
Abstract
The failure of animal models to predict therapeutic responses in humans is a major problem that also brings into question their use for basic research. Organ-on-a-chip (organ chip) microfluidic devices lined with living cells cultured under fluid flow can recapitulate organ-level physiology and pathophysiology with high fidelity. Here, I review how single and multiple human organ chip systems have been used to model complex diseases and rare genetic disorders, to study host–microbiome interactions, to recapitulate whole-body inter-organ physiology and to reproduce human clinical responses to drugs, radiation, toxins and infectious pathogens. I also address the challenges that must be overcome for organ chips to be accepted by the pharmaceutical industry and regulatory agencies, as well as discuss recent advances in the field. It is evident that the use of human organ chips instead of animal models for drug development and as living avatars for personalized medicine is ever closer to realization.
Similar content being viewed by others

Introduction
There is great interest in finding alternatives to animal testing (Box 1), because in addition to being costly, time-consuming and ethically questionable, data from animals frequently fail to predict the results obtained in human clinical trials1,2,3,4,5. The lack of human-relevant preclinical models and the resulting high failure rates of therapeutics in the clinic has led to an unsustainable rise in healthcare costs as well as fewer effective drugs reaching patients. There is also increasing pressure from society and governments to find alternatives to animal testing, as evidenced by the US Food and Drug Administration (FDA) Modernization Act of 2021 and the Humane Research and Testing Act (HR 1744), which are currently moving through the US Congress. Importantly, these observations bring into question the current heavy reliance upon genetically engineered mice and other animal models that dominate basic research as well as drug development today.
Organ-on-a-chip (organ chip) microfluidic culture devices represent one of the recent successes in the search for in vitro human microphysiological systems that can recapitulate organ-level and even organism-level functions. This microfluidic form of microphysiological system comes in various sizes and shapes (Fig. 1), but they all contain hollow channels lined by living cells and tissues cultured under dynamic fluid flow. Some devices recreate organ-level structures (for example, tissue–tissue interfaces) as well as provide relevant mechanical cues (for example, breathing and peristalsis-like motions) that are necessary to faithfully model organ physiology and disease states. By fluidically coupling two or more organ chips, human ‘body-on-chips’ multi-organ systems can be created that mimic whole-body physiology as well as drug distribution and disposition. Advances in stem cell technology, such as induced pluripotent stem (iPS) cells and organoids, have enabled sourcing of patient-specific stem cells that can now be integrated and differentiated within organ chips to create patient-specific preclinical models.

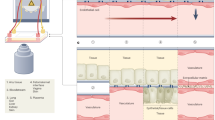
a | An optically clear, two-channel, mechanically actuable, organ chip fabricated from polydimethylsiloxane (PDMS) using soft lithography with two parallel channels separated by a flexible microporous membrane (now sold by Emulate). Different tissue cells are cultured on the top and bottom of the central extracellular matrix (ECM)-coated membrane with micrometre-sized pores to recreate a tissue–tissue interface that permits cell transmigration, and air can be introduced above the epithelium to create an air–liquid interface (for example, in lung) or fluid can be perfused through this channel. Application of cyclic suction to hollow side chambers results in rhythmic distortion of the flexible membrane and attached tissues, thereby mimicking organ-level mechanical distortions (such as breathing motions). b | A multiplexed array of three-channel plastic (polystyrene) chips that contain a thick ECM gel in the central channel, which lacks solid sidewalls and instead restrains the gel using a phase guide. Cells can be cultured in one or both of the flow channels as well as within the ECM gel (now sold by Mimetas). c | One or more hollow channels are created within a thick 3D ECM gel material by removing cylindrical mandrels after gelling has occurred, and cells can be cultured on the inner surface of the channels as well as within the ECM gel in these plastic devices (now sold by Nortis). d | A multiplexed PDMS microfluidic device containing two endothelium-lined channels separated by a third diamond-shaped chamber filled with ECM gel that can be used to support capillary ingrowth and 3D microvascular network formation surrounded by cells such as tumour cells in the gel (now sold by Aracari Biosciences). e | A plastic, multiwell-format organ chip system that incorporates multiple bioreactor chambers, each with a rigid porous membrane and lower microfluidic chamber linked to a fluid reservoir that can be cultured individually under flow or fluidically linked together through the lower compartment. Tissue–tissue interfaces are created by plating different cell types on either side of the membrane, and air or fluid can be included in the upper chamber (now sold by CN Bio Innovations). f | A higher-throughput (384-well) format plastic organ chip that includes two parallel channels separated by a rigid microporous membrane, and air or fluid can be introduced into the upper channel. g | Organ chips created using 3D printing to deposit sacrificial material in a cylindrical form in any desired pattern within an ECM gel with or without embedded cells. Once gelation is complete, the material is removed and epithelial or endothelial cells are cultured on the inner surface of the channel. h | A plastic multi-chamber organ chip system in which multiple mini-bioreactor chambers positioned on a flat plate can be cultured individually or fluidically coupled through a shared underlying fluidic channel. The cells may be cultured at bottom of the chamber in the flow path (left) or on top of the rigid porous membrane within a Transwell insert that placed within a chamber (right) so that they are separated from the flow path (now sold by TissUse). Part a adapted with permission from ref.11, AAAS. Part e adapted with permission from ref.20, RSC. Part g adapted with permission from ref.22, CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/). Part h adapted with permission from ref.29 RSC.
Previously, the organ chip field focused on the design and engineering of these microfluidic devices, and on the experimental demonstration of their ability to replicate relevant tissue and organ functions. However, the challenge now is to move to the next level by demonstrating equivalence or superiority relative to animal models. If human organ chips are found to be superior, then in addition to reducing animal testing, they may be used to develop or select therapeutics that are personalized for individual patients, distinct genetic subpopulations or even subgroups with particular disease comorbidities, which could revolutionize clinical trials design.
In this Review, I first introduce the different types of in vitro human models and provide a brief overview of the range of microfluidic organ chip designs, including different multi-organ human body-on-chips formats. I then describe recent advances in the organ chip field that demonstrate clinical mimicry by replicating human patient responses or that have applied this technology to advance drug development and personalized medicine. I review the major hurdles that must be overcome to enable replacement of animal testing by human organ chips (as well as any other microphysiological system) in industry and academia. Finally, I assess future opportunities and challenges for the field. For additional details on microfluidic systems engineering design or materials and the entire range of healthy and diseased organ chip models that have been developed using cells from humans or other species, I refer readers to previous reviews6,7.
Emerging human in vitro models
For the past century, living mammalian cells have been primarily cultured in nutrient medium under static conditions on 2D substrates coated with serum or extracellular matrix (ECM) molecules that were initially optimized to promote cell proliferation. Unfortunately, stimulation of cell growth is often accompanied by loss of tissue-specific functions8, and thus, many in the field have questioned the physiological relevance of results from in vitro experiments9. For this reason, there has been a constant interest in the design of culture systems that can better sustain tissue functionality for extended times in vitro, which has led to the emergence of the microphysiological systems field.
Microphysiological systems development has progressed through combined advances in microsystems engineering, tissue engineering and stem cell biology. As a result, many microscale culture system designs have been developed that reconstitute tissue and organ functions at levels not attainable in the past. Two broad approaches to microphysiological systems development have been pursued to attain improved biological mimicry: (1) the creation of static 3D culture systems with greater structural complexity, and (2) engineering of microfluidic 3D culture devices that also incorporate dynamic fluid flow, which have come to be referred to as organ chips.
Static microphysiological systems
Static microphysiological systems models, including microengineered tissues and organoids grown within 3D ECM gels, have demonstrated an impressive ability to recapitulate tissue histogenesis and many biological functions, including drug metabolism and cytotoxicity responses, as well as to model various disease states at the cell and tissue levels7,10. Thus, they provide powerful tools with which to dissect the molecular basis of developmental control as well as disease pathogenesis and, hence, could help to replace animal studies for particular applications. Nonetheless, they do not replicate tissue–tissue interfaces, vascular perfusion, interstitial flow, circulating immune cells or organ-specific mechanical cues that are critical for accurate mimicry of the delivery and absorption, distribution, metabolism and excretion (ADME) of pharmaceutical compounds or their pharmacokinetics and pharmacodynamics (PK/PD). Thus, static microphysiological systems are not optimal for accurate assessment of drug disposition, efficacy and toxicity within the human body. By contrast, microfluidic organ chips can provide all these functions.
Microfluidic organ chips
The first microfluidic culture device referred to as an organ chip recapitulated the organ-level (that is, multi-tissue) structures and functions of a major functional unit of a human organ, the lung alveolus11. It was fabricated using a soft lithography-based manufacturing approach adapted from the computer microchip industry. This approach was inspired by a simpler device that contained a hollow channel the size of a small lung airway, which enabled the recreation of acoustically detectable sounds when liquid plugs were flowed through the channel, nearly identical to the respiratory ‘crackle’ sounds physicians listen for through a stethoscope and use to diagnose fluid on the lungs12.
The lung alveolus chip is a computer memory stick-sized device composed of an optically clear, elastomeric, polydimethylsiloxane (PDMS) material containing two parallel hollow channels separated by a porous ECM-coated membrane lined with human lung alveolar epithelial cells on one side and human vascular endothelium on the other, thereby recreating the alveolar–capillary interface11 (Fig. 1a). Culture medium is perfused through the endothelium-lined channel to mimic vascular perfusion, and air is introduced into the epithelial channel to mimic the air–liquid interface of the lung, which is critical for lung differentiation and function, while application of cyclic suction to hollow side chambers enables application of cyclic tissue deformations to the flexible tissue–tissue interface to mimic breathing motions. This two-channel chip design has since been modified to provide open access to one channel and to enable formation of a thicker tissue construct while still exerting cyclic mechanical strain (for example, for a skin chip)13,14, and it has been used by multiple groups to create microfluidic models of various organ types with fluids flowing through both channels (Table 1). Other single device and multiplexed versions have been fabricated using various methods (for example, soft lithography, physical removal of sacrificial cylindrical mandrels, injection moulding) or that replace the porous membrane with an ECM gel to support tissue–ECM, tissue–tissue interface formation or cellular ingrowth to study dynamic 3D morphogenetic processes (for example, tumour angiogenesis15,16,17,18 and lymphangiogenesis19) (Fig. 1b–d); however, these do not enable application of mechanical deformations.
Another organ chip design developed at about the same time used a more conventional, plastic, transwell-like, multiwell (12-well) format in which tissue–tissue interfaces are recreated by culturing two different human cell types (for example, hepatocytes and liver sinusoidal endothelial cells) on opposite sides of a rigid porous membrane within minibioreactor chambers20 (Fig. 1e). A streamlined plastic 384-well format organ chip design also was described recently and used to create an air–liquid interface21 (Fig. 1f). 3D printing technology has been adapted to create dynamically perfused tubular structures lined by human organ-specific cells that can be positioned precisely within printed ECM gels to recreate, for example, a proximal tubule (Fig. 1g) or the proximal tubule–microvessel interface that is responsible for solute reabsorption in the kidney22,23. Organ chips have also been created in which some cell layers are 3D printed and others are assembled using microengineering approaches24. In addition, plastic plates have been created containing a flow channel beneath open chambers that can be used alone or in combination, and cells can be either cultured directly in the chamber or on top of porous membranes within transwell inserts that are placed in the chambers (Fig. 1h).
Multi-organ human body-on-chip systems have been created to study multi-organ physiology and whole-body level drug responses by adding fluidic coupling to culture chamber bioreactors contained within the multiwell system25,26,27,28 (Figs 1e,2a,b) or that use robotic liquid control systems that may more easily integrate with pharmaceutical robotic pipelines to sequentially transfer fluids between multiple soft lithography-fabricated two-channel chips13. An arteriovenous (AV) mixing reservoir is integrated into the latter system to provide mixing of medium, thereby mimicking blood mixing in the central circulation; this reservoir enables fluid sampling that is more analogous to sampling peripheral blood in a patient (rather than measuring drug or metabolite levels in outflows of individual organ chips) (Fig. 2c). An alternative approach fluidically links multiple micro-organ bioreactor chambers within the same plate (Fig. 1e,h), with each chamber or transwell insert containing a different tissue type (for example, liver, skin, marrow, tumour, and so on), and these devices may be configured differently depending on the number of organs to be coupled29,30,31,32 (Fig. 2a,b). In some versions of these multi-organ systems, medium is flowed directly from one parenchymal cell type to another (for example, from liver cells in chamber 1 to skin cells in chamber 2) without or with an intervening porous membrane26,27,28,29,30,31,32,33 (Figs 1h,2a). More recently, body-on-chip systems have perfused medium through channels lined by endothelium that interfaces across a porous membrane with organotypic tissues in adjacent channels or chambers to better replicate vascular perfusion and transendothelial transport of drugs and metabolites in the human body13,31,32,34,35 (Fig. 2b,c).

a | A simple fluidic coupling between multiple chambers lined by different organotypic cell types and a common flow chamber in two-chamber organ chip designs; a porous membrane within each chamber separates the overlying cell type from the fluid flow path or transwell inserts containing cells may be placed within open chambers as shown in Fig. 1h. To mimic intravenous (IV) adminstration, the drug can be introduced into the lower flow path. b | A similar multiwell configuration, except that the surface of the shared flow chamber is lined by endothelium. c | A diagram of linked two-channel organ chips containing both parenchymal cells and an endothelium-lined flow channel that are fluidically coupled using a robotic liquid handler to transfer fluids drop-by-drop between the chips and an arteriovenous mixing reservoir. The reservoir is integrated into the system to mimic blood mixing in the central circulation, and it also allows fluid sampling that is more analogous to sampling peripheral blood in a patient. Red arrows indicate the directional fluid flow or transfer path, and the circled ‘I’ depicts points in the circuit where a liquid-handling robot is used to move fluid into or out of the organ models or the arteriovenous reservoir; small blue arrows indicate independent transfers of fluids to and from the parenchymal channels of each chip. In this configuration, IV adminstration is modelled by injecting the drug into the arteriovenous reservoir, whereas oral adminstration is accomplished by introducing the drug into the lumenal channel of an intestine chip. Part c adapted from ref.35, Springer Nature Limited.
With the explosion of interest in this field, all of these approaches and even simpler single channel microfluidic devices that only contain a single cell type have come to be referred to as organ chips. Most of these devices are optically clear and enable real-time high-resolution microscopic imaging; however, some that contain fluidically coupled mini-reactors or incorporate thick 3D-printed or engineered tissue constructs do not. Importantly, regardless of the device design, the presence of dynamic fluid flow and organ-specific mechanical cues has been repeatedly shown to promote higher levels of tissue-specific differentiation and to enhance organ-level functions, even above those displayed in static 3D organoid cultures when organoid-derived cells are used to line the chips36,37. In addition, because these chips are actively perfused, immune cells can be flowed through the vascular endothelium-lined channel as they are in circulating blood11,28,37,38,39,40,41,42,43. Resident immune cells can also be placed within 3D ECM gels on-chip to recreate lymphoid and haematopoietic microenvironments and, thereby, to reconstitute specialized lymphoid follicle44 and bone marrow45,46 structures and functions in vitro.
A unique advantage of microfluidic organ chips over static microphysiological systems is that oxygen gradients can be established across the tissue–tissue interface so that complex living microbiome composed of both anaerobic and aerobic bacteria can be co-cultured directly on top of human epithelium (for example, gut microbiome on intestinal epithelium), and viable co-cultures can be sustained for multiple days in vitro47. This is in contrast to static microphysiological systems, including organoid cultures, in which oxygen gradients can be established, but bacterial cultures can only be maintained for about 24 h before microbial overgrowth and cell death result10,48. Because organ chips are usually fabricated using microengineering approaches, it is also possible to integrate various types of in-line sensors for monitoring of tissue viability and function, including real-time monitoring of oxygen levels47, changes in tissue barrier integrity (for example, using integrated electrodes to measure transepithelial and/or transendothelial electrical resistance49 or impedance spectroscopy50) and cellular electrical activity (for example, using multi-electrode arrays)51.
Importantly, over the past 12 years, organ chips have been used to model a broad range of human disorders and diseases across virtually all organ systems, to garner new insights into the molecular and cellular basis of various physiological and pathophysiological processes, to model various types of drug delivery approaches (for example, adeno-associated viral vectors and nanocarriers), and to recapitulate clinical responses to therapeutics, radiation, toxins and surgical implants seen in human patients (Table 1). Below, I summarize results from key human organ chip studies, focusing on those that provide the strongest evidence of clinical mimicry, particularly in the context of drug development and personalized medicine.
Clinical mimicry in organ chips
Lung
The human lung alveolus chip with two parallel microchannels separated by a microporous membrane (Fig. 1a) garnered great attention because it was the first organ chip to replicate complex integrated organ-level physiological and pathophysiological responses, rather than to simply demonstrate retention of cell- or tissue-level differentiated functions11. This chip utilized an established human lung alveolar epithelial cell line (NCI-H441 or A549) interfaced with human umbilical vein endothelial cells. The study revealed that introduction of either living bacteria to mimic infection or nanoparticles to simulate inhalation of airborne smog nanoparticulates into the airspace of the epithelial channel of the two-channel chip resulted in recruitment of circulating immune cells and secretion of inflammatory cytokines, as occurs in vivo, and these processes could be visualized in real-time at high resolution. Perfusion of the cancer drug interleukin-2 (IL-2) at a clinically relevant dose through the endothelium-lined channel of this chip also led to vascular leakage and filling of the air channel with interstitial fluid and thrombi, replicating the pulmonary oedema toxicity it causes in some patients52. Moreover, the ability to replicate physiological mechanical cues revealed that breathing motions enhance inflammatory reactions and increase nanoparticle absorption across the alveolar–capillary interface; these findings were not observed in static transwell cultures11. Moreover, mechanical stimulation was required for IL2-induced pulmonary oedema as well52. Importantly, in vivo studies carried out in parallel confirmed that all of these responses are also seen in whole lung in vivo. Another lung alveolus chip lined with human lung alveolar A549 epithelial cells was used to assess cytotoxicity responses to zinc oxide nanoparticles, which are used in biopharmaceutical preparations, drug delivery and biomedical imaging53. This study revealed that these nanoparticles exhibited lower toxicity when administered under dynamic flow in the lung chip compared to static conditions.
A lung alveolus chip lined with primary human lung alveolar epithelium interfaced with primary pulmonary microvascular endothelial cells has been used to model pulmonary thrombosis by flowing human whole blood through its vascular channel. This approach enabled quantitative analysis of organ-level contributions to inflammation-induced thrombosis, and recapitulated complex responses, including platelet–endothelial dynamics within individual thrombi that were nearly identical to those visualized within thrombi that formed in vivo54. Analysis of thrombosis formation on-chip induced by lipopolysaccharide endotoxin also revealed that it acts indirectly by stimulating the alveolar epithelium to produce other inflammatory molecules that induce endothelium activation, such as IL-6, rather than acting directly on endothelium.
Healthy and diseased human lung airways have been modelled by populating chips with primary bronchial or bronchiolar epithelial cells obtained from healthy donors or patients with chronic obstructive pulmonary disease (COPD) and culturing them under an air–liquid interface39,55. Chips lined with COPD epithelial cells exhibited selective cytokine hypersecretion, increased neutrophil recruitment and mimicked clinical exacerbation by exposure to viral and bacterial infections as well as cigarette smoke39,55. Transcriptomic profiles of healthy airway chips exposed to cigarette smoke closely resembled those obtained in past clinical studies. In addition, models of the asthmatic lung airway were developed by exposing healthy airway chips to IL-13, which induced goblet cell hyperplasia, inflammatory cytokine secretion and endothelial activation, while reducing cilia beating frequency, all of which are observed in asthmatic patients39,56. The IL-13-stimulated airway chips also recruited increased numbers of circulating neutrophils under flow, which could be inhibited by administering an anti-inflammatory drug (bromodomain-containing protein 4 inhibitor), and this recruitment response was greater than observed in a static transwell microphysiological system39.
More recently, lung airway chips have been used to model the cystic fibrosis airway by incorporating primary epithelium isolated from patients with cystic fibrosis40. These chips accurately replicate many features of the human cystic fibrosis airway, including increases in cilia density, mucus accumulation, ciliary beating frequency and IL-8 secretion, leading to greater adhesion of circulating immune cells to the endothelium and their transmigration into the airway compartment compared to healthy chips. The cystic fibrosis chips also provided a more favourable environment for growth of Pseudomonas aeruginosa bacteria and an enhanced inflammatory response, both of which are major contributors to morbidity in patients with cystic fibrosis.
The ability of lung alveolus and airway chips lined by primary cells to support growth of human non-small-cell lung adenocarcinoma cells has also been studied57. The cancer cells grew much more rapidly in the alveolus chip, which faithfully recapitulated the preferential growth at this site seen in patients with this type of cancer. Interestingly, the ability to control mechanical cues revealed that breathing motions suppress cancer growth and invasion in the lung, and that they alter the efficacy of a first-line anticancer drug (rociletinib). A more sophisticated version of this device, containing three parallel channels with an ECM gel lined with endothelial cells and fibroblasts that supports formation of 3D branching microvascular networks, enabled analysis of the response of human lung adenocarcinoma cells to clinically relevant concentrations of the chemotherapy drug paclitaxel and demonstrated both tumour and endothelial cell toxicities58.
A human immunocompetent lung alveolus chip containing primary lung cells and peripheral blood mononuclear cells was used to assess the safety of T cell bispecific antibody cancer therapies37. These chips effectively reproduced non-human primate (NHP) toxicities of T cell bispecific antibodies targeting folate receptor 1 that are currently under clinical development. Importantly, when a lower-affinity T cell bispecific antibody that was found to exhibit lower toxicity in the lung chip was tested in NHPs, none of the animals experienced the lung inflammation observed with the original higher-affinity antibody, thus confirming the safer profile predicted by this human organ chip. Currently, one of the most clinically relevant uses of human lung chips is in the study of respiratory virus infections. For example, infection of a lung alveolus chip lined by an established alveolar epithelial cell (HPAEpiC) line interfaced with a lung endothelial cell (HULEC‐5a) line with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) revealed preferential infection of the epithelium and stimulation of distinct innate immune responses in these two tissues, as well as recruitment of circulating immune cells, which led to endothelium detachment and further increased inflammatory cytokine release41. Treatment with remdesivir inhibited viral replication and alleviated barrier compromise on-chip. Human lung airway chips containing primary bronchial epithelium and pulmonary endothelium also recapitulated viral infection, cytokine production and recruitment of circulating immune cells, as well as virus strain-dependent virulence observed in human patients when infected with various influenza A virus strains42. Use of the model led to the discovery that co-administration of the anticoagulant drug nafamostat with the first-line antiviral drug oseltamivir doubled oseltamivir’s treatment-time window. Moreover, when these chips were infected with a pseudotyped SARS-CoV-2 virus and perfused with clinically relevant doses of the antimalarial drugs chloroquine or hydroxychloroquine, these drugs were found not to be active, thus predicting the negative results seen in clinical studies. By contrast, a related drug, amodiaquine, was shown to actively inhibit SARS-CoV-2 pseudovirus virus entry on-chip, as well as infection by native infectious SARS-CoV-2 in a hamster model. Importantly, these results contributed to the repurposing of this drug and its entry into clinical trials for COVID-19 in Africa, where this drug is used widely for antimalarial prophylaxis.
Another recent example of drug repurposing comes from use of the mechanical control capabilities of the primary lung alveolus chip, which revealed that physical forces associated with cyclic breathing motions influence lung innate immune responses to viral infection59. This work led to the discovery that receptor for advanced glycation end products (RAGE), an inflammatory mediator that is most highly expressed in the lung alveolus in vivo, has a central role in this response. Moreover, when a RAGE inhibitor drug (azeliragon) was tested, it potently suppressed production of inflammatory cytokines and synergized with the antiviral drug molnupiravir in this model. These results were included in a recent submission of a pre-IND (Investigational New Drug) meeting application to the FDA to request initiation of clinical trials to test whether azeliragon can suppress the cytokine storm in patients with COVID-19. Use of lung alveolus and airway chips also led to the discovery of a new class of broad-spectrum RNA therapeutics that induce a potent type I interferon response and inhibit infection by SARS-CoV2, SARS-CoV, MERS-CoV, HCoV-NL63 (a common cold virus) and multiple influenza A virus strains60.
Virus evolution through both mutation and gene reassortment as seen in patients with influenza has also been reconstituted in vitro by sequentially passaging infected mucus droplets between multiple human lung airway chips infected with influenza H1N1 in the continued presence of the antiviral drugs amantadine or oseltamivir (also known as Tamiflu)61. This selective pressure led to the spontaneous emergence of clinically prevalent resistance mutations as well as previously unobserved strains. Interestingly, strains that were resistant to both drugs also emerged; however, studies revealed that they were still sensitive to the anticoagulant nafamostat, which targets host serine proteases.
Liver
Multiple liver chip designs have been developed and used to model drug metabolism, drug–drug interactions, hepatotoxicity, inflammation and infection (Table 1). A microfluidic liver chip with the multiwell bioreactor design (Fig. 1e) lined by primary human hepatocytes and Küpffer cells replicated breakdown of the glucocorticoid hydrocortisone to phase I and phase II metabolites, and the intrinsic clearance values measured on-chip correlated with human data62. A similar liver chip system was used to show that sustained stimulation of inflammation by IL-6 suppresses cytochrome P450 3A4 isoform (CYP3A4) activity, increases C-reactive protein secretion and decreases shedding of soluble IL-6 receptor63. Treatment with a therapeutic anti-IL-6 receptor monoclonal antibody (tocilizumab) that is used to treat rheumatoid arthritis modulated CYP3A4 enzyme activities and altered metabolism of the small-molecule CYP3A4 substrate simvastatin hydroxy acid in this model, thus replicating the drug–drug interactions observed in patients, which was not possible using static 2D culture models.
Another interesting study explored the effects of human population variability on hepatic drug metabolism using multiple liver chips each lined by hepatocytes from a different donor64. Analysis of metabolic depletion profiles of six drugs confirmed the existence of substantial inter-donor variability with respect to gene expression levels, drug metabolism and other hepatocyte functions. Importantly, clearance values predicted on-chip correlated well with those observed in vivo, and a physiologically based pharmacokinetics model developed for lidocaine successfully predicted the observed clinical concentration–time profiles and associated population variability.
One of the most valuable uses of liver chips has been to model human-specific hepatotoxicities (Table 1), which are often missed in preclinical animal models such as rodents or dogs. An early liver chip lined only by primary human hepatocytes but separated from flow by a microengineered porous barrier (to mimic the endothelial permeability barrier) exhibited mass transport properties similar to the liver sinusoid and reproduced the metabolism-specific hepatotoxicity of the anti-inflammatory drug diclofenac65. Various cross-species liver toxicities (such as hepatocellular injury, steatosis, cholestasis and fibrosis) were also successfully reproduced in vitro by perfusing drugs with known species-specific toxicities through liver chips lined with multiple primary liver cell types (such as hepatocytes, liver sinusoidal endothelium, Küpffer cells and hepatic stellate cells) isolated from humans, dog or rat66. The ability to visualize these responses at high resolution yielded insights into a novel toxicity mechanism in which one drug was found to unexpectedly target the endothelium, rather than a parenchymal cell.
As in the case of the lung, liver chips have been used to model viral infection in vitro. For example, infection of a liver chip lined by human hepatocytes with hepatitis B virus (HBV) reproduced all steps of the HBV life cycle, including replication of patient-derived HBV and maintenance of HBV covalently closed circular DNA67. Moreover, innate immune and cytokine responses detected on-chip mimicked those observed in HBV-infected patients.
Heart
A heart chip was developed by culturing human iPS-cell-derived cardiomyocytes on flexible ECM gels overlaid on multi-electrode arrays in a single channel microfluidic device, which supported laminar cardiac tissue formation and enabled recording of tissue-level electrophysiological responses in real time51. This chip mimicked the difference in safety profiles between the cardiotoxic pro-drug terfenadine and its non-toxic metabolite fexofenadine seen in patients. Another heart chip created by combining microfabrication and 3D printing that contains human iPS-cell-derived cardiomyocytes interfaced with endothelial cells reproduced the toxic effects of the cancer drug doxorubicin on heart myocardium observed clinically24.
Intestine
Numerous organ chip models of the small and large intestine lined by intestinal epithelial cells with or without underlying endothelium have been created to model various diseases as well as to study drug metabolism and toxicities (Table 1). The presence of dynamic fluid flow has been found to be necessary and sufficient to promote villi formation in small intestine chips36,37,68 as well as production of high numbers of goblet cells and accumulation of a thick mucus bilayer in colon chips69, which closely resemble those in living intestine using the two-channel chip design (Fig. 1a); however, additional application of peristalsis-like mechanical motions is required for optimal differentiation68. Moreover, when lined with duodenal or colon organoid-derived epithelial cells, these mechanically active microfluidic chips more closely resemble human small and large intestine than organoids cultured in static microphysiological systems (either 3D ECM gel or transwell cultures) based on transcriptomic and histological characterization36,37,68. In a study on infection with an enteric coronavirus (NL63), organoid-derived intestinal epithelial cells were found to also dramatically increase their ACE2 receptor levels when cultured under flow in the presence of peristalsis-like mechanical deformations in two-channel microfluidic intestine chips compared to when cultured statically as organoids in 3D gels or in transwell inserts70. A two-channel human intestine chip lined by Caco-2 intestinal epithelial cells interfaced with endothelium was used to study SARS-CoV-2 infection and to reconstitute the morphological, structural and inflammatory changes consistent with gastrointestinal symptoms observed in patients with COVID-19 (ref.71).
The human intestine–microorganism interface was modelled in a two-channel microfluidic chip lined by established intestinal epithelial cell lines (Caco-2 or CRL-1459 cells), which recreates an oxygen gradient supporting the growth of a single commensal obligate anaerobe (Lactobacillus rhamnosus GG); this co-culture modified transcriptional, metabolic and immunological host responses in a manner similar to that previously observed in vivo72. However, the microorganisms had to be separated from the host epithelium by a nanoporous membrane in this chip and so only the effects of transported soluble mediators could be studied.
More recently, a living complex gut microbiome containing more than 200 different living bacterial species with similar complexity to a human stool microbiome was co-cultured in direct contact with human intestinal epithelium and its naturally secreted mucus layer for 5 days within two-channel intestine chips lined by either Caco-2 cells or primary human organoid-derived epithelium interfaced with primary intestinal microvascular endothelium47. This was accomplished by establishing a hypoxia gradient across the epithelial–endothelial interface that mimics the one existing in vivo, which was confirmed using integrated on-chip oxygen sensors, and intestinal barrier function was either maintained or enhanced by the presence of living bacteria. By contrast, exposure of small intestine chips lined by Caco-2 intestinal cells interfaced with primary microvascular endothelium to pathogens can induce inflammation, cause cell death and disrupt the intestinal permeability barrier, as observed clinically in patients with inflammatory bowel disease43. Use of a human colon chip lined by organoid-derived epithelium in combination with metabolomics was also used to identify specific microbial metabolites that mediate species-specific (human versus mouse) differences in sensitivity to enterohaemorrhagic Escherichia coli infection73. Interestingly, cessation of cyclic mechanical deformations results in bacterial overgrowth and epithelial injury in the small intestine chip, hence mimicking the clinic disorder known as ileus, which is caused by intestinal paralysis (for example, due to prolonged anaesthesia)43. By contrast, the presence of dynamic fluid flow and active peristalsis-like cyclic deformations enhance Shigella infection and invasion in a colon chip74. It is important to note that none of these mechanobiological behaviours can be detected in static microphysiological systems, and bacteria cannot be co-cultured in these static models for more than about 24 h without inducing cell death10,48.
An intestine chip model of acute radiation injury also replicated the same radiation dose sensitivity observed in humans, which differs from that observed in animal models75. Radiation exposure induced reactive oxygen species production primarily by the endothelium, which resulted in epithelial cytotoxicity, apoptosis and DNA fragmentation, as well as disruption of tight junctions, villus blunting and compromise of the intestinal barrier; pre-treatment with a potential prophylactic radiation countermeasure drug (dimethyloxaloylglycine) suppressed these responses in this model.
An inflammatory condition of the intestine that is endemic in children in low-resource nations, known as environmental enteric dysfunction (EED), was recently modelled in human intestine chips76. Key features of this disease, including villus blunting, compromised intestinal barrier function and reduced nutrient absorption, as well as impairment of fatty acid uptake and amino acid transport, were reconstituted in two-channel chips lined by intestinal epithelial cells isolated from EED patient-derived organoids when the cells were cultured in nutrient-deficient medium. These culture conditions led to transcriptional changes similar to those observed in clinical EED patient samples from multiple low-resource nations. Human intestine chips lined by organoid-derived cells and perfused with peripheral blood mononuclear cells also have been used to reproduce and predict clinical toxicities of a T cell bispecific antibody currently under clinical development that targets human carcinoembryonic antigen so specifically that neither its safety nor efficacy can be evaluated in animal models37.
Kidney
Kidney chips lined by human renal tubular or glomerular cells have been used for studies on drug and molecule transport, reabsorption and toxicity as well as disease modelling (Table 1). For example, a two-channel kidney chip lined by primary proximal tubular epithelium that expressed high P-glycoprotein efflux transporter activity replicated a transporter-specific cisplatin toxicity that is observed in patients but not in static 2D cultures or animal models77. Albumin reabsorption and cyclosporine toxicity were also recapitulated in a 3D-printed kidney chip containing human proximal tubular epithelium deposited within tiny cylindrical structures surrounded by a thick ECM gel22. This approach was extended further by printing closely apposed kidney tubules and vessels lined by proximal tubular epithelium and kidney endothelium within an ECM gel, which exhibited active reabsorption via tubular–vascular exchange of solutes similar to that observed in vivo23. Hyperglycaemia-induced endothelial cell dysfunction was replicated in this model, as well as its reversal by administration of a glucose transport inhibitor drug. Moreover, a kidney distal tubule chip was used to explore pathogenesis of Pseudorabies virus-induced renal dysfunctions78. Virus infection resulted in altered sodium reabsorption, disruption of the reabsorption barrier and changes in microvilli, which may contribute to the serum electrolyte abnormalities observed in virus-infected patients.
Importantly, a kidney proximal tubule chip also replicated phenotypic features of two rare X-linked monogenetic diseases (Lowe syndrome and Dent II disease), which are characterized by renal reabsorption defects caused by mutations in the OCRL gene79. Knocking down OCRL expression in the cells revealed that proximal tubule cells lacking OCRL upregulate collagen deposition, which can contribute to the interstitial fibrosis and disease progression seen in these disorders.
Use of a human kidney glomerulus chip containing closely opposed layers of immortalized kidney podocytes and glomerular endothelial cells revealed that glomerular mechanical forces play a crucial part in cell damage that leads to increased glomerular leakage, as observed in patients with hypertensive nephropathy80. Another glomerulus chip lined by human iPS-cell-derived podocytes interfaced with glomerular endothelium reconstituted in vivo levels of urinary clearance and mimicked the toxic effects of the anticancer drug adriamycin on kidney podocytes81. More recently, a personalized version of this chip was developed using both human iPS-cell-derived kidney glomerular endothelial cells and podocytes from a single patient82. In addition, the renal effects of autoimmunity have been studied using a human kidney glomerulus chip that recapitulates the permselectivity of the glomerulus83. When exposed to patient sera containing anti-podocyte autoantibodies, the chips developed albuminuria proportional to patients’ proteinuria, and this phenomenon was not seen using sera from healthy controls or individuals with primary podocyte defects.
Brain
Parkinson disease is a disorder of the substantia nigra region of the brain that is characterized by abnormal accumulation of α-synuclein aggregates, loss of dopaminergic neurons and proliferation of glial cells. A two-channel human brain chip representative of this region was created containing iPS-cell-derived dopaminergic neurons, astrocytes, microglia and pericytes interfaced with iPS cell-derived microvascular brain endothelium84. Transcriptomic analysis revealed that gene expression levels on-chip were much closer to those seen in the mature adult substantia nigra than in conventional culture systems. Importantly, introduction of α-synuclein fibrils, key components of Lewy bodies, resulted in their accumulation and phosphorylation, which is also observed in vivo. This phenotype was accompanied by an increase in reactive oxygen species production, mitochondrial dysfunction, cell death and neuroinflammation, which are all key features of Parkinson disease.
Many human blood–brain barrier (BBB) chip models of the neurovascular unit have been developed (Table 1), because the BBB serves as a key barrier to the delivery of many neuroactive therapeutics and plays a central part in many neurological diseases. These two-channel chips contain a brain endothelium interfaced with either brain pericytes and astrocytes, or astrocytes and neurons; some are lined with one or more cell types derived from iPS cells84,85,86, whereas others use primary cells87. BBB chips have been shown to reconstitute a human in vivo-like permeability barrier (for example, when analysed using transepithelial electrical resistance), which is critical for modelling healthy and diseased functions of neurovascular units84,85,86. They have also been used to model neuroinflammation84,87 and protection of neurons from injury when the vascular channel is perfused with human whole blood86, as well as shuttling of drugs, hormones, monoclonal antibodies, nanocarriers and tumour extracellular vesicles across the BBB85,86,88,89,90. Moreover, when BBB chips were created using brain endothelium from iPS cells derived from a patient with Huntingdon disease, inter-individual variability in BBB permeability could be detected, whereas this was not observed in chips made with cells from healthy donors86. This study used a similar approach to model monocarboxylate transporter 8 (MCT8) deficiency, a severe form of psychomotor retardation associated with a reduction in thyroid hormone transport across the brain endothelium, and this clinical phenotype could be replicated on-chip.
In a recent study, the human neurovascular unit was modelled in a microfluidic chip containing a perfused channel lined by endothelium interfaced with pericytes adjacent to a 3D ECM gel containing astrocytes and neurons derived by in situ differentiation of human neural stem cells91. This chip was used to model brain infection by the fungus Cryptococcus neoformans and revealed that clusters of the fungal cells penetrate the BBB without altering tight junctions, suggesting a transcytosis-mediated mechanism as well as providing a testbed in which to develop novel therapeutics.
Eye
An eye chip that reconstituted the outer retinal–choroid barrier containing human retinal pigmented epithelium adjacent to a perfusable 3D blood vessel network was able to recreate the choroidal neovascularization associated with wet macular degeneration by demonstrating penetration of the retinal pigmented epithelial monolayer by angiogenic sprouts that extended from pre-existing choroidal vessels92. This pathological angiogenesis was inhibited by the monoclonal therapeutic antibody bevacizumab, which is used clinically for this condition.
Furthermore, a retina chip was developed containing more than seven different retinal cell types, all derived from human iPS cells, which provided vascular perfusion and recapitulated interactions of mature photoreceptor segments with retinal pigmented epithelium93. In addition to mimicking the formation of outer segment-like structures and establishing in vivo-like physiological processes of the eye (for example, outer segment phagocytosis and calcium dynamics), this organ chip reproduced the clinical retinopathic toxicities of chloroquine and gentamicin.
Bone
A bone chip containing human mesenchymal stromal cells, osteoblasts and bone marrow mononuclear cells embedded within human de-cellularized bone was used to model the release of dissolved metals (cobalt and chromium) from arthroplasty implants, which can lead to peri-implant bone loss46. Indeed, both metals were found to integrate into bone matrix in this 3D model in a manner also seen in patient bone samples.
A bone marrow chip was created by incorporating human donor-derived CD34+ haematopoietic cells from marrow or blood and bone-marrow-derived stromal cells in a 3D ECM gel separated from a neighbouring endothelium-lined channel45. This chip reconstituted differentiation and maturation of multiple blood cell lineages over 1 month in culture and replicated myeloerythroid toxicities in response to clinically relevant exposures to the cancer therapeutic 5-fluorouracil as well as to γ-radiation. Even more impressively, it was able to replicate regimen-specific toxicities of a cancer drug observed in human clinical trials when the drug exposure (pharmacokinetics) profiles previously measured in patients were recreated on-chip. And again, these responses were not observed when the same haematopoietic cells were placed in static cultures. In addition, personalized bone marrow chip models of a rare genetic disorder, Shwachman–Diamond syndrome (SDS), were created in this study using CD34+ cells obtained from patients with SDS. These chips replicated key haematopoietic defects seen in patients and led to the discovery of a previously unknown neutrophil maturation abnormality in a subpopulation of these patients.
Reproductive organs
A mammary gland chip lined by mammary duct epithelium cultured in an ECM gel in close proximity to a perfused endothelium-lined vessel underwent branching morphogenesis on-chip94. Impressively, this chip was able to reproduce ductal changes associated with invasive cancer progression by amplifying HER2 (also known as ERBB2) receptor expression or expressing constitutively active PI3Kα in non-tumorigenic mammary epithelium using retroviral vectors.
A placenta chip lined by trophoblast cells interfaced with endothelial cells was used to screen drugs for their ability to cross the placenta95. This model reconstituted the efflux transporter-mediated active transport function of the human placental barrier, which serves to limit fetal exposure to maternally administered drugs, using the gestational diabetes mellitus drug glyburide.
Blood vessels
Organ chip models of large and small blood vessels have been used to study various vascular disorders, including inflammation, thrombosis and atherosclerosis, as well as to model vascular contributions to numerous diseases (Table 1). Notably, a blood vessel chip lined by vascular endothelium and perfused with human whole blood reproduced the clinical thrombotic toxicity of a therapeutic monoclonal antibody drug (Hu5c8) that had been previously withdrawn from clinical trials owing to unexpected life-threatening complications that were not detected during preclinical animal testing3. This model was also used to validate the lower thrombotic risk of an improved version of a related antibody that incorporates an Fc domain that does not bind the FcγRIIa receptor.
Hutchinson–Gilford progeria syndrome is a premature ageing disease characterized by accelerated death due to cardiovascular disease, particularly involving mechanically active blood vessels. When a progeria blood vessel chip was developed using vascular smooth muscle cells derived from iPS cells of patients with progeria, it exhibited exacerbated inflammation and DNA damage in response to mechanical strain compared to chips lined with cells from healthy donors96. Interestingly, this physical stress-induced injury could be reversed by administering two drugs, lovastatin and lonafarnib, which are often used clinically for this disease.
An aorta chip was used to study the development of the more common congenital disorder aortic valve disease97. Chips lined by aortic vascular smooth muscle cells isolated from patients exhibited suppressed NOTCH1 expression and impaired mitochondrial dynamics, and genetic knockdown of NOTCH1 in healthy smooth muscle cells suppressed mitochondrial fusion and reduced contractility. Use of this model revealed that the mitochondrial fusion activator drugs leflunomide and teriflunomide can partially rescue the mitochondrial dynamics disorder in these diseased cells.
Some of the vascular applications that have been most pursued using human organ chip technology focus on the study of angiogenesis, inflammation and the role of neovascularization in a wide range of diseases, as well as transendothelial transport and delivery of nanomaterials (Table 1). For example, by incorporating 3D ECM gels into one microfluidic channel, nearby endothelium can be stimulated to extend capillary sprouts in a directional manner; if endothelia are cultured in two parallel channels separated by ECM, the invasive sprouts will meet, differentiate into hollow tubes and form a fully perfused 3D capillary network15,58. Recently, this approach was adapted to demonstrate endothelial sprouting induced by primary human patient-derived renal cell carcinoma cells from multiple donors that exhibited patient-specific patterns of angiogenic factor production18. A similarly vascularized tumour chip revealed that gene expression, tumour heterogeneity and therapeutic responses observed on-chip more closely model colorectal tumour clinicopathology than do current standard drug screening modalities, including 2D cultures and 3D spheroids98. A three-channel microfluidic device that mimics the vascular–tissue–lymphatic interface has also been developed to study lymphangiogenesis associated with cancer metastasis19.
Lymphoid organs
Inflammatory reactions including recruitment and activation of immune cells, release of pro-inflammatory cytokines, tissue barrier disruption and cell injury have been reproduced in many organ chips (Table 1). However, it is possible to model more complex human immune responses using organ chip technology as well. A lymphoid follicle chip was recently developed that cultures peripheral blood-derived primary human blood B and T lymphocytes within a 3D ECM gel in one channel while flowing medium through a parallel channel44. Superfusion in this manner prevents the lymphocyte autoactivation observed in conventional cultures and promotes the formation of germinal centre-like structures that contain B cells that undergo antibody class switching and plasma cell differentiation upon activation with antigens. When autologous monocyte-derived dendritic cells were integrated into the gel and the chips were inoculated with a commercial influenza vaccine, increases in follicle size and number, plasma cell formation, production of anti-haemagglutinin IgG antibodies and secretion of cytokines similar to those observed in vaccinated humans were detected over clinically relevant timescales.
Organ chips have also been used to evaluate immunotherapies, including personalized therapies when used with patient-derived cells. For example, small-molecule inhibitors of cyclin-dependent kinases 4 and 6 (CDK4/6) were identified as potential novel immune checkpoint inhibitors using a chip in which patient-derived tumour cell spheroids containing autologous infiltrating immune cells were cultured within 3D ECM gels surrounded by microfluidic channels99. These CDK4/6 inhibitors augmented the response to PD-1 blockade, thus providing a rationale for combining these agents with other immunotherapies to enhance therapeutic efficacy.
The tumour microenvironment imposes significant constraints on the anticancer efficacy of adoptive T cell immunotherapy, which cannot easily be modelled in conventional cultures. To address this challenge, human liver cancer cells were cultured in an organ chip inside a 3D ECM gel, and human T cells engineered to express tumour-specific T cell receptors (TCRs) T cells were flowed through an adjacent channel100. The TCR T cell ability to migrate and kill tumour targets was analysed, as well as the influence of varying levels of oxygen and inflammatory cytokines. This approach could be used in the future to design and evaluate personalized cellular immunotherapies, for example, using 3D-printed organ chips containing patient-derived tumour biopsy samples that enable real-time monitoring of the responses of resident lymphocyte populations101.
Multi-organ physiological coupling and drug PK/PD modelling
One of the challenges in drug development that most pharmaceutical scientists assume is likely not to be solved in the near term is the need for animal testing to assess drug disposition (for example, ADME) in the whole body and to quantify PK/PD parameters that help to guide clinical trial design. Although animal models may still be required for these purposes for some years to come, recent work suggests that many of these complex whole-body physiological responses may be modelled in vitro using fluidically coupled multi-organ human body-on-chips systems. Multi-organ chip models developed so far, which may have from 2 to 10 different organ chips, have provided impressive mimicry of complex physiological and pathophysiological responses in addition to providing new in vitro tools with which to assess drug ADME and PK/PD (Table 2).
In one impressive example of complex physiological mimicry, dynamic inter-organ hormonal coupling and the 28-day menstrual cycle were reconstituted in vitro by fluidically linking human organ chip models of uterus, cervix, fallopian tube and liver to a mouse ovary chip and recirculating the medium25. Coupled organ chip systems have also been used to model multi-organ regulation of insulin secretion30,102; metastatic spread of cancer between different organs103; targeted immune responses to organ-specific damage104; and the toxic effects of environmental chemicals that required hepatic bioactivation105, including activation of germ cell toxins in linked liver and testis chips31.
The most common application of multi-organ chip systems has been to replicate the absorption (for example, oral or transdermal), distribution and clearance of drugs through metabolism or excretion that naturally occur in the organs of our body, but which are absent in single organ chips or other in vitro models (Table 1). Small-molecule and biologic drug efficacies, as well as both on- and off-target toxicities that can be modified by inter-organ interactions (via hepatic activation or metabolism), have been demonstrated too, and the results generated generally agree with published human data32,34,35,106,107,108,109. In addition, organ chips have been coupled to assess drug efficacy and safety simultaneously. For example, by measuring the effects of anti-EGFR therapeutic monoclonal antibodies in a lung cancer chip while analysing toxicity in a fluidically coupled skin chip, it was possible to replicate the inhibition of keratinocyte growth and altered expression of CXCL8 and CXCL10 seen in patients110. As another example, a clinically relevant drug–drug interaction between the arrhythmogenic gastroprokinetic drug cisapride and the fungicide ketoconazole was reproduced using a liver chip linked to a heart chip when both were lined by cells derived from the same human iPS cell line111, which shows the potential of using organ chips to optimize therapeutics selection in a personalized manner.
Microfluidic systems composed of multiple culture chambers in which medium is flowed directly from one parenchymal cell compartment to another via a single channel have been used to carry out PK/PD modelling in vitro for many years, although established cell lines were usually used in early studies33. Later versions separated the flow chamber from the culture chambers with an intervening porous membrane or used transwell inserts as culture chambers26,27,28,29,30 (Fig. 2a). A body-on-chip system of this type composed of linked liver, kidney, intestine, skeletal muscle and BBB chips generated predictions of drug metabolism and barrier penetrance that were consistent with clinical data112. But it is only recently that this type of modelling has been carried out in organ chips lined by primary human cells that are fluidically linked by flowing or transferring a common blood substitute medium through endothelium-lined vascular channels that are separated from parenchymal channels or chambers, as occurs in our bodies13,31,32,34,35 (Fig. 2b,c). This more physiologically relevant approach has been used in combination with computational modelling to generate predictions of drug PK/PD parameters (Fig. 3) and toxicities in many studies (Table 2). For example, it has been possible to quantitatively predict human clinical pharmacokinetics parameters using a multi-organ chip first-pass model composed of linked two-channel human liver, kidney and intestine chips along with an AV reservoir in combination with computational physiologically based pharmacokinetics modelling35 (Fig. 3b). Successful in vitro to in vivo translation of pharmacokinetics parameters closely mimicked results observed clinically (Fig. 3c) and this was carried out using two drugs (cisplatin and nicotine) administered via two different routes (intravenous versus oral). In addition, when the intestine chip was replaced by a bone marrow chip, predictions of cisplatin pharmacodynamics also matched previously reported patient data35. As these body-on-chip systems are modular, the chips can be linked either in series or in parallel, and the flow can be either unidirectional13,30,34,35,102,103,105,111,112 or recirculated25,27,28,31,32,104,106,107,108,109,110,113.

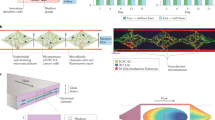
a | Multi-organ chip systems linked by common flow channels can mimic the physiological linking of organs in our bodies, and hence drug absorption, distribution, metabolism and excretion (ADME) that occurs in the human body as a result of whole body-level physiology can be modelled using this approach. Aerosolized, oral and intravenous (IV) delivery of drugs that occurs in our bodies can be modelled by introducing them into the air space of a lung chip, the lumen of an intestine chip or the vascular channel, respectively; however, IV dosing can be complicated by organ chips immediately downstream from the injection site abnormally experiencing higher doses than other chips due to the lack of mixing that normally occurs in human vasculature. Linked liver and kidney chips can be used to quantify drug metabolism and clearance, respectively, and by linking other relevant chips (for example, a bone marrow chip for myelotoxins), efficacy and potency can be measured as well. b | A schematic diagram showing the fluidic linkages among two-channel intestine, liver and kidney chips corresponding to flow through respective in vivo organ-feeding vessels mimicked by robotic fluid transfers (long arrows indicating flow direction) along with an arteriovenous (AV) reservoir that is fluidically linked to the vascular channels of the organ chips to model blood mixing for more physiologically relevant drug exposures across all chips and to enable experimental sampling analogous to peripheral blood sampling. A common blood substitute is flowed through the vascular channels and the AV reservoir while organ-specific medium is flowed through the parenchymal channel of each chip (small arrows). c | Because the drug levels in effluents of both the vascular and parenchymal channels can be measured over time, pharmacokinetics and pharmacodynamics (PK/PD) parameters — such as area under the curve (AUC), maximum drug concentration in blood (Cmax), and time to reach half-maximal levels (t1/2) — can be determined in vitro using computational physiologically based PK modelling along with scaling approaches. d | This approach has been used to quantitatively predict PK/PD parameters observed in humans in vivo, for example, as shown for cisplatin35, using the body-on-chips linking configuration shown in part b. Squares and triangles indicate PK data obtained from patients in which cisplatin was infused for 1 hour or 3 hours, and dotted lines indicate computational PK predictions generated using data obtained from the human body-on-chips model. The vertical error bars represent the standard deviation. Parts a and b are adapted with permission from ref.148, Annual Reviews. Part c is reprinted with permission from ref.148, Annual Reviews. Part d is reprinted with permission from ref.35, Springer Nature Limited.
What will it take to replace animal models?
The question of what will be required to enable eventual acceptance of human organ chips in lieu of preclinical animal models, their integration into drug development pipelines, and inclusion of data generated using these models in FDA regulatory submissions is a complex one114,115. One point that is clear is that this process will probably occur gradually and involve replacement of one particular animal model at a time. To meet this goal, pharmaceutical companies and regulatory agencies will require rigorous demonstration of equivalent or superior performance relative to animal models. This goal will require definition of specific validation methods and performance criteria in addition to successful completion of statistically well powered validation studies that demonstrate that a specific organ chip model can generate human-relevant results in a reproducible and statistically robust manner regardless of where the work is carried out. Because only small numbers of organ chips are used in most academic studies, and different groups use different device configurations and methodologies, true validation of their use as animal replacements will require large-scale evaluation involving hundreds of devices of the same design carried out using the same protocols. Thus, this problem will not be solved by academic researchers publishing more research or even establishing design standards at the community level. Instead, this will probably be decided on the commercial battlefield, which is now possible given that there are many companies manufacturing organ chips as well as automated control systems to run them116.
A key challenge is to identify the critical design criteria and performance parameters that must be met to qualify an organ chip model as functional at a level necessary for adoption by pharmaceutical and biotechnology companies as well as regulatory agencies115. This will require commercial chip suppliers to demonstrate reliability, robustness and consistency in terms of cell sourcing as well as to define performance and quality control expectations precisely for every step in the execution and analysis of these model systems. Importantly, all interested parties recognize that there is no way to pursue replacement of animal models in a generalized way, and instead that each organ chip must be validated for a specific commercial ‘context of use’ (for example, drug safety, drug efficacy, toxicology, ADME characterization, PK/PD modelling). However, each organ chip must also be validated for a particular physiological context of use. For example, a lung alveolus chip with epithelium and endothelium might be useful for studying efficacy of drugs in pharmacology studies relating to pulmonary oedema, but a different chip also containing fibroblasts would be required to model pulmonary fibrosis.
Commercial adoption is likely to occur in a staged or stepwise manner by identifying applications that can overcome existing barriers in the pharmaceutical industry workflow, for example, by validating that human organ chips can successfully replace existing animal models that are known to be suboptimal and lead to failures in the clinic (for example, rodent and dog liver toxicity assays). But as their predictive power is increasingly validated over time, they should be integrated into other early stages of the drug development pipeline, including being used for drug discovery and lead target validation as well as for the optimization and prioritization of lead compounds, or for the identification of alternate leads when safety issues arise (Fig. 4). For example, organ chips lined by patient-derived cells would be particularly useful when toxicities are observed in a particular patient subpopulation in phase I trials (for example, women versus men, or a genetically similar subgroup); if the toxicity could be replicated on chip, this would provide a valuable tool with which to identify other related active compounds in the pipeline that do not exhibit this toxicity. There is also increasing recognition of the need to create organ chip models made with cells from other species (for example, rodents, dogs or NHPs) to generate benchmark comparison data that could help pharmaceutical companies and vaccine developers to validate results obtained in these models relative to results obtained in past in vivo studies, and thus become more comfortable with integrating organ chips into their development processes115. Although the initial goal in these efforts might be to demonstrate equivalence or superiority to a particular animal model, the ultimate benchmark for success should be the replication of existing in vivo human clinical data.

Organ chips lined by patient-derived cells may be used to model rare genetic disorders, to identify toxicities difficult to study clinically (for example, effects of lethal radiation exposure or exposure of pregnant women to potential teratogens) or to compare drug responses in different subpopulations (such as women versus men or young versus old individuals). When multiple chips are created, each lined with cells from a different donor representing a different subpopulation or a patient with a different comorbidity, they might also be used to design and optimize drugs for specific subgroups whose members could be used as participants in future targeted clinical trials to increase the likelihood of success. Individualized single-organ chips and multi-organ chip systems lined by one or more organotypic cell types from the same patient (for example, using induced pluripotent stem (iPS) cell technology), from a population of genetically related individuals, or from patients with similar comorbidities, could also be used to personalize drug selection to optimize drug efficacy, minimize toxicity, determine optimal delivery routes, and when combined with pharmacokinetics and pharmacodynamics (PK/PD) predictions, to design optimal dosing regimens for use in targeted phase I clinical trials.
On a more pragmatic level, organ chips are generally more expensive than conventional cultures, and so to be accepted, the improvement in performance they offer must be equally great. However, given the level of human clinical mimicry that they can potentially provide, animal models might be a more appropriate comparand in terms of costs and complexity of use. In this context, it is important to note that a survey of pharmaceutical companies estimated that microphysiological systems could save their industry up to about 25% in research and development costs117. Another recent analysis suggests that even the replacement of one animal model by an organ chip for a single context of use, such as drug-induced liver toxicity, could generate the industry US$3 billion annually owing to increased productivity118. Importantly, because the organ chip industry is in its infancy, the cost of chips and the instruments required to run them should also progressively decrease over time, owing to economies of scale.
To meet these challenges and harness these benefits, international organizations composed of experts from the pharmaceutical and biotechnology industries, such as the microphysiological systems affiliate of the International Consortium for Innovation and Quality in Pharmaceutical Development (IQ) Consortium, have proposed approaches to be followed to qualify organ chips and other microphysiological systems to ensure their acceptance in lieu of animal models by the industry and, hopefully, by regulatory agencies1. These requirements include obvious ones, such as following good laboratory practice (GLP) regulations and developing strategies to validate and document the functionality, reliability and robustness of cell sources, devices, instruments, experimental protocols and analytical procedures. But they have also established specific performance metrics for particular applications, such as prediction of drug-induced liver injury (DILI), which includes testing of specific drug compounds that have known safety records in animals and humans119. These types of studies are important because they can be used to quantitatively assess the predictive performance of organ chips and compare those predictions to both human clinical data and past animal results. The FDA’s Innovative Science and Technology Approaches for New Drugs (ISTAND) pilot programme, which has established similar qualification programmes for drug development tools, including organ chips, now also offers a defined path to expedite their acceptance for regulatory use. One positive sign for the field is that a recent report describes an analysis involving almost 800 human liver chips created with cells from two different donors that successfully met the IQ consortium’s DILI qualification guidelines across a blinded set of over 27 known hepatotoxic and non-toxic drugs with a sensitivity of up to 87% and a specificity of 100%, demonstrating a major improvement in performance relative to animal models118.
Future opportunities and challenges
The advances described above demonstrate that human organ chips can be used to address many types of question in human biology and medicine that could not be addressed using prior culture systems or even many static microphysiological systems. For example, whereas static 3D organoid cultures have many advantages (for example, they provide patient-specific insights, exhibit highly differentiated behaviours, and are easy to multiplex for higher-throughput studies), they cannot provide many of the critical functions offered by organ chips, including direct quantification of barrier function, digestion and absorption; the creation of an air–liquid interface; the physiological recruitment of circulating immune cells; the application of physiological mechanical cues and fluid shear stress; sustained co-culture with a complex microbiome; replication of tissue responses to clinically relevant drug exposure profiles with delivery of drugs and chemicals across an endothelial barrier; or the prediction of human drug pharmacokinetics parameters. However, these two technologies are clearly synergistic, because organ chips lined by patient organoid-derived cells offer some of the most powerful in vitro preclinical models available today. Even more importantly, as described above, organ chips have been repeatedly shown to mimic human pathophysiology and predict human clinical responses to therapeutics better than animal models do, and hence the bar has been raised when it comes to relying on animal-testing data. Notably, the use of healthy and diseased human organ chip models for toxicity and efficacy testing represents a major advantage over existing testing approaches that use inbred healthy animals (Box 1). Also, exploring the variation of results obtained using organ chips made with cells from different patient donors, from individuals from defined genetic subpopulations, or even patients with different comorbidities (that is, other ongoing chronic disorders), might better replicate the diversity of in vivo clinical data, and may help to both facilitate clinical trial design and improve success. In addition, their ability to support extended co-culture of human cells with a complex living microbiome provides a tool with which to analyse host–microorganism interactions as well as to test potential probiotic or prebiotic therapies in a human-relevant context. Moreover, when fluidically coupled to create human body-on-chips systems, they can potentially provide a testbed for pharmaceutical scientists to analyse complex drug disposition, ADME and PK/PD behaviours, in addition to enabling basic scientists to study complex inter-organ interactions (for example, menstrual cycle or metastatic cancer spread). This could have real clinical value: for example, the ability to quantitatively predict human drug PK/PD parameters in vitro could be used to optimize dose regimens for new therapeutics and thereby shorten phase I clinical trials.
Organ chips can be particularly useful for applications when it is difficult to carry out studies in humans (for example, effects of potentially lethal radiation exposures), when preclinical testing needs to be carried out using biologics that do not recognize non-human targets, or when a genetic disease cannot be modelled using other approaches (Fig. 4). The ability to build chips with patient-derived cells also opens up new approaches for therapeutic development, particularly for patients with rare genetic disorders. Organ chips provide a more human-relevant alternative to animal models such as genetically engineered mice that do not faithfully replicate human physiology or disease states, yet that still dominate basic research and drug development today. Perhaps more importantly, given the difficulty in gathering enough patients with a rare disorder together at a single site for clinical study, it could provide a way to carry out a preliminary human trial on chips using cells obtained from patients around the world to optimize drug candidate selection before initiating full trials. One can also imagine using organ chips lined by female versus male cells to circumvent the current lack of female individuals in human clinical trials, to compare young versus old cells to carry out paediatric trials on chips, or to test drugs on subpopulations of patients with similar comorbidities, given the difficulty in doing this in the clinic (Fig. 4). Also, as it is very difficult to carry out safe clinical trials in pregnant women, the ability to study drug and toxin passage across the placental barrier on-chip could provide a valuable new preclinical model for drug developers.
The COVID-19 pandemic has brought to the fore the need for new ways to accelerate drug repurposing and discovery during viral outbreaks, and human organ chips have been successfully used to confront this challenge. The recently developed lymphoid follicle chip that recapitulates germinal centre formation, plasma cell generation, antigen-specific antibody class switching and relevant cytokine production44 offers a potential human-relevant replacement for the NHP models currently used to test vaccines or drugs, which do not always predict results in humans and are severely limited in availability owing to the COVID-19 pandemic. In addition, the ability to model virus evolution using lung airway chips61 offers the possibility of predicting new strains before they emerge, which could accelerate development of both drugs and seasonal vaccines for various viruses (for example, influenza, SARS-CoV-2 variants) in the future. Given the ability of organ chips to reproduce human bacterial infections in tissues from both healthy individuals and patients with chronic diseases (for example, cystic fibrosis)11,40,43,73,74, and to mimic fungal infections91 as well, similar approaches may be used for the discovery of antibiotics for emerging antibiotic-resistant organisms.
In the short term, the use of organ chips lined by mouse, rat, dog, NHP and other animal cells could help to validate the approach, garner acceptance by pharmaceutical and academic scientists115 and provide a way to confront One Health challenges, for example, by developing animal models to study how disease vectors arise before they reach humans (for example, SARS-CoV-2 infection in bat organ chips). However, based on recent progress in this field using iPS cells (Table 1), it is now possible to imagine a future in which single organ chips or multi-organ chip systems fabricated using organotypic cells from the same patient could be used as personalized living avatars to repurpose or select more effective and less toxic therapeutics, optimize delivery routes and design dose regimens for that specific patient, thereby opening up a new path in precision medicine (Fig. 4). Such an approach will be costly, and so an earlier step might be to create chips lined with primary or stem cells from patients representing unique genetic subpopulations and to use these chips to develop or select new drugs for particular groups, then carry out clinical trials using these patients (Fig. 4). This mid-term approach could shorten the time, decrease the cost and increase the likelihood of success in the drug development process.
Although organ chip technology has much to offer, there are still technical, social and economic hurdles that must be overcome before we see significant numbers and types of animals being replaced. Sourcing of high-quality human cells is always a great challenge, but this is now less of a problem given the availability of iPS cells and organoids. Most organ chips are also low throughput, but high content; this can be valuable at later stages in the drug development pipeline (for example, where selection of lead compounds must be made for drug advancement), but not in the earlier discovery phase. Higher-throughput systems are needed to enable practical implementation of statistically significant studies with many replicates, which is essential for pharmaceutical validation, and indeed, the recent development of higher-throughput organ chips (that is, single devices containing many parallel culture chambers) that still retain many of the key features required for drug developers21,120 (Table 1) suggest that this path should be pursued as well. However, this goal also can be met by developing automated instruments that can culture and analyse multiple lower-throughput organ chips simultaneously.
Human organ chips have been created using primary cells derived from human donors from commercial suppliers, established cell lines and patient-derived organoids or iPS cells (Table 1). Although the prevailing view is that use of established lines is not ideal from the standpoint of in vitro to in vivo correlation and that the use of primary cells or patient-derived cells is clearly preferred, cell lines could be valuable for particular applications as long as the model is experimentally validated by demonstrating successful mimicry of in vivo human responses relevant to the specific context of use. This is because cell lines can provide a virtually unlimited supply of similar cells that can enable higher-throughput studies and potentially enhance the reproducibility of results, which may be extremely valuable for early phases of drug development. While iPS cells offer the same advantage, they can be limited by their frequent inability to exhibit a fully mature differentiated phenotype. However, if chips lined by cell lines are used, confirmatory follow-up studies would then need to be carried out with a smaller set of lead compounds using organ chips lined by primary cells.
Another potential limitation is the prevailing concern that the PDMS polymer used to fabricate many organ chips cannot be used effectively for drug studies owing to drug absorption121; however, this has not turned out to be as serious a concern as originally assumed. Significant absorption by PDMS is observed only with highly hydrophobic drugs, which represent a subset of small-molecule therapeutics, and the reality is that more than 40% of drugs in current development pipelines are biologics (for example, therapeutic antibodies or RNA therapeutics) that do not have these issues. Mitigation strategies, such as computational correction of the absorption effect by quantifying drug levels using mass spectrometry have been described, which circumvent this problem for most drugs (for example, as demonstrated in testing of 27 different small-molecule drugs, including hydrophobic compounds)118. In fact, most of the organ chip studies reviewed here used PDMS devices (Table 1), and this includes those that demonstrated in vivo mimicry in response to clinically relevant drug exposures and that enabled quantitative prediction of human pharmacokinetics parameters35,45. Nevertheless, it is true that for a small number of highly hydrophobic compounds (for example, sex steroids), absorption into PDMS remains a complicating factor. This limitation can be circumvented by coating PDMS with non-absorptive coatings122, using alternative flexible elastomeric materials that are less absorptive (for example, certain polyurethanes, styrenic block copolymers, polycarbonate-thermoplastic elastomer hybrids)123,124 or by using rigid thermoplastics that are non-absorptive (for example, polystyrene or polycarbonate)125.
Perhaps the biggest challenge we face is a conceptual one: many pharmaceutical, regulatory and academic researchers have much invested in the ways they carry out research at present, and they may be wary of changing their ways; convincing data that demonstrates the advantages of human organ chips over animal models will be needed before they accept this new technology into their laboratories. Nonetheless, with the help of groups such as the IQ Consortium and the FDA115, there are now benchmarks that must be met to validate the use of human organ chips as animal replacements, and the first advances are being seen in this area. As a result, we are now at a tipping point in this field, with the possibility of seeing real reductions in the use of animals and the application of more effective approaches for drug development and personalized medicine as well as basic research in the near future.
References
Fabre, K. et al. Introduction to a manuscript series on the characterization and use of microphysiological systems (MPS) in pharmaceutical safety and ADME applications. Lab Chip 20, 1049–1057 (2020).
Golding, H., Khurana, S. & Zaitseva, M. What is the predictive value of animal models for vaccine efficacy in humans? The importance of bridging studies and species-independent correlates of protection. Cold Spring Harb. Persp. Biol. 10, a028902 (2018).
Barrile, R. et al. Organ-on-chip recapitulates thrombosis induced by an anti-CD154 monoclonal antibody: translational potential of advanced microengineered systems. Clin. Pharmacol. Ther. 104, 1240–1248 (2018).
Seok, J. et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl Acad. Sci. USA 110, 3507–3512 (2013).
Franco, R. & Cedazo-Minguez, A. Successful therapies for Alzheimer’s disease: why so many in animal models and none in humans? Front. Pharmacol. 5, 146 (2014).
Zhang, B. et al. Advances in organ-on-a-chip engineering. Nat. Rev. Mater. 3, 257–278 (2018).
Chen, Y. et al. Advances in engineered three-dimensional (3D) body articulation unit models. Drug Des. Dev. Ther. 16, 213–235 (2022).
Victor, I. A., Andem, A. B., Archibong, I. A. & Iwok, E. O. Interplay between cell proliferation and cellular differentiation: a mutually exclusive paradigm. Glob. Sci. J. 8, 1328–1338 (2020).
Ghallab, A. In vitro test systems and their limitations. EXCLI J. 12, 1024–1026 (2013).
Clevers, H. Modeling development and disease with organoids. Cell 165, 1586–1597 (2016).
Huh, D. et al. Reconstituting organ-level lung functions on a chip. Science 328, 1662–1668 (2010).
Huh, D. et al. Acoustically detectable cellular-level lung injury induced by fluid mechanical stresses in microfluidic airway systems. Proc. Natl Acad. Sci. USA 104, 18886–18891 (2007).
Novak, R. et al. Robotic fluidic coupling and interrogation of multiple vascularized organ chips. Nat. Biomed. Eng. 4, 407–420 (2020).
Varone, A. et al. A novel organ-chip system emulates three-dimensional architecture of the human epithelia and the mechanical forces acting on it. Biomaterials 275, 120957 (2021).
Nguyen, D. H. et al. Biomimetic model to reconstitute angiogenic sprouting morphogenesis in vitro. Proc. Natl Acad. Sci. USA 110, 6712–6717 (2013).
Trietsch, S. J., Israëls, G. D., Joore, J., Hankemeier, T. & Vulto, P. Microfluidic titer plate for stratified 3D cell culture. Lab Chip 13, 3548–3554 (2013).
Phan, D. T. T. et al. A vascularized and perfused organ-on-a-chip platform for large-scale drug screening applications. Lab Chip 17, 511–520 (2017).
Miller, C. P., Tsuchida, C., Zheng, Y., Himmelfarb, J. & Akilesh, S. A 3D human renal cell carcinoma-on-a-chip for the study of tumor angiogenesis. Neoplasia 20, 610–620 (2018).
Cho, Y. et al. Three-dimensional in vitro lymphangiogenesis model in tumor microenvironment. Front. Bioeng. Biotechnol. 9, 697657 (2021).
Domansky, K. et al. Perfused multiwell plate for 3D liver tissue engineering. Lab Chip 10, 51–58 (2010).
Gard, A. L. et al. High-throughput human primary cell-based airway model for evaluating influenza, coronavirus, or other respiratory viruses in vitro. Sci. Rep. 11, 14961 (2021).
Homan, K. A. et al. Bioprinting of 3D convoluted renal proximal tubules on perfusable chips. Sci. Rep. 6, 34845 (2016).
Lin, N. Y. C. et al. Renal reabsorption in 3D vascularized proximal tubule models. Proc. Natl Acad. Sci. USA 116, 5399–5404 (2019).
Zhang, Y. S. et al. Bioprinting 3D microfibrous scaffolds for engineering endothelialized myocardium and heart-on-a-chip. Biomaterials 110, 45–59 (2016).
Xiao, S. et al. A microfluidic culture model of the human reproductive tract and 28-day menstrual cycle. Nat. Commun. 8, 14584 (2017).
Tsamandouras, N. et al. Integrated gut and liver microphysiological systems for quantitative in vitro pharmacokinetic studies. AAPS J. 19, 1499–1512 (2017).
Edington, C. D. et al. Interconnected microphysiological systems for quantitative biology and pharmacology studies. Sci. Rep. 8, 4530 (2018).
Chen, W. L. K. et al. Integrated gut/liver microphysiological systems elucidates inflammatory inter-tissue crosstalk. Biotechnol. Bioeng. 114, 2648–2659 (2017).
Wagner, I. et al. A dynamic multi-organ-chip for long-term cultivation and substance testing proven by 3D human liver and skin tissue co-culture. Lab Chip 13, 3538–3547 (2013).
Bauer, S. et al. Functional coupling of human pancreatic islets and liver spheroids on-a-chip: towards a novel human ex vivo type 2 diabetes model. Sci. Rep. 7, 14620 (2017).
Baert, Y. et al. A multi-organ-chip co-culture of liver and testis equivalents: a first step toward a systemic male reprotoxicity model. Hum. Reprod. 35, 1029–1044 (2020).
Maschmeyer, I. et al. Chip-based human liver-intestine and liver-skin co-cultures — a first step toward systemic repeated dose substance testing in vitro. Eur. J. Pharm. Biopharm. 95, 77–87 (2015).
Shuler, M. L., Ghanem, A., Quick, D., Wong, M. C. & Miller, P. A self-regulating cell culture analog device to mimic animal and human toxicological responses. Biotechnol. Bioeng. 52, 45–60 (1996).
Maoz, B. M. et al. A linked organ-on-chip model of the human neurovascular unit reveals the metabolic coupling of endothelial and neuronal cells. Nat. Biotechnol. 36, 865–874 (2018).
Herland, A. et al. Quantitative prediction of human pharmacokinetic responses to drugs via fluidically coupled vascularized organ chips. Nat. Biomed. Eng. 4, 421–436 (2020).
Kasendra, M. et al. Development of a primary human small intestine-on-a-chip using biopsy-derived organoids. Sci. Rep. 8, 2871 (2018).
Kerns, S. J. et al. Human immunocompetent organ-on-chip platforms allow safety profiling of tumor-targeted T-cell bispecific antibodies. eLife 10, e67106 (2021).
Mondadori, C. et al. Recapitulating monocyte extravasation to the synovium in an organotypic microfluidic model of the articular joint. Biofabrication 13, 115–128 (2021).
Benam, K. H. et al. Small airway-on-a-chip enables analysis of human lung inflammation and drug responses in vitro. Nat. Methods 13, 151–157 (2016).
Plebani R. et al. Modeling pulmonary cystic fibrosis in a human lung airway-on-a-chip: cystic fibrosis airway chip. J. Cyst. Fibrosis https://doi.org/10.1016/j.jcf.2021.10.004 (2021).
Zhang, M. et al. Biomimetic human disease model of SARS-CoV-2 induced lung injury and immune responses on organ chip system. Adv. Sci. 8, 2002928 (2020).
Si, L. et al. A human-airway-on-a-chip for the rapid identification of candidate antiviral therapeutics and prophylactics. Nat. Biomed. Eng. 5, 815–829 (2021).
Kim, H. J., Li, H., Collins, J. J. & Ingber, D. E. Contributions of microbiome and mechanical deformation to intestinal bacterial overgrowth and inflammation in a human gut-on-a-chip. Proc. Natl Acad. Sci. USA 113, E7–E15 (2016).
Goyal, G. et al. Ectopic lymphoid follicle formation and human seasonal influenza vaccination responses recapitulated in an organ-on-a-chip. Adv. Sci. https://doi.org/10.1002/advs.202103241 (2022).
Chou, D. B. et al. On-chip recapitulation of clinical bone marrow toxicities and patient-specific pathophysiology. Nat. Biomed. Eng. 4, 394–406 (2020).
Schoon, J. et al. Metal-specific biomaterial accumulation in human peri-implant bone and bone marrow. Adv. Sci. 7, 2000412 (2020).
Jalili-Firoozinezhad, S. et al. A complex human gut microbiome cultured in an anaerobic intestine-on-a-chip. Nat. Biomed. Eng. 3, 520–531 (2019).
Kim, R. et al. An in vitro intestinal platform with a self-sustaining oxygen gradient to study the human gut/microbiome interface. Biofabrication 12, 015006 (2019).
Odijk, M. et al. Measuring direct current trans-epithelial electrical resistance in organ-on-a-chip microsystems. Lab Chip 15, 745–752 (2015).
van der Helm, M. W. et al. Non-invasive sensing of transepithelial barrier function and tissue differentiation in organs-on-chips using impedance spectroscopy. Lab Chip 19, 452–463 (2019).
Kujala, V. J., Pasqualini, F. S., Goss, J. A., Nawroth, J. C. & Parker, K. K. Laminar ventricular myocardium on a microelectrode array-based chip. J. Mater. Chem. B. 4, 3534–3543 (2016).
Huh, D. et al. A human disease model of drug toxicity-induced pulmonary edema in a lung-on-a-chip microdevice. Sci. Transl Med. 4, 159ra147 (2012).
Arathi, A., Joseph, X., Akhil, V. & Mohanan, P. V. l-Cysteine capped zinc oxide nanoparticles induced cellular response on adenocarcinomic human alveolar basal epithelial cells using a conventional and organ-on-a-chip approach. Colloids Surf. B 211, 112300 (2021).
Jain, A. et al. Primary human lung alveolus on a chip model of intravascular thrombosis for assessment of therapeutics. Clin. Pharmacol. Ther. 103, 332–340 (2018).
Benam, K. H. et al. Matched-comparative modeling of normal and diseased human airway responses using a microengineered breathing lung chip. Cell Syst. 3, 456–466.e4 (2016).
Nawroth, J. C. et al. A microengineered airway lung chip models key features of viral-induced exacerbation of asthma. Am. J. Respir. Cell Mol. Biol. 63, 591–600 (2020).
Hassell, B. et al. Human organ chip models recapitulate orthotopic lung cancer growth, therapeutic responses and tumor dormancy in vitro. Cell Rep. 21, 508–516 (2017).
Paek, J. et al. Microphysiological engineering of self-assembled and perfusable microvascular beds for the production of vascularized three-dimensional human microtissues. ACS Nano 13, 7627–7643 (2019).
Bai, H. et al. Mechanical control of innate immune responses against viral infection revealed in a human lung alveolus chip. Preprint at bioRxiv https://doi.org/10.1101/2021.04.26.441498 (2021).
Si, L. et al. Self-assembling short immunostimulatory duplex RNAs with broad spectrum antiviral activity. Preprint at bioRxiv https://doi.org/10.1101/2021.11.19.469183 (2021).
Si, L. et al. Clinically relevant influenza virus evolution reconstituted in a human lung airway-on-a-chip. Microbiol. Spectr. 9, e0025721 (2021).
Sarkar, U. et al. Metabolite profiling and pharmacokinetic evaluation of hydrocortisone in a perfused three-dimensional human liver bioreactor. Drug Metab. Dispos. 43, 1091–1099 (2015).
Long, T. J. et al. Modeling therapeutic antibody-small molecule drug–drug interactions using a three-dimensional perfusable human liver coculture platform. Drug Metab. Dispos. 44, 1940–1948 (2016).
Tsamandouras, N. et al. Quantitative assessment of population variability in hepatic drug metabolism using a perfused three-dimensional human liver microphysiological system. J. Pharmacol. Exp. Ther. 360, 95–105 (2017).
Lee, P. J., Hung, P. J. & Lee, L. P. An artificial liver sinusoid with a microfluidic endothelial-like barrier for primary hepatocyte culture. Biotechnol. Bioeng. 97, 1340–1346 (2007).
Jang, K.-J. et al. Reproducing human and cross-species toxicities using a liver-chip. Sci. Transl Med. 11, eaax5516 (2019).
Ortega-Prieto, A. M. et al. 3D microfluidic liver cultures as a physiological preclinical tool for hepatitis B virus infection. Nat. Commun. 9, 682 (2018).
Kim, H. J. & Ingber, D. E. Gut-on-a-chip microenvironment induces human intestinal cells to undergo villus differentiation. Integr. Biol. 5, 1130–1140 (2013).
Sontheimer-Phelps, A. et al. Human colon-on-a-chip enables continuous in vitro analysis of colon mucus layer accumulation and physiology. Cell Mol. Gastroenterol. Hepatol. 9, 507–526 (2020).
Bein, A. et al. Enteric coronavirus infection and treatment modeled with an immunocompetent human intestine-on-a-chip. Front. Pharmacol. 12, 718484 (2021).
Guo, Y. et al. SARS-CoV-2 induced intestinal responses with a biomimetic human gut-on-chip. Sci. Bull. 66, 783–793 (2021).
Shah, P. et al. A microfluidics-based in vitro model of the gastrointestinal human-microbe interface. Nat. Commun. 7, 11535 (2016).
Tovaglieri, A. et al. Species-specific enhancement of enterohemorrhagic E. coli pathogenesis mediated by microbiome metabolites. BMC Microbiome 7, 43 (2019).
Grassart, A. et al. Bioengineered human organ-on-chip reveals intestinal microenvironment and mechanical forces impacting Shigella infection. Cell Host Microbe 26, 435–444.e4 (2019).
Jalili-Firoozinezhad, S. et al. Modeling radiation injury and countermeasure drug responses in a human gut-on-a-chip. Cell Death Dis. 9, 223 (2018).
Bein, A. et al. Nutritional deficiency recapitulates intestinal injury associated with environmental enteric dysfunction in patient-derived Organ Chips. Preprint at medRxiv https://doi.org/10.1101/2021.10.11.21264722 (2021).
Jang, K. J. et al. Human kidney proximal tubule-on-a-chip for drug transport and nephrotoxicity assessment. Integr. Biol. 5, 1119–1129 (2013).
Wang, J. et al. A virus-induced kidney disease model based on organ-on-a-chip: pathogenesis exploration of virus-related renal dysfunctions. Biomaterials 219, 119367 (2019).
Naik, S. et al. A 3D renal proximal tubule on chip model phenocopies lowe syndrome and Dent II disease tubulopathy. Int. J. Mol. Sci. 22, 5361 (2021).
Zhou, M. et al. Development of a functional glomerulus at the organ level on a chip to mimic hypertensive nephropathy. Sci. Rep. 6, 31771 (2016).
Musah, S. et al. Mature induced-pluripotent-stem-cell-derived human podocytes reconstitute kidney glomerular-capillary-wall function on a chip. Nat. Biomed. Eng. 1, 0069 (2017).
Roye, Y. et al. A personalized glomerulus chip engineered from stem cell-derived epithelium and vascular endothelium. Micromachines 12, 967 (2021).
Petrosyan, A. et al. A glomerulus-on-a-chip to recapitulate the human glomerular filtration barrier. Nat. Commun. 10, 3656 (2019).
Pediaditakis, I. et al. Modeling α-synuclein pathology in a human brain-chip to assess blood–brain barrier disruption. Nat. Commun. 12, 5907 (2021).
Park, T.-E. et al. Hypoxia-enhanced blood–brain barrier chip recapitulates human barrier function and shuttling of drugs and antibodies. Nat. Commun. 10, 2621 (2019).
Vatine, G. D. et al. Human iPSC-derived blood–brain barrier chips enable disease modeling and personalized medicine applications. Cell Stem Cell 24, 995–1005.e6 (2019).
Brown, J. A. et al. Metabolic consequences of inflammatory disruption of the blood–brain barrier in an organ-on-chip model of the human neurovascular unit. J. Neuroinflammation 13, 306 (2016).
Sahtoe, D. D. et al. Transferrin receptor targeting by de novo sheet extension. Proc. Natl Acad. Sci. USA 118, e2021569118 (2021).
Lee, S. W. L. et al. Modeling nanocarrier transport across a 3D in vitro human blood–brain-barrier microvasculature. Adv. Health. Mater. 9, e1901486 (2020).
Morad, G. et al. Tumor-derived extracellular vesicles breach the intact blood–brain barrier via transcytosis. ACS Nano. 13, 13853–13865 (2019).
Kim, J. et al. Fungal brain infection modelled in a human-neurovascular-unit-on-a-chip with a functional blood–brain barrier. Nat. Biomed. Eng. 5, 830–846 (2021).
Chung, M. et al. Wet-AMD on a chip: modeling outer blood-retinal barrier in vitro. Adv. Health. Mater. 7, 1700028 (2018).
Achberger, K. et al. Merging organoid and organ-on-a-chip technology to generate complex multi-layer tissue models in a human retina-on-a-chip platform. eLife 8, e46188 (2019).
Kutys, M. L. et al. Uncovering mutation-specific morphogenic phenotypes and paracrine-mediated vessel dysfunction in a biomimetic vascularized mammary duct platform. Nat. Commun. 11, 3377 (2020).
Blundell, C. et al. Placental drug transport-on-a-chip: a microengineered in vitro model of transporter-mediated drug efflux in the human placental barrier. Adv. Health. Mater. 7, 1700786 (2018).
Ribas, J. et al. Biomechanical strain exacerbates inflammation on a progeria-on-a-chip model. Small 13, 1603737 (2017).
Abudupataer, M. et al. Aorta smooth muscle-on-a-chip reveals impaired mitochondrial dynamics as a therapeutic target for aortic aneurysm in bicuspid aortic valve disease. eLife 10, e69310 (2021).
Hachey, S. J. et al. An in vitro vascularized micro-tumor model of human colorectal cancer recapitulates in vivo responses to standard-of-care therapy. Lab Chip 21, 1333–1351 (2021).
Deng, J. et al. CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation. Cancer Discov. 8, 216–233 (2018).
Pavesi, A. et al. A 3D microfluidic model for preclinical evaluation of TCR-engineered T cells against solid tumors. JCI Insight 2, e89762 (2017).
Beckwith, A. L., Velásquez-García, L. F. & Borenstein, J. T. Microfluidic model for evaluation of immune checkpoint inhibitors in human tumors. Adv. Health. Mater. 8, e1900289 (2019).
Tao, T. et al. Microengineered multi-organoid system from hiPSCs to recapitulate human liver-islet axis in normal and type 2 diabetes. Adv. Sci. 9, 2103495 (2021).
Skardal, A., Devarasetty, M., Forsythe, S., Atala, A. & Soker, S. A reductionist metastasis-on-a-chip platform for in vitro tumor progression modeling and drug screening. Biotechnol. Bioeng. 113, 2020–2032 (2016).
Sasserath, T. et al. Differential monocyte actuation in a three-organ functional innate immune system-on-a-chip. Adv. Sci. 7, 2000323 (2020).
Chang, S. Y. et al. Human liver–kidney model elucidates the mechanisms of aristolochic acid nephrotoxicity. JCI Insight 2, e95978 (2017).
Oleaga, C. et al. Multi-organ toxicity demonstration in a functional human in vitro system composed of four organs. Sci. Rep. 6, 20030 (2016).
Skardal, A. et al. Multi-tissue interactions in an integrated three-tissue organ-on-a-chip platform. Sci. Rep. 7, 8837 (2017).
McAleer, C. W. et al. Multi-organ system for the evaluation of efficacy and off-target toxicity of anticancer therapeutics. Sci. Transl Med. 11, eaav1386 (2019).
Chramiec, A. et al. Integrated human organ-on-a-chip model for predictive studies of anti-tumor drug efficacy and cardiac safety. Lab Chip 20, 4357–4372 (2020).
Hübner, J. et al. Simultaneous evaluation of anti-EGFR-induced tumour and adverse skin effects in a microfluidic human 3D co-culture model. Sci. Rep. 8, 15010 (2018).
Lee-Montiel, F. T. et al. Integrated isogenic human induced pluripotent stem cell-based liver and heart microphysiological systems predict unsafe drug–drug interaction. Front. Pharmacol. 12, 667010 (2021).
Vernetti, L. et al. Functional coupling of human microphysiology systems: intestine, liver, kidney proximal tubule, blood–brain barrier and skeletal muscle. Sci. Rep. 7, 42296 (2017).
Ramme, A. P. et al. Autologous induced pluripotent stem cell-derived four-organ-chip. Future Sci. OA 5, FSO413 (2019).
Hargrove-Grimes, P., Low, L. A. & Tagle D. A. Microphysiological systems: stakeholder challenges to adoption in drug development. Cells Tissues Organs 211, 1–13 (2022).
Baran, S. W. et al. Perspectives on the evaluation and adoption of complex in vitro models in drug development: workshop with the FDA and the pharmaceutical industry (IQ MPS Affiliate). ALTEX https://doi.org/10.14573/altex.2112203 (2022).
Zhang, B. & Radisic, M. Organ-on-a-chip devices advance to market. Lab Chip 17, 2395–2420 (2017).
Franzen, N. et al. Impact of organ-on-a-chip technology on pharmaceutical R&D costs. Drug Discov. Today 24, 1720–1724 (2019).
Ewart, L. et al. Qualifying a human liver-chip for predictive toxicology: performance assessment and economic implications. Preprint at bioRxiv https://doi.org/10.1101/2021.12.14.472674 (2021).
Baudy, A. R. et al. Liver microphysiological systems development guidelines for safety risk assessment in the pharmaceutical industry. Lab Chip 20, 215–225 (2020).
Bircsak, K. M. et al. A 3D microfluidic liver model for high throughput compound toxicity screening in the OrganoPlate®. Toxicology 450, 152667 (2021).
Toepke, M. W. & Beebe, D. J. PDMS absorption of small molecules and consequences in microfluidic applications. Lab Chip 6, 1484–1486 (2006).
van Meer, B. J. et al. Small molecule absorption by PDMS in the context of drug response bioassays. Biochem. Biophys. Res. Commun. 482, 323–328 (2017).
Domansky, K. et al. Clear castable polyurethane elastomer for fabrication of microfluidic devices. Lab Chip 13, 3956–3964 (2013).
Domansky, K. et al. SEBS elastomers for fabrication of microfluidic devices with reduced drug absorption by injection molding and extrusion. Microfluid. Nanofluid. 21, 107 (2017).
Schneider, S., Brás, E. J. S., Schneider, O., Schlünder, K. & Loskill, P. Facile patterning of thermoplastic elastomers and robust bonding to glass and thermoplastics for microfluidic cell culture and organ-on-chip. Micromachines 12, 575 (2021).
Bagley, A. F. et al. Endothelial thermotolerance impairs nanoparticle transport in tumors. Cancer Res. 75, 3255–3267 (2015).
Cho, M. & Park, J. K. Modular 3D in vitro artery-mimicking multichannel system for recapitulating vascular stenosis and inflammation. Micromachines 12, 1528 (2021).
Achberger, K. et al. Human stem cell-based retina on chip as new translational model for validation of AAV retinal gene therapy vectors. Stem Cell Rep. 16, 2242–2256 (2021).
Rogal, J. et al. WAT-on-a-chip integrating human mature white adipocytes for mechanistic research and pharmaceutical applications. Sci. Rep. 10, 6666 (2020).
Villenave, R. et al. Human gut-on-a-chip supports polarized infection of coxsackie B1 virus in vitro. PLoS ONE 12, e0169412 (2017).
Beaurivage, C. et al. Development of a human primary gut-on-a-chip to model inflammatory processes. Sci. Rep. 10, 21475 (2020).
Šuligoj, T. et al. Effects of human milk oligosaccharides on the adult gut microbiota and barrier function. Nutrients 12, 2808 (2020).
Apostolou, A. et al. A novel microphysiological colon platform to decipher mechanisms driving human intestinal permeability. Cell Mol. Gastroenterol. Hepatol. 12, 1719–1741 (2021).
Vormann, M. K. et al. Implementation of a human renal proximal tubule on a chip for nephrotoxicity and drug interaction studies. J. Pharm. Sci. 110, 1601–1614 (2021).
Ehrlich, A. et al. Microphysiological flux balance platform unravels the dynamics of drug induced steatosis. Lab Chip 18, 2510–2522 (2018).
Rowe, C. et al. Perfused human hepatocyte microtissues identify reactive metabolite-forming and mitochondria-perturbing hepatotoxins. Toxicol. Vitr. 46, 29–38 (2018).
Freag, M. S. et al. Human nonalcoholic steatohepatitis on a chip. Hepatol. Commun. 5, 217–233 (2020).
Li, J. et al. AAV-mediated gene therapy targeting TRPV4 mechanotransduction for treatment of pulmonary vascular leakage. APL Bioeng. 3, 046103 (2019).
Rivera-Burgos, D. et al. Glucocorticoid clearance and metabolite profiling in an in vitro human airway epithelium lung model. Drug Metab. Dispos. 44, 220–226 (2016).
Sances, S. et al. Human iPSC-derived endothelial cells and microengineered organ-chip enhance neuronal development. Stem Cell Rep. 10, 1222–1236 (2018).
Spijkers, X. M. et al. A directional 3D neurite outgrowth model for studying motor axon biology and disease. Sci. Rep. 11, 2080 (2021).
Glieberman, A. L. et al. Synchronized stimulation and continuous insulin sensing in a microfluidic human islet on a chip designed for scalable manufacturing. Lab Chip 19, 2993–3010 (2019).
Zhang, J. et al. Construction of a high fidelity epidermis-on-a-chip for scalable in vitro irritation evaluation. Lab Chip 21, 3804–3818 (2021).
França, C. M. et al. The tooth on-a-chip: a microphysiologic model system mimicking the biologic interface of the tooth with biomaterials. Lab Chip 20, 405–413 (2020).
Rodrigues et al. Biomaterial and biofilm interactions with the pulp-dentin complex-on-a-chip. J. Dent. Res. 100, 1136–1143 (2021).
Gnecco, J. S. et al. Compartmentalized culture of perivascular stroma and endothelial cells in a microfluidic model of the human endometrium. Ann. Biomed. Eng. 45, 1758–1769 (2017).
Ahn, J. et al. Three-dimensional microengineered vascularised endometrium-on-a-chip. Hum. Reprod. 36, 2720–2731 (2021).
Prantil-Baun, R. et al. Physiologically based pharmacokinetic and pharmacodynamic analysis enabled by microfluidically linked organs-on-chips. Annu. Rev. Pharmacol. Toxicol. 58, 37–64 (2018).
Bjornson-Hooper, Z. B. et al. A comprehensive atlas of immunological differences between humans, mice and non-human primates. Preprint at bioRxiv https://doi.org/10.1101/574160 (2019).
Cho, H. W. & Eom, Y. B. Forensic analysis of human microbiome in skin and body fluids based on geographic location. Front. Cell Infect. Microbiol. 11, 695191 (2021).
Fogel, D. B. Factors associated with clinical trials that fail and opportunities for improving the likelihood of success: a review. Contemp. Clin. Trials Commun. 11, 156–164 (2018).
Acknowledgements
The author thanks all of the research team members, trainees, collaborators and other members of the scientific community for contributing to the development of the organ chip field. The author’s work discussed here was funded by grants from DARPA, FDA, NIH, BARDA, Cancer Research United Kingdom and the Gates Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
D.E.I. holds equity in Emulate, chairs its scientific advisory board and is a member of its board of directors.
Peer review
Peer review information
Nature Reviews Genetics thanks Jeffrey T. Borenstein, YongTae Kim and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Aracari Biosciences: https://aracaribio.com
CN Bio Innovations: https://cn-bio.com
Emulate: https://emulatebio.com
Mimetas: https://www.mimetas.com
Nortis: https://nortisbio.com
TissUse: https://www.tissuse.com
Glossary
- Microfluidic
-
A miniaturized device containing one or more channels or chambers through which fluids flow.
- Organoids
-
Self-assembled hollow clusters of cells derived from stem cells that exhibit tissue-specific structures and functions when placed in 3D cultures.
- Clinical mimicry
-
Recapitulation of physiological, pathophysiological or therapeutic responses detected in the human body.
- Personalized medicine
-
A practice of medicine that uses data obtained from an individual patient’s cells to guide decisions regarding prevention, diagnosis or treatment of disease.
- Extracellular matrix
-
(ECM). A structural scaffold composed of multiple macromolecules that orients and supports cells in living tissues.
- Soft lithography
-
A technique for fabricating microstructures or micropatterning materials using flexible stamps moulded on surfaces etched using photolithography.
- Minibioreactor
-
A small-volume (<10 ml) cell chamber in which cells are cultured to carry out a biological reaction or process.
- Cilia
-
Moving microscopic hair-like projections found on the surfaces of cells, such as lung airway epithelial cells, that cause motions in overlying fluids or mucus.
- One Health
-
A public health approach that recognizes the importance of animal health as a key component of global disease prevention, surveillance, control and mitigation.
Rights and permissions
About this article

Cite this article
Ingber, D.E. Human organs-on-chips for disease modelling, drug development and personalized medicine. Nat Rev Genet 23, 467–491 (2022). https://doi.org/10.1038/s41576-022-00466-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41576-022-00466-9
This article is cited by
-
Microfluidic high-throughput 3D cell culture
Nature Reviews Bioengineering (2024)
-
Modelling and targeting mechanical forces in organ fibrosis
Nature Reviews Bioengineering (2024)
-
Neuropathogenesis-on-chips for neurodegenerative diseases
Nature Communications (2024)
-
Facilitating long-term cell examinations and time-lapse recordings in cell biology research with CO2 mini-incubators
Scientific Reports (2024)
-
Tetralol derivative NNC-55-0396 targets hypoxic cells in the glioblastoma microenvironment: an organ-on-chip approach
Cell Death & Disease (2024)
