
Abstract
The human voltage-gated potassium channel KCNQ2/KCNQ3 carries the neuronal M-current, which helps to stabilize the membrane potential. KCNQ2 can be activated by analgesics and antiepileptic drugs but their activation mechanisms remain unclear. Here we report cryo-electron microscopy (cryo-EM) structures of human KCNQ2-CaM in complex with three activators, namely the antiepileptic drug cannabidiol (CBD), the lipid phosphatidylinositol 4,5-bisphosphate (PIP2), and HN37 (pynegabine), an antiepileptic drug in the clinical trial, in an either closed or open conformation. The activator-bound structures, along with electrophysiology analyses, reveal the binding modes of two CBD, one PIP2, and two HN37 molecules in each KCNQ2 subunit, and elucidate their activation mechanisms on the KCNQ2 channel. These structures may guide the development of antiepileptic drugs and analgesics that target KCNQ2.
Similar content being viewed by others

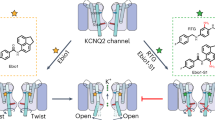

Introduction
The voltage-gated potassium channel KCNQ family contains five members, among which KCNQ1 carries the cardiac slow delayed-rectifier (IKs) current and repolarizes the cardiac action potential1,2,3 whereas the KCNQ2-5 channels modulate the neuronal excitability4. Among the neuronal KCNQ channels, KCNQ2 and KCNQ3 form homo- and hetero-tetrameric channels and are responsible for the M-current, which contributes to the stability of the membrane potential5,6,7. Mutations in the KCNQ2 or KCNQ3 lead to hyper-excitation of neurons and the occurrence of seizures, such as benign familial neonatal seizures (BFNS or BFNE)8,9 and epileptic encephalopathy (EE)10,11,12, making KCNQ2 an important target of antiepileptic drugs13,14,15. Besides, modulation of KCNQ2 is a potential therapeutic strategy for the treatment of some other neuronal diseases such as pain16,17,18,19, Parkinson’s disease20,21, and schizophrenia22, which are also characterized by hyper-excitation of neurons.
To develop new antiepileptic drugs and analgesics, a number of natural or synthetic compounds targeting KCNQ2 have been identified. Retigabine (RTG) was an approved antiepileptic drug that potentiates the activation of potassium channels23,24. The non-opioid analgesic flupirtine, which has been used for the treatment of acute and chronic pain in Europe, can also activate KCNQ225,26. Cannabidiol (CBD) is one of the main effective compounds in cannabis and a clinically effective antiepileptic drug. Pharmacological and structural studies reveled that multiple ionotropic receptors contributing to the CBD’s antiepileptic activity include blockade of Nav and desensitization of TRPV channels27,28,29,30. Recently, CBD was shown to directly activate the KCNQ2/KCNQ3 channel31,32. We previously developed a series of KCNQ2 activators, exemplified by ztz240 and HN37 (pynegabine)33,34, the latter has been in the clinical trial phase I34. Because many of these KCNQ2 activators including RTG can cause side effects due to their poor target selectivity35,36,37, revealing the structural basis for the ligand recognition and activation of KCNQ2 will advance the development of KCNQ2-selective activators and overcome the side effects of current analgesics and antiepileptic drugs.
Recently, structures of KCNQ channels revealed the overall architecture and gating mechanism of KCNQs38,39,40,41,42,43,44. We determined structures of KCNQ2 in complex with its activators RTG and ztz240, both in the closed state38. KCNQ2 adopts the canonical structural organization of the tetrameric KV channels in a domain-swapped arrangement38. Each KCNQ2 subunit contains six transmembrane segments (S1–S6): S1–S4 form the voltage-sensing domain (VSD) and the S5–S6 constitute the pore domain (PD). The C-terminal domain (CTD), which contains three helices, namely HA, HB, and HC, interacts with the calcium-modulated protein calmodulin (CaM). Upon membrane depolarization, the VSD activates and induces conformational changes to open the PD via the coupling of the VSD and the PD (electro-mechanical coupling, E–M coupling). The lipid phosphatidylinositol 4,5-bisphosphate (PIP2) is required for the E–M coupling and depleting PIP2 results in a closed pore with normal VSD activation45. Moreover, many epilepsy-associated mutations in KCNQ2 disrupt E-M coupling and RTG can restore the E–M coupling in some cases46. Since all the current KCNQ2 structures are in decoupled conformation with activated VSDs and a closed PD38, how PIP2 and antiepileptic drugs activate KCNQ2 by enhancing the E–M coupling remains largely unknown.
In this report, we use cryo-electron microscopy (cryo-EM) and electrophysiology to study the modulation mechanisms of KCNQ2 E–M coupling by the activators PIP2, CBD, and HN37. We present 2.5–3.5 Å resolution structures of human KCNQ2-CaM complex in the CBD-bound closed, HN37-bound closed, and CBD-PIP2-bound open states (Table 1). This study not only elucidates the molecular basis for the activation of the KCNQ2 by different small molecules but also may guide the development of KCNQ2-selective activators as potential analgesics and antiepileptic drugs.
Results
Structure determination of KCNQ2-CaM in the presence of PIP2
Previously, we determined the apo KCNQ2-CaM structure (KCNQ2-CaMapo, PDB: 7CR3)38, which was in decoupled conformation with activated VSDs and a closed gate. Because PIP2 can potentiate the activation of KCNQ channels by enhancing the E–M coupling, to capture an open-state KCNQ2, we solved the structure of the KCNQ2-CaM complex in the presence of 1 mM PIP2 (KCNQ2-CaMPIP2(-), where (-) indicates that PIP2 was added in the protein sample but not observed in the structure throughout this paper) at 2.7 Å resolution (Supplementary Fig. 1a–h, Table 1). Unfortunately, we were unable to capture the open-state KCNQ2 structure with the presence of PIP2 in the sample, nor PIP2 molecule was observed in the map, suggesting that PIP2 does not tightly bind to KCNQ2 in vitro under our structure determination conditions. The low affinity of PIP2 bound to KCNQ2 was reported in a previous electrophysiological study which showed that the half-maximal value (EC50) for diC8-PIP2 on KCNQ2 was ~205 µM47. By contrast, in the case of the human KCNQ1-CaM-KCNE3 complex, with the presence of PIP2, the open-state KCNQ1 structure was readily obtained41.
The KCNQ2-CaMPIP2(-) structure is similar to the KCNQ2-CaMapo structure, with activated VSDs and a closed gate38. In the VSD, the side chain of Arg210 (R5) points to the gating charge transfer center formed by Phe137, Glu140, and Asp172 (Supplementary Fig. 1i). The activation gate is closed by two layers of constriction formed by residues Ser314 and Leu318, with the shortest diagonal atom-to-atom distance of 4–6 Å at the activation gate (Supplementary Fig. 1j). In the CTD, HA and HB helices are wrapped by CaM. As no inter-subunit interactions are observed in the CaM, the conformation of HA/HB/CaM is likely stabilized by interactions between the linker connecting the fifth and sixth helix of CaM (H5-H6 linker) and S0 and the S2–S3 linker from the VSD (Supplementary Fig. 1k).
Structural basis for the CBD recognition
To reveal the molecular mechanism of CBD activation on KCNQ2-CaM, we next determined the CBD-bound KCNQ2-CaM structure (KCNQ2-CaMCBD) at 3.3 Å resolution (Fig. 1a, Table 1, Supplementary Figs. 2a–e, 3a). In the TMD, the high-quality map of KCNQ2-CaMCBD allows us to clearly resolve two CBD molecules located in the hydrophobic cleft formed by S6 from the first subunit (S6I), S5, pore helix (PH), and S6 from the second subunit (S5II, PHII, and S6II), and S1 from the third subunit (S1III) (Fig. 1a, b). The configurations of CBDs were assigned based on their large cyclohexenyl head group and the short hydrophobic tail of the pentylphenyl group (Fig. 1b). One CBD molecule (CBDA) sits deep in the classical fenestration pocket of voltage-gated ion channels lined by S6I, S5II, and S6II (Fig. 1c). CBDA mainly forms hydrophobic interactions with residues Leu299, Ile300 and Phe304 in S6I, Leu232, Trp236, and Phe240 in S5II, and Phe305, Pro308, and Leu312 in S6II (Fig. 1c). In addition, it also forms hydrogen bonds with main-chain carbonyls of Leu299 in S6I and Leu232 in S5II (Fig. 1c). The other CBD molecule (CBDB) is localized at an upper, shallow pocket formed by S5II, PHII, and S1III, covering the upper part of CBDA (Fig. 1d). Similarly, CBDB also mainly forms hydrophobic interactions with surrounding residues such as Leu299 and Ile300 in S6I, Trp236, Phe240, and Leu243 in S5II, Leu268 and Leu272 in PHII, and Phe104 in S1III (Fig. 1d). Specifically, CBDB forms a hydrogen bond with the side chains of the conserved Trp236 in S5II, as well as Thr296 in S6I (Fig. 1d).

a The 3D reconstruction and the cartoon model of KCNQ2-CaMCBD. b The density maps of two CBD molecules in different orientations at the contour level of 3.6 σ. c Interactions between CBDA and KCNQ2. The dashed lines indicate hydrogen bonds. d Interactions between CBDB and KCNQ2. The dashed lines indicate hydrogen bonds. e Representative traces of KCNQ2 currents in the absence (black) and presence of 10 μM CBD (red) or 100 nM HN37 (blue). (Inset) The recording protocol. The cell was clamped at −80 mV and the KCNQ2 currents were elicited by a depolarizing voltage step of −40 mV for 1500 ms, followed by a hyperpolarizing step to −120 mV for 500 ms. f The dose-response curve of CBD on the current amplitude of KCNQ2 at −40 mV. Data are presented as mean ± SEM; n = 3 (0.1 μM), 3 (1 μM), 5 (3 μM), 5 (10 μM), and 3 (30 μM) individual cells. g Voltage-dependent activation curves of KCNQ2 before (black) and after (red) application of 10 μM CBD. Data are presented as mean ± SEM; n = 5 individual cells. h The half maximal activation voltage shift (ΔV1/2) of WT KCNQ2 and mutants by 10 μM CBD. Data are presented as mean ± SEM. An unpaired two-tailed t test was performed to compare WT and each mutant. **p = 0.0036 for F104A, *p = 0.0307 for W236A, **p = 0.0083 for L272A, and p = 0.3123 for T296A. For W236A, n = 3 individual cells; For WT and other mutants, n = 5 individual cells. i CBDs induce a rotamer change of Trp236 in S5. j Structural comparison of VSDs from KCNQ2-CaMapo and KCNQ2-CaMCBD when the whole channels are aligned. k The closed activation gate of KCNQ2-CaMCBD. The dashed lines show diagonal atom-to-atom distance (in Å) at the constriction-lining residues Ser314 and Leu318. For (f–h), source data are provided in the Source Data file.
To confirm the two CBD binding sites revealed by the structure data, we expressed KCNQ2 in Chinese hamster ovary (CHO) cells and recorded the current using the whole-cell patch-clamp technique (Fig. 1e). Under −40 mV, 10 μM CBD enhances the outward current of KCNQ2 by an average factor of 1.55 ± 0.07 (Fig. 1f). CBD enlarges the current amplitudes in a dose-dependent manner with an EC50 value of 4.50 ± 1.79 μM (n = 5) at −40 mV (Fig. 1f). 10 μM CBD also potentiates the voltage activation of KCNQ2 by left-shifting the conductance-voltage (G–V) curve, with an average half maximal activation voltage shift (∆V1/2) of −10.11 ± 1.08 mV (Fig. 1g). Thus, CBD increases the voltage sensitivity of KCNQ2, similar to the previous report31. We next examined the CBD potentiation effect on KCNQ2 channels with mutations on the CBD-interacting residues. Among the nine mutants we tested, five (L232A, L268A, A295L, F304A, and F305A) were non-functional and one (T296A) showed similar CBD sensitivity as WT (Fig. 1h, Supplementary Fig. 4, Supplementary Data 1). The rest three, namely F104A, W236A, and L272A, all displayed a significantly reduced potentiation activity of CBD by attenuating the left shift of the G–V curve (∆V1/2) (Fig. 1h, Supplementary Fig. 4). While Phe104 and Leu272 specifically interact with CBDB, Trp236 forms interactions with both CBD molecules. These results collectively support the binding sites of CBDs in KCNQ2.
Structural comparison of KCNQ2-CaMCBD and KCNQ2-CaMapo reveals that the two CBDs induce a rotamer change of Trp236 in S5II, which eliminates the potential clash with CBDA on one hand and facilitates the formation of a hydrogen bond with CBDB on the other hand (Fig. 1i). In addition, a 2–3 Å shift of the whole VSD is observed (Fig. 1j). However, this disturbance of VSD does not completely destroy interactions between VSD and the attached CaM (Fig. 1a). The gate in KCNQ2-CaMCBD remains closed in the absence of PIP2 (Fig. 1k), hindering the elucidation of the activation mechanism of CBD.
The CBD- and PIP2-bound open-state structure of KCNQ2-CaM
To decipher the CBD and PIP2 activation mechanism, we then determined the PIP2-CBD-bound KCNQ2-CaM structure (KCNQ2-CaMCBD-PIP2) at 3.1 Å resolution (Fig. 2a, Table 1, Supplementary Figs. 2f–i,3b). Like those in KCNQ2-CaMCBD, two CBD molecules are clearly observed in the map of KCNQ2-CaMCBD-PIP2 (Fig. 2a). In addition, between VSD and PD, one PIP2 molecule is assigned, with its fatty acid chains switched by S1III/S4III and S5II in the transmembrane region and its inositol 1,4,5-trisphosphate head group pointing to S6III helix in the cytosol (Fig. 2b). The inositol 1,4,5-trisphosphate group interacts with positively charged residues Arg87 in S0III, Arg89 in S1III, Arg214 in S4III, Lys230 in S5II, and Lys327 in S6III (Fig. 2b). This PIP2 binding site is similar to the P2 site of PIP2 in the structure of KCNQ4 in complex with PIP2 and the agonist ML21343. Sequence alignment of KCNQ1-5 show that these PIP2-interacting residues are highly conserved across KCNQ2-5 but not in KCNQ1 (Supplementary Fig. 5), which may explain why this PIP2-binding site is not observed in KCNQ1 structures39,41.

a The 3D reconstruction and the cartoon model of KCNQ2-CaMCBD-PIP2. b The binding site of PIP2 in KCNQ2. Side chains of residues in KCNQ2 involved in the interactions with PIP2 are shown as sticks. c Voltage-dependent activation curves of WT KCNQ2 and mutants. Data are presented as mean ± SEM. n values are indicated in (d). d V1/2 values of WT KCNQ2 and mutants. Data are presented as mean ± SEM. One-way ANOVA with Dunnett’s multiple comparisons test was applied. p and n values compared to WT (n = 12) are p = 0.9379 and n = 7 for R87A, ***p = 0.0003 and n = 7 for R89A, p = 0.1484 and n = 9 for R214A, and **p = 0.0015 and n = 10 for R327A. n represents the number of experiments from individual cells. e Different structure arrangements of CTD and CaM in KCNQ2-CaMapo and KCNQ2-CaMCBD-PIP2. S6, S6-HA linker, HA, HB, and HC helices of KCNQ2 are colored in blue, red, cornflower-blue, green, and yellow, respectively. The N-lobe and C-lobe of CaM are shown in wheat and pink, respectively. f Interactions between S6 and HB-HC linker. The dashed lines indicate the salt bridge Arg332-Asp566 and the hydrogen bonds Arg325-Asp563 and His328-Pro561. g Structural comparison of the HA, HB, and HC helices in KCNQ2-CaMapo (wheat) and KCNQ2-CaMCBD-PIP2 (salmon) in the top view with TMD and HA/HB from the other three subunits omitted for clarity. The N-terminal residue Val564 of HC rotates by ∼40° from KCNQ2-CaMapo to KCNQ2-CaMCBD-PIP2. h Overlay of the pore domains of KCNQ2-CaMapo (wheat) and KCNQ2-CaMCBD-PIP2 (salmon) structures showing the conformational change in the ion-conducting pathway. The S6 helix bends outward upon PIP2 binding at the point of PAG segment. i The open activation gate of KCNQ2-CaMCBD-PIP2. The dashed lines show diagonal atom-to-atom distance (in Å) at the constriction-lining residues Gly310, Ser314, and Leu318. For (c, d), source data are included in the Source Data file.
To test whether these five positively charged residues are essential for the voltage activation of KCNQ2 via the PIP2, we then introduced mutations at the PIP2 binding site in KCNQ2 and recorded the currents of CHO cells expressing WT and mutant KCNQ2. While the mutations R87A and R214A did not change the voltage activation of KCNQ2, K327A reduced the voltage sensitivity of KCNQ2 by right-shifting the G–V curve (Fig. 2c, d). In addition, the mutant K230A lost the activity, emphasizing its essential role in the PIP2 binding. The mutation R89A left-shifts the G–V curve, whose mechanism remains unknown (Fig. 2c, d).
The binding of CBD and PIP2 induces a large structural arrangement of the CTD and CaM, as well as S6 (Fig. 2e–i). HA and HB, together with their surrounding CaM, rotate by almost 180°. HB which sits on the right side of HA in KCNQ2-CaMapo moves to the left side of HA in KCNQ2-CaMCBD-PIP2 (Fig. 2e). Along with this rotation, S6 and HA, which were connected by a loop linker of residues 330EKR332, now form a continuous straight helix (Fig. 2e, Supplementary Movies 1 and 2). This re-folding of the 330EKR332 motif is likely energy-favorable, as multiple interactions including the salt bridge and hydrogen bonds are introduced at the interface of S6/HA and the HB-HC linker (Fig. 2f). Consequently, the coiled-coil-forming helix HC, which is directly connected to HB, rotates by ~40° (Fig. 2g). This structural arrangement of CTD and S6 causes the bending of the S6 C-terminal half at the conserved “PAG” segment away from the central axis of the channel pore, resulting in the opening of the activation gate with the shortest diagonal atom-to-atom distance extending to 8–9 Å at the activation gate (Fig. 2h, i, and Supplementary Movie 3). Therefore, bound with both CBD and PIP2, KCNQ2-CaMCBD-PIP2 is in an open state. This KCNQ2-CaMCBD-PIP2 structure resembles the previously reported PIP2-bound open-state structures of KCNQ1 and KCNQ439,41,43.
The CBD and PIP2 activation mechanism
How do CBD and PIP2 potentiate the voltage activation of KCNQ2? First, the structure alignment of KCNQ2-CaMapo and KCNQ2-CaMCBD shows that the CBD induces a 2–3 Å shift of VSD, which tends to disturb interactions of VSD and CaM (Fig. 1j). Second, structure alignment KCNQ2-CaMCBD and KCNQ2-CaMCBD-PIP2 shows that PIP2 induces a further 3–4 Å shift of VSD (Fig. 3a), which completely destroys interactions of VSD and CaM, resulting in the release of CaM and HA/HB from the VSD, followed by the rotation of CaM and HA/HB, as well as the folding of S6 and HA into one continuous helix (Fig. 2d). In addition, by interaction with the Lys327 in S6, PIP2 “pulls” S6 up and stabilizes the open-state conformation (Fig. 3b).

a Structural comparison of VSDs from KCNQ2-CaMCBD and KCNQ2-CaMCBD-PIP2 when the whole channels are aligned. b PIP2 “pulls” S6 up by interaction with the Lys327 in S6. c The proposed CBD and PIP2 activation mechanism of KCNQ2 in vivo. d The proposed CBD and PIP2 activation model of KCNQ2 in vitro.
These CBD and PIP2 activation insights are obtained from comparisons of the KCNQ2-CaM structures with or without ligands. In fact, in vivo the channel activation is initiated by the depolarization of membrane potential instead of the binding of PIP2 because the PIP2 is already bound in KCNQ2. Then what the activation process is in vivo? Based on the above analyses, we propose that in the absence of CBD, upon membrane potential depolarization, S4 moves up, which would change the conformation and (or) the location of PIP2. PIP2 then disturbs interactions between VSD and CaM, resulting in the release of CaM and HA/HB from the VSD on one hand, and pulls S6 up to stabilize the open-state conformation on the other hand (Fig. 3c). However, in vitro the binding of PIP2 in the activated VSD is not that strong and PIP2 is easy to dissociate, which can explain why we were unable to capture the PIP2-bound open-state KCNQ2-CaM structure in the presence of PIP2 alone (KCNQ2-CaMPIP2(-)) (Fig. 3d). The binding of CBDs likely stabilizes the PIP2-bound state by pushing VSD away to ensure a sufficient hydrophobic pocket between VSD and PD for PIP2, enabling us to capture the CBD-PIP2-bound open-state structure (Fig. 3d). Therefore, both PIP2 and CBD potentiate the voltage activation of KCNQ2 by enhancing the E–M coupling.
Different roles of the two CBDs
While CBDA sits deep in the classical fenestration pocket and coincides with the RTG in the structure of KCNQ238, CBDB is localized at the pocket surface and binds to KCNQ2 relatively loosely (Fig. 1a, i). We then ask whether these two sites are both essential for the potentiation of the KCNQ2 voltage activation. To probe the role of CBDB, we introduced an alanine substitution on the residue Phe104 in S1, which directly interacts with CBDB. Electrophysiological studies show that the activation of CBD is largely affected in the mutant F104A (Fig. 1h). We next collected cryo-EM data using the F104A mutant sample incubated with both PIP2 and CBD. Interestingly, two different KCNQ2-CaMF104A maps were reconstructed from one dataset, which were designated KCNQ2-CaMF104A-CBD-PIP2(-)-I and KCNQ2-CaMF104A-CBD-PIP2-II at 2.7 Å and 3.5 Å resolutions, respectively (Fig. 4a, Table 1, Supplementary Figs. 2j–q, 3c, d). In both maps, CBDA molecules maintain strong densities but those for CBDB are barely observed (Fig. 4a–c). Interestingly, KCNQ2-CaMF104A-CBD-PIP2(-)-I remains in a closed state with no PIP2 bound while KCNQ2-CaMF104A-CBD-PIP2-II is in an open state with the PIP2 bound (Fig. 4c–e). The number ratio of particles used for the final reconstruction of KCNQ2-CaMF104A-CBD-PIP2(-)-I and KCNQ2-CaMF104A-CBD-PIP2-II is ~4: 1 (Fig. 4a). In comparison, only the open-state 3D reconstruction KCNQ2-CaMPIP2-CBD was obtained from the WT sample with both PIP2 and CBD bound. The differential distribution of the KCNQ2-CaMF104A-CBD-PIP2 particles with only CBDA bound in closed and open states indicates that with the presence of PIP2, (i) CBDA alone is able to stabilize some KCNQ2 channels in the open conformation, which accounts for ~20% of total channels, and (ii) CBDB further enhances this stabilization and dramatically increases the ratio of the open-conformation channels. Therefore, both CBDA and CBDB contribute to the potentiation of KCNQ2 voltage activation.

a Two different KCNQ2-CaMF104A maps were reconstructed from one dataset for KCNQ2 F104A mutant with both CBD and PIP2. b The density maps of one CBD molecule in different orientations at the contour level of 3.7 σ. c The density maps of one CBD molecule and one PIP2 molecule in different orientations at the contour level of 3.7 σ and 3.5 σ, respectively. d The closed activation gate of KCNQ2-CaMF104A-CBD-PIP2(-)-I. The dashed lines show diagonal atom-to-atom distance (in Å) at the constriction-lining residues Gly310, Ser314, and Leu318. e The open activation gate of KCNQ2-CaMF104A-CBD-PIP2-II. The dashed lines show diagonal atom-to-atom distance (in Å) at the constriction-lining residues Gly310, Ser314, and Leu318.
Structural basis for the HN37 recognition
Previously we developed an effective KCNQ2 activator named HN37, which is now under clinical trial in phase I as a promising anti-epileptic drug candidate. Compared with RTG, HN37 has several advantages, such as higher activation potency to neuronal Kv7 channels, improved chemical stability, and better safety margin34. To reveal the activation mechanism of HN37 on KCNQ2, we then determined the HN37-bound KCNQ2 structure (KCNQ2-CaMHN37) at 2.5 Å resolution (Fig. 5a, Table 1, Supplementary Fig. 6). In KCNQ2-CaMHN37, to our surprise, there are also two HN37 molecules bound in each KCNQ2 subunit (Fig. 5a, b). One HN37 molecule (HN37A) binds in the same pocket with RTG and CBDA. HN37A mainly forms hydrophobic interactions with residues Leu299, Ile300 and Phe304 in S6I, Trp236 and Phe240 in S5II, Phe305, Pro308 and Leu312 in S6II, and Phe104 in S1III (Fig. 5c). In addition, HN37A forms two hydrogen bonds with residues Trp236 and Leu299, as well as the π-π interaction with the Trp236 side chain. The other HN37 molecule (HN37B) is located at the interface between VSD and PD and switched by S1III/S4III and S5II (Fig. 5d). HN37B also mainly forms hydrophobic interactions with surrounding residues such as Val233, Trp236, and Tyr237 in S5II, Phe100 in S1III, and Met208 and Met211 in S4III. Besides, one π-π interaction is observed between the fluorophenyl group of HN37A and HN37B (Fig. 5d).

a The 3D reconstruction and the cartoon model of KCNQ2-CaMHN37. b The density maps of two HN37 molecules in different orientations at the contour level of 4.5 σ. c Interactions between HN37A and KCNQ2. The dashed lines indicate hydrogen bonds and π-π interaction. d Interactions between CBDB and KCNQ2. The dashed lines indicate π-π interaction. e, f Dose-response curve of the activation efficacy (IDrug / IControl) and the half maximal activation voltage shift (ΔV1/2) for HN37 on KCNQ2 channels. The EC50 values were 28.52 ± 2.99 nM and 27.97 ± 5.32 nM, respectively. Data are presented as mean ± SEM. For Idrug / IControl, n = 3 (0.1 μM), 5 (1 μM), 4 (10 μM), 4 (30 μM), 4 (100 μM), and 4 (1000 μM) individual cells; for ΔV1/2, n = 3 (0.1 μM), 3 (1 μM), 5 (10 μM), 3 (30 μM), 3 (100 μM), and 5 (1000 μM) individual cells. g The activation effects of 100 nM HN37 on KCNQ2 WT and mutants. Data are presented as mean ± SEM. An unpaired two-tailed t test was performed. p and n values compared to WT (n = 6) are p = 0.0727 and n = 6 for F100A, p = 0.2335 and n = 3 for M211A, ***p = 0.0001 and n = 6 for W236A, p = 0.5599, n = 6 for F240A, and ***p = 0.0001 and n = 5 for L299A. n represents the number of experiments from individual cells. h The ΔV1/2 of KCNQ2 WT and mutants caused by 100 nM HN37. Data are presented as mean ± SEM. An unpaired two-tailed t test was used. p and n values compared to WT (n = 5) are p = 0.4114 and n = 4 for F100A, ***p = 0.0001 and n = 4 for M211A, ***p = 0.0001 and n = 4 for W236A, **p = 0.0065 and n = 5 for F240A, and ***p = 0.0001 and n = 3 for L299A. n represents the number of experiments from individual cells. i The activated VSD of KCNQ2-CaMHN37. For clarity purpose, S1 is not shown. Side chains of positively charged (or polar) residues in S4 and residues forming gating charge transfer center are displayed as sticks. j The closed activation gate of KCNQ2-CaMHN37. The dashed lines show diagonal atom-to-atom distance (in Å) at the constriction-lining residues Ser314 and Leu318. k Structural comparison of VSDs from KCNQ2-CaMapo and KCNQ2-CaMHN37 when the whole channels are aligned. For (e–h), source data are included in the Source Data file.
To validate the two HN37 binding modes revealed by the structure data, we then introduced mutations at the two HN37 binding sites in KCNQ2 and recorded the currents of CHO cells expressing WT and mutant KCNQ2 using the whole-cell patch clamp. Under −10 mV, HN37 increases the current amplitudes of WT KCNQ2 in a dose-dependent manner with an EC50 value of 28.52 ± 2.99 nM (Fig. 5e). In particular, 100 nM HN37 increases the current of KCNQ2 by an average factor of 2.74 ± 0.10. Meanwhile, HN37 enhances the voltage sensitivity of KCNQ2 by left-shifting the G–V curve, with an EC50 of 27.97 ± 5.32 nM fitted from data points of ΔV1/2 (Fig. 5f). Specifically, 100 nM HN37 induces a shift of half maximal activation voltage (V1/2) by −23.49 ± 0.91 mV. Among the six mutants we tested, V233A was non-functional. The mutation on Trp236 which interacts with both HN37 molecules, abolished the sensitivity of KCNQ2 to HN37 in both current amplitude and voltage activation (Fig. 5g, h, and Supplementary Fig. 7). For the HN37A-specific interacting residues, mutation on Leu299 significantly decreased the potentiation activity of HN37 by both reducing the increase of outward current amplitude and attenuating the left-shift of the G–V curve (ΔV1/2) whereas mutation on Phe240 only attenuated the left-shift of the G–V curve (Fig. 5g, h). For the HN37B-specific interacting residues, mutation on Met211 attenuated the left-shift of the G–V curve but did not change the current amplitude; in contrast, mutation on Phe100 did not alter HN37 sensitivity in either current amplitude or voltage activation (Fig. 5g, h). These data suggest that HN37A is more potent than HN37B in the activation of KCNQ2.
Like CBD, the HN37 alone does not allow us to capture an open-state channel. KCNQ2-CaMHN37 is also in decoupled conformation, with activated VSDs and a closed gate (Fig. 5i, j). Although the two HN37 molecules induce a 1–2 Å shift of the VSD (Fig. 5k), the activation gate remains unchanged, with the shortest diagonal atom-to-atom distance of 4–6 Å at the activation gate (Fig. 5j).
The HN37 and PIP2 activation mechanism
To reveal the HN37 activation mechanism, we determined two KCNQ2-CaM structures in the presence of both HN37 and PIP2, one with HN37 added to the protein sample first (KCNQ2-CaMHN37-PIP2(-), Table 1, Supplementary Fig. 8a–h) and the other one with PIP2 added first (KCNQ2-CaMPIP2-HN37, Table 1, Supplementary Fig. 9). In the map of KCNQ2-CaMHN37-PIP2(-), no PIP2 molecule was assigned, and consequently, the KCNQ2-CaMHN37-PIP2(-) structure is in a closed state with two HN37 bound (Supplementary Fig. 8i, j), similar to the KCNQ2-CaMHN37 structure (Fig. 5b). In contrast, the KCNQ2-CaMPIP2-HN37 is in an open state with one HN37 (HN37A) and one PIP2 bound (Fig. 6a–c). In KCNQ2-CaMPIP2-HN37, PIP2 binds in the cleft between VSD and PD and directly interacts with Lys327 in S6 (Fig. 6d), similar to the observations in KCNQ2-CaMCBD-PIP2 (Fig. 2b).

a The 3D reconstruction and the cartoon model of KCNQ2-CaMPIP2-HN37. b The binding site of PIP2 and HN37A in KCNQ2. Side chains of residues in KCNQ2 involved in the interactions with PIP2 are shown as sticks. c The open activation gate of KCNQ2-CaMPIP2-HN37. The dashed lines show diagonal atom-to-atom distance (in Å) at the constriction-lining residues Gly310, Ser314, and Leu318. d Overlay of the pore domains of KCNQ2-CaMHN37 (green) and KCNQ2-CaMPIP2-HN37 (orange) structures showing the conformational change in the ion-conducting pathway. The S6 helix bends outward upon PIP2 binding at the point of PAG segment. e HN37B and the hydrophobic tails of PIP2 occupy the same site in KCNQ2 by overlay of KCNQ2-CaMPIP2-HN37 (orange) and KCNQ2-CaMHN37 (green). f Structural comparison of VSDs from KCNQ2-CaM PIP2-HN37 and KCNQ2-CaMHN37 (green) when the whole channels are aligned. g The proposed HN37 and PIP2 activation mechanism of KCNQ2 in vivo. h The proposed HN37 and PIP2 activation model of KCNQ2 in vitro.
To analyze why HN37B does not bind in the structure of KCNQ2-CaMPIP2-HN37, we compare structures of KCNQ2-CaMPIP2-HN37 and KCNQ2-CaMHN37 (Fig. 6e). Interestingly, HN37B and the hydrophobic tails of PIP2 occupy the same site in KCNQ2 (Fig. 6e). With the presence of PIP2 in KCNQ2-CaMPIP2-HN37, HN37B was unable to bind. Because PIP2 is essential for the activation of KCNQ channels, the clash of HN37B and PIP2 in KCNQ2 suggests that HN37B may be only observed without PIP2 in vitro and not exist in vivo due to the presence of PIP2 in the membrane. However, we could not rule out the possibility that the two-HN37-bound KCNQ structures (KCNQ2-CaMHN37 and KCNQ2-CaMHN37-PIP2(-)) may present an in vivo transient state after which HN37B will be replaced by PIP2 easily.
HN37A and CBDA bind in the same classical fenestration pocket and likely share similar activation mechanisms on KCNQ2. Like CBD, the binding of HN37 induces a 1–2 Å shift of the VSD (Fig. 6f), which would likely favor the binding of PIP2 in vivo45. The binding of PIP2 further destroys interactions between VSD and CaM and causes structural rearrangements of S6, CTD, and CaM, along with the opening of the activation gate (Fig. 6g). Then what is the role of HN37B if it does exist in vivo? First, by forming π-π interaction, HN37B stabilizes the binding of HN37A. Second, HN37B inserts into the cleft between PD and VSD and directly interacts with S4. Thus, the HN37B likely carries part of PIP2’s role by stabilizing the activated VSD before PIP2 binds (Fig. 6h).
Discussion
While here we use the homo-tetramer KCNQ2 as a model to probe the activation mechanisms of CBD and HN37, KCNQ2 and KCNQ3 can form hetero-tetrameric channels in vivo. To test whether the homo-tetramer KCNQ2 and hetero-tetrameric KCNQ2/KCNQ3 share similar ligand activations, the activation effects of 10 µM CBD and 100 nM HN37 on the activation efficacy (IDrug / IControl) and the half maximal activation voltage shift (ΔV1/2) of wild type KCNQ2/KCNQ3 and KCNQ2(W236L)/KCNQ3 hetero-tetramers were separately examined. The sensitivity of the wild type KCNQ2/KCNQ3 to CBD and HN37 were similar to that of the KCNQ2 homo-tetramer (Figs. 1e–g and 5e, f, Supplementary Fig. 10). Consistent to the key role of Trp236 in the binding of CBD and HN37, the activation effects of the two molecules on the KCNQ2(W236L)/KCNQ3 hetero-tetramers were dramatically decreased compared to the wild type (Supplementary Fig. 10). Thus, the binding sites of CBD and HN37 are conserved among KCNQ2 and KCNQ2/KCNQ3.
Comparisons of the binding sites of RTG, CBDA, and HN37A in KCNQ2 reveal a conserved agonist binding pocket in KCNQ channels, which is located in a side fenestration in TMD and lined by S5, S6, pore helix, and S6 from the adjacent subunit. While ligands bound in this pocket mainly form hydrophobic interactions with surrounding residues, specific hydrogen bonds are observed between ligands and main-chain carbonyls or side chains of nearby residues, such as Trp236, Lue299, and Ser303 (Supplementary Fig. 11). In addition, the residue Trp236, a characteristic residue for the RTG binding site in KCNQ2-5, adopts a different rotamer upon the binding of RTG and HN37 and forms π-π stacking with them. Interestingly, in the CBD-bound KCNQ2 structures, the Trp236 side chain adopts a third rotamer with no π-π stacking involved (Supplementary Fig. 11).
While CBDA and HN37A bind in the same pocket, it is interesting to speculate whether HN37B and CBD can simultaneously bind to KCNQ2. We first align structures of KCNQ2-CaMHN37 and KCNQ2-CaMCBD, which shows that HN37B clashes with CBDA and Trp236 side chain, but not CBDB. Thus, this HN37B is unlikely to co-bind with two CBDs (Supplementary Fig. 12a). To functionally verify whether CBD and HN37 can simultaneously bind to KCNQ2, the activation effects of HN37 were tested alone or in combination with 30 µM CBD, a saturated concentration according to Fig. 1f. We found that 30 µM CBD potentiated the KCNQ2 current amplitude measured at −10 mV by 1.35 ± 0.08 folds and addition of 1 µM HN37 conferred a further increase (Supplementary Fig. 12b). The EC50 values obtained from the dose-response curves of HN37 in the absence and presence of 30 µM CBD were 28.75 ± 6.76 nM and 178.41 ± 46.11 nM, respectively (Supplementary Fig. 12d). The Hill coefficient was slightly changed, from 1.42 (without CBD) to 1.13 (with CBD). These data showed that CBD and HN37 exert their modulations on KCNQ2 competitively. Moreover, the maximal activation efficacy (IDrug / IControl) and the half-maximal activation voltage shift (ΔV1/2) of saturated HN37 + CBD on KCNQ2 were comparable to those of HN37 alone (Supplementary Fig. 12c, e, f). The data suggested that the activation effect of saturated CBD + HN37 is dominated by HN37. Taken together, these structural and electrophysiological data suggest that HN37 and CBD can not simultaneously bind to KCNQ2.
Although PIP2 is essential for the activation of KCNQ channels, it is difficult to capture the PIP2-bound structures of KCNQ channels with PIP2 added alone in the protein sample. So far, all the PIP2-bound KCNQ structures were determined in the presence of either the auxiliary subunit KCNE41 or exogenous ligands such as ML27739, ML21343, CBD (Fig. 2), and HN37 (Fig. 6), suggesting low binding affinity of PIP2 bound to KCNQs in vitro47. Structural alignments of KCNQs in the apo state and KCNE3-bound or these exogenous ligand-bound states reveals a possible consensus potentiation mechanism. KCNE3 and all these exogenous ligands induce lateral shifts of VSD relative to PD, which likely favor the binding of PIP2 (Supplementary Fig. 13), although their binding sites are different among KCNQ channels.
In summary, in this study, we present structures of KCNQ2 in complex with two exogenous activators, namely CBD and HN37, with or without the endogenous lipid PIP2, in either closed or open state. These structures, along with electrophysiological analyses, reveal conserved ligand activation mechanisms of KCNQ2. Both HN37 and CBD bind one KCNQ2 subunit in a 2:1 ratio – HN37A and CBDA molecules bind in the same classical fenestration pocket and HN37B and CBDB molecules in different nearby pockets. The bindings of HN37A and CBDA induce disturbance of VSD and likely favor the binding of PIP2 in vivo, which transfers the conformational change of S4 upon the depolarization of the membrane potential to the CTD/CaM and S6, leading to the opening of the activation gate. The B molecules of both ligands likely stabilize the bindings of A molecules and enhance the potentiation effect. The two binding sites of both CBD and HN37 not only explain their high activation potency to KCNQ2 compared with RTG which has only one site in KCNQ238, but also provides clues for the development KCNQ2 activators with higher activation potency.
Previously, Mackinnon lab determined the PIP2-bound open-state human KCNQ1-CaM-KCNE3 structure41. Similarly, we determined the activator ML277- and PIP2-bound open-state structures of KCNQ1-CaM39. In the open-state KCNQ1, PIP2 binds to S0 and the S2–S3 linker and does not directly interact with S6, suggesting that PIP2 may only involve in disrupting interactions of CaM and VSD. Here the CBD- and PIP2-bound open-state structure of KCNQ2-CaM reveals a different binding site and a different role of PIP2. In the open-state KCNQ2, PIP2 is located in the VSD and PD interface where it not only pushes VSD away from CaM but also pulls S6 up to stabilize the open-state conformation. These two different PIP2 binding sites were also observed in the same channel of KCNQ443, suggesting the conservation and divergence of the PIP2’s role in the E–M coupling of KCNQ channels.
Methods
Protein expression and purification
DNAs encoding human KCNQ2 (Homo sapiens: NP_004509.2) and CaM (Homo sapiens: NP_001734.1) were synthesized by GenScript. To improve the biochemical and thermal stability of KCNQ2, the N-terminal and C-terminal regions were truncated, yielding a construct containing residues 64–674. NheI and XhoI sites were used for cloning the KCNQ2 construct into the modified pEZT-BM vector with a C-terminal strep tag. The human CaM gene was cloned into pEZT-BM vector with a C-terminal Histidine tag. KCNQ2 and CaM complex were heterologously expressed in Human Embryonic Kidney (HEK) 293S suspension cells (Life Technologies, ATCC, #CRL-3022) maintained at 30 °C in SMM 293-TI complete medium (Sino Biological Inc.) supplemented with 2% fetal bovine serum (FBS, Yeasen Biotechnology (Shanghai) Co., Ltd.). The P2 baculovirus was generated via the BacMam system (Thermo Fisher Scientific) and used for expression when cell density reached 3.5 × 106 cells/mL. P2 baculovirus mixture of KCNQ2: CaM (6:1) was used for transduction of HEK293S cells for protein expression. To boost protein expression, 10 mM sodium butyrate was added 12 h post-transduction. Cells were harvested after 48 h, then flash-frozen in liquid nitrogen and stored at −80 °C until needed.
Cells were resuspended and lysed by sonication in buffer A (20 mM Tris, pH 8.0, 150 mM KCl) supplemented with a protease inhibitor cocktail (2 μg/mL DNase I, 0.5 μg/mL pepstatin, 2 μg/mL leupeptin, 1 μg/mL aprotinin, and 1 mM PMSF). The lysate was then solubilized with 1.5% n-dodecyl-β-D-maltoside (DDM, Anatrace) and 0.3% cholesteryl hemisuccinate tris salt (CHS, Anatrace) at 4 °C for 3 h. The insoluble cell fragment was removed by centrifugation at 48,000 g for 50 min at 4 °C. The supernatant was incubated with Strep-Tactin Sepharose resin (IBA) at 4 °C for 1.5 h with gentle rotation. Beads were loaded onto a gravity column and washed with buffer B (buffer A supplemented with 0.05% DDM and 0.01% CHS) for 4 CVs (column volumes), followed by washing with buffer C (buffer A supplemented with 0.02% GDN) for 16 CVs. The protein was then eluted with buffer C plus 10 mM d-Desthiobiotin (Sigma) and further purified on a Superose 6 gel filtration column (GE Healthcare) in buffer C. The peak fraction was collected and concentrated for cryo-EM sample preparation. For KCNQ2-CaM bound to CBD and/or PIP2, the purified protein was incubated with 0.25 mM CBD (Sigma-Aldrich) and/or 1 mM PIP2. For KCNQ2-CaM bound to HN37 and/or PIP2, the purified protein was incubated with 0.15 mM HN37 and/or 1 mM PIP2. HN37 was synthesized by the laboratory of Professor Fajun Nan (Shanghai Institute of Material Medica). The PIP2 we used is 1,2-dioctanoyl-sn-glycero-3-phospho-(1’-myo-inositol-4’,5’-bisphosphate) (ammonium salt) purchased from Avanti.
Cryo-EM data acquisition
For grids preparation, 3 μL concentrated protein was loaded onto glow-discharged R1.2/1.3 Quantifoil grids at 4 °C under 100% humidity. Grids were blotted for 4.5 s and plunge-frozen in liquid ethane using a Vitrobot Mark IV (FEI). Micrographs were acquired on a Titan Krios microscope (FEI) operating at a voltage of 300 kV. During one collection session (KCNQ2-CaMHN37, Supplementary Fig. 6) micrographs were collected at a magnification of 49,310× (calibrated pixel size 1.014 Å, defocus range −1.1 μm to −1.3 μm, 40 frames, and a total dose 64 e−/Å2) using a Gatan K2 Summit direct electron detection camera via SerialEM48 following standard procedure. After the microscope was upgraded with a Gatan Falcon 4 camera equipped with energy filter (Thermo Fisher Scientific) with slit width set to 10 eV, additional collection sessions (Supplementary Figs. 1, 2, 8, 9) were conducted at a magnification of 130,000× (calibrated pixel size 0.93 Å, defocus range −0.8 μm to −1.5 μm, 40 frames, and a total dose 52 e−/Å2) using EPU software (Thermo Fisher Scientific).
Image processing
Motion correction and CTF parameters estimation were performed with the MotionCorr249 and the GCTF50 programs, respectively. All image processing steps were carried out with RELION 3.151.
For KCNQ2-CaMCBD, 2849 micrographs were collected and 1,034,485 particles were auto-picked and extracted with a binning factor of 3 for 2D classification. The following two rounds of 3D classification with 940,410 selected particles were performed using the map of human KCNQ2-CaM complex (PDB: 7CR3)38 as a reference. After 3D classification, selected particles were combined and re-extracted to the pixel size of 0.93 Å for 3D refinement with a C4 symmetry and Bayesian polishing via RELION 3.1. The final resolution of the EM map by 3D reconstruction of 99,018 particles was 3.0 Å. For KCNQ2-CaMPIP2(-), KCNQ2-CaMCBD-PIP2, KCNQ2-CaMF104A-CBD-PIP2(-)-I, KCNQ2-CaMF104A-CBD-PIP2-II, KCNQ2-CaMHN37, KCNQ2-CaMHN37-PIP2(-), and KCNQ2-CaMPIP2-HN37 dataset, data processing was performed following similar procedures (Supplementary Figs. 1, 2, 6, 8, 9).
The resolution was estimated by applying a soft mask around the protein density and the gold-standard Fourier shell correlation (FSC) = 0.143 criterion. Local resolution maps were calculated with RELION 3.1.
Model building, refinement, and validation
De novo atomic models were built in Coot52 based on the 2.5 Å resolution map of KCNQ2-CaMHN37. The amino acid assignment was achieved based on the clearly defined density for bulky residues (Phe, Trp, Tyr, and Arg) and the model of KCNQ2-CaM complex (PDB: 7CR3)38 was used as a reference. PHENIX53 was utilized for model refinement against cryo-EM maps using real-space refinement, with secondary structure restraints and non-crystallography symmetry applied. The models of KCNQ2-CaMPIP2(-), KCNQ2-CaMCBD, KCNQ2-CaMCBD-PIP2, KCNQ2-CaMF104A-CBD-PIP2(-)-I, KCNQ2-CaMF104A-CBD-PIP2-II, KCNQ2-CaMHN37-PIP2(-), and KCNQ2-CaMPIP2-HN37 were built using the model of KCNQ2-CaMHN37 as a template. The MolProbity54 was used for model geometry statistics generation (Supplementary Table 1). Structural figures were prepared in PyMoL (The PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC.) and UCSF Chimera X55.
Mutagenesis
The wild-type human KCNQ2 cDNAs were subcloned into pcDNA5/FRT vector. Mutations were introduced into the WT cDNA constructs using the QuickChange II XL site-directed mutagenesis kit from Agilent Technologies, according to the manufacturer’s manual. The mutants were verified by fully sequencing before use. Sequences of primers for mutagenesis study have been provided in the separate Supplementary Data 1.
Cell culture and transfection
Chinese hamster ovary (CHO) cells (CCL-61) were grown in 50/50 DMEM/F-12 (Gibco) supplemented with 10% fetal bovine serum (FBS) and 2 mM L-glutamine (Life Technology). To transiently express the isoforms for electrophysiological studies, cells were seeded into 6-well plate and then transfected with 3.6 µg of the cDNA using the Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s guideline. A GFP cDNA construct (0.4 µg, Amaxa) was co-transfected to aid identification of transfected cells by fluorescence microscopy. For electrophysiological recordings, the cells were plated onto glass coverslips coated with poly-D-lysine and cultured in wells of sterile 6-well tissue culture plates in a humidified incubator at 37 °C, 5% CO2, until use.
Electrophysiological recording
Standard whole-cell voltage-clamp recording was conducted at room temperature with the Axopatch-700B amplifier. Pipettes were pulled from borosilicate glass capillaries (World Precision Instruments) with tip resistances of 3–5 MΩ when filled with the intracellular solution. The intracellular solution contained (in mM): 145 KCl, 1 MgCl2, 5 EGTA, 10 HEPES (pH 7.3 adjusted by KOH); bath or extracellular solution contained (in mM): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose (pH 7.3 adjusted by NaOH). The data were filtered at 2 kHz and digitized using a DigiData 1440 A with pClamp 10.3 software (Molecular Devices). Series resistance compensation was used and set to 60%. To elicit currents, cells were stimulated by a series of 1500 ms depolarizing steps from −90 mV to +70 mV in 10 mV increments at a holding potential of −80 mV (testing CBD) or −100 mV (testing HN37). During the recordings, the bath solution was continuously perfused by a BPS perfusion system (ALA Scientific Instruments).
Data analysis
Patch-clamp data were processed using Clampfit 10.3 (Molecular Devices, Sunnyvale, CA) and then analyzed using GraphPad Prism 5 (GraphPad Software, San Diego, CA). Voltage dependent activation curves were fitted with the Boltzmann equation, G = Gmin + (Gmax − Gmin)/(1 + exp [V–V1/2]/S), where Gmax is the maximum conductance, Gmin is the minimum conductance, V1/2 is the voltage for reaching 50% of maximum conductance, and S is the slope factor. Dose-response curves were fitted with the Hill equation, Y = Bottom + (Top − Bottom)/(1 + 10 ^ ((LogEC50 − X) * P)), where X is log of drug concentration, Y is the response, Top and Bottom are corresponding to the maximum and minimum of the responses, EC50 is the drug concentration producing the half-maximum response, and P is the Hill coefficient. The data are presented as mean ± SEM. The significance was estimated using unpaired two-tailed Student’s t test or one-way ANOVA with Tukey’s post hoc procedure. Statistical significance: *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support this study are available from the corresponding authors upon request. The cryo-EM maps have been deposited in the Electron Microscopy Data Bank (EMDB) under accession codes EMD-35884 (KCNQ2-CaMPIP2(-)), EMD-35879 (KCNQ2-CaMCBD), EMD-35880 (KCNQ2-CaMCBD-PIP2), EMD-35882 (KCNQ2-CaMF104A-CBD-PIP2(-)-I), EMD-35881 (KCNQ2-CaMF104A-CBD-PIP2-II), EMD-35877 (KCNQ2-CaMHN37), EMD-35883 (KCNQ2-CaMHN37-PIP2(-)), EMD-37270 (KCNQ2-CaMPIP2-HN37). The coordinates have been in the Protein Data Bank (PDB) under accession codes 8J05 (KCNQ2-CaMPIP2(-)), 8J00 (KCNQ2-CaMCBD), 8J01 (KCNQ2-CaMCBD-PIP2), 8J03 (KCNQ2-CaMF104A-CBD-PIP2(-)-I), 8J02 (KCNQ2-CaMF104A-CBD-PIP2-II), 8IZY (KCNQ2-CaMHN37), 8J04 (KCNQ2-CaMHN37-PIP2(-)), 8W4U (KCNQ2-CaMPIP2-HN37). Additional datasets used in this study include Protein Data Bank accession codes 7CR3, 8SIN, 6UZZ, 6V00, 7XNI, 7XNK, 7BYL, and 7VNQ. The source data underlying Figs. 1f–h, 2c, d, 5e–h, and Supplementary Figs. 1a, 2a, j, 4a–e, 6a, 7a–k, 8a, 9a, 10b, c,12c–f are provided as a Source Data file. Source data are provided with this paper.
References
Wang, Q. et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat. Genet. 12, 17–23 (1996).
Sanguinetti, M. C. et al. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 384, 80–83 (1996).
Barhanin, J. et al. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature 384, 78–80 (1996).
Brown, D. A. & Adams, P. R. Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature 283, 673–676 (1980).
Wang, H. S. et al. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282, 1890–1893 (1998).
Cooper, E. C. Made for “anchorin”: Kv7.2/7.3 (KCNQ2/KCNQ3) channels and the modulation of neuronal excitability in vertebrate axons. Semin. Cell Dev. Biol. 22, 185–192 (2011).
Schroeder, B. C., Kubisch, C., Stein, V. & Jentsch, T. J. Moderate loss of function of cyclic-AMP-modulated KCNQ2/KCNQ3 K+ channels causes epilepsy. Nature 396, 687–690 (1998).
Singh, N. A. et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat. Genet. 18, 25–29 (1998).
Charlier, C. et al. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat. Genet. 18, 53–55 (1998).
Kato, M. et al. Clinical spectrum of early onset epileptic encephalopathies caused by KCNQ2 mutation. Epilepsia 54, 1282–1287 (2013).
Weckhuysen, S. et al. KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann. Neurol. 71, 15–25 (2012).
Lauritano, A. et al. A novel homozygous KCNQ3 loss-of-function variant causes non-syndromic intellectual disability and neonatal-onset pharmacodependent epilepsy. Epilepsia Open 4, 464–475 (2019).
Orhan, G. et al. Dominant-negative effects of KCNQ2 mutations are associated with epileptic encephalopathy. Ann. Neurol. 75, 382–394 (2014).
Wulff, H., Castle, N. A. & Pardo, L. A. Voltage-gated potassium channels as therapeutic targets. Nat. Rev. Drug Discov. 8, 982–1001 (2009).
Jentsch, T. J. Neuronal KCNQ potassium channels: physiology and role in disease. Nat. Rev. Neurosci. 1, 21–30 (2000).
Zheng, Q. et al. Suppression of KCNQ/M (Kv7) potassium channels in dorsal root ganglion neurons contributes to the development of bone cancer pain in a rat model. Pain 154, 434–448 (2013).
Zhang, F. et al. Suppression of KCNQ/M potassium channel in dorsal root ganglia neurons contributes to the development of osteoarthritic pain. Pharmacology 103, 257–262 (2019).
Blackburn-Munro, G. & Jensen, B. S. The anticonvulsant retigabine attenuates nociceptive behaviours in rat models of persistent and neuropathic pain. Eur. J. Pharmacol. 460, 109–116 (2003).
Munro, G. & Dalby-Brown, W. Kv7 (KCNQ) channel modulators and neuropathic pain. J. Med. Chem. 50, 2576–2582 (2007).
Chen, X. et al. Potassium channels: a potential therapeutic target for Parkinson’s disease. Neurosci. Bull. 34, 341–348 (2018).
Liu, H., Jia, L., Chen, X., Shi, L. & Xie, J. The Kv7/KCNQ channel blocker XE991 protects nigral dopaminergic neurons in the 6-hydroxydopamine rat model of Parkinson’s disease. Brain Res. Bull. 137, 132–139 (2018).
Peng, H., Bian, X. L., Ma, F. C. & Wang, K. W. Pharmacological modulation of the voltage-gated neuronal Kv7/KCNQ/M-channel alters the intrinsic excitability and synaptic responses of pyramidal neurons in rat prefrontal cortex slices. Acta Pharmacol. Sin. 38, 1248–1256 (2017).
Wickenden, A. D., Yu, W., Zou, A., Jegla, T. & Wagoner, P. K. Retigabine, a novel anti-convulsant, enhances activation of KCNQ2/Q3 potassium channels. Mol. Pharmacol. 58, 591–600 (2000).
Main, M. J. et al. Modulation of KCNQ2/3 potassium channels by the novel anticonvulsant retigabine. Mol. Pharmacol. 58, 253–262 (2000).
Szelenyi, I. Flupirtine, a re-discovered drug, revisited. Inflamm. Res. 62, 251–258 (2013).
Friedel, H. A. & Fitton, A. Flupirtine. A review of its pharmacological properties, and therapeutic efficacy in pain states. Drugs 45, 548–569 (1993).
Franco, V. & Perucca, E. Pharmacological and therapeutic properties of cannabidiol for epilepsy. Drugs 79, 1435–1454 (2019).
Pumroy, R. A. et al. Structural insights into TRPV2 activation by small molecules. Nat. Commun. 13, 2334 (2022).
Huang, J. et al. Cannabidiol inhibits Na(v) channels through two distinct binding sites. Nat. Commun. 14, 3613 (2023).
Pumroy, R. A. et al. Molecular mechanism of TRPV2 channel modulation by cannabidiol. Elife 8, e48792 (2019).
Zhang, H. B. et al. Cannabidiol activates neuronal Kv7 channels. Elife 11, e73246 (2022).
Pisanti, S. et al. Cannabidiol: State of the art and new challenges for therapeutic applications. Pharmacol. Ther. 175, 133–150 (2017).
Bai, F. et al. A Statistical thermodynamic model for ligands interacting with ion channels: theoretical model and experimental validation of the KCNQ2 channel. Front. Pharmacol. 9, 150 (2018).
Zhang, Y. M. et al. Discovery of HN37 as a potent and chemically stable antiepileptic drug candidate. J. Med. Chem. 64, 5816–5837 (2021).
Ciliberto, M. A., Weisenberg, J. L. & Wong, M. Clinical utility, safety, and tolerability of ezogabine (retigabine) in the treatment of epilepsy. Drug Healthc. Patient Saf. 4, 81–86 (2012).
Daniluk, J., Cooper, J. A., Stender, M. & Kowalczyk, A. Survey of physicians’ understanding of specific risks associated with retigabine. Drugs Real World Outcomes 3, 155–163 (2016).
Groseclose, M. R. & Castellino, S. An investigation into retigabine (ezogabine) associated dyspigmentation in rat eyes by MALDI imaging mass spectrometry. Chem. Res. Toxicol. 32, 294–303 (2019).
Li, X. et al. Molecular basis for ligand activation of the human KCNQ2 channel. Cell Res. 31, 52–61 (2021).
Ma, D. et al. Structural mechanisms for the activation of human cardiac KCNQ1 channel by electro-mechanical coupling enhancers. Proc. Natl. Acad. Sci. USA 119, e2207067119 (2022).
Sun, J. & MacKinnon, R. Cryo-EM structure of a KCNQ1/CaM complex reveals insights into congenital long QT syndrome. Cell 169, 1042–1050.e1049 (2017).
Sun, J. & MacKinnon, R. Structural basis of human KCNQ1 modulation and gating. Cell 180, 340–347.e349 (2020).
Li, T. et al. Structural basis for the modulation of human KCNQ4 by small-molecule drugs. Mol. Cell. 81, 25–37.e24 (2021).
Zheng, Y. et al. Structural insights into the lipid and ligand regulation of a human neuronal KCNQ channel. Neuron 110, 237–247 e234 (2022).
Willegems, K. et al. Structural and electrophysiological basis for the modulation of KCNQ1 channel currents by ML277. Nat. Commun. 13, 3760 (2022).
Kim, R. Y., Pless, S. A. & Kurata, H. T. PIP2 mediates functional coupling and pharmacology of neuronal KCNQ channels. Proc. Natl. Acad. Sci. USA 114, E9702–E9711 (2017).
Yang, N. D. et al. Electro-mechanical coupling of KCNQ channels is a target of epilepsy-associated mutations and retigabine. Sci. Adv. 8, eabo3625 (2022).
Li, Y., Gamper, N., Hilgemann, D. W. & Shapiro, M. S. Regulation of Kv7 (KCNQ) K+ channel open probability by phosphatidylinositol 4,5-bisphosphate. J. Neurosci. 25, 9825–9835 (2005).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Zhang, K. Gctf: real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, e42166 (2018).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sec. D-Struct. Biol. 66, 213–221 (2010).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallog. Sec. D-Struct. Biol. 66, 12–21 (2010).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Acknowledgements
Single-particle cryo-electron microscopy data were collected at Center of Cryo-Electron Microscopy at Zhejiang University. We thank Dr. Xing Zhang and Dr. Shenghai Chang for support in facility access and data acquisition. This work was supported in part by the National Key Research and Development Program of China (2020YFA0908501 and 2018YFA0508100 to J.G.), Zhejiang Provincial Natural Science Foundation (LR19C050002 to J.G.), the National Science Fund for Distinguished Young Scholars (81825021 to Z.G.), National Science and Technology Innovation 2030 Major Program (2021ZD0200900 to Z.G.), the National Natural Science Foundation of China (92169202 to Z.G.), and the Youth Innovation Promotion Association of the Chinese Academy of Sciences (2020284 to Y.Z.). J.G. is supported by MOE Frontier Science Center for Brain Science & Brain-Machine Integration, Zhejiang University and Fundamental Research Funds for the Central Universities.
Author information
Authors and Affiliations
Contributions
J.G. and Z.G. conceived and supervised the project. D.M., X.L., Y.Zhang., Z.Y. and N.S. performed sample preparation, data acquisition, and structure determination. J.G., Z.G., W.Y., N.S., F.N., P.H., J.F. and G.Z. performed structure data analysis. Y.Zheng, X.Z., L.W. and W.Z. performed electrophysiological studies. All authors participated in the manuscript preparation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Guanghui Yang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ma, D., Zheng, Y., Li, X. et al. Ligand activation mechanisms of human KCNQ2 channel. Nat Commun 14, 6632 (2023). https://doi.org/10.1038/s41467-023-42416-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-42416-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
