cell Biol. 217, 2633–2645 (2018).
Bas, L. et al. Reconstitution reveals Ykt6 as the autophagosomal SNARE in autophagosome–vacuole fusion. J. Cell Biol. 217, 3656–3669 (2018).
Gao, J., Reggiori, F. & Ungermann, C. A novel in vitro assay reveals SNARE topology and the role of Ykt6 in autophagosome fusion with vacuoles. J. Cell Biol. 217, 3670–3682 (2018).
Zhao, Y. G. & Zhang, H. Autophagosome maturation: an epic journey from the ER to lysosomes. J. Cell Biol. 218, 757–770 (2019).
Ramya, V. & Rajasekharan, R. ATG15 encodes a phospholipase and is transcriptionally regulated by YAP1 in Saccharomyces cerevisiae. FEBS Lett. 590, 3155–3167 (2016).
Yu, L. et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465, 942–926 (2010).
Rong, Y. et al. Clathrin and phosphatidylinositol-4,5-bisphosphate regulate autophagic lysosome reformation. Nat. Cell Biol. 14, 924–934 (2012).
Du, W. et al. Kinesin 1 drives autolysosome tubulation. Dev. Cell 37, 326–336 (2016).
Zhou, C. et al. Recycling of autophagosomal components from autolysosomes by the recycler complex. Nat. Cell Biol. 24, 497–512 (2022).
Galluzzi, L. & Green, D. R. Autophagy-independent functions of the autophagy machinery. Cell 177, 1682–1699 (2019).
Wijshake, T. et al. Tumor-suppressor function of Beclin 1 in breast cancer cells requires E-cadherin. Proc. Natl. Acad. Sci. USA 118, e2020478118 (2021).
Heckmann, B. L. & Green, D. R. LC3-associated phagocytosis at a glance. J. Cell Sci. 132, jcs222984 (2019).
Nieto-Torres, J. L., Leidal, A. M., Debnath, J. & Hansen, M. Beyond autophagy: the expanding roles of ATG8 proteins. Trends Biochem. Sci. 46, 673–686 (2021).
Durgan, J. & Florey, O. Many roads lead to CASM: diverse stimuli of noncanonical autophagy share a unifying molecular mechanism. Sci. Adv. 8, eabo1274 (2022).
Fletcher, K. et al. The WD40 domain of ATG16L1 is required for its non-canonical role in lipidation of LC3 at single membranes. EMBO J. 37, e97840 (2018). This paper reports that the C-terminal WD40 repeat domain of ATG16L1 is required for ATG8 lipidation on endocytic single membranes but not on autophagosomes.
Xu, Y. et al. A bacterial effector reveals the V-ATPase–ATG16L1 axis that initiates xenophagy. Cell 178, 552–566 (2019).
Fischer, T. D., Wang, C., Padman, B. S., Lazarou, M. & Youle, R. J. STING induces LC3B lipidation onto single-membrane vesicles via the V-ATPase and ATG16L1–WD40 domain. J. Cell Biol. 219, e202009128 (2020).
Ulferts, R. et al. Subtractive CRISPR screen identifies the ATG16L1/vacuolar ATPase axis as required for non-canonical LC3 lipidation. Cell Rep. 37, 109899 (2021).
Hooper, K. M. et al. V-ATPase is a universal regulator of LC3-associated phagocytosis and non-canonical autophagy. J. Cell Biol. 221, e202105112 (2022).
Durgan, J. et al. Non-canonical autophagy drives alternative ATG8 conjugation to phosphatidylserine. Mol. Cell 81, 2031–2040 e2038 (2021).
Solvik, T. A. et al. Secretory autophagy maintains proteostasis upon lysosome inhibition. J. Cell Biol. 221, e202110151 (2022).
Choi, Y., Bowman, J. W. & Jung, J. U. Autophagy during viral infection — a double-edged sword. Nat. Rev. Microbiol. 16, 341–354 (2018).
Joo, J. H. et al. The noncanonical role of ULK/ATG1 in ER-to-Golgi trafficking is essential for cellular homeostasis. Mol. Cell 62, 491–506 (2016).
Claude-Taupin, A. et al. ATG9A protects the plasma membrane from programmed and incidental permeabilization. Nat. Cell Biol. 23, 846–858 (2021).
Kumar, S. et al. Mammalian Atg8 proteins and the autophagy factor IRGM control mTOR and TFEB at a regulatory node critical for responses to pathogens. Nat. Cell Biol. 22, 973–985 (2020).
Nakamura, S. et al. LC3 lipidation is essential for TFEB activation during the lysosomal damage response to kidney injury. Nat. Cell Biol. 22, 1252–1263 (2020).
Goodwin, J. M. et al. GABARAP sequesters the FLCN–FNIP tumor suppressor complex to couple autophagy with lysosomal biogenesis. Sci. Adv. 7, eabj2485 (2021).
Walczak, M., Ganesan, S. M., Niles, J. C. & Yeh, E. ATG8 is essential specifically for an autophagy-independent function in apicoplast biogenesis in blood-stage malaria parasites. mBio 9, e02021–02017 (2018).
Fu, J. et al. Apicoplast biogenesis mediated by ATG8 requires the ATG12–ATG5–ATG16L and SNAP29 complexes in Toxoplasma gondii. Autophagy https://doi.org/10.1080/15548627.2022.2123639 (2022).
Lee, I. H. et al. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science 336, 225–228 (2012).
Rubinstein, A. D., Eisenstein, M., Ber, Y., Bialik, S. & Kimchi, A. The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol. Cell 44, 698–709 (2011).
Sasai, M. et al. Essential role for GABARAP autophagy proteins in interferon-inducible GTPase-mediated host defense. Nat. Immunol. 18, 899–910 (2017).
Marzella, L., Ahlberg, J. & Glaumann, H. Autophagy, heterophagy, microautophagy and crinophagy as the means for intracellular degradation. Virchows Arch. B 36, 219–234 (1981).
Mortimore, G. E., Hutson, N. J. & Surmacz, C. A. Quantitative correlation between proteolysis and macro- and microautophagy in mouse hepatocytes during starvation and refeeding. Proc. Natl. Acad. Sci. USA 80, 2179–2183 (1983).
Oku, M. & Sakai, Y. Three distinct types of microautophagy based on membrane dynamics and molecular machineries. Bioessays 40, e1800008 (2018).
Wang, L., Klionsky, D. J. & Shen, H. M. The emerging mechanisms and functions of microautophagy. Nat. Rev. Mol. Cell Biol. https://doi.org/10.1038/s41580-022-00529-z (2022).
van Zutphen, T. et al. Lipid droplet autophagy in the yeast Saccharomyces cerevisiae. Mol. Biol. Cell 25, 290–301 (2014).
Wang, C. W., Miao, Y. H. & Chang, Y. S. A sterol-enriched vacuolar microdomain mediates stationary phase lipophagy in budding yeast. J. Cell Biol. 206, 357–366 (2014).
Tsuji, T. et al. Niemann–Pick type C proteins promote microautophagy by expanding raft-like membrane domains in the yeast vacuole. eLife 6, e25960 (2017).
Strømhaug, P. E., Bevan, A. & Dunn, W. A. Jr. GSA11 encodes a unique 208-kDa protein required for pexophagy and autophagy in Pichia pastoris. J. Biol. Chem. 276, 42422–42435 (2001).
Mukaiyama, H. et al. Modification of a ubiquitin-like protein Paz2 conducted micropexophagy through formation of a novel membrane structure. Mol. Biol. Cell 15, 58–70 (2004).
Kissova, I. et al. Selective and non-selective autophagic degradation of mitochondria in yeast. Autophagy 3, 329–336 (2007).
Krick, R. et al. Piecemeal microautophagy of the nucleus requires the core macroautophagy genes. Mol. Biol. Cell 19, 4492–4505 (2008).
Otto, F. B. & Thumm, M. Mechanistic dissection of macro- and micronucleophagy. Autophagy 17, 626–639 (2021).
Nakamura, S., Hidema, J., Sakamoto, W., Ishida, H. & Izumi, M. Selective elimination of membrane-damaged chloroplasts via microautophagy. Plant Physiol. 177, 1007–1026 (2018).
Soubannier, V. et al. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. 22, 135–141 (2012).
Schulze, R. J. et al. Direct lysosome-based autophagy of lipid droplets in hepatocytes. Proc. Natl. Acad. Sci. USA 117, 32443–32452 (2020).
Zhao, M. et al. CGAS is a micronucleophagy receptor for the clearance of micronuclei. Autophagy 17, 3976–3991 (2021).
Sahu, R. et al. Microautophagy of cytosolic proteins by late endosomes. Dev. Cell 20, 131–139 (2011).
Leidal, A. M. et al. The LC3-conjugation machinery specifies the loading of RNA-binding proteins into extracellular vesicles. Nat. Cell Biol. 22, 187–199 (2020).
Farre, J. C., Burkenroad, A., Burnett, S. F. & Subramani, S. Phosphorylation of mitophagy and pexophagy receptors coordinates their interaction with Atg8 and Atg11. EMBO Rep. 14, 441–449 (2013).
Loi, M., Raimondi, A., Morone, D. & Molinari, M. ESCRT-III-driven piecemeal micro-ER-phagy remodels the ER during recovery from ER stress. Nat. Commun. 10, 5058 (2019).
Schafer, J. A. et al. ESCRT machinery mediates selective microautophagy of endoplasmic reticulum in yeast. EMBO J. 39, e102586 (2020).
Wang, Y. Y. et al. Molecular and structural mechanisms of ZZ domain-mediated cargo selection by Nbr1. EMBO J. 40, e107497 (2021).
Goodwin, J. M. et al. Autophagy-independent lysosomal targeting regulated by ULK1/2-FIP200 and ATG9. Cell Rep. 20, 2341–2356 (2017).
Ohshima, T., Yamamoto, H., Sakamaki, Y., Saito, C. & Mizushima, N. NCOA4 drives ferritin phase separation to facilitate macroferritinophagy and microferritinophagy. J. Cell Biol. 221, e202203102 (2022).
Morozova, K. et al. Structural and biological interaction of hsc-70 protein with phosphatidylserine in endosomal microautophagy. J. Biol. Chem. 291, 18096–18106 (2016).
Mukherjee, A., Patel, B., Koga, H., Cuervo, A. M. & Jenny, A. Selective endosomal microautophagy is starvation-inducible in Drosophila. Autophagy 12, 1984–1999 (2016).
Karsli-Uzunbas, G. et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 4, 914–927 (2014). This paper reports the physiological roles of autophagy in adult mice as well as its role in tumours using acute Atg7 knockout mice.
Yang, Y. et al. Autophagy in PDGFRα+ mesenchymal cells is essential for intestinal stem cell survival. Proc. Natl. Acad. Sci. USA 119, e2202016119 (2022).
Yoshii, S. R. et al. Systemic analysis of Atg5-null mice rescued from neonatal lethality by transgenic ATG5 expression in neurons. Dev. Cell 39, 116–130 (2016).
Kim, M. et al. Mutation in ATG5 reduces autophagy and leads to ataxia with developmental delay. eLife 5, e12245 (2016).
Collier, J. J. et al. Developmental consequences of defective ATG7-mediated autophagy in humans. N. Engl. J. Med. 384, 2406–2417 (2021).
McMichael, G. et al. Whole-exome sequencing points to considerable genetic heterogeneity of cerebral palsy. Mol. Psychiat. 20, 176–182 (2015).
Maddirevula, S. et al. Autozygome and high throughput confirmation of disease genes candidacy. Genet. Med. 21, 736–742 (2019).
Jelani, M. et al. A mutation in the major autophagy gene, WIPI2, associated with global developmental abnormalities. Brain 142, 1242–1254 (2019).
Maroofian, R. et al. Homozygous missense WIPI2 variants cause a congenital disorder of autophagy with neurodevelopmental impairments of variable clinical severity and disease course. Brain Commun. 3, fcab183 (2021).
Najmabadi, H. et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 478, 57–63 (2011).
Anazi, S. et al. Clinical genomics expands the morbid genome of intellectual disability and offers a high diagnostic yield. Mol. Psychiat. 22, 615–624 (2017).
Suleiman, J. et al. WDR45B-related intellectual disability, spastic quadriplegia, epilepsy, and cerebral hypoplasia: a consistent neurodevelopmental syndrome. Clin. Genet. 93, 360–364 (2018).
Almannai, M. et al. El-Hattab–Alkuraya syndrome caused by biallelic WDR45B pathogenic variants: further delineation of the phenotype and genotype. Clin. Genet. 101, 530–540 (2022).
Haack, T. B. et al. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am. J. Hum. Genet. 91, 1144–1149 (2012).
Saitsu, H. et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat. Genet. 45, 445–449 (2013).
Hayflick, S. J. et al. β-propeller protein-associated neurodegeneration: a new X-linked dominant disorder with brain iron accumulation. Brain 136, 1708–1717 (2013).
Nguyen, T. N., Padman, B. S. & Lazarou, M. Deciphering the molecular signals of PINK1/Parkin mitophagy. Trends Cell Biol. 26, 733–744 (2016).
Dawson, T. M., Ko, H. S. & Dawson, V. L. Genetic animal models of Parkinson’s disease. Neuron 66, 646–661 (2010).
McWilliams, T. G. et al. Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell Metab. 27, 439–449.e435 (2018).
Noda, S. et al. Loss of Parkin contributes to mitochondrial turnover and dopaminergic neuronal loss in aged mice. Neurobiol. Dis. 136, 104717 (2020).
Matheoud, D. et al. Intestinal infection triggers Parkinson’s disease-like symptoms in Pink1-/- mice. Nature 571, 565–569 (2019).
Pickrell, A. M. & Youle, R. J. The roles of PINK1, Parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85, 257–273 (2015).
Sliter, D. A. et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 561, 258–262 (2018).
Gajwani, P. et al. Pink1-mediated mitophagy in the endothelium releases proteins encoded by mitochondrial DNA and activates neutrophil responses. Preprint at bioRxiv https://doi.org/10.1101/2022.08.07.503084 (2022).
Haack, T. B. et al. Absence of the autophagy adaptor SQSTM1/p62 causes childhood-onset neurodegeneration with ataxia, dystonia, and gaze palsy. Am. J. Hum. Genet. 99, 735–743 (2016).
Khaminets, A. et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 522, 354–358 (2015).
Nguyen, T. N. et al. Unconventional initiation of PINK1/Parkin mitophagy by optineurin. Preprint at bioRxiv https://doi.org/10.1101/2022.08.14.503930 (2022).
Gao, F. B., Almeida, S. & Lopez-Gonzalez, R. Dysregulated molecular pathways in amyotrophic lateral sclerosis–frontotemporal dementia spectrum disorder. EMBO J. 36, 2931–2950 (2017).
Kim, G., Gautier, O., Tassoni-Tsuchida, E., Ma, X. R. & Gitler, A. D. ALS genetics: gains, losses, and implications for future therapies. Neuron 108, 822–842 (2020).
Maruyama, H. et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465, 223–226 (2010).
Deng, Z. et al. Autophagy receptors and neurodegenerative diseases. Trends Cell Biol. 27, 491–504 (2017).
Harding, O. et al. ALS- and FTD-associated missense mutations in TBK1 differentially disrupt mitophagy. Proc. Natl. Acad. Sci. USA 118, e2025053118 (2021).
Mathieu, C., Pappu, R. V. & Taylor, J. P. Beyond aggregation: pathological phase transitions in neurodegenerative disease. Science 370, 56–60 (2020).
Zaffagnini, G. et al. p62 filaments capture and present ubiquitinated cargos for autophagy. EMBO J. 37, e98308 (2018).
Sun, D., Wu, R., Zheng, J., Li, P. & Yu, L. Polyubiquitin chain-induced p62 phase separation drives autophagic cargo segregation. Cell Res. 28, 405–415 (2018).
Gallagher, E. R. & Holzbaur, E. L. F. The selective autophagy receptor p62 and the heat shock protein HSP27 facilitate lysophagy via the formation of phase-separated condensates. Preprint at bioRxiv https://doi.org/10.1101/2022.07.10.499468 (2022).
O’Loughlin, T. et al. OPTN recruitment to a Golgi-proximal compartment regulates immune signalling and cytokine secretion. J. Cell Sci. 133, jcs239822 (2020).
Laurin, N., Brown, J. P., Morissette, J. & Raymond, V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am. J. Hum. Genet. 70, 1582–1588 (2002).
Gennari, L. et al. Update on the pathogenesis and genetics of Paget’s disease of bone. Front. Cell Dev. Biol. 10, 932065 (2022).
Hampe, J. et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 39, 207–211 (2007).
Rioux, J. D. et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 39, 596–604 (2007).
Lassen, K. G. et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc. Natl. Acad. Sci. USA 111, 7741–7746 (2014).
Lavoie, S. et al. The Crohn’s disease polymorphism, ATG16L1 T300A, alters the gut microbiota and enhances the local Th1/Th17 response. eLife 8, e39982 (2019).
Fujita, N. et al. Differential involvement of Atg16L1 in Crohn disease and canonical autophagy: analysis of the organization of the Atg16L1 complex in fibroblasts. J. Biol. Chem. 284, 32602–32609 (2009).
Martinez, J. et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat. Cell Biol. 17, 893–906 (2015).
Henckaerts, L. et al. Genetic variation in the autophagy gene ULK1 and risk of Crohn’s disease. Inflamm. Bowel Dis. 17, 1392–1397 (2011).
Meddens, C. A. et al. Systematic analysis of chromatin interactions at disease associated loci links novel candidate genes to inflammatory bowel disease. Genome Biol. 17, 247 (2016).
Ellinghaus, D. et al. Association between variants of PRDM1 and NDP52 and Crohn’s disease, based on exome sequencing and functional studies. Gastroenterology 145, 339–347 (2013).
Sazonovs, A. et al. Large-scale sequencing identifies multiple genes and rare variants associated with Crohn’s disease susceptibility. Nat. Genet. 54, 1275–1283 (2022).
Ishibashi, K. et al. Atg16L2, a novel isoform of mammalian Atg16L that is not essential for canonical autophagy despite forming an Atg12-5-16L2 complex. Autophagy 7, 1500–1513 (2011).
Khor, B. et al. Distinct tissue-specific roles for the disease-associated autophagy genes ATG16L2 and ATG16L1. J. Immunol. 203, 1820–1829 (2019).
Yang, S. K. et al. Genome-wide association study of Crohn’s disease in Koreans revealed three new susceptibility loci and common attributes of genetic susceptibility across ethnic populations. Gut 63, 80–87 (2014).
Qi, Y. Y., Zhou, X. J. & Zhang, H. Autophagy and immunological aberrations in systemic lupus erythematosus. Eur. J. Immunol. 49, 523–533 (2019).
Rai, P. et al. IRGM1 links mitochondrial quality control to autoimmunity. Nat. Immunol. 22, 312–321 (2021).
Marchi, S., Guilbaud, E., Tait, S. W. G., Yamazaki, T. & Galluzzi, L. Mitochondrial control of inflammation. Nat. Rev. Immunol. https://doi.org/10.1038/s41577-022-00760-x (2022).
Henault, J. et al. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity 37, 986–997 (2012).
Baselli, G. A. et al. Rare ATG7 genetic variants predispose patients to severe fatty liver disease. J. Hepatol. 77, 596–606 (2022).
Takahashi, S. S. et al. Loss of autophagy impairs physiological steatosis by accumulation of NCoR1. Life Sci. Alliance 3, e201900513 (2020).
Amaravadi, R., Kimmelman, A. C. & White, E. Recent insights into the function of autophagy in cancer. Genes. Dev. 30, 1913–1930 (2016).
Poillet-Perez, L. & White, E. Role of tumor and host autophagy in cancer metabolism. Genes. Dev. 33, 610–619 (2019).
Greer, S. U. et al. Germline variants of ATG7 in familial cholangiocarcinoma alter autophagy and p62. Sci. Rep. 12, 10333 (2022).
Matsuura, A., Tsukada, M., Wada, Y. & Ohsumi, Y. Apg1p, a novel protein kinase required for the autophagic process in Saccharomyces cerevisiae. Gene 192, 245–250 (1997).
Chen, H. H. et al. Fluorescence tomography of rapamycin-induced autophagy and cardioprotection in vivo. Circ. Cardiovasc. Imaging 6, 441–447 (2013).
Mizushima, N. & Murphy, L. O. Autophagy assays for biological discovery and therapeutic development. Trends Biochem. Sci. 45, 1080–1109 (2020).
Zhang, S., Yazaki, E., Sakamoto, H., Yamamoto, H. & Mizushima, N. Evolutionary diversification of the autophagy-related ubiquitin-like conjugation systems. Autophagy 18, 2969–2984 (2022).
Pang, Y. et al. Evolution from covalent conjugation to non-covalent interaction in the ubiquitin-like ATG12 system. Nat. Struct. Mol. Biol. 26, 289–296 (2019).
Noda, N. N. & Mizushima, N. Atg101: not just an accessory subunit in the autophagy-initiation complex. Cell Struct. Funct. 41, 13–20 (2016).
Kraft, C., Peter, M. & Hofmann, K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat. Cell Biol. 12, 836–841 (2010).
Levine, T. P. Remote homology searches identify bacterial homologues of eukaryotic lipid transfer proteins, including Chorein-N domains in TamB and AsmA and Mdm31p. BMC Mol. Cell Biol. 20, 43 (2019).
Liu, J. et al. Bacterial Vipp1 and PspA are members of the ancient ESCRT-III membrane-remodeling superfamily. Cell 184, 3660–3673 e3618 (2021).
Moon, C. W., Kim, Y. & Hyun, J. K. Active electrochemical high-contrast gratings as on/off switchable and color tunable pixels. Nat. Commun. 13, 3391 (2022).
Acknowledgements
The authors thank N. N. Noda, M. Oku, Y. Sakai, Y. Hama, J.-i. Sakamaki and other Mizushima Laboratory members for their help with the manuscript. This work was supported by the Exploratory Research for Advanced Technology (ERATO) research funding programme of the Japan Science and Technology Agency (JST) (JPMJER1702 to N.M.), a Grant-in-Aid for Transformative Research Areas (A) (21H05256 to H.Y.) and a Grant-in-Aid for Specially Promoted Research (22H04919 to N.M.) from the Japan Society for the Promotion of Science (JSPS).
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Genetics thanks Patrice Codogno, Sascha Martens and Federico Pietrocola for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yamamoto, H., Zhang, S. & Mizushima, N. Autophagy genes in biology and disease. Nat Rev Genet 24, 382–400 (2023). https://doi.org/10.1038/s41576-022-00562-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41576-022-00562-w
