
Abstract
Pituitary adenomas are rare in children and young people under the age of 19 (hereafter referred to as CYP) but they pose some different diagnostic and management challenges in this age group than in adults. These rare neoplasms can disrupt maturational, visual, intellectual and developmental processes and, in CYP, they tend to have more occult presentation, aggressive behaviour and are more likely to have a genetic basis than in adults. Through standardized AGREE II methodology, literature review and Delphi consensus, a multidisciplinary expert group developed 74 pragmatic management recommendations aimed at optimizing care for CYP in the first-ever comprehensive consensus guideline to cover the care of CYP with pituitary adenoma. Part 2 of this consensus guideline details 57 recommendations for paediatric patients with prolactinomas, Cushing disease, growth hormone excess causing gigantism and acromegaly, clinically non-functioning adenomas, and the rare TSHomas. Compared with adult patients with pituitary adenomas, we highlight that, in the CYP group, there is a greater proportion of functioning tumours, including macroprolactinomas, greater likelihood of underlying genetic disease, more corticotrophinomas in boys aged under 10 years than in girls and difficulty of peri-pubertal diagnosis of growth hormone excess. Collaboration with pituitary specialists caring for adult patients, as part of commissioned and centralized multidisciplinary teams, is key for optimizing management, transition and lifelong care and facilitates the collection of health-related quality of survival outcomes of novel medical, surgical and radiotherapeutic treatments, which are currently largely missing.
Similar content being viewed by others
Introduction
In children and young people under 19 years of age (hereafter referred to as CYP), pituitary adenomas (sometimes referred to as pituitary neuroendocrine tumours or PitNETs) are very much rarer than in adults, especially before puberty. In CYP, pituitary adenomas can also differ in their characteristics, be more aggressive or more treatment resistant than adenomas in adults and can present as a sign of genetic disease. Thus, their optimal diagnosis and management need multidisciplinary collaboration from both paediatric and adult pituitary specialists. Their rarity precludes a high-quality evidence base for their management. Therefore, the recommendations in this two-part consensus guideline were developed with Appraisal of Guidelines Research and Evaluation Instrument II (AGREE II)1 methodology between 2014 and 2022 to generate a best practice reference document of 74 recommendations for the management of suspected pituitary adenomas in CYP to improve the quality of clinical care and thus health outcomes.
Part 1 of this consensus guideline includes 17 general recommendations on neuroimaging, visual assessment, histopathology, genetics, pituitary surgery and radiotherapy relevant to all types of pituitary adenoma in CYP2. Here, in Part 2, we detail the 57 recommendations with a total of 69 statements (as some of the recommendations included two or more interrelated statements) pertaining to each adenoma type in CYP: prolactinomas, Cushing disease, growth hormone (GH) excess causing gigantism and acromegaly, clinically non-functioning pituitary adenomas (NFPAs), and thyroid stimulating hormone (TSH)-secreting adenomas (TSHomas) and we also mention the rare functioning gonadotroph adenomas (Supplementary Table 1).
Recommendations
Prolactinomas
Epidemiology and aetiology
Prolactinomas are the most common adenoma type in CYP, occurring in approximately 0.1 million children every year3. However, prolactinomas are exceptionally rare before puberty, when corticotrophinomas are more common. In a series of 136 CYP presenting with pituitary adenomas before 20 years of age, 53% had prolactinomas, but 93% of these presented after 12 years of age. Pituitary adenomas were 3 times4,5,6 to 4.5 times7 more common in female patients than in male patients. Although patients can present with prolactinomas within the first decade of life4,5,8, an adolescent presentation is more typical4,6,7,8,9,10. Median duration of symptom history before diagnosis is 12 months3,11. Of note, macroprolactinomas or giant prolactinomas, which can exert secondary mass effects that compromise growth, puberty and vision, also occur more frequently in CYP than in adults6,12. In a study of CYP with macroprolactinomas, 46% had overweight or obesity at diagnosis and, of these, 23% cited weight gain as one of the reasons for seeking medical advice10. A small percentage of paediatric prolactinomas are related to familial isolated pituitary adenoma or syndromic disease (multiple endocrine neoplasia type 1 syndrome (MEN1), MEN1-like or phaeochromocytoma–paraganglioma-related pituitary disease), even without a known family history13. Therefore, genetic testing should be considered (see Part 1: R11 in ref. 2).
Diagnosis: clinical features
-
Part 2: R1. Offer serum prolactin measurement in CYP presenting with one or more of the following signs and symptoms: delayed puberty; galactorrhoea; visual field loss; growth or pubertal arrest; or girls with menstrual disturbance (strong recommendation, moderate-quality evidence).
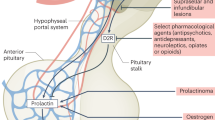
High serum levels of prolactin inhibit gonadotrophin secretion via inhibition of the hypothalamic hormone kisspeptin14. Paediatric patients with hyperprolactinaemia might therefore present with delayed (>2 standard deviations (SD) later than mean population age for sex) or arrested puberty, growth failure or short stature, primary amenorrhoea, galactorrhoea, menstrual disturbance or secondary amenorrhoea (in post-menarcheal girls)3,6. Boys might present with gynaecomastia as a result of hypogonadism. Mass effects, occurring more commonly in boys than girls, include headache and visual field loss15. Obesity, gynaecomastia, constitutional delay in growth and puberty in boys, and menstrual disturbance in girls are common physiological variations that are very rarely caused by prolactinoma. However, the cost of measuring prolactin is offset by the benefits of an early diagnosis and timely treatment.
Diagnosis: biochemical evaluation
-
Part 2: R2. In CYP with signs or symptoms of hyperprolactinaemia, offer prolactin measurement in a single blood sample collected at any time of day (strong recommendation, high-quality evidence).
-
Part 2: R3. Consider investigating modestly elevated serum prolactin levels by serial measurements over time to exclude the effect of stress and prolactin pulsatility (moderate recommendation, low-quality evidence, Delphi 87%).
A single prolactin measurement taken at any time of the day is sufficient to assess hyperprolactinaemia16,17. As prolactin secretion also rises in response to stress, in patients with elevated baseline prolactin (up to five times of the upper limit of normal18), sampling can be repeated on a different day with two or three samples at 20–60 min intervals, using an indwelling cannula, to differentiate stress-related hyperprolactinaemia from organic disease16,18.
-
Part 2: R4. The diagnosis of hyperprolactinaemia in CYP requires age-specific and sex-specific prolactin reference ranges and the exclusion of confounding conditions such as hypothyroidism, renal and/or hepatic impairment, and use of medications that cause hyperprolactinaemia (strong recommendation, moderate-quality evidence).
Serum prolactin concentrations vary with age and sex. They are highest in the first 2 years of life and fall to a nadir in mid-childhood, to rise again in adolescence when they are higher in girls than in boys. Paediatric cohort studies of prolactinomas report diagnostic serum prolactin concentrations usually above 4,000 mU/l (188 µg/l)4,8,10, although lower levels can be seen in patients with microprolactinomas3. To rule out mixed prolactin and GH hypersecretion, age-dependent and sex-dependent insulin-like growth factor 1 (IGF1) evaluation should always accompany prolactin assessment in CYP with prolactinomas.
Unexplained, persistently mildly or moderately elevated prolactin in blood samples taken after rest could be due to the stalk effect (disconnection hyperprolactinaemia, pituitary stalk compression from mass lesions disrupting the dopaminergic inhibition of lactotroph cells). In adult patients with stalk effect, prolactin levels are reported above the normal range but not higher than 2,000 mU/l; 94 µg/l (ref. 19) or six times above the upper limit of normal18. Even if no corresponding symptoms of hyperprolactinaemia, hypopituitarism or a pituitary mass are observed, pituitary imaging should be considered. If hyperprolactinaemia is due to a pituitary mass, baseline and dynamic pituitary assessment can identify a potential lack or excess of other anterior pituitary hormones.
Severe primary hypothyroidism can be accompanied by hyperprolactinaemia, probably due to compensatory thyrotropin-releasing hormone hypersecretion and pituitary hyperplasia; care should be taken to distinguish such pituitary enlargement from a true prolactinoma20,21. Severe and prolonged primary hypothyroidism in children can disrupt kidney and liver function as well as delay growth and puberty. In a large cohort of 2,848 adults, hyperprolactinaemia was reported in 43% of women and 40% of men presenting with frank primary hypothyroidism, in 36% of women and 32% of men with subclinical hypothyroidism, and only in around 2% of euthyroid individuals21. Hyperprolactinaemia is reported in 30–65% of adult patients with chronic kidney disease due to increased prolactin secretion and reduced renal clearance22. Severe liver disease is also associated with hyperprolactinaemia in adults18. Intracranial hypotension can cause hyperprolactinaemia18. Although we could find no parallel data describing the prevalence of hyperprolactinaemia in these clinical scenarios in CYP, the Guideline Development Group (GDG)2 recommends the exclusion of confounding diseases. Up to 80% of patients with tetrahydrobiopterin deficiencies (a group of rare neurometabolic disorders characterized by insufficient synthesis of monoamine neurotransmitters, including dopamine) can have hyperprolactinaemia (10–30 fold elevation of prolactin), usually from the teenage years; development of a microprolactinoma has been also described in case reports23,24. While less likely in the CYP population, pregnancy should not be overlooked as a cause of hyperprolactinaemia18.
Medications are one of the most common causes of hyperprolactinaemia in adults through direct prolactin stimulatory pathways or by antagonizing inhibitory dopaminergic tone25. Medication-induced hyperprolactinaemia is also well described in CYP (Supplementary Table 3). The role of synthetic oral oestrogens (for example, contraceptive pills) in causing mild elevation of prolactin is controversial.
-
Part 2: R5. Assess baseline macroprolactin levels where serum prolactin is found to be mildly or incidentally elevated (strong recommendation, low-quality evidence, GDG consensus).
In addition to monomeric prolactin (23 kDa), dimeric (48–56 kDa) and polymeric (>100 kDa) forms (usually associated with an antibody) can circulate (‘macroprolactin’, which has low biological activity), with or without excess monomeric prolactin. No routine assays distinguish between monomeric prolactin and macroprolactin; therefore, prompt and appropriate secondary analysis should be undertaken to detect the possible presence of macroprolactin in the initial investigation of asymptomatic CYP with hyperprolactinaemia26,27. In large retrospective cohorts of adults with hyperprolactinaemia, macroprolactinaemia was present in 10–40% of individuals with hyperprolactinaemia17,26,27, 20% of whom had galactorrhoea, 45% oligo-amenorrhoea and 20% pituitary adenomas. Few patients with macroprolactinaemia are reported in the paediatric literature. In a cohort of five patients aged 11–18 years with an incidental finding of hyperprolactinaemia due to macroprolactinaemia, none developed clinical features of prolactin excess during an observation period ranging from 3 months to 8 years28. In another report, one of six CYP with macroprolactinaemia was asymptomatic; the other five had either headache, menstrual disturbance, short stature, increased hair growth or early puberty. Four of those with symptoms underwent pituitary MRI and a microadenoma was identified in two (one with headache and one with oligomenorrhoea)29. Given these data and the current widespread clinical practice6, the GDG strengthened R5.
-
Part 2: R6. Perform serial dilutions of serum for prolactin measurement in CYP with large pituitary lesions and normal or mildly elevated prolactin levels (strong recommendation, moderate-quality evidence).
Serum prolactin levels directly correlate with prolactinoma size and are important markers of treatment response. Based on adult data, approximately 5% of patients with macroprolactinomas and a paradoxically modest serum concentration of hyperprolactinaemia have grossly elevated prolactin concentrations following serum dilution30. When prolactin is measured in two-site immunoradiometric assays, very high concentrations of prolactin could saturate the signalling antibody, making it less available for binding to the coupling antibody, resulting in artificially low measurements16. This phenomenon has been described as the ‘high-dose hook effect’30 and is well recognized. Some prolactin assay manufacturers have put specific mitigating factors in place, such as large linear ranges or automatic dilution steps, in many modern assays. However, the potential remains for this effect to be a source of anomalous results31. Thus, contact with the clinical biochemist to request manual dilution is advised when a discrepancy exists between a large pituitary adenoma on imaging and only modestly elevated prolactin concentrations on initial biochemistry. Of note, inconsistent symptoms and laboratory results can occasionally arise due to biotin exposure or heterophilic anti-animal antibodies18.
Treatment
-
Part 2: R7. In CYP with prolactinoma, offer a dopamine agonist as first-line therapy to reduce serum prolactin concentrations and induce tumour shrinkage; cabergoline is the dopamine agonist of choice given its superior effectiveness and lower adverse effect profile (strong recommendation, moderate-quality evidence).
-
Part 2: R8. In CYP with prolactinoma, offer cabergoline as first-line therapy, even in the presence of visual disturbance and pituitary apoplexy, while carefully monitoring for any deterioration in vision, pituitary function or general status (strong recommendation, low-quality evidence, Delphi 100%).
Dopamine agonists reduce pituitary-origin hyperprolactinaemia of any cause18. In adults with prolactinoma, dopamine agonists induce normalization of the prolactin level (median: 68% of patients; range: 40–100%), tumour shrinkage (62%; 20–100%), resolution of visual field defects (67%; 33–100%), normalization of menses (78%; 40–100%), fertility (53%; 10–100%) and sexual function (67%; 6–100%), and resolution of galactorrhoea (86%; 33–100%)11,32,33. In both adults and CYP with prolactinoma, cabergoline is the dopamine agonist of choice3,11,12. Cabergoline has a longer half-life and greater affinity for the dopamine receptor than other dopamine agonists. In a randomized controlled trial of adult women with prolactinoma, cabergoline was superior to bromocriptine in normalizing prolactin (83% versus 59%), resuming ovulatory cycles or achieving pregnancy. Adverse events were more commonly reported with bromocriptine than with cabergoline (72% versus 52%)34.
In studies of CYP with prolactinomas, dopamine agonists lower prolactin concentrations in 60–70% of patients3,4,8,10,11,35, reduce tumour size by 80–88%3,11, improve visual deficits36, resolve pubertal delay and eliminate headache11. In an observational study of 28 paediatric patients, CYP with prolactinomas smaller than 13.5 mm in diameter (13 patients) achieved normalization of prolactin levels without surgery, using conventional cabergoline doses (up to 2 mg/week)12. Moreover, another series of 22 CYP with prolactinomas reported that all tumours of >20 mm diameter required surgery6. Although successful dopamine agonist discontinuation has been achieved in CYP, younger patients and those with high serum prolactin concentrations at diagnosis (a marker of adenoma size) are less likely to achieve complete remission and euprolactinaemia3,11,37.
Medication-induced shrinkage of prolactinomas that have invaded sphenoid bone can cause rhinorrhoea after a few months of drug administration (mean 3.3 months, range 3 days–17 months) due to a cerebrospinal fluid leak38, but this adverse effect can also occur during long-term treatment. Detection of β2-transferrin or β-trace protein (specific to cerebrospinal fluid) in nasal secretions confirms a cerebrospinal fluid leak39. Cerebrospinal fluid leak can require urgent intervention (for example, lumbar drain or surgical repair), with or without a temporary cessation in dopamine agonist therapy40. Apoplexy has been described during cabergoline therapy both in adults and CYP6.
-
Part 2: R9. For CYP resistant to standard doses of cabergoline, offer graduated dose increments of up to 3.5 mg per week or up to 7 mg per week in exceptional cases (strong recommendation, moderate-quality evidence, Delphi 100%).
Evidence indicates that adult patients with prolactinoma who are unresponsive to standard dopamine agonist doses (up to 1.5–2 mg of cabergoline per week) might respond to higher doses (3.5–7 mg per week), whilst even higher doses (up to 12 mg per week41,42 but below the 21 mg per week dose used for Parkinson disease) have been tried. High-dose cabergoline is reportedly well tolerated and doses of up to 7 mg per week have been used to successfully treat CYP with prolactinoma10,11. However, others report little benefit of cabergoline doses above 3.5 mg per week in adults41. Patients with cabergoline resistance or intolerance will require adjuvant therapy with surgery or radiotherapy42.
-
Part 2: R10.1. Following multidisciplinary discussion, for CYP with prolactinomas offer surgery when the patient is unable to tolerate or is resistant to high-dose cabergoline (strong recommendation, low-quality evidence, Delphi 95%).
-
Part 2: R10.2. Following multidisciplinary discussion, for CYP with prolactinomas offer surgery when the patient develops deteriorating vision on cabergoline (strong recommendation, low-quality evidence, Delphi 90%).
-
Part 2: R10.3. Following multidisciplinary discussion, for CYP with prolactinomas offer radiotherapy if surgery is not an option (strong recommendation, low-quality evidence, Delphi 100%).
Small nocturnal dose increments of cabergoline can effectively diminish the adverse effects of gastrointestinal intolerance and postural hypotension, thereby avoiding trials of less effective dopamine agonists (bromocriptine or quinagolide). Dose-independent psychological intolerance (mood changes, depression, aggression, hypersexuality and impulse control disorder) is similar between agents and described in adults as well as CYP37,43, but the frequency of these adverse events might be higher in CYP than in adults11,44. Dopamine agonist resistance is usually defined in adults and CYP as failure to achieve normoprolactinaemia (biochemical resistance) and less than 50% reduction in tumour area in the coronal plane and/or less than 30% reduction of the longest diameter of the tumour (tumour size resistance, assessed by Response Evaluation Criteria In Solid Tumours (RECIST) criteria) after 3–6 months of maximally tolerated dopamine agonist doses (at least 2 mg per week)18,41,45,46. In a paediatric macroprolactinoma cohort of patients who were unresponsive to 3 months of 15 mg per day bromocriptine, 600 µg per day quinagolide or 3.5 mg per week cabergoline, 26% were biochemically resistant and 24% were tumour-shrinkage resistant10. This resistance directly correlated with tumour size and prolactin levels (which in turn were closely correlated) but was independent of MEN1 mutation status10.
In CYP with prolactinoma, neurosurgical intervention should be considered if vision deteriorates or does not improve on medical therapy or if dopamine agonist resistance, escape or intolerance occurs. Careful multidisciplinary discussion is needed if the patient expresses a preference for surgery rather than long-term medication or is non-adherent to the latter.
Transsphenoidal surgery induced remission in 30–50% of adults with prolactinomas and any residual post-operative hyperprolactinaemia was subsequently more responsive to dopamine agonists than pre-operatively47. Tumour size negatively predicted surgical remission rates, with smaller adenomas being more often cured by surgery alone than larger ones47,48. Paediatric series report lower surgical remission rates than in adults, most likely due to the higher incidence of proportionately larger prolactinomas in CYP, as well as a possible higher frequency of new and permanent pituitary hormone deficiencies after surgery43,49,50,51,52. In adults with microprolactinomas or intrasellar macroprolactinomas, surgery is a viable option with an excellent cure rate (83% in microprolactinomas and 60% in macroprolactinomas), especially in high-volume surgical centres, and is certainly an alternative to long-term cabergoline therapy18,53.
Radiotherapy should be reserved for exceptional patients with a growing prolactinoma and where other treatment modalities are not available or have been exhausted; the main indication for radiotherapy is control of tumour growth, whereas normalization of prolactin levels is a secondary objective2. After radiotherapy, initially 6-monthly and later 12-monthly follow-up should monitor for the development of hypopituitarism or recurrence. The detailed assessment and treatment of hypopituitarism in CYP is beyond the scope of these guidelines.
-
Part 2: R11. In CYP with prolactinoma, offer an echocardiogram at the start of treatment with a dopamine agonist; offer yearly surveillance echocardiography for patients receiving >2 mg per week cabergoline and every 5 years if on ≤2 mg per week (moderate recommendation, moderate-quality evidence).
High-dose and long-term use of dopamine agonists in Parkinson disease pose a recognized risk of cardiac valve regurgitation; however, the doses used in treating prolactinomas are notably lower. A meta-analysis that identified an increased prevalence of echocardiographic tricuspid regurgitation in adults was heavily influenced by a single study, with no reports of increased clinical valvular disease54. A subsequent population-based, matched control cohort study in adults failed to identify an excess in hard clinical cardiac endpoints55. Nevertheless, the long-term cardiac safety of ergot-based dopamine agonists in CYP requires a balanced judgement against the increasing background rate of cardiac valvulopathy that occurs with age and the often more aggressive nature of prolactinomas in the paediatric age group, who require longer treatment durations and higher cumulative doses than adults. The relative contributions of peak versus cumulative doses of dopamine agonists in the aetiology of valvulopathy are unknown. In adults with Parkinson disease and moderate-to-severe valvulopathy, the mean cumulative cabergoline dose was 4,015 mg, with one SD below the mean being 720 mg (ref. 56), yet a critical cumulative dose threshold could not be established56. Similar respective cumulative doses in CYP with prolactinoma would require 39 years (4,015 mg) or 7 years (720 mg) of 2 mg per week cabergoline treatment. To date, valvulopathy in CYP treated with dopamine agonists for hyperprolactinaemia has not been reported. A 2019 position statement for adults with prolactinomas treated with dopamine agonists recommends pre-treatment baseline and annual echocardiography with cardiac auscultation for those on >2 mg per week of cabergoline, reduced to 5-yearly echocardiographic surveillance if the cabergoline dose is ≤2 mg per week57. Until data specific to CYP emerge, following these recommendations (endorsed by three relevant UK professional societies56) seems prudent.
-
Part 2: R12. Temozolomide treatment might need consideration for CYP with aggressive pituitary tumours resistant to medical, surgical and radiation therapy (weak recommendation, low-quality evidence, Delphi 88%).
Temozolomide treatment for aggressive pituitary tumours and pituitary carcinomas is well described in the adult pituitary literature58. These entities are extremely rare in CYP: four patients with pituitary carcinomas are reported to have had disease commencing in childhood59,60,61,62. The 2017 European Society of Endocrinology guideline recommends first-line temozolomide monotherapy for aggressive pituitary tumours and pituitary carcinomas unresponsive to standard therapies, with evaluation of responders and non-responders after three cycles of 150 mg/m2 per day for 5 days in every 28 days, with dose increases to 200 mg/m2 per day in patients with good tolerance63. A minimum of 6 months of treatment is recommended for responding patients. Five paediatric patients receiving temozolomide treatment for pituitary tumours were identified in the literature, with two more paediatric-onset patients receiving temozolomide as adults (Table 1).
Follow-up and surveillance
-
Part 2: R13. If the serum level of prolactin has been normalized for at least 2 years on medical therapy and there is no visible residual prolactinoma on MRI, consider gradual cabergoline dose reduction to maintain normoprolactinaemia and eventual treatment discontinuation, with continued serum prolactin monitoring for at least 2 more years (moderate recommendation, low-quality evidence, Delphi 100%).
The relapse rates of prolactinomas in CYP treated with dopamine agonists are not reported. The Endocrine Society guideline for the treatment of hyperprolactinaemia in adults recommends a trial of therapy discontinuation in those patients with no tumour remnant on MRI and normoprolactinaemia after 2 or more years of medical treatment18. Studies report variable (26–89%) hyperprolactinaemia recurrence rates under these conditions, largely within the first 2 years of treatment withdrawal18,64. Discontinuation might also be attempted with normoprolactinaemia and a small tumour remnant51.
Two meta-analyses have examined factors associated with relapse following treatment withdrawal in patients with prolactinoma treated with dopamine agonists. The first (19 studies, 743 patients) concluded that both the use of cabergoline and treatment for more than 2 years were associated with a decreased relapse rate65. The second (11 studies, 637 patients) found that tapering doses prior to withdrawal reduced the risk of relapse but that treatment beyond 2 years had no further beneficial effect66.
CYP with prolactinomas should be monitored clinically (including assessment of growth, puberty, galactorrhoea, menstrual history, gynaecomastia or loss of libido in puberty) and biochemically by measurement of serum prolactin. For macroprolactinomas, MRI should be repeated 3–6 months after starting cabergoline treatment; for microprolactinomas, re-imaging depends on clinical and biochemical follow-up; imaging is suggested before considering cabergoline withdrawal18. Longer-term imaging frequency depends on symptoms, biochemical control and the closeness of the pituitary mass to the optic chiasm18. One observational study (including 11 CYP) reported low bone mineral density (BMD) at diagnosis with modest degrees of recovery after 2 years of dopamine agonist therapy. Assessment of BMD 2 years after diagnosis might be important in patients with prolactinoma67; however, the inevitable negative impact of delayed growth and puberty on peak bone mineral accrual will confound definitions of ‘osteopenia’ in CYP. Thus, repeated longitudinal assessments require interpretation not only alongside cure rates but also with clinical pubertal staging, additional sex and adrenal hormone replacement, and at growth completion (epiphyseal fusion) and full pubertal maturation (age 25 years).
The optimal frequency of MRI imaging following cessation of treatment is unknown. Prolactin levels, assessed at 3–6-monthly intervals initially, can be used as markers of tumour relapse, although biochemical relapse is not always accompanied by radiological MRI change.
Cushing disease
Epidemiology and aetiology
Cushing disease is caused by an adrenocorticotropin (ACTH)-secreting pituitary adenoma and is the most common form of ACTH-dependent Cushing syndrome, yet it is rare in CYP. The incidence is approximately 10% of that in adults, of ~0.5 new patients per million individuals per year68. Cushing disease accounts for 75–80% of CYP with Cushing syndrome, compared with 49–71% of adults69,70. In fact, corticotroph adenomas are the most common pituitary adenoma diagnosed in early childhood (55% of pituitary adenomas in those aged 0–11 years; 30% in those aged 12–17 years)7, with mean age at presentation of 12.3 ± 3.5 years (mean ± SD; range 5.7–17.8)71. An overall male predominance exists in CYP, with 63% of patients with paediatric Cushing disease being boys compared with 79% of patients being female in adult series. This discrepancy is driven by prepubertal male predominance (71%)72. Boys with Cushing disease tend to have more aggressive disease with elevated BMI, shorter height and higher plasma ACTH levels than girls73. At all ages, microadenomas are the most common cause of Cushing disease, accounting for 98% of cases in CYP, with the adenoma diameter frequently being ≤2 mm (refs. 7,68,72,74). Macroadenomas, often showing invasion of the cavernous sinus, are rare in CYP (2–5% CYP versus 10% of adults with Cushing disease)75. Genetic associations are described in Part 1 (ref. 2).
Diagnosis: clinical features
-
Part 2: R14. Offer screening for Cushing syndrome in CYP with obesity but only if weight gain is inexplicable and combined with either a decrement in height SD score (SDS) or height velocity (strong recommendation, moderate-quality evidence).
The clinical features of Cushing disease in CYP are well documented69,73,74,76,77,78 and demonstrate interesting differences compared with adult patients72. CYP might show growth failure (subnormal growth velocity), with respective short stature and weight gain (height SDS below and BMI SDS above the mean for age and sex)69,72,74,78,79. Yet, not all CYP with Cushing syndrome have obesity and few patients with obesity prove to have Cushing syndrome80. Consensus statements advise that only CYP with unexplained weight gain and either growth rate deceleration or decrement in height centile over time require investigation, as this combination of features has a high sensitivity and specificity for Cushing syndrome in CYP81,82. The presence of growth failure sensitively discriminates simple obesity from Cushing syndrome in prepubertal CYP83 but is an unreliable indicator in post-pubertal CYP, who require assessment according to adult guidelines82.
Diagnosis: biochemical investigations
-
Part 2: R15. In CYP with suspected Cushing syndrome, offer investigations using established algorithms, first to determine the diagnosis of Cushing syndrome (the presence of hypercortisolaemia), followed by investigations to ascertain its aetiology (strong recommendation, moderate-quality evidence).
The biochemical investigation of children with suspected Cushing syndrome has been extensively reviewed69,76,81,84,85. The algorithms for testing consist initially of confirmation or exclusion of the diagnosis of hypercortisolaemia and then investigations to determine its aetiology81.
-
Part 2: R16. Suspected Cushing syndrome in CYP is effectively excluded by either two normal 24-h urinary free cortisol (UFC) measurements and a normal low-dose dexamethasone suppression test (LDDST; 0.5 mg 6-hourly for 48 h or, if patient weight is <40 kg, 30 µg/kg per day for 48 h); or a midnight sleeping serum cortisol concentration of <50 nmol/l (strong recommendation, high-quality evidence).
-
Part 2: R17. Two late-night salivary cortisol tests could be a useful alternative for the midnight serum cortisol test as a means of excluding Cushing syndrome, but age-specific and assay-specific normal ranges are not currently available and need to be carefully characterized (moderate recommendation, moderate-quality evidence).
Diagnosis of hypercortisolism usually includes three tests: dexamethasone suppression testing, 24-h UFC, and late-night salivary or sleeping midnight serum cortisol level (Table 2). None of these tests has 100% diagnostic accuracy and each test has some limitations86. It is important to eliminate the effect of exogenous glucocorticoids before biochemical testing.
Dexamethasone suppression testing can be either standard 48-h LDDST or overnight dexamethasone test (25 μg/kg at 11:00 h or midnight, maximum dose 1 mg). The 1 mg overnight dexamethasone suppression test is now routine in adults82 but considerably less data are available in children, where this test has lower sensitivity86,87. Both the 48-h and the overnight dexamethasone tests can be influenced by diarrhoea or coeliac disease, or by medications that increase cortisol-binding globulin levels or influence CYP3A4 activity. Measurements of serum dexamethasone can be used to ensure appropriate blood levels, but data for children are lacking and dexamethasone assays are not widely available. Repeated 24 h UFC measurements in the normal range (corrected for body surface area, micrograms per metre squared per 24 h) can support the lack of hypercortisolism. Physiological increases in UFC excretion can occur in girls in the peri-menarcheal phase. Limitations of the UFC test include difficulties in accurately and repeatedly collecting urinary samples in young children and lower sensitivity in milder cases of hypercortisolism and in severe kidney dysfunction. A sleeping midnight serum cortisol measurement has high sensitivity (94–100%) and specificity (100%) for Cushing disease. The test needs an overnight stay in the hospital and the patient needs to be asleep at the initiation of sampling. By contrast, a late-night salivary cortisol test is easy and cost-efficient, with high sensitivity (93–100%) and specificity (95–100%), but lacks age-specific and assay-specific normal ranges.
Clinical suspicion of Cushing syndrome but normal biochemical test results could rarely be due to periodic Cushing syndrome. In these patients, multiple, periodic, sequential late-night salivary cortisol tests can be helpful in detecting episodes of cortisol excess longitudinally82.
-
Part 2: R18.1. In CYP with confirmed Cushing syndrome, Cushing disease can be confirmed by its ACTH dependency, which is supported by a normal or elevated 09:00 h plasma ACTH (strong recommendation, moderate-quality evidence).
-
Part 2: R18.2. In CYP with confirmed Cushing syndrome, the diagnosis of pituitary-origin ACTH excess is supported by >20% increase in cortisol from baseline during a corticotrophin-releasing hormone (CRH) test (moderate recommendation, low-quality evidence, Delphi 92%).
Following confirmation of hypercortisolism, the priority is to determine its cause. Cushing disease is most easily confirmed by determination of basal (morning, 08:00–09:00 h) plasma ACTH. In all patients with Cushing disease, ACTH is detectable (>5 ng/l (>1.1 pmol/l); Table 2). In the presence of confirmed hypercortisolism, using a cut-off value of 29 ng/l (6.4 pmol/l), ACTH has a 70% sensitivity and 100% specificity for diagnosing Cushing disease85. Based on adult data and guidelines, in ACTH-independent Cushing syndrome, ACTH is always low and usually undetectable.
High-dose dexamethasone suppression tests (HDDST; 80–120 μg/kg, maximum 8 mg) are no longer necessary in the routine investigation of Cushing disease given that, in CYP (as in adults), >30% cortisol suppression during LDDST (but still above 50 nmol/l) correlates with HDDST results and strongly supports the diagnosis of Cushing disease. Furthermore, CYP with ectopic ACTH-secreting tumours might show cortisol suppression on HDDST, while the HDDST itself can induce transient hypertension and hyperglycaemia in CYP76,86,88,89,90. Therefore, many centres have abandoned HDDST. A CRH test using human sequence CRH (1 µg/kg intravenously) is recommended to support the suspected diagnosis of Cushing disease; in 92% of paediatric patients with Cushing disease (36 of 39 patients), serum cortisol level increased by >20% (range 2–454%) in response to CRH72. Ectopic ACTH syndrome is so rare in children that the need for a CRH test is questionable, although a cortisol increase of >20% to CRH has a 97.5% sensitivity and 100% specificity for Cushing disease and can contribute to the diagnosis85. As the availability of CRH is not universal, desmopressin (10 µg intravenous) has been used in CYP for bilateral simultaneous inferior petrosal sinus sampling (BSIPSS), with similar cut-off ratios to the CRH test91,92. A recommended protocol for the diagnosis of Cushing disease in CYP is shown in Table 2. All tests need to be interpreted in the light of the pre-test probability of the disease being present.
Diagnosis: neuroimaging and BSIPSS
In CYP with suspected Cushing disease, pre-operative MRI to localize the pituitary adenoma is strongly recommended, although only 50–63% of corticotroph adenomas were identified on post-contrast images in several large paediatric series72,85,93. This poor visualization rate in children could be explained by the limited spatial resolution of MRI, making small lesions within a small pituitary gland inconspicuous. Therefore, pituitary MRI imaging alone cannot reliably predict the adenoma position or confirm the diagnosis of Cushing disease in CYP.
-
Part 2: R19.1. Offer BSIPSS to CYP with confirmed ACTH-dependent Cushing syndrome and no identified adenoma on pituitary MRI to confirm a central source of ACTH excess (strong recommendation, low-quality evidence, Delphi 83%).
-
Part 2: R19.2. Offer BSIPSS only in a specialist centre with expertise in such testing and by an experienced interventional radiologist who regularly undertakes this procedure in adults (strong recommendation, moderate-quality evidence).
-
Part 2: R19.3. Consider confirming hypercortisolaemia immediately prior to BSIPSS to ensure the patient is in an active disease phase (moderate recommendation, moderate-quality evidence).
-
Part 2: R19.4. During BSIPSS, a pituitary source of ACTH excess is confirmed by a ≥2:1 ratio of central-to-peripheral ACTH before CRH or desmopressin and ≥3:1 ratio after CRH or desmopressin stimulation (strong recommendation, low-quality evidence, Delphi 100%).
-
Part 2: R19.5. BSIPSS could provide some information on tumour lateralization if the inter-petrosal sinus ACTH gradient after CRH or desmopressin stimulation is ≥1.4 between the two sides (moderate recommendation, moderate-quality evidence, Delphi 67% and GDG consensus).
BSIPSS was initially piloted in adults at the National Institute of Health (NIH)94 to enable distinction between Cushing disease and ectopic ACTH syndrome. In adult practice, BSIPSS has become routine unless the MRI unequivocally shows a pituitary adenoma that is unlikely to be an incidentaloma. A pituitary source of ACTH excess confirmed during BSIPSS has a high sensitivity for Cushing disease in experienced centres72,84,93,94,95. Hypercortisolaemia can be confirmed on the morning of the BSIPSS procedure to ensure the few patients with cyclical Cushing disease80 are in an active phase. BSIPSS using desmopressin as the stimulant has been reported in five paediatric series, with similar accuracy to CRH stimulation91,92. The reliability of the results of BSIPSS and the incidence of adverse events are related to the experience of the radiology team84. To enable the accurate interpretation of the results, medical therapy for Cushing disease (steroidogenesis inhibitors) must be stopped before undertaking BSIPSS; the length of time of treatment discontinuation depends on the half-life of the agent used.
BSIPSS might also help to lateralize pituitary ACTH secretion, where no lesion is visible on MRI81. If, on BSIPSS, the ACTH gradient between the two sides is greater than or equal to 1.4 after CRH (or desmopressin) stimulation, this finding might indicate lateralization of the tumour84,93,94,96,97,98 with possibly greater accuracy in CYP than in adults with Cushing disease75,96. The first paediatric data were reported in a large NIH series, where the predictive value for lateralization was 75–80%69. Similar values have been reported in other smaller series, with surgical concordance of adenoma site in 87–91% of patients75,96. Another NIH study of BSIPSS in 94 paediatric patients reported only 58% concurrence of ACTH lateralization with site of adenoma at surgery, which increased to 70% (51 out of 73) after the exclusion of 18 centrally located and 4 bilateral lesions98. Data from 2021 and 2022 confirm similar95 or higher (87.5%) percentages93. Based on these data, the GDG strengthened R19.5. Theoretically, false lateralization could occur due to altered pituitary blood flow94 but, in adult patients with Cushing disease, despite asymmetric internal petrosal sinuses in 11 of 38 patients (39%), both symmetric (100%) and asymmetric (93%) petrosal sinuses gave good lateralization99. Prolactin measurements during BSIPSS have been reported to be a useful marker of accurate catheterization, but two studies in adult Cushing disease suggested that prolactin-corrected ACTH concentrations did not substantially increase the accuracy of lateralization100,101 and this protocol has not been studied in CYP. Thus, no robust data exist to demonstrate, unequivocally, that lateralization of the tumour on BSIPSS improves surgical outcomes or preservation of residual pituitary function. However, observational studies of CYP suggest that improvements in rates of surgical cure might be related to the introduction of BSIPSS75,96.
Treatment: pituitary surgery
-
Part 2: R20.1. Offer selective adenomectomy as first-line treatment of choice for CYP with Cushing disease (strong recommendation, moderate-quality evidence).
-
Part 2: R20.2. Consider repeat surgery for CYP with persistent or recurrent disease (moderate recommendation, low-quality evidence, Delphi 100%).
Optimal treatment for CYP with Cushing disease is surgical resection by selective removal of the adenoma, performed by a surgeon experienced in paediatric transsphenoidal surgery102. Selective removal of the adenoma is now considered first-line therapy, maximizing the potential for normal pituitary tissue to remain in situ7,74,102. Low rates of post-operative hypopituitarism have been reported in several large studies in CYP103,104. However, selective microadenomectomy can be technically very difficult in children and surgeon experience is a predictor of success102,105.
Early post-operative remission in children was associated with identification of the adenoma at surgery, whilst long-term remission correlated with a younger age, a smaller adenoma, the absence of cavernous sinus or dural invasion, and a morning serum cortisol level of <1 µg/dl (<28 nmol/l) after surgery74. Repeat surgery for paediatric Cushing disease resulted in early biochemical remission in 93% of 27 patients74. However, recurrence of Cushing disease in adults has been reported up to 15 years after apparent surgical cure, even in individuals who had very low or undetectable post-operative cortisol levels106,107. Therefore, lifelong follow-up for children treated for Cushing disease is essential.
Treatment: pituitary radiotherapy
-
Part 2: R21. Offer radiotherapy to CYP with recurrent Cushing disease not amenable to curative surgery (strong recommendation, moderate-quality evidence, Delphi 93%).
A proportion of paediatric patients who undergo transsphenoidal surgery for Cushing disease do not achieve post-operative cure or remission87,108,109. The options for second-line therapy are repeat transsphenoidal surgery, radiotherapy, long-term medical therapy to control hypercortisolaemia and bilateral adrenalectomy. Focal external beam radiotherapy is more rapidly effective in children with Cushing disease than in adults108,109,110 and is often initiated 2–4 weeks after unsuccessful transsphenoidal surgery, when it is clear from circulating cortisol levels that a complete cure has not been achieved102.
Stereotactic radiotherapy, fractionated proton beam and gamma knife approaches have been proposed and utilized in adult Cushing disease. By contrast, over the past decade, fractionated proton beam radiotherapy with ongoing safety data monitoring has become the standard for focal cranial radiation in CYP with brain tumours generally111; however, experience is limited, particularly in children112,113. For fractionated treatment, a total radiation dose of 45 Gy in 25 fractions over 35 days seems effective108,114. A gamma knife stereotactic radiosurgery study in CYP with Cushing disease used a maximum dose of 50 Gy (range, 33–80) and a margin dose of 25 Gy (range, 12.90–27.1)115. The rapid effectiveness of these treatments is shown in Table 3; however, long-term data on adverse late effects are needed.
Treatment: medical therapies
-
Part 2: R22.1. Offer oral medical therapies, such as metyrapone or ketoconazole, to reduce the cortisol burden in CYP with Cushing disease awaiting definitive surgery or the effect of pituitary radiotherapy (strong recommendation, low-quality evidence, Delphi 100%).
-
Part 2: R22.2. Due to their adverse effects, metyrapone and ketoconazole have a limited role in the long-term treatment of Cushing disease in CYP (strong recommendation, low-quality evidence, Delphi 60% and GDG consensus).
Adrenal steroidogenesis inhibitors, such as metyrapone and ketoconazole, are well tolerated and can be highly effective at reducing cortisol levels, either alone or in combination102. In CYP, these drugs should be prescribed by experienced clinical teams with careful titration (metyrapone: 15 mg/kg every 4 h for 6 doses, alternatively 300 mg/m2 every 4 h, usual dose 250–750 mg every 4 h; ketoconazole for patients over 12 years: initially 400–600 mg per day in 2–3 divided doses, increased to 800–1,200 mg per day until cortisol levels normalize and then reaching a maintenance dose of 400–800 mg per day in 2–3 divided doses). However, hypercortisolaemia control can be lost due to the hypersecretion of ACTH and treatment might not be effective in the long term. Common adverse effects of metyrapone include hirsutism, dizziness, arthralgia, fatigue, hypokalaemia and nausea82. Prolonged usage can lead to hyperandrogenism and advanced bone age in children. Ketoconazole is associated with hepatotoxicity and liver function should be monitored on therapy. Gastrointestinal disturbance and adrenal insufficiency are recognized adverse effects of both therapies.
The GDG refined R22.2 after three rounds of an international Delphi consensus, in which only four individuals had the expertise to respond (voting details are provided in Part 1 (ref. 2)). Definitive treatments, which allow rapid normalization of subsequent growth and puberty, such as surgery and/or radiotherapy, are currently recommended for the management of paediatric Cushing disease, whilst medical therapies are currently limited87 and not well studied. Nevertheless, given the rarity of this condition in CYP, the effect of these medical agents on growth, and the importance of normalizing childhood growth and puberty, the GDG felt that, in contrast to adults, where long-term medical therapy might have a place after other treatments have failed82, their use in CYP should be confined to normalizing cortisol levels in preparation for surgery or while awaiting a biochemical response to radiotherapy.
The effects and safety of osilodrostat, an inhibitor of 11β-hydroxylase (CYP11B1), are currently being evaluated in a small phase II trial in CYP (NCT03708900)116, while the effects of cabergoline117, mifepristone and pasireotide in children are very limited or unknown. Temozolomide treatment has been used in an adult patient with childhood-onset Cushing disease (Table 1).
-
Part 2: R23. Offer intravenous etomidate treatment in CYP with Cushing disease in an intensive care setting only for the emergency control of severe cortisol excess (strong recommendation, moderate-quality evidence, Delphi 100%).
Intravenous administration of etomidate has successfully controlled hypercortisolaemia in children with severe Cushing disease, who were either too unwell for transsphenoidal surgery or presented with acute unmanageable symptoms, for example, respiratory failure or severe psychosis118. To enable the accurate assessment of surgical response, oral therapies, if feasible, should be stopped before surgery, depending on the half-life of the treatment; for example, for metyrapone, 48 h discontinuation should be sufficient.
Treatment: bilateral adrenalectomy
-
Part 2: R24. Reserve bilateral adrenalectomy only for CYP with severe refractory Cushing disease or for life-threatening emergencies (strong recommendation, low-quality evidence, Delphi 80% and GDG consensus).
Bilateral adrenalectomy remains a therapeutic option for Cushing disease in life-threatening situations or where transsphenoidal surgery is not possible or not available119. However, corticotroph tumour progression after bilateral adrenalectomy (Nelson syndrome, a potentially life-threatening secondary consequence of bilateral adrenalectomy, in which the pituitary adenoma continues to grow and secrete ACTH) seems to be more frequent in children than in adults and often requires pituitary surgery or radiotherapy120,121.
Follow-up and surveillance
-
Part 2: R25. Consider dynamic testing for GH deficiency soon after definitive therapy in all CYP in remission from Cushing disease who have not completed linear growth, and closely monitor pubertal progression to identify hypogonadotrophic hypogonadism (moderate recommendation, moderate-quality evidence).
-
Part 2: R26. Offer prompt initiation of GH replacement to CYP in remission from Cushing disease who are proven GH deficient or fail to show catch-up growth (strong recommendation, moderate-quality evidence, GDG consensus).
-
Part 2: R27. Consider BMD assessment prior to adult transition in patients at high risk for bone fragility (weak recommendation, low-quality evidence, Delphi 86%).
Growth failure and resultant short stature are almost always present at diagnosis in paediatric patients with Cushing disease69. Virilization from adrenal androgens can lead to pubarche and gonadotrophin-independent pubertal development, accelerating skeletal maturity and further compromising adult height potential122. After normalization of cortisol, CYP with growth retardation should be evaluated for GH deficiency with appropriate dynamic testing and receive early GH replacement therapy given the limited window of opportunity to normalize lean-to-adipose mass ratio, promote catch-up growth and attain their normal adult height84,123,124,125,126,127. Given these data and the current clinical practice86, a randomized controlled trial of early GH treatment in CYP in remission from Cushing disease is unlikely, and the GDG therefore strengthened R26. GH deficiency is well recognized following transsphenoidal surgery124 and pituitary irradiation106,108,109. The challenge is to reverse these adverse effects and maximize growth potential to achieve normal adult height and body composition. One approach is to routinely assess for the possibility of GH deficiency soon (up to 3 months)86 after surgery or radiotherapy and substitute GH at conventional replacement doses (0.025 mg/kg per day), if deficient. Gonadotrophin-releasing hormone analogue therapy can be added to delay puberty and epiphyseal closure. Results demonstrate that this regimen usually enables adequate catch-up growth and adult height within the range of target height for the majority of patients106,128. Combined treatment with GH and aromatase inhibitors to reduce bone maturation induced by oestradiol could also be a therapeutic alternative in pubertal patients129. Normal body composition and BMD are more difficult to achieve128,130. Many CYP with Cushing disease have disturbed timing or progression through puberty and require sex steroid replacement to enhance growth velocity and reverse the suppressant effect of cortisol excess on gonadotrophins122.
Pituitary function following remission
-
Part 2: R28. To assess possible recurrence, offer to all CYP in remission from Cushing disease 6-monthly clinical examination, 24 h UFC, electrolytes and morning serum cortisol for at least 2 years and lifelong annual clinical assessment (strong recommendation, low-quality evidence, Delphi 100%).
Pituitary hormone deficiencies are common after surgical or radiotherapeutic cure of Cushing disease106,109,131. GH deficiency is the most frequent pituitary deficit (see previous section), although recovery in adult life has been reported106,132. During long-term follow-up of patients in remission, gonadotrophin secretion is generally preserved, with normal or true gonadotrophin-dependent early puberty occurring, the latter being well recognized after cranial radiotherapy. After radiotherapy, additional anterior pituitary deficiencies (TSH and permanent ACTH deficits) can rarely develop, typically occurring in combination106,132, with the risk of anterior pituitary hormone deficiencies potentially increasing over time84.
Psychiatric and neurocognitive effects
-
Part 2: R29. In addition to lifelong follow-up for endocrinopathies, consider long-term monitoring for psychiatric and neurocognitive co-morbidities following remission of Cushing disease in CYP (moderate recommendation, moderate-quality evidence).
Studies of adult patients with Cushing syndrome have reported brain atrophy, cognitive impairment and psychological disease, most commonly depression, associated with excess endogenous circulating glucocorticoids133. A study in 11 patients134 also found considerable cerebral atrophy in children with Cushing disease at diagnosis but without IQ differences between patients and control individuals. Interestingly, an almost complete reversal occurred of the cerebral atrophy 1 year after cure with transsphenoidal surgery but with a paradoxical decline in cognitive function. Another study shows notable improvements in severe psychiatric and behavioural symptoms after cure; however, long-term cognitive and memory problems were identified in ~25% of patients106 and this observation is consistent with similar cognitive and memory deterioration in adults135. Furthermore, the grey matter volume loss of active Cushing disease reverses 3 months after remission, except in the frontal and temporal lobes, strongly associated with cognition and memory136. Children with Cushing disease experience impaired health-related quality of life, which is not fully resolved at 1 year post-treatment137.
Relapse of Cushing disease
-
Part 2: R30. In suspected recurrence of Cushing disease in CYP, offer the same stepwise investigations as at first presentation (strong recommendation, low-quality evidence, GDG consensus).
Reported recurrence rates after remission of Cushing disease in CYP vary considerably, from 6% to 40%74,79,106,138, and usually occur within 5 years following definitive treatment. However, relapse can occur later106, and the percentage of patients who relapse increases with time107, suggesting that lifelong follow-up is required. This observation is consistent with data from long-term follow-up in adult patients with Cushing disease107; therefore, the GDG strengthened R30.
GH excess: gigantism and acromegaly
Epidemiology and aetiology
A GH-secreting pituitary adenoma arising from somatotroph cells (somatotrophinoma) is the most common cause of acromegaly in CYP and is usually detectable on a contrast-enhanced pituitary MRI. Rarely, CYP with somatotroph hyperplasia due to McCune–Albright syndrome139, Carney complex140, X-linked acrogigantism141 or GH-releasing hormone-secreting tumours (usually associated with MEN1 syndrome in CYP142,143) have been described. GH excess is rare in CYP. Annual incidence rates of GH excess are estimated to be 3–8 cases per million person-years among children aged 0–17 years old, while annual prevalence rates are estimated to be 29–37 patients per million children aged 0–17 years144. Among surgically treated paediatric pituitary adenomas, 8.8%145 to 21%146 are associated with GH excess, with an age-related increase (6.4% at 0–11 years, 9.1% at 12–16 years and 11.8% at 18–19 years). While there are more male patients with gigantism overall145,147,148, more girls (62%) than boys are diagnosed with gigantism in the CYP age group149.
If GH excess occurs before epiphyseal fusion, the patient develops tall stature or gigantism. The definition of pituitary gigantism is arbitrary150,151 (Box 1). Gigantism can be exacerbated by delayed puberty due to gonadotrophin inhibition by co-secretion of prolactin from the adenoma, hyperprolactinaemia from stalk compression or mass effects causing gonadotrophin deficiency. Gigantism has an identifiable genetic basis in almost 50% of patients currently, so genetic assessment and testing are particularly important in CYP (see Part 1, Genetics section2).
Diagnosis: clinical features
-
Part 2: R31. Offer testing for GH excess to CYP with excess height (more than 2 SDS) or consistently elevated height velocity and acromegalic features, with or without delayed or arrested puberty or family history of pituitary adenoma (strong recommendation, moderate-quality evidence, Delphi 100%).
The most prominent clinical feature of GH excess in CYP before epiphyseal closure is increased growth velocity. Serial heights and photographs are useful for timing the onset of disease152.
Ethnicity-adjusted height of >2 SDS above age-adjusted and sex-adjusted normal values or 2 SDS above mid-parental target height, persistently elevated growth velocity (>2 SDS), acral enlargement, headache, visual field defects, pubertal delay, delayed bone age and joint pain are common signs of GH excess in CYP. Other features, typical of adult-onset GH excess, can also develop, including coarsened facial features, prognathism, dental malocclusion, teeth separation, frontal bossing, sweating, kyphosis, insulin resistance and occasionally secondary diabetes mellitus, hypertension, sleep disturbance, sleep apnoea, carpal tunnel syndrome, galactorrhoea, pituitary apoplexy, left ventricular hypertrophy and diastolic dysfunction105,145,149,153,154,155,156,157,158,159,160, while joint hypermobility has occasionally been observed161.
X-linked acrogigantism is characterized by tall stature onset before 5 years (usually before 2 years) in all patients141,147,148,162,163 and disproportionally enlarged hands, feet, teeth separation, acanthosis nigricans, increased BMI and increased appetite141,162. In McCune–Albright syndrome139, early-onset (3 years onwards) GH excess, café-au-lait pigmentation, fibrous dysplasia and precocious puberty (or other hormone excess conditions) are prominent clinical features, while in Carney complex140 typical skin pigmentation, myxomas, testicular and adrenal disease are characteristic, in addition to other pleiotropic manifestations.
Diagnosis: biochemical investigations
-
Part 2: R32. A diagnosis of GH excess is supported by an elevated serum IGF1 level in relation to the age-adjusted, sex-adjusted and Tanner stage-matched normal range (strong recommendation, moderate-quality evidence).
Elevated Tanner stage-matched and age-adjusted circulating serum IGF1 concentration is a reliable marker for GH excess, but marginal or mild elevation in adolescence, during the peak growth spurt, needs cautious interpretation. IGF1 values might be falsely normal or low in CYP with a GH-secreting adenoma and concurrent severe hypothyroidism, malnutrition or severe infection164,165,166, or might be falsely elevated in CYP without GH-secreting adenoma in poorly controlled diabetes mellitus, hepatic and/or renal failure. Oral oestrogens can also confound detection accuracy by reducing IGF1 generation by the liver167, whilst local Tanner stage-matched, sex-matched and age-matched normal ranges for the IGF1 assay must be established to avoid notable inter-assay variability168.
-
Part 2: R33. Consider the diagnosis of GH excess in CYP whose serum GH levels fail to suppress below 1 µg/l in response to an oral glucose load (cut-off based on adult population); however, complete suppression of GH can be difficult to achieve in normal adolescence (moderate recommendation, moderate-quality evidence, Delphi 100%).
Algorithms for assessing GH excess in CYP are based on those established in adult patients150,169. In healthy adults, serum levels of GH should be suppressed after an oral glucose load: cut-off GH nadirs are 1 µg/l or, using sensitive GH assays, 0.4 µg/l (refs. 170,171). GH nadir after glucose load in CYP undergoing puberty is sex and pubertal stage specific: the highest levels were observed in mid-puberty (Tanner stage 2–3) in girls more than in boys (mean ± 2 SD GH nadir after 2.35 g/kg, maximum 100 g glucose load: in girls 0.22 µg/l ± 0.03–1.57, in boys 0.21 µg/l ± 0.09–0.48)172. However, an earlier study reported a lack of GH suppression (defined as <1 µg/l following 1.75 g/kg, maximum 75 g glucose load) in approximately 30% of children with tall stature (+3.1 ± 0.8 height SDS)173, rendering the diagnosis of GH excess by these criteria challenging in this patient group. An elevated serum IGF1 concentration, with apparently normal serum GH values, might reflect early disease174. We suggest that biochemical results should be interpreted within a clinical assessment of phenotype that includes height velocity, pubertal stage and bone age.
From adult data, serum IGF1 levels correlate linearly with GH levels only up to 4 µg/l and plateau at about 10 µg/l (refs. 175,176). Baseline GH levels are predictive of surgical outcome (higher GH levels predict a lower likelihood of remission after surgery)177 and are key to monitoring the hormone-producing activity of the adenoma176. Thus, both GH and IGF1 should be monitored at baseline and during follow-up in CYP with GH excess.
-
Part 2: R34. In CYP with GH excess, offer dynamic pituitary assessment of possible hypofunction and hyperfunction of other anterior pituitary hormones (strong recommendation, high-quality evidence).
Hypofunction of other pituitary hormones caused by tumour mass compression or prolactin co-secretion has been noted in 25–35% of patients with somatotrophinomas105,149. Hypogonadism and consequent bone age delay are particularly relevant to GH excess as they increase the time window for longitudinal growth. Co-secretion of other anterior pituitary hormones is common in both adults and CYP with GH excess. A review of 137 published cases of CYP with acromegaly found that 65% had hyperprolactinaemia at presentation, while half showed both GH and prolactin immunostaining in the adenoma tissue105. Furthermore, 34–36% of CYP with gigantism had prolactin co-secretion in two large cohorts147,149. TSH can also be co-secreted by somatotrophinomas but less frequently than prolactin. Patients require assessment and treatment of complications of GH excess such as glucose intolerance and hypertension.
-
Part 2: R35.1. Pituitary adenomas can be associated with syndromic diseases; offer clinical evaluation for associated syndromic causes of somatotrophinomas to CYP with GH excess (strong recommendation, low-quality evidence, Delphi 93%).
-
Part 2: R35.2. Offer biochemical screening for pituitary hormone excess to all CYP with Carney complex, McCune–Albright syndrome, and patients with MEN1 or MEN1-like disease (strong recommendation, high-quality evidence, Delphi 100%).
Several syndromes, including McCune–Albright syndrome, Carney complex, MEN1 or, rarely, MEN1-like diseases (MEN4, MEN5) and phaeochromocytoma–paraganglioma-related pituitary disease2 can be associated with childhood-onset pituitary adenomas, including GH excess. The biochemical diagnosis of GH excess in the context of such syndromes is identical to sporadic cases of acromegaly139,141,147,149,151,178,179. In CYP with Carney complex, biochemical alterations of the GH axis without overt clinical acromegaly can often be observed. GH-releasing hormone-secreting pancreatic tumours should also be considered as a cause of GH excess in CYP with MEN1 syndrome142,143. GH hypersecretion, usually transient, occurs in 10% of infants and children with neurofibromatosis type 1 and optic pathway hypothalamic gliomas180; some of these patients have been treated temporarily with somatostatin analogues or pegvisomant180,181,182. GH excess is probably a result of hypothalamic dysregulation of the GH axis. Data on the aetiology of temporary GH excess in CYP with optic glioma, therapeutic interventions, the effects of GH excess on glioma growth itself and the observed evolution from GH excess to GH deficiency remain unexplained and poorly understood. The GDG suggested the establishment of a collaborative neuroendocrine oncology research study to gather evidence for these questions.
Treatment: pituitary surgery
-
Part 2: R36. Offer surgery to reduce GH burden as the treatment of choice in the majority of CYP with GH-secreting adenomas, even where surgical cure is unlikely (strong recommendation, moderate-quality evidence, Delphi 93%).
-
Part 2: R37.1. Consider pre-operative medical therapy with somatostatin analogues and/or GH receptor antagonists to rapidly control signs and symptoms and support perioperative airway management (weak recommendation, low-quality evidence, Delphi 75%, GDG consensus).
-
Part 2: R37.2. Consider pre-operative medical therapy with somatostatin analogues and/or GH receptor antagonists to reduce height velocity, particularly if pituitary surgery is delayed (weak recommendation, low-quality evidence, Delphi 100%).
The goals of any therapy for GH excess include normalization of growth velocity, prevention of excessive height and suppression of IGF1 into the normal range; adenoma shrinkage and visual preservation with potential restoration of other hormone function; and prevention of long-term morbidity. Surgery performed by an experienced neurosurgeon offers the prospect of complete remission and is therefore recommended as the first-line treatment for CYP with gigantism or acromegaly159,183,184,185, as for adults150,186,187,188. Even if remission is thought to be unlikely following surgery, tumour debulking can reduce the circulating GH burden and facilitate more successful medical therapy and/or radiotherapy. In an experienced pituitary neurosurgical unit between 2003 and 2016, the surgical success rate in 17 CYP with GH excess was ~50%189. Re-operation could be considered in patients with notable residual tumour and inadequate response to somatostatin analogue therapy or if considerable tumour re-growth occurs.
A role for somatostatin analogues in safely reducing pre-operative GH levels in adults with macroadenomas, with favourable effects on short-term cure, has been suggested by a systematic review, although long-term outcomes require further study190. Patients with severe cardiac and/or respiratory complications from GH excess might benefit from pre-operative reduction of GH levels. Pre-operatively, patients should be assessed for enlargement of the upper airway soft-tissue structures, including the tongue and epiglottis as well as jaw deformity, to prevent perioperative airway difficulties191,192.
In the absence of prospective trials, factors predicting outcome of surgery in CYP with gigantism or acromegaly are limited, although retrospective studies and case reports suggest that a rapid reduction in growth velocity might be a favourable predictor105,149,153.
In patients with genetic causes of acromegaly, the whole gland could be affected (typically in McCune–Albright syndrome, Carney complex and X-linked acrogigantism). Selective adenomectomy193, radical surgery162 or hypophysectomy139,194 have all been described as surgical approaches in these conditions.
Treatment: medical therapies
-
Part 2: R38. Offer monotherapy or combination medical therapy in CYP with GH excess and post-operative residual disease (strong recommendation, moderate-quality evidence, Delphi 100%).
-
Part 2: R39. Assess efficacy of medical treatment in CYP with GH excess by both auxological measurements and serum levels of GH and IGF1 (strong recommendation, moderate-quality evidence, Delphi 100%).
Studies of medical therapy in patients with post-operative residual GH excess have been conducted primarily in adults, although case series and case reports also confirm their utility in CYP105,139,147,149,153,154,155,160,162,195. Adjunctive post-operative long-acting somatostatin analogues reduce serum concentrations of GH to ‘safe’ levels and normalize serum levels of IGF1 in approximately 35% of adult patients with GH-secreting adenoma196 with less predictable tumour shrinkage in up to 40%. Retrospective studies have also confirmed the tumour-shrinking effect of somatostatin analogues in adolescents with gigantism139,147,149,153,162. While no formal dosing recommendation exists for children, the dose of long-acting somatostatin analogues can be titrated to normalize IGF1 levels. In adults, the starting monthly dose of lanreotide autogel is 60 mg and of octreotide-LAR 10 mg whereas, for very young children, the dose needs to be individualized. In CYP with acromegaly, numerous reports of the failure of somatostatin analogues to normalize IGF1, satisfactorily reduce GH levels or initiate tumour shrinkage suggest that treatment resistance is more common in CYP than in adults147,149,156,160,162. CYP with AIP or GPR101 mutations have more aggressive disease and are less responsive to first-generation somatostatin analogue therapy147,149,162, whereas the response to medical treatment in patients with Carney complex and McCune–Albright syndrome is more promising139. The second-generation somatostatin analogue pasireotide has also been used in adult patients with childhood-onset GH excess, with mixed results197,198.
Regardless of the presence or absence of prolactin co-secretion, dopamine agonists (primarily cabergoline) can be used alone in CYP with mild GH excess, co-administered with somatostatin analogues where GH hypersecretion is not adequately controlled, or substituted for the latter where poorly tolerated199,200,201,202. However, the GH-lowering effect of dopamine agonists is only modest105,155,156,160 and doses required in acromegaly are often high (see R11 on cabergoline use).
Several studies and case reports have been published of the GH receptor antagonist pegvisomant normalizing serum IGF1 levels in CYP with acromegaly or gigantism when used in adequate doses105,139,147,149,154,160,162,203,204,205. This medication can suppress growth velocity, a clinical priority in CYP with gigantism, suggesting that earlier introduction could be beneficial. The dose of pegvisomant should be started at 10 mg daily and titrated until serum IGF1 levels normalize. Patients treated with pegvisomant require only IGF1 measurements during follow-up. As pegvisomant has no direct pituitary action, careful radiological tumour monitoring is required. Tumour expansion described during pegvisomant treatment105,152 might represent the natural history of the tumour rather than being secondary to reduced negative feedback from reductions in IGF1. Combination therapy with pegvisomant and somatostatin analogues has also been successfully tried in CYP with GH excess149,203,204,205. Temozolomide therapy has been reported in a CYP patient with a GH-secreting tumour and another with a GH–prolactin co-secreting tumour (Table 1).
Treatment: pituitary radiotherapy
-
Part 2: R40.1. Offer pituitary radiotherapy to CYP with GH-secreting adenoma and uncontrolled tumour growth and incomplete surgical and medical response, except for patients with skull base fibrous dysplasia (strong recommendation, low-quality evidence, Delphi 75%).
-
Part 2: R40.2. After radiotherapy in CYP with GH-secreting adenoma, offer intermittent dose reduction or withdrawal of medical therapy to assess radiation efficacy on GH hypersecretion (strong recommendation, low-quality evidence, Delphi 100%).
Radiotherapy might be useful for CYP with gigantism or acromegaly who have post-operative residual tumour bulk and in whom surgery and medical therapy have failed to control GH–IGF1 levels (see Part 1, Radiotherapy section2). Conventional conformal, stereotactic and proton beam radiotherapy have all been used successfully to control tumour growth and lower serum GH levels in CYP105,115,149,152, although adult data indicate that it might take up to 10 years for radiotherapy to be fully effective in suppressing GH206. Therefore, medical therapy is probably required, at least as a temporary measure, in CYP who receive pituitary radiotherapy. Intermittent interruption of medical therapy for 1–3 months (to allow clearance of the drug) might be required to allow biochemical assessment of the efficacy of radiotherapy. After radiotherapy, at first 6-monthly and later 12-monthly follow-ups should monitor the patient for the development of hypopituitarism or recurrence.
GH excess in the context of McCune–Albright syndrome is typically accompanied by fibrous dysplasia of the craniofacial bones. In one study of patients with this syndrome, 6 of 112 patients, 3 of whom had prior pituitary irradiation, developed a skull base sarcoma139. While a causal link to pituitary radiation is not proven, the risk of sarcomatous transformation is higher in McCune–Albright syndrome than in isolated fibrous dysplasia207. Almost half of the patients with McCune–Albright syndrome are CYP (46%) at the time of diagnosis of GH excess. Given the uncertain transforming effect of GH–IGF1 on dysplastic bone208,209,210, alternative medical and surgical treatments, rather than radiotherapy, might be considered in this condition to control GH excess and preserve vision.
Follow-up and surveillance
-
Part 2: R41. There is no evidence to suggest that CYP with GH excess require routine screening for colonic polyps during childhood (strong recommendation, low-quality evidence, Delphi 92%).
-
Part 2: R42. Consider avoiding corrective surgery for jaw, spine and joint abnormalities in CYP with gigantism or acromegaly until GH and IGF1 are at safe levels (weak recommendation, low-quality evidence, GDG consensus).
While good evidence indicates an increased risk of colonic polyps in adults with acromegaly, such data are lacking in CYP, and the Delphi panel returned a 92% consensus against routine colonoscopy in CYP with GH excess.
Bone overgrowth induced by GH excess does not reverse with successful treatment of acromegaly and, while jaw and dental deformity can be troublesome, corrective surgery should be deferred until GH–IGF1 levels are adequately controlled and jaw changes have stabilized211.
Late recurrence of GH excess, up to 10 years after apparent remission, has been described212. Thus, long-term annual serum IGF1 monitoring is necessary with repeat biochemical (oral glucose tolerance tests) and radiological (MRI) assessment if recurrence is suspected. Due to the high rates of persistent post-operative disease in CYP with GH excess, many will receive long-term medical therapy or radiotherapy. Monitoring of their efficacy and adverse effects, together with management of secondary pituitary deficits and medical complications, requires experienced specialist support within a dedicated pituitary multidisciplinary team. Close interaction between paediatric and adult endocrine services is required to coordinate long-term medical care and the transition to adult services.
TSHoma
Epidemiology
TSH-secreting adenomas are extremely rare in children and the literature is limited to case reports. In adults with TSHoma, the incidence has been estimated to be 0.26 cases per million per year; co-secretion of other pituitary hormones occurs in 25–42% of cases213. Co-secretion of GH was also noted in an adolescent with gigantism and secondary hyperthyroidism214.
Diagnosis
-
Part 2: R43. Consider assessment for TSHoma in CYP with hyperthyroxinaemia and an unsuppressed TSH, particularly in the presence of clinical thyrotoxicosis and neurological or visual deterioration (moderate recommendation, moderate-quality evidence).
-
Part 2: R44. Consider assessment for thyroid hormone resistance and euthyroid hyperthyroxinaemia in the differential diagnosis of TSHoma in CYP (moderate recommendation, low-quality evidence, Delphi 100%).
Patients with TSHomas present with elevated thyroid hormone levels and unsuppressed TSH, differentiating them from primary hyperthyroidism. Usually, children have symptoms of hyperthyroidism but some are reported to be asymptomatic215. Most TSHomas reported in children have presented as macroadenomas; thus, mass effects can cause optic nerve compression and deficiency of other pituitary hormones215,216,217. Following the diagnosis of a TSHoma, investigation for pituitary hypofunction and hyperfunction with baseline and dynamic pituitary testing is recommended. Prompt treatment of any cortisol, GH or sex steroid deficiency is recommended to optimize surgical, growth and well-being outcomes.
An unsuppressed TSH in the setting of hyperthyroxinaemia can occur due to assay interference and due to genetic causes of euthyroid hyperthyroxinaemia such as familial dysalbuminaemic hyperthyroxinaemia and resistance to thyroid hormone. Of note, a case of TSH-secreting microadenoma and resistance to thyroid hormone in a child has been reported218.
Treatment
-
Part 2: R45. Consider pre-operative somatostatin analogue treatment to normalize thyroid function in CYP with confirmed TSHoma (moderate recommendation, low-quality evidence, Delphi 73% and GDG consensus).
-
Part 2: R46. Offer transsphenoidal surgery as the treatment of choice in CYP with TSHomas (moderate recommendation, low-quality evidence, Delphi 93%).
In adults with TSHoma, somatostatin analogues improve the symptoms of thyrotoxicosis, decrease serum-free T4 and TSH concentrations, and can cause pre-operative tumour shrinkage219. In one paediatric patient, somatostatin analogues reduced thyroid hormone levels and tumour size, thereby postponing definitive surgery215,220. Out of 44 patients aged 11–73 years on pre-operative somatostatin analogue treatment, 84% had normalization of thyroid function levels and 61% showed tumour shrinkage221.
Surgery offers a potentially definitive cure. Even partial tumour debulking can be worthwhile in reducing TSH and free T4 levels, decompressing the optic apparatus or improving the effectiveness of subsequent medical therapy216,217,221,222.
Follow-up and surveillance
-
Part 2: R47. Disease monitoring with regular thyroid function tests and regular MRI surveillance, similar to the protocol described for NFPA, is suggested in CYP with confirmed TSHomas (weak recommendation, low-quality evidence, GDG consensus).
There is no evidence base to inform a surveillance schedule in CYP with TSHoma and the natural history of disease progression is unknown. We therefore suggest a pragmatic approach to biochemical and MRI surveillance to detect secondary hyperthyroidism and to identify those with rapid tumour progression or recurrence early, even if asymptomatic. We suggest monthly thyroid function tests for 6 months after initial treatment but, given the uncertainty regarding tumour progression, biochemical and MRI surveillance should be individualized.
-
Part 2: R48. Consider pituitary radiotherapy in CYP with post-operative tumour remnant and resistance to medical therapy or relapsing TSHomas if re-operation is not an option (moderate recommendation, low-quality evidence, Delphi 92% and GDG consensus).
If surgery is unsuccessful or contraindicated and the paediatric patient with TSHoma fails to achieve normal thyroid function or shows tumour growth despite somatostatin analogue therapy, radiotherapy could be considered222,223 (see Part 1, Radiotherapy section2). After radiotherapy, at first 6-monthly and later 12-monthly follow-ups should monitor for the development of hypopituitarism or recurrence.
Incidentalomas and NFPAs
Epidemiology and aetiology
Clinically non-secreting pituitary adenomas account for 4–6% of all paediatric pituitary adenomas224,225 and for 10% of cases in a surgical series220, in stark difference to adult pituitary patients where NFPAs account for 15–30% of clinically relevant pituitary adenomas and 50–60% of cases in a surgical series226. Depending on case selection criteria, NFPAs represent 13–56% of incidentally discovered pituitary lesions (incidentalomas) in large retrospective radiological series of CYP227,228,229. NFPAs also represent 25% of MEN1-associated pituitary lesions in CYP, and small lesions detected by screening can be seen in patients with AIP variants151,230,231. NFPAs in CYP usually present in the second decade of life, with increasing incidence thereafter9,228,230,232. There is no consistent male or female predisposition9,230,232,233,234,235,236, although a 2019 surgical series in CYP reported a slight female predominance220.
Presentation
CYP with NFPAs, by definition, do not have features of a hypersecretory syndrome. Symptomatic NFPAs are macroadenomas, whereas microadenomas are usually discovered as a coincidental finding on an MRI scan. The clinical features of macroadenomas at presentation result from mass effects on surrounding structures. These include headache, visual field defects, pituitary apoplexy (probably more common than in adult NFPA235,236) with or without mild prolactin elevation, or hypopituitarism manifesting as secondary amenorrhoea, delayed puberty, hypothyroidism, hypocortisolaemia, growth failure and/or hyperprolactinaemia9,220,228,230,232,233,234. Headache, visual impairment and hypogonadism were the most common presenting features in a surgical series in CYP236. Immunohistochemistry for pituitary hormones in 14 NFPAs, from CYP aged 12–19 years, revealed 6 silent plurihormonal tumours, 3 silent lactotrophs, 1 each of a silent gonadotroph and a corticotroph, and 3 hormone-negative tumours (transcription factor staining not reported)236. These proportions are different from those in adult patients, where silent gonadotrophinomas represent the majority of NFPAs. Non-functioning pituitary microadenomas do not usually cause hypopituitarism or visual abnormalities227.
Diagnosis and investigation
-
Part 2: R49. Identification of a pituitary incidentaloma or NFPA requires exclusion of clinical or laboratory evidence of pituitary hormone hypersecretion (except mild hyperprolactinaemia from pituitary stalk disruption); exclusion of elevation of serum level of α-fetoprotein (AFP) and β-human chorionic gonadotrophin (βHCG) and the absence of other suprasellar and intracranial lesions (strong recommendation, low-quality evidence, Delphi 100%).
-
Part 2: R50. Offer baseline and dynamic evaluation of pituitary function to assess hypopituitarism and exclude hormone excess in all CYP with suspected NFPA (strong recommendation, low-quality evidence, Delphi 100%).
If a pituitary mass is suspected on MRI, baseline and dynamic pituitary investigations should be undertaken as indicated clinically to confirm the absence of pituitary hormone excess from a functioning adenoma or the presence of hormone deficiency. The MRI appearance of a small Rathke cleft cyst or pars intermedia cyst can present a differential diagnosis for a clinically non-functioning microadenoma. Stalk compression from macroadenomas, or rarely microadenomas237, can cause mild hyperprolactinaemia, usually below 2,000 mU/l (94 µg/l)19.
Intracranial germ-cell tumours, more common in the adolescent age group than NFPAs, can present with similar MRI appearances238,239. AFP and βHCG (markers of germ-cell tumours) should be measured in the serum and, if clinically or radiologically indicated, also in the cerebrospinal fluid240.
Hypopituitarism is common in children with symptomatic non-functioning pituitary macroadenoma9,220,228,230,232,233. Basal and dynamic pituitary assessment is recommended following diagnosis of an NFPA together with prompt replacement therapy of any cortisol, thyroid hormone, sex steroid and GH deficiency to optimize surgical, growth and well-being outcomes. Arginine vasopressin deficiency (also known as central diabetes insipidus) is an extremely infrequent finding at diagnosis of an NFPA unless the tumour has undergone apoplexy9,220,228,230,232,233, and its presence strongly suggests an alternative diagnosis such as a craniopharyngioma, histiocytosis or germ-cell tumour111,240.
Visual deterioration is a well-described presentation of NFPA in CYP9,228,232,233 and an urgent indication for surgery to decompress the optic apparatus if sight is threatened. Thus, early assessment of visual acuity and visual fields is mandated111.
-
Part 2: R51. There is no evidence to suggest a benefit of routine diagnostic biopsy in CYP of incidental pituitary masses whose radiological features are typical of a pituitary adenoma with no other intracranial abnormalities (strong recommendation, low-quality evidence, GDG consensus).
A pituitary biopsy is not required in clinically non-functioning lesions with MRI appearance strongly suggestive of a pituitary adenoma and no other intracranial abnormality, which remain asymptomatic, without visual or pituitary function compromise, unchanged in size over time, and negative for serum βHCG and AFP. In these patients, the risk of a biopsy harming pituitary function is greater than the likelihood of achieving a ‘tissue’ diagnosis or a diagnosis requiring alternative management240. MRI surveillance should be instituted if the optic apparatus is not compressed.
Treatment
-
Part 2: R52. Offer treatment to CYP with NFPA only if the patient is symptomatic (hypopituitarism), the visual pathway is threatened or there is interval tumour growth on MRI (strong recommendation, low-quality evidence, Delphi 94%).
-
Part 2: R53. Offer transsphenoidal surgery as the treatment of choice in CYP with NFPA needing surgical intervention (strong recommendation, high-quality evidence, Delphi 100%).
The majority of non-functioning microadenomas in CYP are asymptomatic and detected incidentally or as part of a screening programme, with little data on their frequency, natural history or complication rates. By contrast, macroadenomas enlarge in over 40% of adult patients at 5 years241, while there are no equivalent data in CYP. Surgical removal of a growing macroadenoma is an effective intervention to prevent further damage to surrounding tissues and cranial nerves9,228,229,230,232,233,234.
-
Part 2: R54. There is insufficient evidence to recommend medical therapy, including cabergoline, in CYP with NFPA (moderate recommendation, low-quality evidence, Delphi 80%).
No data are available to suggest that medical therapy (for example, dopamine agonists or somatostatin analogues) can induce tumour shrinkage in CYP with NFPA, and these medications are not recommended in adult guidelines. Some inconsistent reports of effectiveness in adults with NFPAs receiving somatostatin analogues242 or dopamine agonists243,244,245 suggest that dopamine agonists might reduce tumour growth in some recurrent cases244,245.
-
Part 2: R55. Consider second surgery or radiotherapy for CYP with recurrent or symptomatic NFPAs (moderate recommendation, low-quality evidence, Delphi 94% and GDG consensus).
Both surgery and radiotherapy have been used to treat recurrent NFPA9,146,246,247,248 and the optimal treatment modality for CYP is not clear. The specialist age-appropriate pituitary multidisciplinary team should make an individual treatment recommendation, accounting for tumour location and any hypothalamic or cavernous sinus invasion, ease of surgery, and the age of the child. Local tumour control after radiotherapy for NFPA in CYP ranges from 80% to 97%249. After radiotherapy, at first 6-monthly and later 12-monthly follow-ups should monitor for the potential development of hypopituitarism or recurrence. Temozolomide treatment was reported for one childhood-onset hormone-negative tumour (Table 1). Based on results from adults250 and the above data, the GDG strengthened R55.
Follow-up and surveillance
-
Part 2: R56. Following NFPA surgery in CYP, offer post-operative MRI surveillance at a minimum of 3 and 6 months, and 1, 2, 3 and 5 years after surgery (strong recommendation, low-quality evidence, Delphi 89%).
-
Part 2: R57. In CYP with incidental NFPAs, offer MRI surveillance for microincidentalomas: at 12 months and, if stable, at 1–2 year intervals for 3 years with gradual reduction thereafter; for macroincidentalomas: at 6 months and, if stable, annually for 3 years with gradual reduction thereafter (strong recommendation, low-quality evidence, Delphi 100%).
Although evidence in CYP following pituitary surgery for NFPAs is lacking, data in adults show a recurrence rate of non-irradiated operated pituitary adenomas of up to 38% of those with a visible residuum on post-operative MRI over a follow-up period of 5 years. A post-operative MRI at 3 months will assess the extent of residual tumour, with a further scan at 6 months to assess for recurrence and, if stable, a gradual reduction in the annual scanning frequency as above, with screening continuing lifelong251,252.
The natural history of NFPA in CYP is unknown and could vary. In adults, macroadenomas tend to have higher growth rates than microadenomas (12.5 macroadenomas grow over 100 patient-years versus 3.3 microadenomas)253. Microadenomas seem to follow a benign course in children227, and follow-up can be gradually reduced and stopped227,228,229. For macroadenomas, we recommend lifelong clinical surveillance (as in adults), with an individualized radiological (MRI) surveillance strategy to identify those with rapid tumour progression or post-operative recurrence early, even if asymptomatic.
Visual surveillance in patients with either operated or incidental macroadenomas should be adjusted to their individual needs. Retrospective pituitary imaging studies in CYP highlight the need to recognize that physiological pubertal pituitary hypertrophy can occur and draw attention to the lack of progression of microadenomas227,228,229. As more comprehensive data on incidentalomas or non-gonadotroph silent tumours in CYP are lacking, we suggest following adult guidelines for follow-up as above251,252. While the radiological surveillance of stable non-functioning microadenomas can cease after 1–3 years, macroadenomas need to be followed in the long term251,252. We suggest a decreasing scanning interval for adenomas proven stable over the years. Concerns regarding repeated gadolinium administration over prolonged imaging follow-up are discussed in the Radiology section of Part 1 (ref. 2).
Gonadotrophinomas in CYP
Functioning gonadotroph adenomas are exceedingly rare in CYP. They can result in isosexual precocious puberty or ovarian hyperstimulation syndrome254,255. Germ-cell tumours secreting βHCG can represent a differential diagnostic dilemma as symptoms can overlap with those of functioning gonadotrophinomas. Silent gonadotrophinomas are discussed above.
Conclusions
Children and young people are at a disadvantage compared with adults in accessing age-appropriate, highly specialized pituitary care for pituitary adenomas. The incidence, type, symptomatology, aggressiveness and aetiology of their disease not only differ from that of adults but their impact, treatment choice and timely remediation can carry far greater consequences on the developing body and brain as well as on secondary health, intellectual, visual and psychosocial adult outcomes. These can limit the quality of adult relationships and represent challenges for parenting and employment over a long lifespan. The purpose of these recommendations — the first comprehensive consensus guideline of its kind — is to equalize and optimize health care for CYP with pituitary adenomas, raise awareness of diagnostic and treatment delays with potentially life-limiting complications, and improve access to modern pituitary-specific specialist centres, novel therapies, research and monitoring of long-term health and well-being outcomes. The latter must encompass the transition to adult services; closer integration of paediatric and adult pituitary services and data sets can only benefit CYP.
Key developments have occurred over the past few years regarding the diagnosis and treatment of pituitary adenomas — genetic testing, intraoperative MRI and the introduction of pegvisomant are just a few examples. Due to the rarity of pituitary adenomas in CYP, data have necessarily been gathered from retrospective studies, case series, anecdotal case reports and experience from the adult population for most of the management recommendations we present here and in Part 1 (ref. 2) (Supplementary Table 1). Facilitating referral and service pathways so that CYP can access centralized speciality pituitary multidisciplinary advisory panels across the age range and establishing national and international data registries of morbidity outcomes are in the best interest of CYP and vital to informing future best practice recommendations and to improving the long-term quality of life of these children and young people.
Change history
19 February 2024
In the version of the article initially published, an earlier, incorrect version of the Supplementary Information was posted. This has now been amended, and the correct Supplementary Information file now accompanies the online article.
References
Brouwers, M. C. et al. AGREE II: advancing guideline development, reporting and evaluation in health care. Can. Med. Assoc. J. 182, E839–842 (2010).
Korbonits, M. et al. Consensus guideline for the diagnosis and management of pituitary adenomas in childhood and adolescence: part 1, general recommendations. Nat. Rev. Endocrinol. https://doi.org/10.1038/s41574-023-00948-8 (2024).
Hoffmann, A., Adelmann, S., Lohle, K., Claviez, A. & Muller, H. L. Pediatric prolactinoma: initial presentation, treatment, and long-term prognosis. Eur. J. Pediatr. 177, 125–132 (2018).
Colao, A. et al. Prolactinomas in children and adolescents. Clinical presentation and long-term follow-up. J. Clin. Endocrinol. Metab. 83, 2777–2780 (1998).
Fideleff, H. L., Boquete, H. R., Suarez, M. G. & Azaretzky, M. Prolactinoma in children and adolescents. Horm. Res. 72, 197–205 (2009).
Arya, V. B. et al. Prolactinoma in childhood and adolescence-Tumour size at presentation predicts management strategy: single centre series and a systematic review and meta-analysis. Clin. Endocrinol. 94, 413–423 (2021).
Kunwar, S. & Wilson, C. B. Pediatric pituitary adenomas. J. Clin. Endocrinol. Metab. 84, 4385–4389 (1999).
Acharya, S. V. et al. Clinical profile and long term follow up of children and adolescents with prolactinomas. Pituitary 12, 186–189 (2009).
Cannavo, S. et al. Clinical presentation and outcome of pituitary adenomas in teenagers. Clin. Endocrinol. 58, 519–527 (2003).
Salenave, S. et al. Macroprolactinomas in children and adolescents: factors associated with the response to treatment in 77 patients. J. Clin. Endocrinol. Metab. 100, 1177–1186 (2015).
Breil, T. et al. Clinical features and response to treatment of prolactinomas in children and adolescents: a retrospective single-centre analysis and review of the literature. Horm. Res. Paediatr. 89, 157–165 (2018).
Alikasifoglu, A., Celik, N. B., Ozon, Z. A., Gonc, E. N. & Kandemir, N. Management of prolactinomas in children and adolescents; which factors define the response to treatment? Pituitary 25, 167–179 (2022).
Denes, J. & Korbonits, M. The clinical aspects of pituitary tumour genetics. Endocrine 71, 663–674 (2021).
Sonigo, C. et al. Hyperprolactinemia-induced ovarian acyclicity is reversed by kisspeptin administration. J. Clin. Invest. 122, 3791–3795 (2012).
Eren, E. et al. Clinical and Laboratory characteristics of hyperprolactinemia in children and adolescents: National survey. J. Clin. Res. Pediatr. Endocrinol. 11, 149–156 (2019).
Casanueva, F. F. et al. Guidelines of the Pituitary Society for the diagnosis and management of prolactinomas. Clin. Endocrinol. 65, 265–273 (2006).
Vilar, L. et al. Diagnosis and management of hyperprolactinemia: results of a Brazilian multicenter study with 1234 patients. J. Endocrinol. Invest. 31, 436–444 (2008).
Petersenn, S. et al. Diagnosis and management of prolactin-secreting pituitary adenomas: a Pituitary Society International Consensus Statement. Nat. Rev. Endocrinol. 19, 722–740 (2023).
Karavitaki, N. et al. Do the limits of serum prolactin in disconnection hyperprolactinaemia need re-definition? A study of 226 patients with histologically verified non-functioning pituitary macroadenoma. Clin. Endocrinol. 65, 524–529 (2006).
Sharma, L. K., Sharma, N., Gadpayle, A. K. & Dutta, D. Prevalence and predictors of hyperprolactinemia in subclinical hypothyroidism. Eur. J. Intern. Med. 35, 106–110 (2016).
Honbo, K. S., van Herle, A. J. & Kellett, K. A. Serum prolactin levels in untreated primary hypothyroidism. Am. J. Med. 64, 782–787 (1978).
Sievertsen, G. D., Lim, V. S., Nakawatase, C. & Frohman, L. A. Metabolic clearance and secretion rates of human prolactin in normal subjects and in patients with chronic renal failure. J. Clin. Endocrinol. Metab. 50, 846–852 (1980).
Opladen, T. et al. Consensus guideline for the diagnosis and treatment of tetrahydrobiopterin (BH(4)) deficiencies. Orphanet J. Rare Dis. 15, 126 (2020).
Vitturi, N. et al. High prolactin levels in dihydropteridine reductase deficiency: A sign of therapy failure or additional pathology? JIMD Rep. 61, 48–51 (2021).
Soto-Pedre, E., Newey, P. J., Bevan, J. S., Greig, N. & Leese, G. P. The epidemiology of hyperprolactinaemia over 20 years in the Tayside region of Scotland: the Prolactin Epidemiology, Audit and Research Study (PROLEARS). Clin. Endocrinol. 86, 60–67 (2017).
Donadio, F. et al. Patients with macroprolactinaemia: clinical and radiological features. Eur. J. Clin. Invest. 37, 552–557 (2007).
McKenna, T. J. Should macroprolactin be measured in all hyperprolactinaemic sera? Clin. Endocrinol. 71, 466–469 (2009).
Fideleff, H. L. et al. Macroprolactinemia in childhood and adolescence: a cause of asymptomatic hyperprolactinemia. Horm. Res. 53, 16–19 (2000).
Tutunculer, F., Darendeliler, F., Aygun, M. & Hekim, N. Macroprolactinemia in childhood and adolescence: a cause of hyperprolactinemia. Turk. J. Pediatr. 48, 143–147 (2006).
St-Jean, E., Blain, F. & Comtois, R. High prolactin levels may be missed by immunoradiometric assay in patients with macroprolactinomas. Clin. Endocrinol. 44, 305–309 (1996).
Boesten, L. S. M., Krabbe, J. G. & de Rijke, Y. B. The high dose hook effect in prolactinomas; following the guidelines? Ned. Tijdschr. Klin. Chem. Labgeneesk 40, 230–233 (2015).
Webster, J. et al. Dose-dependent suppression of serum prolactin by cabergoline in hyperprolactinaemia: a placebo controlled, double blind, multicentre study. European Multicentre Cabergoline Dose-finding Study Group. Clin. Endocrinol. 37, 534–541 (1992).
Colao, A. et al. Macroprolactinoma shrinkage during cabergoline treatment is greater in naive patients than in patients pretreated with other dopamine agonists: a prospective study in 110 patients. J. Clin. Endocrinol. Metab. 85, 2247–2252 (2000).
Webster, J. et al. A comparison of cabergoline and bromocriptine in the treatment of hyperprolactinemic amenorrhea. Cabergoline Comparative Study Group. N. Engl. J. Med. 331, 904–909 (1994).
Catli, G. et al. Hyperprolactinemia in children: clinical features and long-term results. J. Pediatr. Endocrinol. Metab. 25, 1123–1128 (2012).
Cuevas, J. L. et al. Visual outcome in patients with macroprolactinoma treated with dopamine agonists [Spanish]. Rev. Med. Chil. 141, 687–694 (2013).
Bulwer, C. et al. Cabergoline-related impulse control disorder in an adolescent with a giant prolactinoma. Clin. Endocrinol. 86, 862–864 (2017).
Lam, G., Mehta, V. & Zada, G. Spontaneous and medically induced cerebrospinal fluid leakage in the setting of pituitary adenomas: review of the literature. Neurosurg. Focus. 32, E2 (2012).
Vilar, L. et al. Controversial issues in the management of hyperprolactinemia and prolactinomas — an overview by the Neuroendocrinology Department of the Brazilian Society of Endocrinology and Metabolism. Arch. Endocrinol. Metab. 62, 236–263 (2018).
Molitch, M. E. et al. Prolactinoma management. Endotext. https://www.ncbi.nlm.nih.gov/pubmed/25905397 (2022).
Maiter, D. Management of dopamine agonist-resistant prolactinoma. Neuroendocrinology 109, 42–50 (2019).
Ono, M. et al. Prospective study of high-dose cabergoline treatment of prolactinomas in 150 patients. J. Clin. Endocrinol. Metab. 93, 4721–4727 (2008).
de Castro, L. F. et al. Beneficial effects of high doses of cabergoline in the treatment of giant prolactinoma resistant to dopamine agonists: a case report with a 21-year follow-up. Horm. Res. Paediatr. 89, 63–70 (2018).
Brichta, C. M., Wurm, M., Krebs, A., Schwab, K. O. & van der Werf-Grohmann, N. Start low, go slowly — mental abnormalities in young prolactinoma patients under cabergoline therapy. J. Pediatr. Endocrinol. Metab. 32, 969–977 (2019).
Spinks, J. J. & Ryan, F. J. Cabergoline resistance in pediatric prolactinomas. J. Pediatr. Hematol. Oncol. 31, 377–379 (2009).
Huang, C., Ezzat, S., Asa, S. L. & Hamilton, J. Dopaminergic resistant prolactinomas in the peripubertal population. J. Pediatr. Endocrinol. Metab. 19, 951–953 (2006).
Kreutzer, J. et al. Operative treatment of prolactinomas: indications and results in a current consecutive series of 212 patients. Eur. J. Endocrinol. 158, 11–18 (2008).
Babey, M., Sahli, R., Vajtai, I., Andres, R. H. & Seiler, R. W. Pituitary surgery for small prolactinomas as an alternative to treatment with dopamine agonists. Pituitary 14, 222–230 (2011).
Abe, T. & Ludecke, D. K. Transnasal surgery for prolactin-secreting pituitary adenomas in childhood and adolescence. Surg. Neurol. 57, 369–378 (2002).
Perry, A. et al. Pediatric pituitary adenoma: case series, review of the literature, and a skull base treatment paradigm. J. Neurol. Surg. B Skull Base 79, 91–114 (2018).
Cozzi, R. et al. Italian Association of Clinical Endocrinologists (AME) and International Chapter of Clinical Endocrinology (ICCE). Position statement for clinical practice: prolactin-secreting tumors. Eur. J. Endocrinol. 186, P1–P33 (2022).
Yang, A. et al. Clinical, hormonal, and neuroradiological characteristics and therapeutic outcomes of prolactinomas in children and adolescents at a single center. Front. Endocrinol. 11, 527 (2020).
Zamanipoor Najafabadi, A. H. et al. Surgery as a viable alternative first-line treatment for prolactinoma patients. A systematic review and meta-analysis. J. Clin. Endocrinol. Metab. 105, e32–e41 (2020).
Stiles, C. E., Tetteh-Wayoe, E. T., Bestwick, J., Steeds, R. P. & Drake, W. M. A meta-analysis of the prevalence of cardiac valvulopathy in hyperprolactinemic patients treated with cabergoline. J. Clin. Endocrinol. Metab. 104, 523–538 (2019).
Stiles, C. E. et al. Incidence of cabergoline-associated valvulopathy in primary care patients with prolactinoma using hard cardiac endpoints. J. Clin. Endocrinol. Metab. 106, e711–e720 (2021).
Stiles, C. E., Steeds, R. P. & Drake, W. M. Monitoring patients receiving dopamine agonist therapy for hyperprolactinaemia. Ann. Endocrinol. 82, 182–186 (2020).
Steeds, R. et al. Echocardiography and monitoring patients receiving dopamine agonist therapy for hyperprolactinaemia: a joint position statement of the British Society of Echocardiography, the British Heart Valve Society and the Society for Endocrinology. Clin. Endocrinol. 90, 662–669 (2019).
McCormack, A. et al. Treatment of aggressive pituitary tumours and carcinomas: results of a European Society of Endocrinology (ESE) survey 2016. Eur. J. Endocrinol. 178, 265–276 (2018).
Holthouse, D. J., Robbins, P. D., Kahler, R., Knuckey, N. & Pullan, P. Corticotroph pituitary carcinoma: case report and literature review. Endocr. Pathol. 12, 329–341 (2001).
Guzel, A. et al. Pituitary carcinoma presenting with multiple metastases: case report. J. Child. Neurol. 23, 1467–1471 (2008).
Graf, C. J., Blinderman, E. E. & Terplan, K. L. Pituitary carcinoma in a child with distant metastases. J. Neurosurg. 19, 254–259 (1962).
Kovacs, G. L. et al. ACTH-secreting Crooke cell carcinoma of the pituitary. Eur. J. Clin. Invest. 43, 20–26 (2013).
Raverot, G. et al. European Society of Endocrinology Clinical Practice Guidelines for the management of aggressive pituitary tumours and carcinomas. Eur. J. Endocrinol. 178, G1–G24 (2018).
Barber, T. M. et al. Recurrence of hyperprolactinaemia following discontinuation of dopamine agonist therapy in patients with prolactinoma occurs commonly especially in macroprolactinoma. Clin. Endocrinol. 75, 819–824 (2011).
Dekkers, O. M. et al. Recurrence of hyperprolactinemia after withdrawal of dopamine agonists: systematic review and meta-analysis. J. Clin. Endocrinol. Metab. 95, 43–51 (2010).
Hu, J., Zheng, X., Zhang, W. & Yang, H. Current drug withdrawal strategy in prolactinoma patients treated with cabergoline: a systematic review and meta-analysis. Pituitary 18, 745–751 (2015).
Colao, A. et al. Prolactinomas in adolescents: persistent bone loss after 2 years of prolactin normalization. Clin. Endocrinol. 52, 319–327 (2000).
Newell-Price, J., Bertagna, X., Grossman, A. B. & Nieman, L. K. Cushing’s syndrome. Lancet 367, 1605–1617 (2006).
Magiakou, M. A. & Chrousos, G. P. Cushing’s syndrome in children and adolescents: current diagnostic and therapeutic strategies. J. Endocrinol. Invest. 25, 181–194 (2002).
Weber, A. et al. Investigation, management and therapeutic outcome in 12 cases of childhood and adolescent Cushing’s syndrome. Clin. Endocrinol. 43, 19–28 (1995).
Verges, B. et al. Pituitary disease in MEN type 1 (MEN1): data from the France-Belgium MEN1 multicenter study. J. Clin. Endocrinol. Metab. 87, 457–465 (2002).
Storr, H. L. et al. Comparisons in the epidemiology, diagnostic features and cure rate by transsphenoidal surgery between paediatric and adult-onset Cushing’s disease. Eur. J. Endocrinol. 164, 667–674 (2011).
Libuit, L. G. et al. A gender-dependent analysis of Cushing’s disease in childhood: pre- and postoperative follow-up. Clin. Endocrinol. 83, 72–77 (2015).
Lonser, R. R. et al. Outcome of surgical treatment of 200 children with Cushing’s disease. J. Clin. Endocrinol. Metab. 98, 892–901 (2013).
Storr, H. L. et al. Factors influencing cure by transsphenoidal selective adenomectomy in paediatric Cushing’s disease. Eur. J. Endocrinol. 152, 825–833 (2005).
Storr, H. L. & Savage, M. O. Management Of endocrine disease: paediatric Cushing’s disease. Eur. J. Endocrinol. 173, R35–45 (2015).
Shah, N. S. et al. Cushing disease in children and adolescents: twenty years’ experience in a tertiary care center in India. Endocr. Pract. 17, 369–376 (2011).
Shah, N. S. & Lila, A. Childhood Cushing disease: a challenge in diagnosis and management. Horm. Res. Paediatr. 76, 65–70 (2011).
Shah, S., Waldman, A. D. & Mehta, A. Advances in pituitary imaging technology and future prospects. Best Pract. Res. Clin. Endocrinol. Metab. 26, 35–46 (2012).
Wedrychowicz, A. et al. Cushing disease in children and adolescents - assessment of the clinical course, diagnostic process, and effects of the treatment — experience from a single paediatric centre. Pediatr. Endocrinol. Diabetes Metab. 25, 127–143 (2019).
Nieman, L. K. et al. The diagnosis of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 93, 1526–1540 (2008).
Fleseriu, M. et al. Consensus on diagnosis and management of Cushing’s disease: a guideline update. Lancet Diabetes Endocrinol. 9, 847–875 (2021).
Greening, J. E. et al. Linear growth and body mass index in pediatric patients with Cushing’s disease or simple obesity. J. Endocrinol. Invest. 29, 885–887 (2006).
Arnaldi, G. et al. Diagnosis and complications of Cushing’s syndrome: a consensus statement. J. Clin. Endocrinol. Metab. 88, 5593–5602 (2003).
Batista, D. L., Riar, J., Keil, M. & Stratakis, C. A. Diagnostic tests for children who are referred for the investigation of Cushing syndrome. Pediatrics 120, e575-86 (2007).
Ferrigno, R. et al. Paediatric Cushing’s disease: epidemiology, pathogenesis, clinical management and outcome. Rev. Endocr. Metab. Disord. 22, 817–835 (2021).
Stratakis, C. A. Cushing syndrome in pediatrics. Endocrinol. Metab. Clin. North Am. 41, 793–803 (2012).
Dias, R. et al. The discriminatory value of the low-dose dexamethasone suppression test in the investigation of paediatric Cushing’s syndrome. Horm. Res. 65, 159–162 (2006).
Isidori, A. M. et al. Discriminatory value of the low-dose dexamethasone suppression test in establishing the diagnosis and differential diagnosis of Cushing’s syndrome. J. Clin. Endocrinol. Metab. 88, 5299–5306 (2003).
Guemes, M. et al. Management of Cushing syndrome in children and adolescents: experience of a single tertiary centre. Eur. J. Pediatr. 175, 967–976 (2016).
Viru-Loza, M. A. & Quispe, A. V. Diagnostic power of bilateral inferior petrosal sinus sampling with desmopressin in paediatric Cushing’s disease. J. Clin. Res. Pediatr. Endocrinol. 14, 334–338 (2022).
Zheng, X. et al. Diagnosis, manifestations, laboratory investigations, and prognosis in pediatric and adult Cushing’s disease in a large center in China. Front. Endocrinol. 12, 749246 (2021).
Turan, H. et al. Diagnostic value of bilateral petrosal sinus sampling in children with Cushing disease: a multi-center study. J. Clin. Res. Pediatr. Endocrinol. 14, 29–36 (2022).
Oldfield, E. H. et al. Petrosal sinus sampling with and without corticotropin-releasing hormone for the differential diagnosis of Cushing’s syndrome. N. Engl. J. Med. 325, 897–905 (1991).
Moszczynska, E. et al. The effects of sampling lateralization on bilateral inferior petrosal sinus sampling for pediatric Cushing’s disease — a single endocrinology centre experience and review of the literature. Front. Endocrinol. 12, 650967 (2021).
Lienhardt, A. et al. Relative contributions of inferior petrosal sinus sampling and pituitary imaging in the investigation of children and adolescents with ACTH-dependent Cushing’s syndrome. J. Clin. Endocrinol. Metab. 86, 5711–5714 (2001).
Kaltsas, G. A. et al. A critical analysis of the value of simultaneous inferior petrosal sinus sampling in Cushing’s disease and the occult ectopic adrenocorticotropin syndrome. J. Clin. Endocrinol. Metab. 84, 487–492 (1999).
Batista, D. et al. An assessment of petrosal sinus sampling for localization of pituitary microadenomas in children with Cushing disease. J. Clin. Endocrinol. Metab. 91, 221–224 (2006).
Andereggen, L. et al. Influence of inferior petrosal sinus drainage symmetry on detection of adenomas in Cushing’s syndrome. J. Neuroradiol. 48, 10–15 (2021).
Jarial, K. D. S. et al. Prolactin-adjusted ACTH ratio in predicting lateralization of ACTH source during simultaneous bilateral inferior petrosal sinus sampling in patients with Cushing’s disease. Indian J. Endocrinol. Metab. 23, 56–59 (2019).
De Sousa, S. M. C., McCormack, A. I., McGrath, S. & Torpy, D. J. Prolactin correction for adequacy of petrosal sinus cannulation may diminish diagnostic accuracy in Cushing’s disease. Clin. Endocrinol. 87, 515–522 (2017).
Biller, B. M. et al. Treatment of adrenocorticotropin-dependent Cushing’s syndrome: a consensus statement. J. Clin. Endocrinol. Metab. 93, 2454–2462 (2008).
Knappe, U. J. & Ludecke, D. K. Transnasal microsurgery in children and adolescents with Cushing’s disease. Neurosurgery 39, 484–492 (1996).
Linglart, A. & Visot, A. Cushing’s disease in children and adolescents [French]. Neurochirurgie 48, 271–280 (2002).
Personnier, C. et al. Clinical features and treatment of pediatric somatotropinoma: case study of an aggressive tumor due to a new AIP mutation and extensive literature review. Horm. Res. Paediatr. 75, 392–402 (2011).
Yordanova, G. et al. Long-term outcomes of children treated for Cushing’s disease: a single center experience. Pituitary 19, 612–624 (2016).
Alexandraki, K. I. et al. Long-term remission and recurrence rates in Cushing’s disease: predictive factors in a single-centre study. Eur. J. Endocrinol. 168, 639–648 (2013).
Storr, H. L. et al. Clinical and endocrine responses to pituitary radiotherapy in pediatric Cushing’s disease: an effective second-line treatment. J. Clin. Endocrinol. Metab. 88, 34–37 (2003).
Acharya, S. V. et al. Radiotherapy in paediatric Cushing’s disease: efficacy and long term follow up of pituitary function. Pituitary 13, 293–297 (2010).
Jennings, A. S., Liddle, G. W. & Orth, D. N. Results of treating childhood Cushing’s disease with pituitary irradiation. N. Engl. J. Med. 297, 957–962 (1977).
Gan, H. W. et al. National UK guidelines for the management of paediatric craniopharyngioma. Lancet Diabetes Endocrinol. 11, 694–706 (2023).
Sheehan, J. P., Xu, Z. & Lobo, M. J. External beam radiation therapy and stereotactic radiosurgery for pituitary adenomas. Neurosurg. Clin. N. Am. 23, 571–586 (2012).
Loeffler, J. S. & Shih, H. A. Radiation therapy in the management of pituitary adenomas. J. Clin. Endocrinol. Metab. 96, 1992–2003 (2011).
Almeldin, D., Fersht, N. & Kosmin, M. Radiotherapy for pituitary tumors. Endotext. https://www.ncbi.nlm.nih.gov/pubmed/25905190 (2023).
Shrivastava, A. et al. Outcomes after gamma knife stereotactic radiosurgery in pediatric patients with Cushing disease or acromegaly: a multi-institutional study. World Neurosurg. 125, e1104–e1113 (2019).
Groselj, U., Sikonja, J. & Battelino, T. Osilodrostat for Cushing disease and its role in pediatrics. Horm. Res. Paediatr. 96, 573–580 (2023).
Guven, A., Baltacioglu, F., Dursun, F., Cebeci, A. N. & Kirmizibekmez, H. Remission with cabergoline in adolescent boys with Cushing’s disease. J. Clin. Res. Pediatr. Endocrinol. 5, 194–198 (2013).
Chan, L. F. et al. Use of intravenous etomidate to control acute psychosis induced by the hypercortisolaemia in severe paediatric Cushing’s disease. Horm. Res. Paediatr. 75, 441–446 (2011).
McArthur, R. G., Hayles, A. B. & Salassa, R. M. Childhood Cushing disease: results of bilateral adrenalectomy. J. Pediatr. 95, 214–219 (1979).
Barber, T. M., Adams, E. & Wass, J. A. Nelson syndrome: definition and management. Handb. Clin. Neurol. 124, 327–337 (2014).
Reincke, M. et al. Corticotroph tumor progression after bilateral adrenalectomy (Nelson’s syndrome): systematic review and expert consensus recommendations. Eur. J. Endocrinol. 184, P1–P16 (2021).
Dupuis, C. C. et al. Abnormal puberty in paediatric Cushing’s disease: relationship with adrenal androgen, sex hormone binding globulin and gonadotrophin concentrations. Clin. Endocrinol. 66, 838–843 (2007).
Gourgari, E. et al. Post-operative growth is different in various forms of pediatric Cushing’s syndrome. Endocr. Relat. Cancer 21, L27–31 (2014).
Magiakou, M. A., Mastorakos, G. & Chrousos, G. P. Final stature in patients with endogenous Cushing’s syndrome. J. Clin. Endocrinol. Metab. 79, 1082–1085 (1994).
Magiakou, M. A., Mastorakos, G., Gomez, M. T., Rose, S. R. & Chrousos, G. P. Suppressed spontaneous and stimulated growth hormone secretion in patients with Cushing’s disease before and after surgical cure. J. Clin. Endocrinol. Metab. 78, 131–137 (1994).
Acharya, S. V. et al. Bone age and factors affecting skeletal maturation at diagnosis of paediatric Cushing’s disease. Pituitary 13, 355–360 (2010).
Peters, C. J. et al. Factors influencing skeletal maturation at diagnosis of paediatric Cushing’s disease. Horm. Res. 68, 231–235 (2007).
Davies, J. H. et al. Final adult height and body mass index after cure of paediatric Cushing’s disease. Clin. Endocrinol. 62, 466–472 (2005).
Boronat, M. et al. Combined treatment with GH and anastrozole in a pubertal boy with Cushing’s disease and postsurgical GH deficiency. Eur. J. Endocrinol. 166, 1101–1105 (2012).
Leong, G. M. et al. Effects of child- and adolescent-onset endogenous Cushing syndrome on bone mass, body composition, and growth: a 7-year prospective study into young adulthood. J. Bone Miner. Res. 22, 110–118 (2007).
Estrada, J. et al. The long-term outcome of pituitary irradiation after unsuccessful transsphenoidal surgery in Cushing’s disease. N. Engl. J. Med. 336, 172–177 (1997).
Chan, L. F. et al. Long-term anterior pituitary function in patients with paediatric Cushing’s disease treated with pituitary radiotherapy. Eur. J. Endocrinol. 156, 477–482 (2007).
Dorn, L. D. et al. The longitudinal course of psychopathology in Cushing’s syndrome after correction of hypercortisolism. J. Clin. Endocrinol. Metab. 82, 912–919 (1997).
Merke, D. P. et al. Children experience cognitive decline despite reversal of brain atrophy one year after resolution of Cushing syndrome. J. Clin. Endocrinol. Metab. 90, 2531–2536 (2005).
Ragnarsson, O., Berglund, P., Eder, D. N. & Johannsson, G. Long-term cognitive impairments and attentional deficits in patients with Cushing’s disease and cortisol-producing adrenal adenoma in remission. J. Clin. Endocrinol. Metab. 97, E1640–1648 (2012).
Hou, B. et al. Reversibility of impaired brain structures after transsphenoidal surgery in Cushing’s disease: a longitudinal study based on an artificial intelligence-assisted tool. J. Neurosurg. 134, 512–521 (2020).
Keil, M. F. et al. Quality of life in children and adolescents 1-year after cure of Cushing syndrome: a prospective study. Clin. Endocrinol. 71, 326–333 (2009).
Batista, D. L., Oldfield, E. H., Keil, M. F. & Stratakis, C. A. Postoperative testing to predict recurrent Cushing disease in children. J. Clin. Endocrinol. Metab. 94, 2757–2765 (2009).
Salenave, S., Boyce, A. M., Collins, M. T. & Chanson, P. Acromegaly and McCune-albright syndrome. J. Clin. Endocrinol. Metab. 99, 1955–1969 (2014).
Kirschner, L. S. et al. Mutations of the gene encoding the protein kinase A type I-α regulatory subunit in patients with the Carney complex. Nat. Genet. 26, 89–92 (2000).
Trivellin, G. et al. Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation. N. Engl. J. Med. 371, 2363–2374 (2014).
Srirangam Nadhamuni, V. et al. GHRH secretion from a pancreatic neuroendocrine tumor causing gigantism in a patient with MEN1. Endocrinol. Diabetes Metab. Case Rep. 67, 20-0208 (2021).
Borson-Chazot, F. et al. Acromegaly induced by ectopic secretion of GHRH: a review 30 years after GHRH discovery. Ann. Endocrinol. 73, 497–502 (2012).
Burton, T., Le Nestour, E., Neary, M. & Ludlam, W. H. Incidence and prevalence of acromegaly in a large US health plan database. Pituitary 19, 262–267 (2016).
Mindermann, T. & Wilson, C. B. Pediatric pituitary adenomas. Neurosurgery 36, 259–268 (1995).
Pandey, P., Ojha, B. K. & Mahapatra, A. K. Pediatric pituitary adenoma: a series of 42 patients. J. Clin. Neurosci. 12, 124–127 (2005).
Iacovazzo, D. et al. Germline or somatic GPR101 duplication leads to X-linked acrogigantism: a clinico-pathological and genetic study. Acta Neuropathol. Commun. 4, 56 (2016).
Naves, L. A. et al. Aggressive tumor growth and clinical evolution in a patient with X-linked acro-gigantism syndrome. Endocrine 51, 236–244 (2016).
Rostomyan, L. et al. Clinical and genetic characterization of pituitary gigantism: an international collaborative study in 208 patients. Endocr. Relat. Cancer 22, 745–757 (2015).
Katznelson, L. et al. Acromegaly: an Endocrine Society clinical practice guideline. J. Clin. Endocrinol. Metab. 99, 3933–3951 (2014).
Hernandez-Ramirez, L. C. et al. Landscape of familial isolated and young-onset pituitary adenomas: prospective diagnosis in AIP mutation carriers. J. Clin. Endocrinol. Metab. 100, E1242–1254 (2015).
Ali, O., Banerjee, S., Kelly, D. F. & Lee, P. D. Management of type 2 diabetes mellitus associated with pituitary gigantism. Pituitary 10, 359–364 (2007).
Colao, A. et al. Growth hormone excess with onset in adolescence: clinical appearance and long-term treatment outcome. Clin. Endocrinol. 66, 714–722 (2007).
Goldenberg, N. et al. Treatment of pituitary gigantism with the growth hormone receptor antagonist pegvisomant. J. Clin. Endocrinol. Metab. 93, 2953–2956 (2008).
Maheshwari, H. G. et al. Long-acting peptidomimergic control of gigantism caused by pituitary acidophilic stem cell adenoma. J. Clin. Endocrinol. Metab. 85, 3409–3416 (2000).
Nozieres, C. et al. Sporadic and genetic forms of paediatric somatotropinoma: a retrospective analysis of seven cases and a review of the literature. Orphanet J. Rare Dis. 6, 67 (2011).
Otsuka, F. et al. Long-term effects of octreotide on pituitary gigantism: its analgesic action on cluster headache. Endocr. J. 51, 449–452 (2004).
Rhee, N., Jeong, K., Yang, E. M. & Kim, C. J. Gigantism caused by growth hormone secreting pituitary adenoma. Ann. Pediatr. Endocrinol. Metab. 19, 96–99 (2014).
Abe, T., Tara, L. A. & Ludecke, D. K. Growth hormone-secreting pituitary adenomas in childhood and adolescence: features and results of transnasal surgery. Neurosurgery 45, 1–10 (1999).
Bergamaschi, S. et al. Eight-year follow-up of a child with a GH/prolactin-secreting adenoma: efficacy of pegvisomant therapy. Horm. Res. Paediatr. 73, 74–79 (2010).
Prezio, J. A., Griffin, J. E. & O’Brien, J. J. Acromegalic gigantism. The Buffalo giant. Am. J. Med. 31, 966–976 (1961).
Beckers, A. et al. X-linked acrogigantism syndrome: clinical profile and therapeutic responses. Endocr. Relat. Cancer 22, 353–367 (2015).
Daly, A. F. et al. GHRH excess and blockade in X-LAG syndrome. Endocr. Relat. Cancer 23, 161–170 (2016).
Clayton, K. L. et al. Loss of the normal relationships between growth hormone, growth hormone-binding protein and insulin-like growth factor-I in adolescents with insulin-dependent diabetes mellitus. Clin. Endocrinol. 41, 517–524 (1994).
Weber, M. M., Auernhammer, C. J., Lee, P. D., Engelhardt, D. & Zachoval, R. Insulin-like growth factors and insulin-like growth factor binding proteins in adult patients with severe liver disease before and after orthotopic liver transplantation. Horm. Res. 57, 105–112 (2002).
Haspolat, K. et al. Relationships between leptin, insulin, IGF-1 and IGFBP-3 in children with energy malnutrition. Clin. Biochem. 40, 201–205 (2007).
Svan, H., Ritzen, E. M., Hall, K. & Johansson, L. Estrogen treatment of tall girls: dose dependency of effects on subsequent growth and IGF-I levels in blood. Acta Paediatr. Scand. 80, 328–332 (1991).
Pokrajac, A. et al. Variation in GH and IGF-I assays limits the applicability of international consensus criteria to local practice. Clin. Endocrinol. 67, 65–70 (2007).
Fleseriu, M. et al. A Pituitary Society update to acromegaly management guidelines. Pituitary 24, 1–13 (2021).
Freda, P. U., Reyes, C. M., Nuruzzaman, A. T., Sundeen, R. E. & Bruce, J. N. Basal and glucose-suppressed GH levels less than 1 microg/L in newly diagnosed acromegaly. Pituitary 6, 175–180 (2003).
Schilbach, K. et al. Determinants of the growth hormone nadir during oral glucose tolerance test in adults. Eur. J. Endocrinol. 181, 55–67 (2019).
Misra, M., Cord, J., Prabhakaran, R., Miller, K. K. & Klibanski, A. Growth hormone suppression after an oral glucose load in children. J. Clin. Endocrinol. Metab. 92, 4623–4629 (2007).
Holl, R. W. et al. Suppression of growth hormone by oral glucose in the evaluation of tall stature. Horm. Res. 51, 20–24 (1999).
Dimaraki, E. V., Jaffe, C. A., DeMott-Friberg, R., Chandler, W. F. & Barkan, A. L. Acromegaly with apparently normal GH secretion: implications for diagnosis and follow-up. J. Clin. Endocrinol. Metab. 87, 3537–3542 (2002).
Barkan, A. L., Beitins, I. Z. & Kelch, R. P. Plasma insulin-like growth factor-I/somatomedin-C in acromegaly: correlation with the degree of growth hormone hypersecretion. J. Clin. Endocrinol. Metab. 67, 69–73 (1988).
Oldfield, E. H. et al. Correlation between GH and IGF-1 during treatment for acromegaly. J. Neurosurg. 126, 1959–1966 (2017).
Nomikos, P., Buchfelder, M. & Fahlbusch, R. The outcome of surgery in 668 patients with acromegaly using current criteria of biochemical ‘cure’. Eur. J. Endocrinol. 152, 379–387 (2005).
Stratakis, C. A., Kirschner, L. S. & Carney, J. A. Clinical and molecular features of the Carney complex: diagnostic criteria and recommendations for patient evaluation. J. Clin. Endocrinol. Metab. 86, 4041–4046 (2001).
Daly, A. F. et al. Pheochromocytomas and pituitary adenomas in three patients with MAX exon deletions. Endocr. Relat. Cancer 25, L37–L42 (2018).
Cambiaso, P. et al. Growth hormone excess in children with neurofibromatosis type-1 and optic glioma. Am. J. Med. Genet. A 173, 2353–2358 (2017).
Drimmie, F. M. et al. Gigantism due to growth hormone excess in a boy with optic glioma. Clin. Endocrinol. 53, 535–538 (2000).
Main, K. M., Sehested, A. & Feldt-Rasmussen, U. Pegvisomant treatment in a 4-year-old girl with neurofibromatosis type 1. Horm. Res. 65, 1–5 (2006).
Ahmed, S. et al. Outcome of transphenoidal surgery for acromegaly and its relationship to surgical experience. Clin. Endocrinol. 50, 561–567 (1999).
Dyer, E. H., Civit, T., Visot, A., Delalande, O. & Derome, P. Transsphenoidal surgery for pituitary adenomas in children. Neurosurgery 34, 207–212 (1994).
Bhansali, A. et al. Adolescent acromegaly: clinical parameters and treatment outcome. J. Pediatr. Endocrinol. Metab. 23, 1047–1054 (2010).
Jallad, R. S., Musolino, N. R., Kodaira, S., Cescato, V. A. & Bronstein, M. D. Does partial surgical tumour removal influence the response to octreotide-LAR in acromegalic patients previously resistant to the somatostatin analogue? Clin. Endocrinol. 67, 310–315 (2007).
Petrossians, P. et al. Gross total resection or debulking of pituitary adenomas improves hormonal control of acromegaly by somatostatin analogs. Eur. J. Endocrinol. 152, 61–66 (2005).
Karavitaki, N. et al. Surgical debulking of pituitary macroadenomas causing acromegaly improves control by lanreotide. Clin. Endocrinol. 68, 970–975 (2008).
Nagata, Y. et al. Growth hormone-producing pituitary adenomas in childhood and young adulthood: clinical features and outcomes. Pituitary 21, 1–9 (2018).
Yang, C., Li, G., Jiang, S., Bao, X. & Wang, R. Preoperative somatostatin analogues in patients with newly-diagnosed acromegaly: a systematic review and meta-analysis of comparative studies. Sci. Rep. 9, 14070 (2019).
Khan, Z. H. & Rasouli, M. R. Intubation in patients with acromegaly: experience in more than 800 patients. Eur. J. Anaesthesiol. 26, 354–355 (2009).
Seidman, P. A., Kofke, W. A., Policare, R. & Young, M. Anaesthetic complications of acromegaly. Br. J. Anaesth. 84, 179–182 (2000).
Cuny, T. et al. Acromegaly in Carney complex. Pituitary 22, 456–466 (2019).
Moran, A. et al. Gigantism due to pituitary mammosomatotroph hyperplasia. N. Engl. J. Med. 323, 322–327 (1990).
Daniel, A., d’Emden, M. & Duncan, E. Pituitary gigantism treated successfully with the growth hormone receptor antagonist, pegvisomant. Intern. Med. J. 43, 345–347 (2013).
Espinosa-de-los-Monteros, A. L., Gonzalez, B., Vargas, G., Sosa, E. & Mercado, M. Octreotide LAR treatment of acromegaly in “real life”: long-term outcome at a tertiary care center. Pituitary 18, 290–296 (2015).
Daly, A. F. et al. AIP-mutated acromegaly resistant to first-generation somatostatin analogs: long-term control with pasireotide LAR in two patients. Endocr. Connect. 8, 367–377 (2019).
Tahara, S., Murakami, M., Kaneko, T. & Shimatsu, A. Efficacy and safety of long-acting pasireotide in Japanese patients with acromegaly or pituitary gigantism: results from a multicenter, open-label, randomized, phase 2 study. Endocr. J. 64, 735–747 (2017).
Cozzi, R., Attanasio, R., Lodrini, S. & Lasio, G. Cabergoline addition to depot somatostatin analogues in resistant acromegalic patients: efficacy and lack of predictive value of prolactin status. Clin. Endocrinol. 61, 209–215 (2004).
Gatta, B., Hau, D. H., Catargi, B., Roger, P. & Tabarin, A. Re-evaluation of the efficacy of the association of cabergoline to somatostatin analogues in acromegalic patients. Clin. Endocrinol. 63, 477–478 (2005).
Sandret, L., Maison, P. & Chanson, P. Place of cabergoline in acromegaly: a meta-analysis. J. Clin. Endocrinol. Metab. 96, 1327–1335 (2011).
Abs, R. et al. Cabergoline in the treatment of acromegaly: a study in 64 patients. J. Clin. Endocrinol. Metab. 83, 374–378 (1998).
Tessaris, D. et al. Growth hormone-insulin-like growth factor 1 axis hyperactivity on bone fibrous dysplasia in McCune-Albright syndrome. Clin. Endocrinol. 89, 56–64 (2018).
Dutta, P. et al. Surgery, octreotide, temozolomide, bevacizumab, radiotherapy, and pegvisomant treatment of an AIP mutation positive child. J. Clin. Endocrinol. Metab. 104, 3539–3544 (2019).
Rodd, C. et al. Somatic GPR101 duplication causing X-Linked acrogigantism (XLAG)-diagnosis and management. J. Clin. Endocrinol. Metab. 101, 1927–1930 (2016).
Minniti, G. et al. The long-term efficacy of conventional radiotherapy in patients with GH-secreting pituitary adenomas. Clin. Endocrinol. 62, 210–216 (2005).
Schwartz, D. T. & Alpert, M. The malignant transformation of fibrous dysplasia. Am. J. Med. Sci. 247, 1–20 (1964).
Galland, F. et al. McCune-Albright syndrome and acromegaly: effects of hypothalamopituitary radiotherapy and/or pegvisomant in somatostatin analog-resistant patients. J. Clin. Endocrinol. Metab. 91, 4957–4961 (2006).
Hansen, M. R. & Moffat, J. C. Osteosarcoma of the skull base after radiation therapy in a patient with McCune-Albright syndrome: case report. Skull Base 13, 79–83 (2003).
Ruggieri, P., Sim, F. H., Bond, J. R. & Unni, K. K. Osteosarcoma in a patient with polyostotic fibrous dysplasia and Albright’s syndrome. Orthopedics 18, 71–75 (1995).
Hampton, R. E. Acromegaly and resulting myofascial pain and temporomandibular joint dysfunction: review of the literature and report of case. J. Am. Dent. Assoc. 114, 625–631 (1987).
Biermasz, N. R., van Dulken, H. & Roelfsema, F. Ten-year follow-up results of transsphenoidal microsurgery in acromegaly. J. Clin. Endocrinol. Metab. 85, 4596–4602 (2000).
Onnestam, L. et al. National incidence and prevalence of TSH-secreting pituitary adenomas in Sweden. J. Clin. Endocrinol. Metab. 98, 626–635 (2013).
Pereira, B. D. et al. Monomorphous plurihormonal pituitary adenoma of pit-1 lineage in a giant adolescent with central hyperthyroidism. Endocr. Pathol. 27, 25–33 (2016).
Rabbiosi, S. et al. Asymptomatic thyrotropin-secreting pituitary macroadenoma in a 13-year-old girl: successful first-line treatment with somatostatin analogs. Thyroid 22, 1076–1079 (2012).
Kessler, M., David, R., Pawelczak, M., Hanono, A. & Shah, B. Thyrotropin-secreting pituitary adenoma in an adolescent boy: challenges in management. Pediatrics 126, e474–478 (2010).
Nakayama, Y. et al. Thyroid-stimulating hormone (thyrotropin)-secretion pituitary adenoma in an 8-year-old boy: case report. Pituitary 15, 110–115 (2012).
Teng, X. et al. A patient with a thyrotropin-secreting microadenoma and resistance to thyroid hormone (P453T). J. Clin. Endocrinol. Metab. 100, 2511–2514 (2015).
Amlashi, F. G. & Tritos, N. A. Thyrotropin-secreting pituitary adenomas: epidemiology, diagnosis, and management. Endocrine 52, 427–440 (2016).
Barzaghi, L. R. et al. Pediatric pituitary adenomas: early and long-term surgical outcome in a series of 85 consecutive patients. Neurosurgery 85, 65–74 (2019).
Fukuhara, N. et al. Short-term preoperative octreotide treatment for TSH-secreting pituitary adenoma. Endocr. J. 62, 21–27 (2015).
Avramides, A. et al. TSH-secreting pituitary macroadenoma in an 11-year-old girl. Acta Paediatr. 81, 1058–1060 (1992).
Mazerkina, N. et al. Thyrotropin-secreting pituitary adenoma in an 11-year-old boy with type 1 autoimmune polyglandular syndrome. J. Pediatr. Endocrinol. Metab. 29, 237–240 (2016).
Yamaguchi-Okada, M., Inoshita, N., Nishioka, H., Fukuhara, N. & Yamada, S. Clinicopathological analysis of nonfunctioning pituitary adenomas in patients younger than 25 years of age. J. Neurosurg. Pediatr. 9, 511–516 (2012).
Guaraldi, F., Storr, H. L., Ghizzoni, L., Ghigo, E. & Savage, M. O. Paediatric pituitary adenomas: a decade of change. Horm. Res. Paediatr. 81, 145–155 (2014).
Saeger, W. et al. Pathohistological classification of pituitary tumors: 10 years of experience with the German Pituitary Tumor Registry. Eur. J. Endocrinol. 156, 203–216 (2007).
Thaker, V. V., Lage, A. E., Kumari, G., Silvera, V. M. & Cohen, L. E. Clinical course of nonfunctional pituitary microadenoma in children: a single-center experience. J. Clin. Endocrinol. Metab. 104, 5906–5912 (2019).
Souteiro, P. et al. Pituitary incidentalomas in paediatric age are different from those described in adulthood. Pituitary 22, 124–128 (2019).
Shareef, M. et al. Pituitary incidentalomas in paediatric population: incidence and characteristics. Clin. Endocrinol. 94, 269–276 (2021).
Daly, A. F. et al. Clinical characteristics and therapeutic responses in patients with germ-line AIP mutations and pituitary adenomas: an international collaborative study. J. Clin. Endocrinol. Metab. 95, E373–383 (2010).
Goudet, P. et al. MEN1 disease occurring before 21 years old: a 160-patient cohort study from the Groupe d’etude des Tumeurs Endocrines. J. Clin. Endocrinol. Metab. 100, 1568–1577 (2015).
Nishio, S. et al. Pituitary tumours in adolescence: clinical behaviour and neuroimaging features of seven cases. J. Clin. Neurosci. 8, 231–234 (2001).
Steele, C. A. et al. Pituitary adenomas in childhood, adolescence and young adulthood: presentation, management, endocrine and metabolic outcomes. Eur. J. Endocrinol. 163, 515–522 (2010).
De Menis, E. et al. Pituitary adenomas in childhood and adolescence. Clinical analysis of 10 cases. J. Endocrinol. Invest. 24, 92–97 (2001).
Zhang, N., Zhou, P., Meng, Y., Ye, F. & Jiang, S. A retrospective review of 34 cases of pediatric pituitary adenoma. Childs Nerv. Syst. 33, 1961–1967 (2017).
Wang, H. et al. Nonfunctioning pituitary adenomas in pediatric and adolescent patients: a clinical analysis of a series of 14 patients. J. Neurooncol. 148, 179–186 (2020).
Kruse, A., Astrup, J., Gyldensted, C. & Cold, G. E. Hyperprolactinaemia in patients with pituitary adenomas. The pituitary stalk compression syndrome. Br. J. Neurosurg. 9, 453–457 (1995).
Sautner, D., Saeger, W. & Ludecke, D. K. Tumors of the sellar region mimicking pituitary adenomas. Exp. Clin. Endocrinol. 101, 283–289 (1993).
Kidooka, M., Okada, T., Nakajima, M. & Handa, J. Intra- and suprasellar germinoma mimicking a pituitary adenoma-case report. Neurol. Med. Chir. 35, 96–99 (1995).
Cerbone, M. et al. Management of children and young people with idiopathic pituitary stalk thickening, central diabetes insipidus, or both: a national clinical practice consensus guideline. Lancet Child. Adolesc. Health 5, 662–676 (2021).
Karavitaki, N. et al. What is the natural history of nonoperated nonfunctioning pituitary adenomas? Clin. Endocrinol. 67, 938–943 (2007).
Warnet, A. et al. The effect of somatostatin analogue on chiasmal dysfunction from pituitary macroadenomas. J. Neurosurg. 71, 687–690 (1989).
Pivonello, R. et al. Dopamine receptor expression and function in clinically nonfunctioning pituitary tumors: comparison with the effectiveness of cabergoline treatment. J. Clin. Endocrinol. Metab. 89, 1674–1683 (2004).
Greenman, Y. et al. Treatment of clinically nonfunctioning pituitary adenomas with dopamine agonists. Eur. J. Endocrinol. 175, 63–72 (2016).
Batista, R. L. et al. Cabergoline in the management of residual nonfunctioning pituitary adenoma: a single-center, open-label, 2-year randomized clinical trial. Am. J. Clin. Oncol. 42, 221–227 (2019).
Wilson, P. J., De-Loyde, K. J., Williams, J. R. & Smee, R. I. A single centre’s experience of stereotactic radiosurgery and radiotherapy for non-functioning pituitary adenomas with the linear accelerator (Linac). J. Clin. Neurosci. 19, 370–374 (2012).
Keshavarzi, S. et al. Initial clinical experience with frameless optically guided stereotactic radiosurgery/radiotherapy in pediatric patients. Childs Nerv. Syst. 25, 837–844 (2009).
Mehrazin, M. Pituitary tumors in children: clinical analysis of 21 cases. Childs Nerv. Syst. 23, 391–398 (2007).
Becker, G. et al. Radiation therapy in the multimodal treatment approach of pituitary adenoma. Strahlenther. Onkol. 178, 173–186 (2002).
Tampourlou, M. et al. Outcome of nonfunctioning pituitary adenomas that regrow after primary treatment: a study from two large UK centers. J. Clin. Endocrinol. Metab. 102, 1889–1897 (2017).
Freda, P. U. et al. Pituitary incidentaloma: an Endocrine Society clinical practice guideline. J. Clin. Endocrinol. Metab. 96, 894–904 (2011).
Deutschbein, T. et al. First German guideline on diagnostics and therapy of clinically non-functioning pituitary tumors. Exp. Clin. Endocrinol. Diabetes 129, 250–264 (2021).
Fernandez-Balsells, M. M. et al. Natural history of nonfunctioning pituitary adenomas and incidentalomas: a systematic review and metaanalysis. J. Clin. Endocrinol. Metab. 96, 905–912 (2011).
Gryngarten, M. G. et al. Spontaneous ovarian hyperstimulation syndrome caused by a follicle-stimulating hormone-secreting pituitary macroadenoma in an early pubertal girl. Horm. Res. Paediatr. 73, 293–298 (2010).
Ntali, G., Capatina, C., Grossman, A. & Karavitaki, N. Clinical review: functioning gonadotroph adenomas. J. Clin. Endocrinol. Metab. 99, 4423–4433 (2014).
Whitelaw, B. C. et al. Temozolomide in the management of dopamine agonist-resistant prolactinomas. Clin. Endocrinol. 76, 877–886 (2012).
Felker, J., Patterson, B., Wrubel, D. & Janss, A. Successful treatment of a child with a prolactin secreting macroadenoma with temozolomide. J. Pediatr. Endocrinol. Metab. 29, 1413–1415 (2016).
Lasolle, H. et al. Temozolomide treatment can improve overall survival in aggressive pituitary tumors and pituitary carcinomas. Eur. J. Endocrinol. 176, 769–777 (2017).
Chentli, F., Yaker, F. A., Azzoug, S. & Belhimer, F. Temozolomide: anti-tumor effect on giant, invasive and resistant pediatric prolactinoma. Indian J. Endocrinol. Metab. 17, 1136–1138 (2013).
Bickler, S. W. et al. Preoperative diagnostic evaluation of children with Cushing’s syndrome. J. Pediatr. Surg. 29, 671–676 (1994).
Shapiro, L. et al. Investigation for paediatric Cushing’s syndrome using twenty-four-hour urinary free cortisol determination. Horm. Res. Paediatr. 86, 21–26 (2016).
Martinelli, C. E. Jr., Sader, S. L., Oliveira, E. B., Daneluzzi, J. C. & Moreira, A. C. Salivary cortisol for screening of Cushing’s syndrome in children. Clin. Endocrinol. 51, 67–71 (1999).
Thoren, M. et al. Treatment of Cushing’s disease in childhood and adolescence by stereotactic pituitary irradiation. Acta Paediatr. Scand. 75, 388–395 (1986).
Acknowledgements
The development of this consensus guideline was sponsored by unrestricted grants from professional societies (The Society of British Neurosurgical Surgeons, Children’s Cancer and Leukaemia Group (CCLG) and British Society for Paediatric Endocrinology and Diabetes), patient support groups (Association of Multiple Endocrine Neoplastic Disorders (AMEND), Success Charity and The Pituitary Foundation) and from Sandoz Pharmaceuticals. All, except Sandoz, were stakeholders. Sandoz supported administrative and travel expenses after the initial seed funding ended and had no role in instigating the endeavour, the development of guideline methodology or the final recommendations. The CCLG provided administrative support throughout the development of these consensus statements and the Royal College of Paediatrics and Child Health (RCPCH) provided advice and appraisal to ensure the process met rigorous AGREE II guideline standards. The CCLG, British Society for Paediatric Endocrinology and Diabetes, AMEND, Success Charity, Society of British Neurosurgical Surgeons or The Pituitary Foundation did not influence the decisions of the Guideline Development Group (GDG) nor the recommendations other than through their roles as stakeholders, as described in the main text. We would like to thank all of the stakeholders who contributed comments to this consensus guideline at various stages of development, including members of the Society for Endocrinology, Society of British Neurological Surgeons, British Paediatric Neurosurgical Group, Royal College of Physicians, British Society for Paediatric Endocrinology and Diabetes, British Society of Paediatric Radiology, RCPCH, British Neuropathology Society, and the Association for Clinical Biochemistry and Laboratory Medicine as well as patient organizations AMEND, Success Charity and the Pituitary Foundation. The GDG would like to thank the Project Board members and Delphi panellists (Supplementary Table 2) and our external peer reviewers for their input into this consensus guideline. The GDG would also like to thank Rosa Nieto Hernandez (Clinical Guidelines Manager) and Helen McElroy (Quality Improvement Committee Clinical Lead for Evidence Base Medicine and Appraisals), both at the Research and Quality Improvement Directorate of the RCPCH, for their advice and appraisal of this consensus guideline at different stages. We also wish to thank Stephen Shalet (University of Manchester UK), William Drake (Queen Mary University of London, London UK), Mark Gurnell (University of Cambridge, Cambridge, UK), Luis Syro (Queen Mary University of London, London, UK), Di Zou (London, UK), Lucas Castro (Queen Mary University of London, London, UK), Sarah Farndon (Great Ormond Street Hospital for Children, London, UK), Mary Dang (Queen Mary University of London, London UK), Amy Ronaldson (Queen Mary University of London, London, UK), Rezvan Salehidoost (Queen Mary University of London, London, UK), Gabriela Mihai (Queen Mary University of London, London, UK) and Benjamin Loughrey (Queen’s University Belfast, Belfast, UK) for their help in updating the literature search, grading the papers from the literature search, reviewing the manuscript and providing administrative support.
Author information
Authors and Affiliations
Contributions
All authors researched data for the article. M.K., J.C.B., J. A., J.H.D., J.E., D.F., C.E.H., T.S.J., S.S., I.S., M.R.D., N.G., N.T., F.M.S., H.L.S. and H.A.S. contributed substantially to discussion of the content. M.K., J.C.B., A.B., J.H.D., J.E., T.S.J., S.S., I.S., N.T., H.L.S. and H.A.S. wrote the article. All authors reviewed and/or edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
M.K. received grant funding or served on the Advisory Board for Pfizer, Crinetics, Novo Nordisk, Recordati, and ONO Pharmaceuticals and honoraria from Ipsen, Corcept, Sandoz and Novo Nordisk. J.C.B. has received honoraria, funding and travel bursaries from Novo Nordisk and honoraria from Sandoz and Ipsen. J.H.D. has received travel bursaries from Sandoz, Pfizer, and Novo Nordisk and grant funding from Pfizer. T.S.J. is director and shareholder in Repath Ltd. and Neuropath Ltd. and received an honorarium from Bayer. H.L.S. received grant funding from Ipsen and Sandoz and consultancy fees from Novo Nordisk, Pfizer and Springer. H.A.S. has received honoraria from Ferring and Pfizer and grants from Novo Nordisk and Sandoz. The other authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Endocrinology thanks Bing Xing and the other, anonymous, reviewers for their contribution to the peer review of this work.
Additional information
Disclaimer Health-care providers need to use clinical judgment, knowledge and expertise when deciding whether it is appropriate to apply these guidelines. The recommendations cited here are a guide and might not be appropriate for use in all situations. The decision to adopt any of the recommendations cited here is the responsibility of the treating clinician and must be made in the light of individual patient circumstances, the wishes of the patient and their family, clinical expertise, and resources.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Korbonits, M., Blair, J.C., Boguslawska, A. et al. Consensus guideline for the diagnosis and management of pituitary adenomas in childhood and adolescence: Part 2, specific diseases. Nat Rev Endocrinol 20, 290–309 (2024). https://doi.org/10.1038/s41574-023-00949-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41574-023-00949-7
This article is cited by
-
Consensus guideline for the diagnosis and management of pituitary adenomas in childhood and adolescence: Part 1, general recommendations
Nature Reviews Endocrinology (2024)
