
Abstract
Approximately 20% of patients diagnosed with a phaeochromocytoma or paraganglioma carry a germline mutation in one of the succinate dehydrogenase (SDHx) genes (SDHA, SDHB, SDHC and SDHD), which encode the four subunits of the SDH enzyme. When a pathogenic SDHx mutation is identified in an affected patient, genetic counselling is proposed for first-degree relatives. Optimal initial evaluation and follow-up of people who are asymptomatic but might carry SDHx mutations have not yet been agreed. Thus, we established an international consensus algorithm of clinical, biochemical and imaging screening at diagnosis and during surveillance for both adults and children. An international panel of 29 experts from 12 countries was assembled, and the Delphi method was used to reach a consensus on 41 statements. This Consensus Statement covers a range of topics, including age of first genetic testing, appropriate biochemical and imaging tests for initial tumour screening and follow-up, screening for rare SDHx-related tumours and management of elderly people who have an SDHx mutation. This Consensus Statement focuses on the management of asymptomatic SDHx mutation carriers and provides clinicians with much-needed guidance. The standardization of practice will enable prospective studies in the near future.
Similar content being viewed by others

Introduction
Mutations in the SDHx genes (SDHA, SDHB, SDHC and SDHD), which encode the four subunits of the mitochondrial enzyme succinate dehydrogenase (SDH), are associated with a predisposition for developing hereditary phaeochromocytoma and/or paraganglioma (PPGL)1. It is currently recommended that all patients with a newly discovered PPGL should be offered genetic counselling2. Germline mutations in SDHx genes are responsible for approximately 20% of cases of PPGL and can also be associated with the presence of other SDHx-related tumours1,2,3. PPGL detection at an early stage has a positive effect on outcomes, including survival4. First-degree family members can also benefit from genetic testing but it remains to be determined how to screen and then follow-up the newly detected asymptomatic mutation carriers as no consensus has as yet been established.
The management of asymptomatic carriers of SDHx mutations is a true clinical challenge for several reasons. For instance, patients with SDHB mutations are highly predisposed to metastatic PPGL and are at risk of developing multiple tumours, which can be widely distributed from the skull base to the pelvic floor. In addition, PPGLs associated with SDHx mutations can be non-functional and, therefore, their detection by biochemical testing is not viable. Furthermore, cumulative radiation exposure from imaging examinations should be limited for genetically predisposed asymptomatic young individuals as they will need lifelong monitoring. Additionally, psychological issues, such as anxiety or depression, can arise during follow-up, which affect a person’s well-being and overall quality of life. A relevant subset of people with SDHx mutations will probably never develop a tumour related to the mutation and SDHx mutations have been associated with other tumours such as renal cell carcinoma (RCC) and gastrointestinal stromal tumours (GIST)5,6,7,8. Children who are asymptomatic carriers of SDHx mutations should receive special attention as their parents are the decision-makers and undertake a considerable responsibility for the follow-up and outcome of their children9,10.
As SDHx-related PPGLs are rare tumours, current practice is not guided by robust evidence and consequently differs widely among clinical centres based on local experience and opinion. Thus, using the Delphi method, we developed an international consensus on the clinical, biochemical and imaging screening as well as follow-up of asymptomatic adults and children carrying an SDHx mutation.
Methods
This Consensus Statement was compiled following discussions using the Delphi process between December 2018 and November 2019 with four rounds of questionnaires to confront and converge the thoughts and opinions of the expert panel with the objective of coming to a group consensus11.
Experts were identified by their long-standing activity in the field of PPGL management through membership in the European Network for the Study of Adrenal Tumours (ENS@T) and/or the Pheochromocytoma and Paraganglioma Research Support Organization (PRESSOR), a consortium of health science professionals around the world dedicated to research in PPGL.
Of the 39 experts invited to participate, 29 responded to the first-round questionnaire and then completed the second, third and fourth (last) rounds. The Delphi panel included 16 endocrinologists and/or internists, 6 imaging specialists, 2 head and neck surgeons and 5 geneticists. The experts are from 20 centres, representing 12 countries across four continents (22 from Europe, 4 from the USA, 2 from Australia and 1 from Asia).
Online software was used to house the questionnaires and responses. The first questionnaire was designed by the two core group members (L.A. and C.L.L.) and approved by an executive committee (J.W.M.L., K.P. and A.P.G.R.). Experts were invited for the first-round questionnaire by e-mail and three reminders were sent within 3 months. Respondents were included in the consensus process and participated in the subsequent three rounds. The first-round questionnaire asked multiple-choice questions on the screening and follow-up of asymptomatic mutation carriers for each of the four genes (SDHA, SDHB, SDHD and SDHC), recognizing that mutations in each of these genes have differing penetrance, dominant sites of disease and malignant potential12. The questions covered the age at which the first screening should take place and the biochemical and imaging tests to be used for initial tumour screening as well as for follow-up. Two moderators (L.A. and C.L.L.) independently analysed the answers of experts and translated them into a series of statements with a methodologist (O.S.). These statements had to be rated and commented on by each expert independently using a 5-point Likert scale (1, strongly agree; 2, agree; 3, neutral; 4, disagree; 5, strongly disagree) for the second round of the Delphi process. The results of the second round as well as the ratings and comments were sent to the experts who had to once again rate those statements for which no consensus had been previously reached (see next paragraph for the definition of consensus). When needed, statements were reformulated following the comments of the experts. This process was repeated for the third and fourth rounds. The responses and comments remained anonymous, except to the moderators.
Consensus was defined as ≥23 of 29 experts (≥79%) for agreement (Likert scale 1 and 2) or disagreement (Likert scale 4 and 5) for the first and second rounds. For the third and fourth rounds, consensus was defined as ≥20 of 29 experts (≥69%) for agreement (Likert scale 1 and 2) or disagreement (Likert scale 4 and 5) and 80% of agreement (Likert scale 1 and 2) or disagreement (Likert scale 4 and 5) after the exclusion of neutral answers (Likert scale 3). When no consensus was reached after two consecutive rounds due to opposite ratings or a majority of neutral ratings, the statement was removed from the consensus, with the experts’ approval.
Consensus statements were graded as A for statements with ≥23 of 29 experts with agreement or disagreement or as B for statements with ≥20 of 29 experts with agreement or disagreement and ≥80% after exclusion of neutral answers.
The question rounds
The questionnaire in the first round included 47 open questions (Supplementary Table S1) on initial and follow-up tumour screening in asymptomatic mutation carriers for each of the four genes (SDHA, SDHB, SDHD and SDHC), including the minimum age for offering predictive genetic testing and thus initial screening as well as the biochemical and imaging tests to be used for initial and follow-up tumour screenings. The second round contained 69 statements (Supplementary Table S2). Several questions were reformulated and new questions were included in accordance with the experts’ comments during the first round. The third round comprised 45 statements (Supplementary Table S3) and the fourth round included 11 statements (Supplementary Table S4). Overall, 14 expert recommendations were made, as detailed here.
Initial screening
Genetic penetrance
The penetrance of SDHx-related PPGL is incomplete and varies in asymptomatic SDHx mutation carriers (between 8% and 37% for SDHB and 38% and 64% for SDHD) across several studies3,13,14,15,16,17,18,19,20,21,22,23. People with SDHD mutations have the highest penetrance, with multiple tumours most frequently located in the head and neck region (parasympathetic), whereas SDHB mutations predispose carriers primarily to retroperitoneal PPGL (sympathetic). SDHB mutation carriers are at a higher risk of developing metastases than carriers of mutations in any of the other SDHx genes3,13,24,25. However, the risk of developing a head and neck paraganglioma for a person with an SDHB mutation or a phaeochromocytoma in someone with an SDHD mutation is still statistically significant over a lifetime15. Data regarding SDHC mutation carriers are scarce but these carriers seem to present predominantly with non-metastatic head and neck paragangliomas with a lower rate of multiplicity and a lower penetrance than people who have an SDHD mutation3,20,26. Very little data have been published on the penetrance of SDHA mutations yet; one study reported a penetrance of 13%27. Moreover, even if some variants can be associated with specific clinical characteristics or penetrance3,28, the experts considered that the data are not yet strong enough to personalize screening and follow-up regarding the type of variant. The expert panel analysed each gene separately but this resulted in similar recommendations for initial screening and follow-up for all the SDHx genes, except regarding the age of first tumour screening during childhood.
Regarding the particular transmission model of inheritance for the SDHD gene, the recommendations were identical to other SDHx genes when an SDHD mutation is transmitted through the father (SDHD-pi). However, when the SDHD mutation is transmitted through the mother (SDHD-mi), the development of a PPGL seems to be a rare event (≤5%)29.
-
Recommendation 1: Tumour screening should be performed after the identification of an SDHA, SDHB, SDHC or SDHD-pi mutation in an asymptomatic carrier (Grade A).
Timing of initial screening in childhood
Even if the risk of developing a tumour during childhood (<18 years old) is low, a small number of SDHB-related paragangliomas have been reported at age 6 years13,20,30,31,32,33,34,35 and children with an SDHB mutation have a higher risk of developing a metastatic paraganglioma than children with mutations in the other SDHx genes32,36. As surgery remains the only curative treatment, the experts surmised that earlier tumour detection and removal is likely to be associated with improved long-term prognosis. For SDHD-pi and SDHC mutation carriers, some cases have been described during childhood but, in most cases, children were older than 10 years3,33,35. For SDHA mutation carriers, paragangliomas have been diagnosed in 17-year-old adolescents according to the literature37. The experts reported some cases in 13 year olds. Based on this evidence, the experts proposed first screening at an earlier age (6 years old) for asymptomatic SDHB mutation carriers than for carriers of mutations in the other SDHx genes (10 years old).
-
Recommendation 2: During childhood, genetic screening should only be performed if tumour screening would be considered if a mutation was discovered (Grade B).
-
Recommendation 3: During childhood, tumour screening should only be performed following the discovery of a mutation (Grade A).
-
Recommendation 4: First tumour screening should be performed between 6 and 10 years of age for asymptomatic SDHB mutation carriers and between 10 and 15 years of age in asymptomatic SDHA, SDHC and SDHD-pi mutation carriers (Grade A).
Screening methods
Initial screening should include an assessment of symptoms (Box 1), a clinical examination and blood pressure evaluation. As recommended by current blood pressure guidelines, blood pressure evaluation should rely on ambulatory blood pressure monitoring with a diagnostic blood pressure threshold for the diagnosis of hypertension of either 130/80 mmHg for patients younger than 65 years or 135/85 mmHg for patients older than 65 years, following international guidelines38,39.
Biochemical measurements of urinary or plasma levels of metanephrine and normetanephrine should be carried out to detect catecholamine production. Their superiority over catecholamines and vanillylmandelic acid measurements has long been established40,41 and they are currently recommended for the diagnosis of PPGL2. Plasma or urinary free metanephrines analyses seem to be the best diagnostic test, with an even higher sensitivity and specificity than analyses of deconjugated urinary metanephrines42. Although urinary measurement of metanephrines is less convenient, it might confer an advantage in childhood because this method avoids venepuncture. In 2019, age-specific paediatric reference intervals for plasma free metanephrine and normetanephrine were published43.
-
Recommendation 5: Tumour screening in asymptomatic SDHA, SDHB, SDHC and SDHD-pi mutation carriers should include clinical examination: blood pressure measurement (ideally, out-of-office blood pressure measurement during adulthood) and a symptoms and signs questionnaire (Box 1) (Grade A).
-
Recommendation 6: Biochemical testing for tumour screening in asymptomatic SDHA, SDHB, SDHC and SDHD-pi mutation carriers should include measurements of either plasma or urinary metanephrine and normetanephrine (Grade A).
-
a.
During childhood, the choice between plasma or urinary tests should be left to the clinician and local laboratory availability and expertise (Grade A).
-
b.
During adulthood, measurements of plasma free metanephrine and normetanephrine should be preferred over urinary measurements (Grade A).
-
a.
-
Recommendation 7: Biochemical testing should not include either vanillylmandelic acid or catecholamines in addition to metanephrine and normetanephrine (Grade A).
An important proportion of tumours, especially SDHx-related and/or head and neck paragangliomas arising from the parasympathetic system, do not secrete or even produce catecholamines44,45. Thus, the surveillance of patients with non-functional paragangliomas requires the use of imaging; there was a strong consensus on the use of MRI for the head and neck region. Indeed, specific imaging protocols have previously been published in the literature15,46. For imaging of thoracic, abdominal and pelvic regions, either CT scanning or MRI can be useful. However, to limit cumulative ionizing radiation exposure in asymptomatic children, experts agreed to recommend MRI. Ultrasound is an option for first-line imaging only in children who might not tolerate MRI47. For adults, the first-line for imaging of abdominal and pelvic regions is MRI. To increase the detection rate and explore the thoracic region, it was decided to perform functional imaging using PET–CT at the initial screening for adult mutation carriers. PET–CT imaging has some technical advantages over SPECT–CT (single-photon emission CT) as it has shorter uptake times, is a shorter imaging procedure, has less drug interference and a higher resolution48. Nevertheless, consensus was only obtained with grade B agreement (Tables 1,2). As PET–CT involves radiation exposure, it was considered that performing one functional imaging scan is of interest because it provides a 3D whole-body examination with a limited radiation dose exposure (7–8 mSv for 18FDG-PET–CT, which is of the same order of magnitude as a cervical, thoracic and abdominopelvic CT scan). Systematic use of PET–CT was preferred over conventional thoracic CT scanning because the latter has a poor sensitivity of only 46.2% (19.2–74.9%)15. PET–CT was not recommended for first screening during childhood.
-
Recommendation 8: Tumour screening in asymptomatic SDHA, SDHB, SDHC and SDHD-pi mutation carriers should include imaging (Grade A).
-
a.
During childhood, MRI of head and neck and thoracic, abdominal and pelvic regions should be used as first-line imaging for initial tumour screening (Grade A for head and neck and for abdominal and pelvic MRI, Grade B for thoracic MRI).
-
b.
During adulthood, a combination of MRI (head and neck, abdominal and pelvic; Grade A) and PET–CT (Grade B) should be used as first-line imaging for initial tumour screening.
-
c.
The expert panel recommends performing dedicated thoracic cross-sectional imaging only in instances of a PET–CT abnormality (Grade A).
-
d.
Ultrasound should not be used as first-line imaging for initial tumour screening in asymptomatic SDHA, SDHB, SDHC and SDHD-pi mutation carriers (Grade B during childhood, Grade A during adulthood). However, some experts underlined the convenience of ultrasound for some young children who will not tolerate MRI.
-
e.
During childhood, functional imaging should not be used for tumour screening as first-line imaging in asymptomatic SDHA, SDHB, SDHC and SDHD-pi mutation carriers (Grade A).
-
f.
During adulthood, functional imaging should rely on PET–CT. 123I-MIBG and 111In-pentetreotide scintigraphy should not be used as first-line imaging studies for initial tumour screening in asymptomatic SDHA, SDHB, SDHC or SDHD-pi mutation carriers (Grade A).
-
a.
Follow-up after a first negative initial screening
SDHx mutation carriers have a lifelong risk of developing PPGL49. Therefore, one initial screening is not sufficient and these individuals should be followed up. Although people who do not have PPGL at initial screening have a reduced risk of developing new tumours during follow-up, they are still at risk of developing these tumours during their lifetime49. This risk has led experts to consider that follow-up is mandatory in asymptomatic carriers of an SDHx mutation.
-
Recommendation 9: SDHA, SDHB, SDHC and SDHD-pi asymptomatic mutation carriers should be followed up on a regular basis after a negative initial work-up (Grade A).
Screening methods
The expert committee recommends that all SDHx mutation carriers should have an annual outpatient follow-up examination during both childhood and adulthood. Biochemical measurements of plasma or urinary metanephrine and normetanephrine should be conducted every 2 years in children and yearly in adults (Figs 1,2). PET–CT was not recommended for follow-up during childhood.

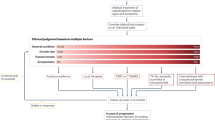
An initial tumour screening should be performed after the discovery of an SDHA, SDHB, SDHC or SDHD-pi mutation relying on blood pressure measurements, a symptoms or signs questionnaire, assessment of metanephrines in plasma or urine, and imaging work-up by MRI of head and neck, thorax, abdomen and pelvis. Even after an initial negative work-up, all asymptomatic mutation carriers should be clinically followed up every year, by biochemical assessments every 2 years and by MRI every 2–3 years.

An initial tumour screening should be performed after the discovery of an SDHA, SDHB, SDHC and SDHD-pi mutation relying on blood pressure measurements, a symptoms or signs questionnaire, assessment of metanephrines in plasma or urine, and imaging work-up by MRI of head and neck, abdomen, pelvis, and a whole-body PET–CT. Even after an initial negative work-up, all asymptomatic mutation carriers should be followed clinically and by biochemistry assessments every year and by MRI every 2–3 years. Thoracic MRI is not mandatory at the first initial work-up if PET–CT does not show any abnormality but is recommended for subsequent follow-up.
-
Recommendation 10: During childhood and adulthood, follow-up of asymptomatic carriers of SDHA, SDHB, SDHC and SDHD-pi mutations should include clinical examination (blood pressure measurement, ideally out-of-office, and a symptom questionnaire), the same biochemical investigations as for the initial screening (for example, metanephrine and normetanephrine) and imaging by MRI (head and neck and thoracic, abdominal and pelvic) (Grade A).
-
a.
Clinical examination should be performed every year (Grade A).
-
b.
Biochemical testing should be performed at least every 2 years during childhood and every year during adulthood (Grade A).
-
c.
MRI should be performed every 2–3 years (Grade A).
-
d.
Ultrasound should not be used as first-line imaging for follow-up in asymptomatic carriers of SDHA, SDHB, SDHC and SDHD-pi mutations (Grade A).
-
e.
Functional imaging should not be used for follow-up in asymptomatic SDHA, SDHB, SDHC and SDHD-pi mutation carriers during childhood (Grade A). Nevertheless, no consensus emerged for or against alternating PET–CT and MRI imaging during adulthood.
-
a.
The experts insisted on the fact that the risk of metastatic progression is more important in SDHB asymptomatic mutation carriers than for the other genes as many retrospective studies showed a higher risk of metastatic disease and shorter survival in patients with an SDHB mutation than in patients with other SDHx mutations3,20,24.
End of follow-up
After a certain age, a carrier of an SDHx mutation who has not developed a PPGL by initial screening or during follow-up has a considerably reduced risk of developing new tumours49.
-
Recommendation 11: If an SDHA, SDHB, SDHC or SDHD-pi mutation carrier never developed any tumour related to SDH deficiency and has been asymptomatic all their life, screening tests should be delayed to every 5 years after 70 years of age (Grade B) and follow-up should be stopped at 80 years of age (Grade A).
Additional expert recommendations
It has been established that a secreting PPGL is a perilous condition during pregnancy, with an elevated risk of pre-eclampsia and gestational diabetes and severe cardiac complications for the mother and prematurity and mortality (including miscarriage, intrauterine fetal loss and death at delivery) for the fetus50. Therefore, the experts considered that screening should be performed before planning a pregnancy to avoid this dangerous situation in an asymptomatic woman carrying an SDHx mutation.
-
Recommendation 12: Screening should not differ between male and female individuals. However, complete screening should be performed before planning a pregnancy (Grade A).
Since the discovery of the SDHx genes, family screening has been widely performed. There is currently only weak evidence indicating that genotype predicts the underlying phenotype. In the same family with all individuals carrying the same genetic variant, it is possible to see lifelong asymptomatic carriers, patients with a single head and/or neck paraganglioma, and others with metastatic PPGL21. Evidence suggests that metastatic progression in SDHx-mutated PPGL is due to immortalization-related mechanisms (TERT activation or ATRX mutations) occurring in the primary tumour51.
-
Recommendation 13: Initial screening and follow-up should not differ for asymptomatic mutation carriers whose family members developed metastatic SDHx-related PPGL or those with non-metastatic PPGL (Grade A).
SDHx mutations have been associated with other tumours with a causal link to RCC and GIST. It remains less clear if SDHx mutations can be associated with other tumours that could be found in affected carriers or their relatives such as pituitary adenomas52,53,54,55.
The majority of GIST in adults are secondary to somatic mutations in the KIT and PDGFRA genes56,57. However, 15% of GIST in adults and 85% of GIST developing during childhood have SDHx mutations58,59. The majority of SDHx mutations identified in GIST are germline SDHA point mutations followed by SDHB and SDHC point mutations5. Recurrent epimutations of SDHC have been identified in GIST and Carney triad (a syndrome characterized by paraganglioma, GIST and pulmonary chondroma). Most of the epimutations are somatic events without risk of familial transmission60. Regarding RCC, SDHx mutations appear to be implicated in 0.05–0.20% of cases, mainly affecting SDHB61,62. In the most recent WHO classification, SDHx-related RCC were identified as a new subtype of RCC63. In large cohorts of SDHx mutation carriers, the penetrance of RCC is estimated at 2–3% of patients3,33 and the risk of GIST development has not yet been evaluated. Moreover, GIST and RCC are readily detected on MRI64,65,66.
-
Recommendation 14: No additional imaging should be performed for RCC, GIST and pituitary adenoma. Nevertheless, RCC and GIST should be searched for on imaging performed for PPGL screening (Grade A).
Statements without consensus
A consensus was not reached for several statements (Supplementary Table S5).
The screening of people with SDHD-mi mutations lacks a consensus due to limited data in the literature. Except for a few case reports67,68,69, only one prospective study evaluated the risk of developing a tumour in 20 SDHD-mi mutation carriers29. Of these carriers, only one phaeochromocytoma was observed in a 35-year-old woman with a double loss of heterozygosity in the paternally derived q arm and the maternally derived p arm of chromosome 11. This case was considered too rare by experts to form the basis of recommendations as it involved three unique genetic events. Experts were not able to estimate the cost–benefit ratio of screening and surveillance of SDHD-mi mutation carriers and, thus, no consensus was reached.
The consideration of environmental factors (for example, living at a high altitude) was also waived. People living at altitudes above 2,000 m develop head and neck paraganglioma (especially carotid body paraganglioma) with a higher frequency than those at lower altitudes, possibly in response to chronic hypoxia70,71,72,73,74,75. However, no evidence of this effect was found in the context of patients with SDHx mutations76 and therefore no consensus was reached.
No agreement was reached regarding the measurement of plasma levels of dopamine, 3-methoxytyramine (the O-methylated metabolite of dopamine) and chromogranin A in addition to metanephrine and normetanephrine for tumour screening in asymptomatic SDHx mutation carriers. Dopamine production by SDHB-related PPGLs might be associated with the development of metastases; however, the measurement of dopamine is not recommended for first-line screening2,35,77. Plasma measurement of 3-methoxytyramine has prognostic value in patients with SDHB-associated PPGLs77 and it could be useful in the detection of dopamine-producing PPGLs and certain non-functional head and neck paragangliomas78,79. Chromogranin A could add value as a complementary biomarker in SDHB-related sympathetic paraganglioma (which typically secrete normetanephrine); however, it is non-specific and often low in head and neck paragangliomas80. The use of specific hormones and new biomarkers to characterize PPGLs and their behaviour as well as the value of screening in asymptomatic individuals are yet to be established.
The discussion regarding imaging methods focused on the ideal balance between the early detection of tumours (which can improve patient management)4 and reducing radiation exposure. Experts agreed on a comprehensive initial screening in adults to differentiate asymptomatic individuals from patients with an unknown disease as approximately 20% of asymptomatic individuals have one or several tumours upon initial screening15. However, some experts voiced the view that functional imaging should only be performed if MRI shows an abnormality. Thus, no consensus was achieved on the frequency of PET–CT during follow-up. The indication of PET–CT once in a lifetime when a child reaches adulthood was not discussed.
Due to the wide variability in availability and costs of the different tracers, the expert panel decided against a consensus for optimal PET radiopharmaceuticals (68Ga-DOTA-somatostatin analogues (68Ga-DOTA-SSAs), 18F-FDOPA, 18F-FDG). However, in 2019, EANM–SNMMI joint guidelines proposed the use of 68Ga-DOTA-SSAs PET as the first-choice functional imaging modality in SDHx mutation carriers and the use of 18F-FDG and/or 18F-FDOPA PET when 68Ga-DOTA-SSAs PET is not available81.
Conclusions
It is strongly recommended that asymptomatic carriers of SDHx mutations (detected by familial genetic testing) undergo regular biochemical testing and clinical examination. Due to the lack of robust data in the literature, a Delphi process enabled the proposal of an expert consensus statement. This consensus on when to begin screening and end follow-up as well as the appropriate imaging for the management of asymptomatic carriers with different SDHx mutations aims to unify global clinical practice. It also provides guidance for clinicians that can be adapted to their own and their patient’s situations. Many outstanding issues, such as age at first screening during childhood, optimal time period between two assessments or the need for a different approach according to mutation subtypes, support the need for large, international prospective studies in the near future as the effect of an early diagnosis on outcome needs to be balanced with the burden and the costs of screening. This screening protocol based on expert opinion should be regularly reviewed and updated when more conclusive data become available.
References
Ben Aim, L. et al. Targeted next-generation sequencing detects rare genetic events in pheochromocytoma and paraganglioma. J. Med. Genet. 56, 513–520 (2019).
Lenders, J. W. M. et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 99, 1915–1942 (2014).
Andrews, K. A. et al. Tumour risks and genotype-phenotype correlations associated with germline variants in succinate dehydrogenase subunit genes SDHB, SDHC and SDHD. J. Med. Genet. 55, 384–394 (2018).
Buffet, A. et al. Positive impact of genetic test on the management and outcome of patients with paraganglioma and/or pheochromocytoma. J. Clin. Endocrinol. Metab. 104, 1109–1118 (2019).
Boikos, S. A. et al. Molecular subtypes of KIT/PDGFRA wild-type gastrointestinal stromal tumors: a report from the National Institutes of Health Gastrointestinal Stromal Tumor Clinic. JAMA Oncol. 2, 922–928 (2016).
Evenepoel, L. et al. Toward an improved definition of the genetic and tumor spectrum associated with SDH germ-line mutations. Genet. Med. 17, 610–620 (2015).
Mason, E. F. & Hornick, J. L. Conventional risk stratification fails to predict progression of succinate dehydrogenase-deficient gastrointestinal stromal tumors: a clinicopathologic study of 76 cases. Am. J. Surg. Pathol. 40, 1616–1621 (2016).
Ricketts, C. et al. Germline SDHB mutations and familial renal cell carcinoma. J. Natl. Cancer Inst. 100, 1260–1262 (2008).
Tufton, N. et al. An analysis of surveillance screening for SDHB-related disease in childhood and adolescence. Endocr. Connect. 8, 162–172 (2019).
Villani, A. et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: 11 year follow-up of a prospective observational study. Lancet Oncol. 17, 1295–1305 (2016).
Dalkey, N. C., Lewis, R. & Rourke, D. L. Studies in the Quality of Life: Delphi and Decision-making (Lexington Books, 1972).
Burnichon, N. et al. The succinate dehydrogenase genetic testing in a large prospective series of patients with paragangliomas. J. Clin. Endocrinol. Metab. 94, 2817–2827 (2009).
Benn, D. E. et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J. Clin. Endocrinol. Metab. 91, 827–836 (2006).
Eijkelenkamp, K. et al. Calculating the optimal surveillance for head and neck paraganglioma in SDHB-mutation carriers. Fam. Cancer 16, 123–130 (2017).
Gimenez-Roqueplo, A.-P. et al. Imaging work-up for screening of paraganglioma and pheochromocytoma in SDHx mutation carriers: a multicenter prospective study from the PGL.EVA Investigators. J. Clin. Endocrinol. Metab. 98, E162–E173 (2013).
Heesterman, B. L. et al. High prevalence of occult paragangliomas in asymptomatic carriers of SDHD and SDHB gene mutations. Eur. J. Hum. Genet. 21, 469–470 (2013).
Jasperson, K. W. et al. Role of rapid sequence whole-body MRI screening in SDH-associated hereditary paraganglioma families. Fam. Cancer 13, 257–265 (2014).
Jochmanova, I. et al. SDHB-related pheochromocytoma and paraganglioma penetrance and genotype-phenotype correlations. J. Cancer Res. Clin. Oncol. 143, 1421–1435 (2017).
Miederer, M. et al. High incidence of extraadrenal paraganglioma in families with SDHx syndromes detected by functional imaging with [18F]fluorodihydroxyphenylalanine PET. Eur. J. Nucl. Med. Mol. Imaging 40, 889–896 (2013).
Neumann, H. P. H. et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA 292, 943–951 (2004).
Niemeijer, N. D. et al. The phenotype of SDHB germline mutation carriers: a nationwide study. Eur. J. Endocrinol. 177, 115–125 (2017).
Rijken, J. A. et al. The penetrance of paraganglioma and pheochromocytoma in SDHB germline mutation carriers. Clin. Genet. 93, 60–66 (2018).
Tufton, N., Sahdev, A., Drake, W. M. & Akker, S. A. Can subunit-specific phenotypes guide surveillance imaging decisions in asymptomatic SDH mutation carriers? Clin. Endocrinol. 90, 31–46 (2019).
Amar, L. et al. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J. Clin. Endocrinol. Metab. 92, 3822–3828 (2007).
Gimenez-Roqueplo, A.-P. et al. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res. 63, 5615–5621 (2003).
Bourdeau, I. et al. A SDHC founder mutation causes paragangliomas (PGLs) in the French Canadians: new insights on the SDHC-related PGL. J. Clin. Endocrinol. Metab. 101, 4710–4718 (2016).
Bausch, B. et al. Clinical characterization of the pheochromocytoma and paraganglioma susceptibility genes SDHA, TMEM127, MAX, and SDHAF2 for gene-informed prevention. JAMA Oncol. 3, 1204–1212 (2017).
Schiavi, F. et al. The endemic paraganglioma syndrome type 1: origin, spread, and clinical expression. J. Clin. Endocrinol. Metab. 97, E637–E641 (2012).
Burnichon, N. et al. Risk assessment of maternally inherited SDHD paraganglioma and phaeochromocytoma. J. Med. Genet. 54, 125–133 (2017).
Cascón, A. et al. Molecular characterisation of a common SDHB deletion in paraganglioma patients. J. Med. Genet. 45, 233–238 (2008).
Imamura, H. et al. Sporadic paraganglioma caused by de novo SDHB mutations in a 6-year-old girl. Eur. J. Pediatr. 175, 137–141 (2016).
King, K. S. et al. Metastatic pheochromocytoma/paraganglioma related to primary tumor development in childhood or adolescence: significant link to SDHB mutations. J. Clin. Oncol. 29, 4137–4142 (2011).
Ricketts, C. J. et al. Tumor risks and genotype-phenotype-proteotype analysis in 358 patients with germline mutations in SDHB and SDHD. Hum. Mutat. 31, 41–51 (2010).
Timmers, H. J. et al. Clinical presentations, biochemical phenotypes, and genotype-phenotype correlations in patients with succinate dehydrogenase subunit B-associated pheochromocytomas and paragangliomas. J. Clin. Endocrinol. Metab. 92, 779–786 (2007).
Neumann, H. P. H., Young, W. F. & Eng, C. Pheochromocytoma and paraganglioma. N. Engl. J. Med. 381, 552–565 (2019).
Jochmanova, I. et al. Clinical characteristics and outcomes of SDHB-related pheochromocytoma and paraganglioma in children and adolescents. J. Cancer Res. Clin. Oncol. 146, 1051–1063 (2020).
van der Tuin, K. et al. Clinical aspects of SDHA-related pheochromocytoma and paraganglioma: a nationwide study. J. Clin. Endocrinol. Metab. 103, 438–445 (2018).
Williams, B. et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension: the Task Force for the management of arterial hypertension of the European Society of Cardiology and the European Society of Hypertension: the Task Force for the management of arterial hypertension of the European Society of Cardiology and the European Society of Hypertension. J. Hypertens. 36, 1953–2041 (2018).
Whelton, P. K. et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on clinical practice guidelines. Hypertension 71, 1269–1324 (2018).
Manu, P. & Runge, L. A. Biochemical screening for pheochromocytoma. Superiority of urinary metanephrines measurements. Am. J. Epidemiol. 120, 788–790 (1984).
Peaston, R. T. & Lai, L. C. Biochemical detection of phaeochromocytoma: should we still be measuring urinary HMMA? J. Clin. Pathol. 46, 734–737 (1993).
Eisenhofer, G. et al. Biochemical diagnosis of chromaffin cell tumors in patients at high and low risk of disease: plasma versus urinary free or deconjugated O-methylated catecholamine metabolites. Clin. Chem. 64, 1646–1656 (2018).
Peitzsch, M., Mangelis, A., Eisenhofer, G. & Huebner, A. Age-specific pediatric reference intervals for plasma free normetanephrine, metanephrine, 3-methoxytyramine and 3-O-methyldopa: particular importance for early infancy. Clin. Chim. Acta 494, 100–105 (2019).
Eisenhofer, G., Lenders, J. W. & Pacak, K. Biochemical diagnosis of pheochromocytoma. Front. Horm. Res. 31, 76–106 (2004).
Timmers, H. J. L. M. et al. Biochemically silent abdominal paragangliomas in patients with mutations in the succinate dehydrogenase subunit B gene. J. Clin. Endocrinol. Metab. 93, 4826–4832 (2008).
Gravel, G. et al. The value of a rapid contrast-enhanced angio-MRI protocol in the detection of head and neck paragangliomas in SDHx mutations carriers: a retrospective study on behalf of the PGL.EVA investigators. Eur. Radiol. 26, 1696–1704 (2016).
Sargar, K. M., Khanna, G. & Hulett Bowling, R. Imaging of nonmalignant adrenal lesions in children. Radiographics 37, 1648–1664 (2017).
Taïeb, D. et al. Modern nuclear imaging for paragangliomas: beyond SPECT. J. Nucl. Med. 53, 264–274 (2012).
Heesterman, B. L. et al. Clinical progression and metachronous paragangliomas in a large cohort of SDHD germline variant carriers. Eur. J. Hum. Genet. 26, 1339–1347 (2018).
Lenders, J. W. M., Langton, K., Langenhuijsen, J. F. & Eisenhofer, G. Pheochromocytoma and pregnancy. Endocrinol. Metab. Clin. North Am. 48, 605–617 (2019).
Job, S. et al. Telomerase activation and ATRX mutations are independent risk factors for metastatic pheochromocytoma and paraganglioma. Clin. Cancer Res. 25, 760–770 (2019).
Casey, R. T. et al. Translating in vivo metabolomic analysis of succinate dehydrogenase deficient tumours into clinical utility. JCO Precis. Oncol. 2, 1–12 (2018).
López-Jiménez, E. et al. SDHC mutation in an elderly patient without familial antecedents. Clin. Endocrinol. 69, 906–910 (2008).
Xekouki, P. et al. Succinate dehydrogenase (SDH) D subunit (SDHD) inactivation in a growth-hormone-producing pituitary tumor: a new association for SDH? J. Clin. Endocrinol. Metab. 97, E357–E366 (2012).
Xekouki, P. et al. Pituitary adenoma with paraganglioma/pheochromocytoma (3PAs) and succinate dehydrogenase defects in humans and mice. J. Clin. Endocrinol. Metab. 100, E710–E719 (2015).
Heinrich, M. C. et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 299, 708–710 (2003).
Hirota, S. et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 279, 577–580 (1998).
Heinrich, M. C. et al. Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III Trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J. Clin. Oncol. 26, 5360–5367 (2008).
Janeway, K. A. et al. Sunitinib treatment in pediatric patients with advanced GIST following failure of imatinib. Pediatr. Blood Cancer 52, 767–771 (2009).
Killian, J. K. et al. Recurrent epimutation of SDHC in gastrointestinal stromal tumors. Sci. Transl Med. 6, 268ra177 (2014).
Gill, A. J. et al. Succinate dehydrogenase (SDH)-deficient renal carcinoma: a morphologically distinct entity: a clinicopathologic series of 36 tumors from 27 patients. Am. J. Surg. Pathol. 38, 1588–1602 (2014).
Vanharanta, S. et al. Early-onset renal cell carcinoma as a novel extraparaganglial component of SDHB-associated heritable paraganglioma. Am. J. Hum. Genet. 74, 153–159 (2004).
Moch, H., Cubilla, A. L., Humphrey, P. A., Reuter, V. E. & Ulbright, T. M. The 2016 WHO classification of tumours of the urinary system and male genital organs-part A: renal, penile, and testicular tumours. Eur. Urol. 70, 93–105 (2016).
Tufton, N., White, G., Drake, W. M., Sahdev, A. & Akker, S. A. Diffusion-weighted imaging (DWI) highlights SDHB-related tumours: a pilot study. Clin. Endocrinol. 91, 104–109 (2019).
Herzberg, M., Beer, M., Anupindi, S., Vollert, K. & Kröncke, T. Imaging pediatric gastrointestinal stromal tumor (GIST). J. Pediatr. Surg. 53, 1862–1870 (2018).
Kalkmann, J. et al. Consensus report on the radiological management of patients with gastrointestinal stromal tumours (GIST): recommendations of the German GIST Imaging Working Group. Cancer Imaging 12, 126–135 (2012).
Hartzell, L. D., McKelvey, K. D., Van Hemert, R. L. & Dornhoffer, J. Cerebellopontine angle tumor in a patient with a maternally inherited SDHD gene mutation. Int. Tinnitus J. 14, 97–100 (2008).
Pigny, P. et al. Paraganglioma after maternal transmission of a succinate dehydrogenase gene mutation. J. Clin. Endocrinol. Metab. 93, 1609–1615 (2008).
Yeap, P. M. et al. Molecular analysis of pheochromocytoma after maternal transmission of SDHD mutation elucidates mechanism of parent-of-origin effect. J. Clin. Endocrinol. Metab. 96, E2009–E2013 (2011).
Arias-Stella, J. & Bustos, F. Chronic hypoxia and chemodectomas in bovines at high altitudes. Arch. Pathol. Lab. Med. 100, 636–639 (1976).
Arias-Stella, J. & Valcarcel, J. The human carotid body at high altitudes. Pathol. Microbiol. 39, 292–297 (1973).
Nathanson, S. D. & Gaylis, H. Multicentric chemodectomata at high altitude. A case report and review of the literature. S. Afr. Med. J. 48, 1715–1717 (1974).
Pacheco-Ojeda, L., Durango, E., Rodriquez, C. & Vivar, N. Carotid body tumors at high altitudes: Quito, Ecuador, 1987. World J. Surg. 12, 856–860 (1988).
Rodríguez-Cuevas, S., López-Garza, J. & Labastida-Almendaro, S. Carotid body tumors in inhabitants of altitudes higher than 2000 meters above sea level. Head Neck 20, 374–378 (1998).
Saldana, M. J., Salem, L. E. & Travezan, R. High altitude hypoxia and chemodectomas. Hum. Pathol. 4, 251–263 (1973).
Jech, M., Alvarado-Cabrero, I., Albores-Saavedra, J., Dahia, P. L. M. & Tischler, A. S. Genetic analysis of high altitude paragangliomas. Endocr. Pathol. 17, 201–202 (2006).
Eisenhofer, G. et al. Plasma methoxytyramine: a novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur. J. Cancer 48, 1739–1749 (2012).
Rao, D. et al. Plasma methoxytyramine: clinical utility with metanephrines for diagnosis of pheochromocytoma and paraganglioma. Eur. J. Endocrinol. 177, 103–113 (2017).
van Duinen, N. et al. Plasma levels of free metanephrines and 3-methoxytyramine indicate a higher number of biochemically active HNPGL than 24-h urinary excretion rates of catecholamines and metabolites. Eur. J. Endocrinol. 169, 377–382 (2013).
Zuber, S. et al. Clinical utility of chromogranin A in SDHx-related paragangliomas. Eur. J. Clin. Invest. 44, 365–371 (2014).
Taïeb, D., Jha, A., Treglia, G. & Pacak, K. Molecular imaging and radionuclide therapy of pheochromocytoma and paraganglioma in the era of genomic characterization of disease subgroups. Endocr. Relat. Cancer 26, R627–R652 (2019).
Acknowledgements
We want to thank J. Favier and E. Cornu for their advice on the production of the questionnaires. We also want to thank the European Network for the Study of Adrenal Tumours (ENS@T) and the Pheochromocytoma and Paraganglioma Research Support Organization (PRESSOR) for their help with the identification of experts.
Author information
Authors and Affiliations
Contributions
L.A. and C.L.-L. contributed to all aspects of the article. K.P., O.S., S.A.A., S.J.B.A., E.B., A.B., N.B., M.F., A.B.G., P.H., R.J.H., A.J., C.J., H.P.M.K., M.M., M.N. and M.R. researched data for the article, contributed substantially to the discussion of content, and reviewed and/or edited the manuscript before submission. R.J.C.-B. and P.L.M.D. researched data for the article and reviewed and/or edited the manuscript before submission. D.L., D.T., D.R.T., H.J.L.M.T., G.T., N.T., W.F.Y., J.W.M.L. and A.-P.G.-R. researched data for the article, wrote the article, and reviewed and/or edited the manuscript before submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Endocrinology thanks J.P. Bayley and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Amar, L., Pacak, K., Steichen, O. et al. International consensus on initial screening and follow-up of asymptomatic SDHx mutation carriers. Nat Rev Endocrinol 17, 435–444 (2021). https://doi.org/10.1038/s41574-021-00492-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41574-021-00492-3
This article is cited by
-
The evolutionary impact of childhood cancer on the human gene pool
Nature Communications (2024)
-
Management of phaeochromocytoma and paraganglioma in patients with germline SDHB pathogenic variants: an international expert Consensus statement
Nature Reviews Endocrinology (2024)
-
International consensus statement on the diagnosis and management of phaeochromocytoma and paraganglioma in children and adolescents
Nature Reviews Endocrinology (2024)
-
So, you want to get into “total-body” PET/CT scanning? An installation guide for beginners!
Cancer Imaging (2023)
-
A need to tailor surveillance based on family history: describing a highly penetrant familial paraganglioma kindred with an SDHD pathogenic variant
Familial Cancer (2023)
