
Abstract
CO2 hydrogenation to chemicals and fuels is a significant approach for achieving carbon neutrality. It is essential to rationally design the chemical structure and catalytic active sites towards the development of efficient catalysts. Here we show a Ce-CuZn catalyst with enriched Cu/Zn-OV-Ce active sites fabricated through the atomic-level substitution of Cu and Zn into Ce-MOF precursor. The Ce-CuZn catalyst exhibits a high methanol selectivity of 71.1% and a space-time yield of methanol up to 400.3 g·kgcat−1·h−1 with excellent stability for 170 h at 260 °C, comparable to that of the state-of-the-art CuZnAl catalysts. Controlled experiments and DFT calculations confirm that the incorporation of Cu and Zn into CeO2 with abundant oxygen vacancies can facilitate H2 dissociation energetically and thus improve CO2 hydrogenation over the Ce-CuZn catalyst via formate intermediates. This work offers an atomic-level design strategy for constructing efficient multi-metal catalysts for methanol synthesis through precise control of active sites.
Similar content being viewed by others

Introduction
Carbon dioxide (CO2) hydrogenation can be a major enabling technology for establishing a carbon neutral circular economy. Chemicals and fuels such as methanol, light olefins, and gasoline can be produced through CO2 hydrogenation1,2,3,4. Methanol can be used as liquid fuel for transportation, serving as hydrogen carrier for renewable energy to chemicals schemes5. Additionally, methanol is a useful solvent and feedstock for synthesis of other chemicals such as olefins6,7. Thus, the implementation of CO2 hydrogenation to methanol could not only reduce CO2 emission but also generate useful products.
Cu-based catalysts have been widely used for chemical synthesis due to their low-cost and high-performance under intermediate reaction temperatures8,9,10. The commercial methanol synthesis process is based on converting a synthesis gas feed (a mixture of CO, CO2 and H2 typically obtained from fossil fuels) over a Cu/ZnO/Al2O3 catalyst6. However, the poor stability and low methanol selectivity of this catalyst for hydrogenation of pure CO2 have serious impacts on large-scale industrial applications. The methanol selectivity is significantly reduced due to the competing reverse water-gas shift reaction (RWGS). Additionally the high rate of water production poses problems for long term stability11,12,13,14,15, due to the sintering of Cu nanoparticles and oxide compounds (e.g. ZnO and ZrO2) and the restructuring of their interfaces along with Cu oxidation. The pure Cu-based systems often have low catalytic performance because Cu alone interacts very poorly with CO2 and the apparent activation energy for methanol synthesis is high on Cu(111)16. Thus, the multi-metal catalyst system with synergistic effect and abundant interfaces as well as strong metal-support interactions has been extensively developed for addressing this problem3,17. In addition to the commercial CuZnAl catalysts for COx (x = 1, 2) hydrogenation to methanol18,19, other multi-metallic catalysts such as CuZnZr11, CuZnGa20, CuZnCe21, CuCeTi16, and CuZnAlZr have also been reported22. Despite the improvements in the catalytic performance of these catalysts, there is still debate regarding the active sites and reaction mechanism of these multi-metal catalyst systems, with the role of oxide and alloy formation being challenging to fully understand8. Herein, we combine a concerted experimental and theoretical approach to design an advanced CuZnCe catalyst for methanol synthesis and obtain fundamental understanding as to the nature of active sites and their stability.
Extensive efforts have been undertaken to investigate the above issues with experimental and theoretical approaches, and operando characterization methods. In 2014, Graciani et al. reported that the CeOx/Cu(111) and Cu/CeOx/TiO2(110) interfaces with the combination of metal and oxide sites could have complementary chemical properties to regulate the reaction pathways16. In 2017, the active sites over CuZnAl catalyst were further demonstrated to be ZnO/Cu interfacial sites and Zn-Cu bimetallic sites were reconstructed to ZnO/Cu surfaces23. However, the active sites are still under debate because Zn-Cu bimetallic alloy sites have also been reported to catalyze the reaction24,25,26, especially through the formation of Zn-Cu surface alloy active sites in the presence of CO27. Recently, Zabilskiy et al. employed a series of high-pressure operando techniques to further investigate the reaction mechanism on CuZnAl catalyst, and they also observed not only the oxidation of CuZn alloys to ZnO/Cu surfaces, but also the presence of zinc formate as an important reaction intermediate28. Interestingly, Beck et al. also observed the existence of zinc formate under a high pressure of 10 bar, however, the zinc formate is difficult to be detected under lower pressure19. They also demonstrated that the CuZnAl catalyst composition and morphology were sensitive to the applied pressure and temperature. Therefore, the catalytic active sites and reaction mechanism for methanol synthesis remain under debate without comprehensive agreement.
Metal-organic frameworks (MOFs) with tunable chemical components and tailored structures are an ideal platform to engineer the active sites at the atomic and molecular levels29,30. Thus, MOF-based materials can act as templates for the preparation of MOF-derived catalysts at the nanoscale and even at the atomic scale31,32. For example, a site-directed reduction strategy was employed to engineer the MOFs/nanoparticle systems with different structures and size-selective properties for ketone hydrogenation32. A photoactivated Cu-CeO2 catalyst, which has abundant Cu-OV-Ce active sites derived from the substitution of Cu into Ce-MOF precursor33, was fabricated through MOFs crystal engineering for the preferential oxidation of CO. Moreover, Yang et al. have demonstrated that the copper-ceria solid solution with enhanced Cu-OV-Cex active species could improve the CO2 hydrogenation to methanol34. On the basis of these works, we were inspired to engineer the active sites of multi-metal catalysts with more metal incorporated into the support through a MOF crystal engineering strategy. In addition, the hydrogenation of formate species, an important CO2 hydrogenation intermediate, is facilitated by the close contact between the zinc and copper phases28. Thus, the MOFs derived catalysts exhibiting intimate contact may present high catalytic activity.
The conventional multi-metal catalysts are usually prepared by impregnation35, coprecipitation36, sol-gel37, or hydrothermal methods38. Herein, we synthesized a series of CuZnCe catalysts through MOFs crystal engineering method. As the CuZnCe-MOF was difficult to be synthesized by a one-pot method, the CuZnCe catalysts derived from CuZnCe-MOF were prepared step by step. As a result, we found that the order of introduction of metal during MOFs preparation influences the growth of MOFs, thus influencing the active sites, which would also be influenced by the types of metal species and preparation method. The obtained Ce-CuZn catalyst with abundant Cu/Zn-OV-Ce species exhibited high-performance CO2 hydrogenation to methanol. This was because the introduction of Cu and Zn to CeO2 energetically facilitates CO2 hydrogenation over Ce-CuZn catalyst via formate intermediates, which were observed by in-situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) and the proposed mechanism supported by density functional theory (DFT) calculations. The Cu-CeO2 interactions inhibited the RWGS while further introduction of Zn to decorate Cu-OV-Ce active sites promoted CO2 hydrogenation to methanol via Zn-decorated Cu active sites.
Results
Synthetic route
As the ligand of 1,3,5-benzenetricarboxylic acid (1,3,5-BTC) could be coordinated with many metal ions like Cu2+, Zn2+ and Ce3+, we tried to synthesize the CuZnCe-MOF by one-pot method (Route 1 in Supplementary Fig. 1). However, the color was white instead of blue for the dried CuZnCe-MOF sample, indicating that the copper species did not exist in the sample. The X-ray powder diffraction (XRD) patterns of the dried CuZnCe-MOF sample only presented the diffraction peaks of Ce-MOF (namely, Ce-BTC) and the XRD patterns of the calcined CuZnCe sample also only presented the diffraction peaks of CeO2 (Supplementary Fig. 2a, b), proving that the three metals could not grow simultaneously into CuZnCe-MOF sample by one step under these reaction conditions. To solve this problem, we tried to synthesize it by two steps. Firstly, the CuZn-MOF was prepared and then introduced Ce3+ into CuZn-MOF to produce CuZn-Ce-MOF (Route 2 in Supplementary Fig. 1). Or the Ce-MOF was prepared firstly and then Cu2+/Zn2+ into Ce-MOF to produce Ce-CuZn-MOF (Route 3 in Supplementary Fig. 1). The color of the dried CuZn-Ce-MOF and Ce-CuZn-MOF samples were blue and their XRD patterns displayed the diffraction peaks of Cu-MOF. After calcination, the diffraction peaks of CuO and CeO2 could be found, showing the segregation and agglomeration of metal species as well as the decomposition of MOF precursors. The dried Ce-MOF sample lost the solvent molecules at about 150 °C and then lost the 1,3,5-BTC ligand at about 350 °C (Supplementary Fig. 2c). The decomposition temperatures for Ce-CuZn-MOF are lower than Ce-MOF because that Ce-CuZn-MOF was prepared via more steps including solvent washing. Furthermore, the XRD patterns over Ce-CuZn-MOF which originally showed the presence of Ce-MOF, changed upon introduction of Cu2+/Zn2+ into Ce-MOF (Supplementary Fig. 3), indicating transformation to the Cu-MOF crystal structure. Thus, the crystal structure of Ce-MOF was reconstructed into Cu-MOF for the Ce-CuZn sample, indicating that Cu2+/Zn2+ were introduced into the frameworks of Ce-MOF and subsequently doped into the structure of CeO2 after calcination (Fig. 1a). To prepare the control sample with the Cu2+/Zn2+ on the surface of CeO2, the Ce-CuZn-IM was prepared by an impregnation method that involved introduction of Cu2+/Zn2+ after calcination of Ce-MOF (Route 4 in Supplementary Fig. 1).

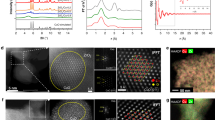
a The samples’ evolution from Ce-MOF to Ce-CuZn. Setp I: Ultrasonication, and Cu2+/Zn2+ ion exchange; Step II: Calcination, reduction, and MOF decomposition to form Cu/Zn-OV-Ce. The purple, green, yellow, red, blue and black balls represent Zn, Ce, Cu, O, OV and C, respectively. b–g The TEM and HRTEM images of reduced Ce-CuZn sample with corresponding elemental mapping.
Morphological and textural properties of the catalysts
To further investigate the micro and nanostructure of the Ce-CuZn with optimized catalytic performance, transmission electron microscopy (TEM) and scanning electron microscopy (SEM) images of Ce-CuZn sample are presented in Fig. 1 and Supplementary Fig. 4, respectively. The original Ce-MOF presents bundles of smooth nanorods (Supplementary Fig. 4a) while the dried Ce-CuZn-MOF show bundles of coarse nanorods with octahedral structure (Supplementary Fig. 4b, d), which was the typical structure of Cu-MOF as demonstrated by the above XRD results. Upon calcination, the morphology of Ce-MOF also changed from nanorods to the mixture of nanorods, nanosheets, and nanoparticles for the Ce-CuZn-MOF sample (Supplementary Fig. 4c).
The Ce-CuZn nanoparticles were also small and highly dispersed from the TEM image (Fig. 1b). The HRTEM image of Ce-CuZn shows that it could expose different crystal phases of CeO2 (Fig. 1c), such as CeO2 (111), CeO2 (200), and CeO2 (220). The crystal phase of copper could not be observed due to the lighter atomic weight of Cu with respect to Ce and the similar contrast of Cu and CeO2, which was also observed in the other reported Cu/CeO2 catalysts33,39. The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images with the corresponding EDS elemental mapping further show that Ce-CuZn sample exhibited homogeneous of dispersion Cu, Zn, Ce metals (Fig. 1d–g). It should be mentioned that the mean Cu nanoparticle sizes were increased from 5.34 nm over the calcined Ce-CuZn to 8.17 nm over the reduced Ce-CuZn, indicating that the Cu nanoparticles were accompanied by slight migratory agglomeration during reduction process (Supplementary Figs. 5 and 6). However, the solid solution could be kept over the reduced Ce-CuZn sample, which would be demonstrated by the following Raman and electron paramagnetic resonance (EPR) results. For the other two CuZn-Ce and Ce-CuZn-IM samples, the nanorods combined with the nanoparticles could also be observed (Supplementary Figs. 7 and 8). In particular, the partial agglomeration of Cu/Zn nanoparticles were found over the surface of CeO2. Also, the control samples of CuZn-Ce and Ce-CuZn-IM exposed similar crystal phases of CeO2 from the HRTEM images analysis.
The physicochemical properties of CuZnCe series samples are presented in Table 1. The actual Cu, Zn, and Ce loading over the CuZnCe series samples were different. The CuZn-Ce and Ce-CuZn possessed similar copper content of about 52 wt.% but the former had not loaded Zn. However, its precursor of CuZn had 3.55 wt.% of Zn, indicating that the Zn was lost during the second step of adding the Ce. For the Ce-CuZn-IM sample, it has much lower Cu loading (22.36 wt.%) and more Zn loading (11.47 wt.%). This is because the Ce-MOF was calcined to obtain the CeO2 powder and then loaded with Cu and Zn without the centrifugation or washing procedures by the impregnation method. Thus, we regulated the mass of nitrates to synthesize the Ce-CuZn-IM-B sample with a similar metal loading as the Ce-CuZn sample. The CuZnCe series samples present a similar hysteresis loop and specific surface area (SBET) of about 41 m2/g, but the Ce-CuZn sample exhibits larger pore size and pore volume (Supplementary Fig. 9 and Table 1). However, the copper surface area and copper dispersion over the Ce-CuZn sample were lower than the control samples (Table 1), indicating that more copper species were doped into the ceria matrix. Although the copper loading over Ce-CuZn-IM was much lower, its surface copper area was similar to that over Ce-CuZn.
Evolution of crystalline phase and surface properties
The evolution of metal species is illustrated by the XRD results. For the calcination of pure CuZn-MOF, the acute diffraction peaks of CuO are observed (Supplementary Fig. 2b). After introduction of Ce into CuZn-MOF, the diffraction peaks of CuO become broader. All the calcined CuZnCe series samples show the CuO and CeO2 species and the Ce-CuZn-IM also presents the ZnO species. After reduction by hydrogen, CuO species can no longer be detected by XRD while the metallic Cu species appear for the CuZn-Ce and Ce-CuZn-IM samples (Fig. 2a). However, the broad diffraction peaks of CuO and sharp diffraction peaks of Cu2O could be detected over the Ce-CuZn sample. Thus, we further operated the in-situ XRD test under the 40%H2-N2 atmosphere for the Ce-CuZn sample (Fig. 2b). The diffraction peaks of CuO and Cu2O could not be detected in the in-situ XRD results, indicating that the reduced Ce-CuZn sample was very active thus would be facile to be oxidized during offline XRD test. The diffraction peaks over Ce-CuZn sample were also stable during in-situ reaction atmosphere (Supplementary Fig. 10). This suggests that the copper species were different over CuZnCe series samples. In addition, the Ce-CuZn-IM sample still showed obvious diffraction peaks of ZnO after reduction.

a Normal XRD patterns of the reduced samples tested offline. b In-situ XRD patterns of Ce-CuZn samples reduced at different temperature under the atmosphere of 40%H2-N2. c Raman spectra. d H2-TPR curves. e CO2-TPD curves. f EPR spectra. K, B, A1, and A2 represent the peak signal, where signal K is ascribed to Cu2+/Zn2+ dimer; signal B is also ascribed to Cu2+ ions; signal A1 has been correlated with isolated Cu2+ in octahedral sites in ceria with a tetragonal distortion; signal A2 has been attributed to isolated Cu2+ localized in surface substitutional sites with a square-pyramidal symmetry56.
From the Raman spectra (Fig. 2c), the intensity of triply degenerate F2g mode of CeO2 became weaker and there was a distinct blue shift (466 to 461 cm−1) from Ce-CuZn-IM to CuZn-Ce and Ce-CuZn samples, suggesting the doping of Cu and Zn to the CeO2 lattice33. Simultaneously, an obvious broad band (550 ~ 650 cm−1) induced from oxygen vacancies was observed over CuZn-Ce and Ce-CuZn samples, which was attributed to the substitutional incorporation of Cu/Zn ions into the CeO2 lattice34. Thus, the Cu/Zn-OV-Ce active sites could be produced from the Cu/Zn substitution into the CeO2 lattice and CuO/ZnO-CeO2 boundary33, which would also be demonstrated by the following X-ray absorption spectra (XAS) results.
The temperature-programmed reduction (H2-TPR) profiles were fitted with three peaks (Fig. 2d and Supplementary Fig. 11), which were attributed to the reduction of dispersed copper species that weakly interact with CeO2 (peak α), bulk CuO and dispersed copper species that strongly interact with CeO2 (peak β), and Cu/Zn-OV-Ce solid solution (peak γ)21,40. It shows that Ce-CuZn and Ce-CuZn-IM samples exhibit higher reduction temperatures than that of CuZn-Ce, which is probably because that the former two samples have higher contents of Zn to decorate the Cu particles21. Moreover, the order of peak γ ratio over CuZnCe catalysts is as follows: Ce-CuZn> CuZn-Ce> Ce-CuZn-IM, indicating the existence of many Cu/Zn-OV-Ce species with strong metal-support interaction over the Ce-CuZn sample. The above characterization results indicate that the Ce-CuZn sample exhibits Cu0, Cu2O and Cu+/Zn-OV-Ce species.
The CO2 temperature-programmed desorption (CO2-TPD) experiments were carried out to determine the surface basicity of the catalysts. Three desorption peaks assigned to weak, moderate, and strong basic sites could be observed over CuZnCe series samples (Fig. 2e). Moreover, the Ce-CuZn sample shows a larger CO2 desorption peak and the fitted peak areas of Ce-CuZn is about three times of CuZn-Ce sample, confirming that its surface exhibited weaker basic sites for CO2 adsorption.
Quasi in-situ X-ray photoelectron spectroscopy (XPS) was further performed to analyze the surface species over the reduced CuZnCe series samples. The binding energy of Cu 2p3/2 over the Ce-CuZn-IM present at 932.8 eV is attributed to Cu0/Cu+ species (Supplementary Fig. 12a). Moreover, the Cu LMM XAES spectra were carried out to determine the specific Cu+/(Cu++Cu0) ratio as shown in Supplementary Fig. 13a and Supplementary Table 1. The Ce-CuZn sample has a higher Cu+ content (56.5%) and that over Ce-CuZn-IM is not available due to the effect of Zn 2p. The Ce-CuZn-IM showed weaker peaks of Cu 2p but stronger peaks of Zn 2p, which were almost absent over the CuZn-Ce sample (Supplementary Fig. 12b). This suggests that the surface of Ce-CuZn-IM exhibits many Zn species while CuZn-Ce has no Zn species, which is consistent with the above XRD and ICP results. Moreover, O1s peaks are deconvoluted into three components (Supplementary Fig. 13b), namely lattice oxygen (Oα), oxygen vacancies (Oβ) and surface oxygen (Oγ)41,42. Similarly, the Ce 3d peaks are deconvoluted into four peaks of 3d104f 1 Ce3+ (u0, u1, u’0, and u’1) and six peaks of 3d104f 0 Ce4+ (υ0, υ1, υ2, υ’0, υ’1, and υ’2) (Supplementary Fig. 13c). The Ce-CuZn catalyst exhibits a higher ratio (29.9%) of Oβ than the other two samples (Supplementary Table 1), indicating that Ce-CuZn catalyst possesses a higher concentration of oxygen vacancies. However, the Ce3+/(Ce3++Ce4+) ratio (34.8%) of Ce-CuZn is slightly lower than that over CuZn-Ce (36.4%), but higher than that over Ce-CuZn-IM (27.4%, Supplementary Table 1). This is because that the catalyst has two types of oxygen vacancies: I) generation from the reduction of Ce4+ to Ce3+, and II) the replacement of Ce4+ by Cu/Zn ions, resulting in the formation of oxygen vacancies43. Thus, the Ce-CuZn catalyst with slightly lower Ce3+/(Ce3++Ce4+) ratio could still exhibit higher Oβ/(Oα + Oβ + Oγ) ratio due to the substitutional incorporation of Cu/Zn ions into the CeO2 lattice. Therefore, the oxygen vacancies results at 531.2 eV in O 1s spectra and the Ce3+/(Ce3++Ce4+) ratio in Ce 3d spectra are consistent with the Raman results.
Moreover, EPR and chemisorption measurements were carried out to confirm the oxygen vacancies. The EPR spectrum of Ce-CuZn sample presents obvious oxygen vacancies peak at g|| value of 1.99, which is much different with the other two samples (Fig. 2f)43. Moreover, the type K peaks at g|| values of 2.29 and 1.83 are ascribed to Cu2+/Zn2+ dimer, which could be observed when two neighboring Ce4+ ions with short separation distance are substituted by Cu2+/Zn2+ ions44. Thus, the appearance of K signals suggests that Cu/Zn-OV-Ce solid solution is indeed generated in Ce-CuZn sample. In addition, the chemisorption measurement to determine the amount of oxygen vacancies (NOV) was developed by Zhu et al.40, and the results are summarized in Table 1. It presents that the Ce-CuZn sample exhibits 18.1 μmol·gcat−1 of oxygen vacancies, which is still much higher than the other two samples (2.9 ~ 6.0 μmol·gcat−1). These results are consistent with the above quasi in-situ XPS and the EPR results.
To further obtain the quantitative information of electron and coordination environment, XAS analysis was conducted. The fingerprint effect of X-ray absorption near edge structure (XANES) ascertains the valence state of the absorption atom. Figure 3a shows the spectra of normalized Cu K-edge XANES and corresponding standard samples. As demonstrated by the XANES of Ce-CuZn and Ce-CuZn-IM of Cu K-edge, the absorption edge slightly shifts to higher values compare with Cu foil, implying the presence of oxidized copper in Ce-CuZn and Ce-CuZn-IM. Meanwhile, the oxidation of Ce-CuZn-IM exhibited a slightly high oxidation state. We then resorted to extended X-ray absorption fine structure (EXAFS) spectra to investigate the local structure. The Fourier transform of Cu K-edge EXAFS result in Fig. 3b reveals that the Ce-CuZn and Ce-CuZn-IM demonstrates a weak path at around 1.4 Å and a predominant peak at 2.2 Å, corresponding to Cu-O and Cu-Cu scattering path, respectively. Least-squares EXAFS fitting analysis was further adopted (Supplementary Fig. 14 and Supplementary Table 2), with best fitting analysis of Ce-CuZn showing the dominance of Cu-Cu bond with coordination number of 10.0 and a low Cu-O contribution was determined as 0.2. For Zn, the normalized XANES showed in Fig. 3c, and the spectrum of Ce-CuZn is in the same position with ZnO; for comparison, the position of Ce-CuZn-IM is between Zn foil and ZnO, implying that the valence state of Zn in Ce-CuZn is Zn2+ and in Ce-CuZn-IM is the mixture of Zn0 and Zn2+, and the specific valence state is 1.6 via linear combination fitting (LCF). The evolution of coordination configuration of Ce-CuZn and Ce-CuZn-IM was identified by EXAFS (Fig. 3d–f). The fitting results of coordination environment of Zn K-edge spectra show more differences (Supplementary Fig. 14 and Supplementary Table 3). The best EXAFS fitting of Ce-CuZn gives 4.5 Zn-O bonds (R = 1.96\(\mathring{\text \AA}\)) and 11.5 Zn-Zn bonds (R = 3.23\(\mathring{\text \AA}\)); and the Ce-CuZn-IM exhibits 3.3 Zn-O bonds (R = 1.98\(\mathring{\text \AA}\)) and 12.0 Zn-Zn bonds (R = 3.24\(\mathring{\text \AA}\)). Since the higher oxidation state of Zn in Ce-CuZn, and almost no CuZn alloy generated in the catalyst, we speculate that some Cu and Zn were incorporated into CeO2 lattice to form Cu/Zn-OV-Ce species while the other Cu and Zn on the catalyst surface combined with CeO2 to form Cu/Zn-ceria interfaces. While the Zn species over the Ce-CuZn-IM samples are mainly ZnO, and also a part of CuZn alloy and Cu/Zn-OV-Ce species.

a Cu K-edge XANES and corresponding standard samples. b Fourier-transformed k2-weight EXAFS of Cu K-edge. c Zn K-edge XANES and corresponding standard samples. d Fourier-transformed k2-weight EXAFS of Zn K-edge. e, f The WT spectroscopy of Ce-CuZn sample.
Catalytic activity and stability
The catalytic performance of CO2 hydrogenation to methanol on CuZnCe series samples was evaluated in a fix-bed reactor (Fig. 4a) and the results are shown in Fig. 4 and Supplementary Table 4. The Ce-CuZn displayed the highest Con.CO2 and space-time yield (STY) of methanol (140.6 g·kgcat−1·h−1) under identical reaction conditions (P = 2.0 MPa, T = 280 °C, GHSV = 10,000 mL·gcat−1·h−1) among CuZnCe series samples (Fig. 4b). While the Ce-Zn, CuZn and Ce-CuZn-IM samples exhibited higher methanol selectivity but much lower CO2 conversion, as a result with lower STY of methanol (5.5 ~ 26.8 g·kgcat−1·h−1, Supplementary Table 4). When compared at similar CO2 conversion, the Ce-CuZn still presented the highest methanol selectivity and STY of methanol (Supplementary Fig. 15). We further calculated the TOF values of CuZnCe series catalysts (Supplementary Table 5), and the results show that the TOF value (19.0 h−1) of Ce-CuZn is 5 ~ 8 times of the other CuZnCe series catalysts (2.3 ~ 3.4 h−1).

a Schematic illustration of the thermal CO2 hydrogenation to methanol. The purple, green, yellow, red, blue, black and white balls represent Zn, Ce, Cu, O, OV, C and H, respectively. b Catalytic performance under the identical reaction conditions. c Catalytic stability of Ce-CuZn catalyst. d Comparison of the Ce-CuZn sample with the reported methanol synthesis catalysts under the reaction conditions of 2–5 MPa, 1500–48,000 mL·gcat−1·h−1, and 220–330 °C (Supplementary Table 6). The reference data is cited below: Cu/SiO2-AE (Cat-1)57, Cu/ZnO@m-SiO2 (Cat-2)58, Cu/ZnO/SiO2 (Cat-3)59, CuZnGa/SiO2 (Cat-4)60, Cu/Ga2O3/ZrO2 (Cat-5)61, Cu/Zn/Al/Y (Cat-6)62, CuZnAl-C-1.25 (Cat-7)63, Cu/ZnO/Al2O3-1 (Cat-8)64, Cu/ZnO/Al2O3-2 (Cat-9)65, Cu/ZnO/Al2O3-3 (Cat-10)62, Cu/ZnO/Al2O3-4 (Cat-11)66, Cu/CeO2 (Cat-12)52, Cu-Pd/CeO2 (Cat-13)67, CuZn@UiO-bpy (Cat-14)66, CuZn-BTC (Cat-15)68, In2O3 (Cat-16)69, and h-In2O3-R (Cat-17)70. e Apparent activation energy (Ea) determined by Arrhenius plots based on CO2 hydrogenation. Reaction conditions: b P = 2.0 MPa, T = 280 °C, GHSV = 10,000 mL·gcat−1·h−1, H2: CO2: N2 = 72: 24: 1; c GHSV = 20,000 mL·gcat−1·h−1, P = 2.8 MPa, T = 260 °C, H2: CO2: N2 = 72: 24: 1; e P = 2.0 MPa, GHSV = 20,000 mL·gcat−1·h−1 for Ce-CuZn and 10,000 mL·gcat−1·h−1 for the others, H2: CO2: N2 = 72: 24: 1. Ea is the apparent activation energy.
To assess the effect on performance of the order in which the ions are introduced, more control samples have been prepared and evaluated, as illustrated in Supplementary Table 4. The pure CeO2 support and the binary system of CuZn, Ce-Cu, and Ce-Zn catalysts have presented poor catalytic performance. Thus, it is necessary to prepare the ternary system. For the order of introducing the Cu, Zn, and Ce elements, they were firstly introduced together, but the CuZnCe sample exhibited only 1.4% of Con.CO2 at 280 °C. The CuZnCe sample only grew Ce-MOF with very low CuZn and showed poor performance. Thus, the three metals could not be added together bringing about the question of which element should be introduced first. When the Cu was introduced first, the CuZn-Ce sample with low content of Zn (0.03%, Table 1) presented low performance as the Zn was lost during the second step of preparation. When the Zn was introduced first, the Zn-CuCe sample could not be prepared because that Zn-MOF was not generated under the similar conditions. When the Ce was introduced first, the CeCu-Zn with low Cu content and CeZn-Cu samples with low Zn content have bad performance because Ce-MOF could not be grown together with Cu/Zn-MOF. However, the Ce-CuZn sample could grow Ce-MOF and Cu/Zn-MOF well with two main steps, and thus it had high Cu/Zn-OV-Ce species and exhibited the best performance. When the Zn content was increased, the Ce2-CuZn2 and Ce1-CuZn4 samples would decrease CO2 conversion although the methanol yield was slightly increased. In addition, we also investigated more complicated preparation procedures with three main steps. It was shown that the Ce-Cu-Zn and Ce-Zn-Cu samples also have good performance, but the preparation procedures are more complicated. Furthermore, although the Sel.MeOH and STYMeOH slightly increased over Ce2-CuZn2, Ce1-CuZn4 and Ce-Cu-Zn samples, they were increased at the expense of CO2 conversion compared with Ce-CuZn sample. When the Con.CO2 over Ce-CuZn sample was also near 10%, the Sel. MeOH and STYMeOH were increased to 45.5% and 154.0 g·kgcat−1·h−1, respectively. Therefore, the optimized Ce-CuZn catalyst has the appropriate metal elements and suitable introduction order to form abundant Cu/Zn-OV-Ce species, thus it presents the best catalytic performance for methanol synthesis compared with the control catalysts.
As the Ce-CuZn-IM sample possessed different Cu and Zn contents compared with Ce-CuZn, we also tested the performance of Ce-CuZn-IM-B sample that possessed similar Cu and Zn contents compared with Ce-CuZn sample. As a result, the Ce-CuZn-IM-B samples still showed much lower CO2 conversion and STY of methanol compared with Ce-CuZn sample (Supplementary Table 4). Furthermore, the actual surface Cu contents of Ce-CuZn and Ce-CuZn-IM were similar (Supplementary Table 1), but their performance was very different. Thus, this indicates that the state of Zn promoter and intimate contact of active species in Cu/Zn-OV-Ce, influenced by the preparation method and the order or method of metal introduction, play an essential role for direct CO2 hydrogenation to methanol.
Supplementary Fig. 16 shows that the optimized reaction temperature for the STY of methanol was 260 °C over the Ce-CuZn catalyst. Thus, the long-term stability test was further carried out at 260 °C. The average Con.CO2, Sel.MeOH, Sel.CO, and STY of methanol over Ce-CuZn sample during time on stream of 170 h were 8.0%, 71.1%, 26.7%, and 400.3 g·kgcat−1·h−1, respectively (Fig. 4c), which were stable without obvious decrease. Compared to the some technical CuZnAl catalysts, the Ce-CuZn have presented lower CO selectivity (Supplementary Table 6). Moreover, the STY of methanol on CuZnAl catalyst was decreased about 17% from 180 to 150 g·kgcat−1·h−1 after about 150 h15. This indicates that the Ce-CuZn sample here is a robust catalyst with excellent stability, outperforming the commercial CuZnAl. Compared with the state-of-the-art catalysts reported in the literature, the Ce-CuZn sample also exhibits comparable STY of methanol under similar reaction conditions (Fig. 4d and Supplementary Table 6).
As the Ce-CuZn catalyst possessed superior CO2 hydrogenation performance, we further operated the CO hydrogenation and apparent activation energy (Ea) tests. As illustrated in Supplementary Fig. 17, the Ce-CuZn catalyst also presented high methanol selectivity (81.0%) and yield (225.6 g·kgcat−1·h−1) at 300 °C during CO hydrogenation, suggesting that the CO produced during CO2 hydrogenation via the RWGS reaction could be recycled and hydrogenated on Ce-CuZn catalyst again. Furthermore, the apparent activation energy on Ce-CuZn (59.3 kJ·mol−1) is much lower than the other CuZnCe series catalysts (Fig. 4e), especially lower than the Ce-Zn and Ce-Cu samples, indicating that Cu and Zn simultaneously doping into CeO2 via the MOFs crystal engineering method can significantly improve the activation of the reactant molecules.
Reaction mechanism and DFT calculations
The above preliminary characterization results show that the Cu/Zn-OV-Ce active sites could be produced from the Cu/Zn substitution into the CeO2 lattice and CuO/ZnO-CeO2 boundary. We further performed in-situ CO-DRIFTS to investigate the surface interactions with the CO probe molecule. The peaks at 2173 cm−1 are attributed to CO adsorbed on the CeO2 surface45, thus it indicates that the Ce-Zn and Ce-Cu samples exhibit some CeO2 surfaces. The main peaks at 2120–2106 cm−1 are resulted from CO adsorbed on Cu+ species while the CO adsorbed on Cu0 species were not obvious at around 2058 cm−1 (Fig. 5a), which was probably because the in-situ CO-DRIFTS tests were operated at room temperature and the CO adsorption on Cu0 species was weak45. It shows that the Ce-CuZn-IM exhibits the largest CO adsorption peak while the Ce-Zn almost has no peak, indicating that the surface of Ce-CuZn-IM possesses abundant copper species. In addition, the Ce-CuZn and CuZn-Ce samples present an obvious shoulder peak at 2138 cm−1, which belongs to single-site Cu+ ions located in constrained environments (Fig. 5a)46,47,48. Therefore, the in-situ CO-DRIFTS confirmed the existence of Cu+ species on the CuZnCe catalysts.

a CO-DRIFTS results of CuZnCe after purged by He for 20 min at 30 °C. b The result of in-situ DRIFTS for CO2 hydrogenation at 260 °C. c Transient in-situ DRIFTS experiments on Ce-CuZn sample, and the peak intensity of HCOO* species at 1585 cm−1 as a function of time when the reaction atmospheres were switched from CO2 to H2, CO2 + H2, and Ar.
To investigate the potential reaction mechanism, in-situ DRIFTS experiments of CO2 hydrogenation were carried out on Ce-CuZn and Ce-CuZn-IM catalysts. As shown in Supplementary Fig. 18 and Fig. 5b, the main adsorption peaks over Ce-CuZn-IM and Ce-CuZn catalysts are similar except the peaks at 3730–3602 cm−1, which are attributed to the terminal, bridged, and triply bridged hydroxyls as well as the Ce3+-OH and bicarbonate (Fig. 5b)49. Both Ce-CuZn and Ce-CuZn-IM catalysts have presented strong peaks of HCOO* species at 2846, 1585, and 1374 cm−1, which were corresponding to the CH stretch mode, symmetric and asymmetric OCO stretching modes12,34. However, the gas phase CO* peak (2178 cm−1) and CO adsorbed peak (2110 cm−1) on Cu+ species were weak39,50,51. The other surface species such as monodentate carbonates (1522 cm−1)52, polydentate carbonates (1330, 847 cm−1)39, and methoxy species (2955, 1075 cm−1) could also be observed from the in-situ DRIFTS spectra12,53. Thus, the in-situ DRIFTS experiments demonstrate that the Ce-CuZn and Ce-CuZn-IM catalysts have the same main reaction intermediates of formate species.
Moreover, the transient in-situ DRIFTS experiments were performed to investigate the role of formate in the mechanism. The Ce-CuZn catalyst was firstly exposed to pure CO2 then the system was switched to other reaction atmospheres (H2, CO2 + H2, Ar), resulting in significant change of the surface species, as shown in Fig. 5c and Supplementary Fig. 19. Firstly, when the CO2 gas was injected to the system, different types of hydroxyl groups at around 3730 ~ 3602 cm−1 appeared and increased with the time. Some other carbonates (1522, 1330 cm−1) and formate (1585, 1374 cm−1) peaks are also shown in Supplementary Fig. 19a. When the gas was switched to H2, the above peaks became weak and peaks of hydroxyl groups disappeared (Supplementary Fig. 19b). In particular, the intensity of formate at 1585 cm−1 increased firstly and then decreased slightly when the system was dosed CO2 (Fig. 5c), which was because that CO2 firstly reacted with hydrogen available on the catalyst surface to generate formate but then decreased due to conversion to methoxy and lack of hydrogen. Thus, when the hydrogen was injected into the system, the intensity of formate increased quickly and then decreased with time.
Secondly, upon switching the reaction atmosphere from hydrogen to CO2 + H2, the OH* peaks became positive but then became negative after 120 min (Supplementary Fig. 19c), which was much different compared with the pure CO2 atmosphere. It was possible that the surface hydroxyl groups reacted with CO2 and the bicarbonates were converted to methanol. The formate peaks also decreased first and then increased to a stable state during reaction. Finally, the gas flow was switched to Ar, and the OH* peaks disappeared and other species peaks became weaker and weaker. However, the formate and carbonates could still be observed after purging for 30 min (Supplementary Fig. 19d), indicating that these surface species were stable. In addition, the CO* peaks at 2178 and 2110 cm−1 could not be observed during the transient in-situ DRIFTS experiments, thus these CO* species were regarded mainly as spectators during the reaction. Therefore, the surface species of formate, carbonates, bicarbonate, and methoxy are proposed as the main reaction intermediates in the mechanism of CO2 hydrogenation to methanol.
DFT calculations were further implemented to investigate the adsorption and activation of H2 and CO2, as well as the potential reaction mechanism over CuZnCe catalysts. Combined with CO-DRIFTS and XPS results, it can be concluded that the Ce-CuZn sample exhibits both the Cu0 and Cu+ species, in which the content of Cu+ species is up to 56.5%, indicating that abundant surface Cu+ species are responsible for the formation of Cu/Zn-OV-Ce active sites on the Ce-CuZn sample. Meanwhile, the EXAFS result (Fig. 3) showed that the Cu-O and Cu-Cu scattering path existed in the Ce-CuZn sample, thus, two Cu atoms are considered to replace the Ce atoms to reflect the Cu-Cu and Cu-O coordination structures. Firstly, we adopt the dominantly exposed (111) facet and further construct three models to stimulate CeO2 sample with oxygen vacancy, Cu doped Ce-Cu sample and Cu/Zn co-doped Ce-CuZn sample (Supplementary Fig. 20), which are named as the OV-CeO2, Cu+-CeO2-x and Zn/Cu+-CeO2-x, respectively. Given that Cu+ acts as the main species in the Ce-CuZn sample, the differential charge density and Bader charge are employed to characterize the electronic effect of dopant Cu and Zn atoms.
As shown in Fig. 6a, the Cu ions have the charge of −0.49/−0.55 e over Cu+-CeO2-x and those are close to bulk Cu2O (−0.53 e), indicating that the Cu ions are correctly displayed to be the +1 valence state. Over Zn/Cu+-CeO2-x, the Cu ions have the similar charge of −0.44/−0.59 e with that over Cu+-CeO2-x, while the Zn ion has much higher charge of −0.68 e than Cu+ ions, indicating that the more electrons are transferred from the neighboring vacancies to Zn ion compared with Cu ions and thus charge accumulation around the Zn-OV sites. Moreover, the previous work reported the promotion effect of Zn species in CO2 hydrogenation to methanol, for example, Liu et al. have demonstrated that defective ZnO1-x/Cu interfaces present superior activity toward methanol synthesis54. Thus, the enriched charge around the Zn-OV sites makes it possible to act as the active regions to facilitate the adsorption and activation of CO2 and H2, as well as CO2 hydrogenation.

a Differential charge density (∆ρ) of Cu+-CeO2-x and Zn/Cu+-CeO2-x catalysts, the dark blue and light blue regions represent the charge accumulation and depletions. The potential energy profiles and the corresponding structures involved in b the dissociation of molecular adsorption H2 and c CO2 activation. d The relationship of CO2 binding energy with the activation barrier of CO2 hydrogenation to HCOO*. Green: Ce, purple: Zn, blue: Cu, black: C, white: H; red represents the surface O and gray represents O in adsorbed molecules. RWGS reverse water-gas shift reaction, TS transition state.
H2 adsorption and activation over OV-CeO2, Cu+-CeO2-x and Zn/Cu+-CeO2-x catalysts are firstly examined, and the corresponding configurations of H2 molecular and dissociative adsorption are presented in Supplementary Fig. 21 and Supplementary Table 7. Over OV-CeO2, H2 dominantly exists in the form of molecular adsorption. Over Cu+-CeO2-x, H2 is molecular adsorption at the Cu+ site, while the adjacent oxygen site of Cu+ site promotes the spontaneous homolytic dissociation of H2. Over Zn/Cu+-CeO2-x, Cu+ site is still dominantly responsible for molecular adsorption H2, however, the Zn-OV active regions induced the polarization of Zn–O bonds, which favor the heterolytic dissociation of H2.
The dissociation of molecular adsorption H2 over OV-CeO2, Cu+-CeO2-x and Zn/Cu+-CeO2-x catalysts are further analyzed, as presented in Fig. 6b and Supplementary Fig. 22. It can be seen that the dissociation of H2 is exothermic over these three surfaces, correspondingly, the activity of H2 dissociation follows the order of OV-CeO2 (133.1 kJ·mol−1) < Cu+-CeO2-x (109.1 kJ·mol−1) < Zn/Cu+-CeO2-x (84.8 kJ·mol−1), namely, compared to CeO2 catalyst, Cu- doped CeO2 favor the dissociation of H2, and the addition of Zn into Cu+-CeO2-x further promotes H2 dissociation.
Also, as presented in Supplementary Fig. 23 and Supplementary Table 7, both the linear and bent adsorption configurations of CO2 over OV-CeO2, Cu+-CeO2-x and Zn/Cu+-CeO2-x catalysts are observed. CO2 dominantly exists in the form of carbonate with the interaction of a surface oxygen atom, which is more energetically favorable compared to the linear CO2 adsorption. Meanwhile, CO2 adsorption energies in the form of carbonate follow the order of OV-CeO2 (−30.6 kJ·mol−1) < Cu+-CeO2-x (−48.2 kJ·mol−1) < Zn/Cu+-CeO2-x (−58.5 kJ·mol−1), suggesting that the stronger CO2 adsorption induced by the doping with Cu and Cu/Zn is attributed to the formation of Cu+ and Zn-OV active sites.
For CO adsorption, as presented in Supplementary Fig. 24 and Supplementary Table 7, CO prefers to adsorb at the OV site on the OV-CeO2, while the most favorable adsorption site over Cu+-CeO2-x and Zn/Cu+-CeO2-x catalysts is Cu+ site, which is consistent with the CO-DRIFTS results. Moreover, CO adsorption energies follow the order of Zn/Cu+-CeO2-x (−87.5 kJ∙mol−1)>Cu+-CeO2-x (−61.3 kJ∙mol−1) > OV-CeO2 (−36.7 kJ∙mol−1), and the C−O bond lengths also follow the same order of Zn/Cu+-CeO2-x (1.171 Å)>Cu+-CeO2-x (1.167 Å)>OV-CeO2 (1.141 Å), indicating that both the doping of Cu and Cu/Zn promote the adsorption of CO molecule at the Cu+ site over Cu+-CeO2-x and Zn/Cu+-CeO2-x catalysts.
Furthermore, previous studies regarded the formate (HCOO*) and carboxylate (COOH*) species as the key intermediates in the formate and RWGS pathways for CO2 conversion11,41. Thus, both HCOO* and COOH* intermediates are considered for CO2 activation over OV-CeO2, Cu+-CeO2-x and Zn/Cu+-CeO2-x catalysts, as shown in Supplementary Table 8. The potential energy profiles and the corresponding structures are presented in Fig. 6c and Supplementary Fig. 25. Over OV-CeO2, CO2 hydrogenation to HCOO* has the activation barrier and reaction energy of 324.8 and 1.6 kJ·mol−1, which is kinetically and thermodynamically superior to COOH* formation (364.2 and 64.6 kJ·mol−1). The same also occurs over Cu+-CeO2-x and Zn/Cu+-CeO2-x. The formate pathway for CO2 activation is dominant over these three catalysts with the activity order of OV-CeO2 (324.8 kJ·mol−1) < Cu+-CeO2-x (164.8 kJ·mol−1) < Zn/Cu+-CeO2-x (56.0 kJ·mol−1).
Overall, DFT calculations unraveled that CO2 hydrogenation to produce HCOO* is more favorable both in kinetics and thermodynamics than COOH* formation over OV-CeO2, Cu+-CeO2-x and Zn/Cu+-CeO2-x catalysts, indicating that the doping of Cu and Zn into CeO2 matrix could inhibit RWGS reaction via COOH* intermediate and hence enhances methanol selectivity via HCOO* intermediate. Meanwhile, CO2 activation over Cu+-CeO2-x catalyst is more kinetically favorable than that over OV-CeO2 catalyst. Moreover, a small amount of Zn added into CuCe catalyst to form Zn/Cu+-CeO2-x catalyst significantly enhances the activity of CO2 activation to HCOO* and therefore promotes subsequent reactions to product CH3OH. As shown in Fig. 6d, CO2 binding energy has a good linear relationship with the activation barrier of CO2 hydrogenation to HCOO* over OV-CeO2, Cu+-CeO2-x and Zn/Cu+-CeO2-x catalysts, in which Zn/Cu+-CeO2-x catalyst exhibits high CO2 binding energy, and greatly lowers the activation barrier of CO2 hydrogenation to HCOO*. Thus, Zn/Cu+-CeO2-x catalyst facilitates the formation of HCOO* intermediate in comparison with the OV-CeO2 and Cu+-CeO2-x catalysts.
Starting from the HCOO* intermediate, the free energy diagram of CO2 hydrogenation to methanol over Zn/Cu+-CeO2-x catalyst is further investigated, as depicted in Fig. 7. HCOO* hydrogenation to H2COO* is more favorable compared with HCOOH* formation in kinetics (63.1 vs. 131.2 kJ·mol−1). Both the HCOOH* and H2COO* intermediates can further be hydrogenated to H2COOH* with the reaction energies of −22.4 and −1.4 kJ·mol−1, respectively. Interestingly, our results show that H2COO* hydrogenation to H2COOH* is a spontaneous reaction without any barrier over Zn/Cu+-CeO2-x catalyst. Subsequently, the decomposition of H2COOH* to H2CO* is exothermic by 18.5 kJ·mol−1, however, H2CO* successive hydrogenation to CH3OH* via CH3O* intermediate is strongly exothermic by 127.3 and 118.0 kJ·mol−1, respectively. Overall, the formation of CH3OH over Zn/Cu+-CeO2-x catalyst mainly undergoes the pathway of HCOO* + H* →H2COO* + H* →H2COOH* →H2CO* + OH* →CH3O* + H* →CH3OH*, which is kinetically and thermodynamically preferred compared to other possible pathways. Thus, DFT calculations well explain the experimental activity results.

The color codes, RWGS, and TS are the same as those in Fig. 6.
Discussion
A MOFs crystal engineering strategy was employed to develop a series of CuZnCe catalysts for methanol synthesis. A ternary metal Ce-CuZn catalyst was shown to possess high STY (400.3 g·kgcat−1·h−1) and selectivity of 71.1% to methanol (comparable to the commercial CuZnAl system). In particularly, the developed catalyst demonstrated long-term stability when tested for 170 h on stream. It was found that the order of introduction of metal during MOFs preparation influences the growth of MOFs and hence the distribution of active Cu phases in the final catalyst. The Ce-MOF was grown first and then in-situ ion exchange of Cu2+ and Zn2+ ions into Ce-MOF was used to derive Ce-CuZn-MOF. After its pyrolysis, the obtained Ce-CuZn catalyst with many active Cu/Zn-OV-Ce species thus presented robust property for CO2 hydrogenation to methanol with excellent stability, which was comparable to that of the industrial CuZnAl catalyst.
In-situ DRIFTS experiment and DFT calculations provided insights into the reaction mechanism of methanol synthesis, proceeding mainly through the formate path. Methanol synthesis is a structure sensitive reaction and CO-DRIFTS was used to show that the surface composition of different Cu species influenced catalytic activity. Compared to OV-CeO2-x catalyst, the Cu+-CeO2-x catalyst with Cu doping into CeO2-x reduces the activation barrier of CO2 hydrogenation to generate HCOO*. Furthermore, doping Zn into Cu+/CeO2-x can largely facilitate H2 dissociation and the formation of HCOO*. A similar Zn promotion effect on CO2 hydrogenation to CH3OH has already been reported21, and the addition of Zn in the Cu-Zn-Ce oxide catalysts was beneficial to inhibit the RWGS reaction. As a result, CO2 hydrogenation to methanol is preferred to occur by the formate pathway, and CH3OH selectivity can be improved.
In summary, the Ce-CuZn catalyst outperformed by large amount the other catalysts is attributed to the following reasons: The Ce-CuZn catalyst from atomic-level substitution of Cu and Zn into Ce-MOF precursor produced many active Cu/Zn-OV-Ce species. Moreover, the Ce-CuZn catalyst has abundant Cu+ species, a large number of oxygen vacancies, and more weaker basic sites for CO2 adsorption. Kumari et al. reported that the number of oxygen vacancies could influence CO2 activation via lower reaction barriers of CO2 dissociation55. Our DFT calculation results have shown that the incorporation of Cu and Zn into CeO2 with abundant oxygen vacancies can facilitate the H2 dissociation and the formation of HCOO*, thus improving CO2 hydrogenation over Ce-CuZn catalyst via formate intermediates. In addition, it is worth noting that oxygen vacancies in Cu/CeO2 are not beneficial for CO hydrogenation because they are poisoned by adsorbed CO2 to form carbonate-like species40. Thus, the role of oxygen vacancies and their stability in CO/CO2 hydrogenation still needs to be investigated in the development of new catalysts.
This work provides an atomic level regulating strategy towards constructing multi-metal catalysts step by step with effective active sites for CO2 hydrogenation to methanol. The synergistic effect of CuZnCe in the Cu/Zn-OV-Ce species catalyses CO2 activation and the hydrogenation of formates to achieve a high yield of methanol. It was illustrated that the active sites for methanol synthesis would be affected by the kind of metal species, preparation method, and the order of introduction of metal. This approach of precisely engineering active sites by controlling the structure and atom vacancies of the catalyst can be extended to other catalytic systems.
In the future, the metal components should be optimized to consider their economy as the Cu loading has exceeded 50%. The methanol yield can be further improved as the CO selectivity is still high over CuZnCe catalysts. Thus, more strategies should be adopted to suppress the reverse water-gas shift reaction. In addition, as the polymetallic Cu/Zn-OV-Ce active sites have presented excellent catalytic performance, the high-entropy alloy-based catalysts with various unique synergistic effects may have potential application in methanol synthesis, but it would be challenging to illustrate the polymetallic interfaces.
Methods
Chemicals
Cerium nitrate hexahydrate (Ce(NO3)3·6H2O, AR), copper nitrate trihydrate (Cu(NO3)2·3H2O, AR), zinc nitrate hexahydrate (Zn(NO3)2·6H2O, AR), methanol (CH3OH, AR), and ethanol (C2H5OH, AR) were purchased from Sinopharm Chemical Reagent Co., Ltd. 1,3,5-benzenetricarboxylic acid (1,3,5-BTC, 98%) was purchased from Aladdin. Deionized water (DI) was prepared in the laboratory. All chemicals were used without further treatment.
Catalyst preparation
The preparation of CuZnCe catalysts is described in Supplementary Fig. 1 and depicted below.
Synthesis of Ce-CuZn (Route 3): The Ce-MOF was synthesized first, and then Ce-CuZn-MOF was prepared. Specifically, 1.736 g of Ce(NO3)3·6H2O and 0.840 g of 1,3,5-BTC were dissolved into a solvent mixture consisting of 40 mL methanol and 40 mL ethanol, respectively. Then the 1,3,5-BTC solution was added into the cerium nitrate solution, followed by the addition of 40 mL H2O. After stirring at RT for 20 min, the mixture was centrifuged. The supernatant was discarded and the solid obtained from centrifugation was washed with ethanol several times. The product was dried at 60 °C in a vacuum oven for 9 h and in a common oven for another 3 h to obtain the dried Ce-MOF. 0.8 g of the above dried Ce-MOF powder was dispersed into 12 mL of methanol, then a solution of 0.484 g of Cu(NO3)2·3H2O and 0.298 g of Zn(NO3)2·6H2O dissolved in 15 mL methanol was added into the above Ce-MOF suspension under ultrasonic conditions. After sonication for 30 seconds, the mixture was allowed to stand for 20 min and then centrifuged and washed with ethanol several times. Finally, the mixture was dried at 60 °C for 12 h in a vacuum oven for 9 h and in a common oven for another 3 h and then calcined at 450 °C for 5 h in air with a heating rate of 2 °C /min. The obtained samples were denoted as Ce-CuZn-MOF and Ce-CuZn, respectively. In addition, the synthesis of Ce-Cu-MOF and Ce-Zn-MOF was similar to that of the Ce-CuZn-MOF except that the zinc nitrate or copper nitrate was not added, respectively, and the final catalysts were named Ce-Cu and Ce-Zn catalysts.
The preparation of other CuZnCe series catalysts is presented in the Supplementary Information.
Catalyst characterization
All the detailed catalyst characterizations are depicted in the Supplementary Information.
DFT calculations
The detailsof the DFT calculations are shown in the Supplementary Information.
Catalyst evaluation
The CuZnCe catalysts were evaluated in a fixed-bed reactor under reaction conditions of 2.0 ~ 2.8 MPa, 10,000 ~ 20,000 mL·gcat−1·h−1, 220 ~ 320 °C, and a H2: CO2: N2 ratio of 72: 24: 1. The detailed catalyst evaluation method is described in the Supplementary Information.
Data availability
The data supporting the findings of this study are available within the main text, the Supplementary Information file, the Source Data files, or from the corresponding authors upon request. Source data are provided with this paper and also deposited in Figshare repository (https://doi.org/10.6084/m9.figshare.25129247). Source data are provided with this paper.
References
Ye, R. P. et al. CO2 hydrogenation to high-value products via heterogeneous catalysis. Nat. Commun. 10, 5698 (2019).
Yang, M. et al. Probing the nature of zinc in copper-zinc-zirconium catalysts by operando spectroscopies for CO2 hydrogenation to methanol. Angew. Chem. Int. Ed. 62, e202216803 (2023).
Pinheiro Araujo, T. et al. Flame-made ternary Pd-In2O3-ZrO2 catalyst with enhanced oxygen vacancy generation for CO2 hydrogenation to methanol. Nat. Commun. 13, 5610 (2022).
Monai, M. et al. Restructuring of titanium oxide overlayers over nickel nanoparticles during catalysis. Science 380, 644–651 (2023).
Kumar, A., Daw, P. & Milstein, D. Homogeneous catalysis for sustainable energy: Hydrogen and methanol economies, fuels from biomass, and related topics. Chem. Rev. 122, 385–441 (2021).
Zhong, J. et al. State of the art and perspectives in heterogeneous catalysis of CO2 hydrogenation to methanol. Chem. Soc. Rev. 49, 1385–1413 (2020).
Tian, P., Wei, Y., Ye, M. & Liu, Z. Methanol to olefins (MTO): From fundamentals to commercialization. ACS Catal. 5, 1922–1938 (2015).
Jiang, X., Nie, X., Guo, X., Song, C. & Chen, J. G. Recent Advances in Carbon Dioxide Hydrogenation to Methanol via Heterogeneous Catalysis. Chem. Rev. 120, 7984–8034 (2020).
Kordus, D. et al. Shape-dependent CO2 hydrogenation to methanol over Cu2O nanocubes supported on ZnO. J. Am. Chem. Soc. 145, 3016–3030 (2023).
Shao, Y., Kosari, M., Xi, S. & Zeng, H. C. Single solid precursor-derived three-dimensional nanowire networks of CuZn-silicate for CO2 hydrogenation to methanol. ACS Catal. 12, 5750–5765 (2022).
Wang, Y. et al. Strong evidence of the role of H2O in affecting methanol selectivity from CO2 hydrogenation over Cu-ZnO-ZrO2. Chem 6, 419–430 (2020).
Wang, J. et al. A highly selective and stable ZnO-ZrO2 solid solution catalyst for CO2 hydrogenation to methanol. Sci. Adv. 3, e1701290 (2017).
Gao, Y. et al. Recent advances in intensified ethylene production-a review. ACS Catal. 9, 8592–8621 (2019).
Prašnikar, A., Pavlišič, A., Ruiz-Zepeda, F., Kovač, J. & Likozar, B. Mechanisms of copper-based catalyst deactivation during CO2 reduction to methanol. Ind. Eng. Chem. Res. 58, 13021–13029 (2019).
Liang, B. et al. Investigation on deactivation of Cu/ZnO/Al2O3 catalyst for CO2 hydrogenation to methanol. Ind. Eng. Chem. Res. 58, 9030–9037 (2019).
Graciani, J. et al. Highly active copper-ceria and copper-ceria-titania catalysts for methanol synthesis from CO2. Science 345, 546–550 (2014).
Rasteiro, L. F. et al. Insights into the alloy-support synergistic effects for the CO2 hydrogenation towards methanol on oxide-supported Ni5Ga3 catalysts: An experimental and DFT study. Appl. Catal. B: Environ. 302, 120842 (2022).
Divins, N. J. et al. Operando high-pressure investigation of size-controlled CuZn catalysts for the methanol synthesis reaction. Nat. Commun. 12, 1435 (2021).
Beck, A. et al. Following the structure of copper-zinc-alumina across the pressure gap in carbon dioxide hydrogenation. Nat. Catal. 4, 488–497 (2021).
Li, M. M. J. et al. CO2 hydrogenation to methanol over catalysts derived from single cationic layer CuZnGa LDH precursors. ACS Catal. 8, 4390–4401 (2018).
Zhu, J. et al. Flame synthesis of Cu/ZnO-CeO2 catalysts: Synergistic metal-dupport interactions promote CH3OH selectivity in CO2 hydrogenation. ACS Catal. 11, 4880–4892 (2021).
Hou, X. X. et al. Improved methanol synthesis from CO2 hydrogenation over CuZnAlZr catalysts with precursor pre-activation by formaldehyde. J. Catal. 379, 147–153 (2019).
Kattel, S., Ramírez, P. J., Chen, J. G., Rodriguez, J. A. & Liu, P. Active sites for CO2 hydrogenation to methanol on Cu/ZnO catalysts. Science 355, 1296–1299 (2017).
Stangeland, K., Li, H. & Yu, Z. CO2 hydrogenation to methanol: the structure–activity relationships of different catalyst systems. Energ. Ecol. Environ. 5, 272–285 (2020).
Kosari, M. et al. Revamping SiO2 spheres by core-shell porosity endowment to construct a mazelike nanoreactor for enhanced catalysis in CO2 hydrogenation to methanol. Adv. Funct. Mater. 31, 2102896 (2021).
Behrens, M. et al. The active site of methanol synthesis over Cu/ZnO/Al2O3 industrial catalysts. Science 336, 893–897 (2012).
Amann, P. et al. The state of zinc in methanol synthesis over a Zn/ZnO/Cu(211) model catalyst. Science 376, 603–608 (2022).
Zabilskiy, M. et al. The unique interplay between copper and zinc during catalytic carbon dioxide hydrogenation to methanol. Nat. Commun. 11, 2409 (2020).
Xia, W., Mahmood, A., Zou, R. & Xu, Q. Metal–organic frameworks and their derived nanostructures for electrochemical energy storage and conversion. Energy Environ. Sci. 8, 1837–1866 (2015).
Ye, R. P. et al. Synthesis of robust MOF-derived Cu/SiO2 catalyst with low copper loading via sol-gel method for the dimethyl oxalate hydrogenation reaction. ACS Catal. 8, 3382–3394 (2018).
Zhang, B. et al. Designing MOF nanoarchitectures for electrochemical water splitting. Adv. Mater. 33, e2006042 (2021).
Bai, X. J., Zhai, X., Zhang, L. Y., Fu, Y. & Wei, Q. Site-directed reduction engineering within bimetal-organic frameworks for efficient size-selective catalysis. Matter 4, 2919–2935 (2021).
Wang, F. et al. A Photoactivated Cu-CeO2 Catalyst with Cu-[O]-Ce Active Species Designed through MOF Crystal Engineering. Angew. Chem. Int. Ed. 59, 8203–8209 (2020).
Yang, B., Deng, W., Guo, L. & Ishihara, T. Copper-ceria solid solution with improved catalytic activity for hydrogenation of CO2 to CH3OH. Chin. J. Catal. 41, 1348–1359 (2020).
Liu, C., Shi, J. W., Gao, C. & Niu, C. M. Manganese oxide-based catalysts for low-temperature selective catalytic reduction of NOx with NH3: A review. Appl. Catal. A: Gen. 522, 54–69 (2016).
Niu, J. et al. Process and mechanism of toluene oxidation using Cu1-yMn2CeyOx/sepiolite prepared by the co-precipitation method. J. Hazard. Mater. 357, 332–340 (2018).
Yu, J. et al. Sulfur poisoning resistant mesoporous Mn-base catalyst for low-temperature SCR of NO with NH3. Appl. Catal. B Environ. 95, 160–168 (2010).
Alvarez, A. et al. Challenges in the greener production of formates/formic acid, methanol, and DME by heterogeneously catalyzed CO2 hydrogenation processes. Chem. Rev. 117, 9804–9838 (2017).
Zhang, Y. et al. Highly efficient Cu/CeO2-hollow nanospheres catalyst for the reverse water-gas shift reaction: Investigation on the role of oxygen vacancies through in situ UV-Raman and DRIFTS. Appl. Surf. Sci. 516, 146035 (2020).
Zhu, J. et al. Mechanism and nature of active sites for methanol synthesis from CO/CO2 on Cu/CeO2. ACS Catal. 10, 11532–11544 (2020).
Liu, S. et al. Producing ultrastable Ni-ZrO2 nanoshell catalysts for dry reforming of methane by flame synthesis and Ni exsolution. Chem Catal. 2, 2262–2274 (2022).
Cheng, X. et al. NO reduction by CO over copper catalyst supported on mixed CeO2 and Fe2O3: Catalyst design and activity test. Appl. Catal. B: Environ. 239, 485–501 (2018).
Jiang, J., Yang, H., Jiang, H., Hu, Y. & Li, C. Boosting catalytic activity of Cu-Ce solid solution catalysts by flame spray pyrolysis with high Cu+ concentration and oxygen vacancies. Chem. Eng. J. 471, 144439 (2023).
Chen, J. et al. Characterization and catalytic performance of Cu/CeO2 and Cu/MgO-CeO2 catalysts for NO reduction by CO. Appl. Catal. A: Gen. 363, 208–215 (2009).
Chen, A. et al. Structure of the catalytically active copper-ceria interfacial perimeter. Nat. Catal. 2, 334–341 (2019).
Mosrati, J. et al. Tiny species with big impact: High activity of Cu single atoms on CeO2–TiO2 deciphered by operando spectroscopy. ACS Catal. 11, 10933–10949 (2021).
Abdel-Mageed, A. M. et al. Highly active and stable single-atom Cu catalysts supported by a metal-organic framework. J. Am. Chem. Soc. 141, 5201–5210 (2019).
Szanyi, J., Kwak, J. H., Zhu, H. & Peden, C. H. Characterization of Cu-SSZ-13 NH3 SCR catalysts: an in situ FTIR study. Phys. Chem. Chem. Phys. 15, 2368–2380 (2013).
Guo, Y. et al. Low-temperature CO2 methanation over CeO2-supported Ru single atoms, nanoclusters, and nanoparticles competitively tuned by strong metal–support interactions and H-spillover effect. ACS Catal. 8, 6203–6215 (2018).
Chen, S., Zou, H., Liu, Z. & Lin, W. DRIFTS study of different gas adsorption for CO selective oxidation on Cu–Zr–Ce–O catalysts. Appl. Surf. Sci. 255, 6963–6967 (2009).
Polster, C. S., Nair, H. & Baertsch, C. D. Study of active sites and mechanism responsible for highly selective CO oxidation in H2 rich atmospheres on a mixed Cu and Ce oxide catalyst. J. Catal. 266, 308–319 (2009).
Wang, W., Qu, Z., Song, L. & Fu, Q. CO2 hydrogenation to methanol over Cu/CeO2 and Cu/ZrO2 catalysts: Tuning methanol selectivity via metal-support interaction. J. Energy Chem. 40, 22–30 (2020).
Ye, R. P. et al. High-performance of nanostructured Ni/CeO2 catalyst on CO2 methanation. Appl. Catal. B: Environ. 268, 118474 (2020).
Liu, X. et al. In situ spectroscopic characterization and theoretical calculations identify partially reduced ZnO1-x/Cu interfaces for methanol synthesis from CO2. Angew. Chem. Int. Ed. 134, e202202330 (2022).
Kumari, N., Haider, M. A., Agarwal, M., Sinha, N. & Basu, S. Role of reduced CeO2(110) surface for CO2 reduction to CO methanol. J. Phys. Chem. C. 120, 16626–16635 (2016).
Chen, J. et al. The synergetic mechanism between copper species and ceria in NO abatement over Cu/CeO2 catalysts. Appl. Catal. A: Gen. 377, 121–127 (2010).
Wang, Z. Q. et al. High-performance and long-lived Cu/SiO2 nanocatalyst for CO2 hydrogenation. ACS Catal. 5, 4255–4259 (2015).
Yang, H. Y. et al. Core–shell structured Cu@m-SiO2 and Cu/ZnO@m-SiO2 catalysts for methanol synthesis from CO2 hydrogenation. Catal. Commun. 84, 56–60 (2016).
Jiang, Y. et al. Slurry methanol synthesis from CO2 hydrogenation over micro-spherical SiO2 support Cu/ZnO catalysts. J. CO2 Util. 26, 642–651 (2018).
Toyir, J., de la Piscina, P. R., Fierro, J. L. G. & Homs, N. Highly effective conversion of CO2 to methanol over supported and promoted copper-based catalysts: influence of support and promoter. Appl. Catal. B: Environ. 29, 207–215 (2001).
Liu, X. M., Lu, G. Q. & Yan, Z. F. Nanocrystalline zirconia as catalyst support in methanol synthesis. Appl. Catal. A: Gen. 279, 241–245 (2005).
Gao, P. et al. Influence of modifier (Mn, La, Ce, Zr and Y) on the performance of Cu/Zn/Al catalysts via hydrotalcite-like precursors for CO2 hydrogenation to methanol. Appl. Catal. A: Gen. 468, 442–452 (2013).
Lei, H., Hou, Z. & Xie, J. Hydrogenation of CO2 to CH3OH over CuO/ZnO/Al2O3 catalysts prepared via a solvent-free routine. Fuel 164, 191–198 (2016).
Tan, Q., Shi, Z. & Wu, D. CO2 hydrogenation to methanol over a highly active Cu−Ni/CeO2-nanotube natalyst. Ind. Eng. Chem. Res. 57, 10148–10158 (2018).
Hu, J. et al. Sulfur vacancy-rich MoS2 as a catalyst for the hydrogenation of CO2 to methanol. Nat. Catal. 4, 242–250 (2021).
An, B. et al. Confinement of ultrasmall Cu/ZnOx nanoparticles in metal-organic frameworks for selective methanol synthesis from catalytic hydrogenation of CO2. J. Am. Chem. Soc. 139, 3834–3840 (2017).
Choi, E. J., Lee, Y. H., Lee, D.-W., Moon, D.-J. & Lee, K.-Y. Hydrogenation of CO2 to methanol over Pd–Cu/CeO2 catalysts. Mol. Catal. 434, 146–153 (2017).
Zhang, C., Liao, P., Wang, H., Sun, J. & Gao, P. Preparation of novel bimetallic CuZn-BTC coordination polymer nanorod for methanol synthesis from CO2 hydrogenation. Mater. Chem. Phys. 215, 211–220 (2018).
Sun, K. H. et al. Hydrogenation of CO2 to methanol over In2O3. Catal. J. CO2 Util. 12, 1–6 (2015).
Dang, S. et al. Rationally designed indium oxide catalysts for CO2 hydrogenation to methanol with high activity and selectivity. Sci. Adv. 6, eaaz2060 (2020).
Acknowledgements
This work was supported financially by the Natural Science Foundation of Jiangxi Province for Distinguished Young Scholars (20232ACB213001, R.Y.), National Natural Science Foundation of China (U23A20132, R.Z.; 22005296, R.Y.), National Key R&D Program of China (2022YFA1504503, J.L.), Science Foundation for Distinguished Young Scholar of Shanxi Province (20210302121005, R.Z.), Natural Science Foundation of Chongqing, China (CSTB2022NSCQ-MSX0231, R.Y.), Natural Science Foundation of Jiangxi Province (20224BAB213015, R.Y.), and the Thousand Talents Plan of Jiangxi Province (jxsq2023101072, R.Y.). The authors would also like to thank Dr. Caixia Meng from Dalian Institute of Chemical Physics, Chinese Academy of Sciences for the quasi in-situ XPS analysis and Shiyanjia Lab (www.shiyanjia.com) for the TEM test. A.G. thanks the support from the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division, Catalysis Science Program to the SUNCAT Center for Interface Science and Catalysis.
Author information
Authors and Affiliations
Contributions
R.Z., M.D., Z.J. and J.L. supervised the project. R.Y. and M.D. conceived and performed the experiments. L.M., B.W. and R.Z. performed DFT calculations and made their interpretations. X.W. carried out the SEM measurement. J.M. and Z.J. performed the EXAFS analysis. X.H. and Y.M. performed in-situ DRIFTS characterizations. A.G. and W.L. helped to interpret the EXAFS, TEM and XPS data. R.Y., L.M. and J.M. wrote the draft and the other authors revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ye, R., Ma, L., Mao, J. et al. A Ce-CuZn catalyst with abundant Cu/Zn-OV-Ce active sites for CO2 hydrogenation to methanol. Nat Commun 15, 2159 (2024). https://doi.org/10.1038/s41467-024-46513-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-46513-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
