
Abstract
Planar chiral [2.2]paracyclophane derivatives are a type of structurally intriguing and practically useful chiral molecules, which have found a range of important applications in the field of asymmetric catalysis and material science. However, access to enantioenriched [2.2]paracyclophanes represents a longstanding challenge in organic synthesis due to their unique structures, which are still highly dependent on the chiral chromatography separation technique and classical chemical resolution strategy to date. In this work, we report on an efficient and versatile kinetic resolution protocol for various substituted amido[2.2]paracyclophanes, including those with pseudo-geminal, pseudo-ortho, pseudo-meta and pseudo-para disubstitutions, using chiral phosphoric acid (CPA)-catalyzed asymmetric amination reaction, which was also applicable to the enantioselective desymmetrization of an achiral diamido[2.2]paracyclophane. Detailed experimental studies shed light on a new reaction mechanism for the electrophilic aromatic C-H amination, which proceeded through sequential triazane formation and N[1,5]-rearrangement. The facile large-scale kinetic resolution reaction and diverse derivatizations of both the recovered chiral starting materials and the C-H amination products showcased the potential of this method.
Similar content being viewed by others
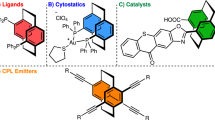

Introduction
Since the first discovery of [2.2]paracyclophane (PCP) by gas-phase pyrolysis of para-xylene1, as well as the practical synthesis through an intramolecular cyclization route2 around 1950, these structurally interesting molecules have drawn considerable research interests due to their unique photophysical and optoelectronic properties3. Moreover, the substituted [2.2]paracyclophanes displayed intriguing planar chirality, and these chiral skeletons have been widely studied in the development of chiral catalysts and ligands, as well as versatile chiral synthons in material science4,5,6,7 (Fig. 1a). Specifically, the amino[2.2]paracyclophanes represent as an important type of substituted [2.2]paracyclophanes, which have found numerous applications in various fields, such as the development of planar chiral bisthiourea catalyst8, N-heterocyclic carbene (NHC) ligands9, thermally activated delayed fluorescence (TADF) circularly polarized luminescence (CPL) emitters10 and others11,12,13,14,15,16,17,18,19,20.

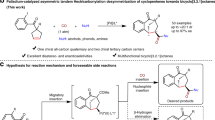
a The applications of chiral substituted and 4-amino [2,2]paracyclophanes. b Previous work on KR of substituted [2.2]paracyclophanes. c This work: KR of various substituted 4-amido [2.2]paracyclophanes via asymmetric amination.
However, the access to enantiomerically pure [2.2]paracyclophanes represents a formidable challenge in organic synthesis7,21, which still largely relied on the chiral chromatography separation or classical chemical resolution by forming diastereomeric salts or covalent adducts with chiral reagents22,23,24,25. On the other hand, catalytic kinetic resolution (KR) has emerged as an attractive and practical approach to afford enantioenriched molecules, which have found widespread applications in both academia and industry26,27,28,29. In the past two decades, a number of elegant catalytic kinetic resolution methods have been developed to access chiral PCP derivatives, including the asymmetric C-N coupling of PCP-based dihalides30, hydrolysis/esterification of PCP-based diphenol derivatives31,32, reduction of the PCP-based aldehydes and ketones33,34,35,36,37, and reduction/addition of PCP-based imines38,39,40 (Fig. 1b). Despite these advances, most of these methods still suffer certain limitations, including unsatisfactory KR performances, limited substrate scope, and the inconvenience of accessing both enantiomers of the planar chiral PCP derivatives. Moreover, to the best of our knowledge, the catalytic kinetic resolution of amino[2.2]paracyclophanes is currently unprecedented, and these valuable chiral skeletons could only be afforded by chemical resolution22 or the derivatizations from other known chiral PCP derivatives11,20.
Recently, our group has developed a highly efficient kinetic resolution method for arylamines bearing central41,42,43 or axial chirality44,45, by the utilization of chiral phosphoric acid46,47,48,49 (CPA) catalyzed asymmetric amination reaction with commercially available azodicarboxylates50,51. Herein, we report on the efficient and versatile KR of a variety of substituted amido[2.2]paracyclophanes, including those with pseudo-geminal, pseudo-ortho, pseudo-meta and pseudo-para disubstitutions, through the CPA-catalyzed asymmetric amination protocol, in which both enantiomers of the substituted amido[2.2]paracyclophanes could be readily afforded by the facile derivatization of the C-H amination products (Fig. 1c).
Results and discussion
Reaction condition optimizations
We began our study by selecting the racemic 4-NHBoc substituted [2.2]paracyclophane 1a as the model substrate, which could be easily prepared from the Pd-catalyzed C-N coupling of 4-bromo substituted PCP and BocNH2 (Table 1). Encouragingly, the KR of racemic 1a with dibenzyl azodicarboxylate 2a (1.0 equiv.) in the presence of CPA A1 (10 mol%) in toluene at 20 °C afforded the desired C-H amination product 3a with 88% ee and the recovered 1a with 32% ee, corresponding to a conversion of 27% and selectivity factor52 (s) of 21 (entry 1). Next, a series of CPA catalysts bearing various ortho-substitutions and varied chiral scaffolds were screened in this reaction (entries 2–8), and we were pleased to find that the BINOL-derived CPA catalyst A5 bearing ortho-(9-phenanthracenyl)-substituents could generate the C-H amination product 3a in 94% ee and the recovered 1a with 90% ee (s = 100, entry 5). It is worth mentioning that the sterically hindered CPA could not provide the desired C-H amination product under the same conditions (entries 6 and 8). A diverse set of solvents suitable for solubilizing substrate 1a was examined, which suggested that the nonpolar solvents gave more superior KR performances (entries 9–13). Satisfyingly, the KR reaction in CCl4 provided 3a with 95% ee and the recovered 1a with 90% ee, corresponding to a selectivity factor of 120 (entry 13). Moreover, it is noteworthy that the reaction conducted in CCl4 exhibited the highest reaction rate, which would enable a decrease in both the reaction temperature and the necessary amount of azodicarboxylate. Consequently, the reaction temperature was investigated as well (entries 14–16), which suggested that −10 °C was the optimal reaction temperature, and both the recovered 1a and C-H amination product 3a could be afforded with 96% ee (s = 194, entry 15). Finally, to facilitate the purification process, the amount of azodicarboxylate 2a was reduced to 0.75 equivalents while increasing the concentration of this reaction, which still provided satisfactory KR performance (s = 127, entry 17). Recognizing the potential toxicity associated with CCl4, the reaction was also explored in toluene at lower temperatures (entries 18–19). Although this resulted in slightly diminished KR performance, it nonetheless offered an alternative set of conditions for the KR of amido[2.2]paracyclophanes.
Substrates scope investigation
With the optimal KR conditions in hand, we set out to investigate the scope of substituted amido[2.2]paracyclophanes (Fig. 2). First, a series of N-substitutions were screened, and we were delighted to find that various N-alkoxycarbonyl groups were well tolerated in this reaction, which could be selectively removed under distinct conditions (1a–1e). Next, we turned our attention to the disubstituted amido[2.2]paracyclophanes. The pseudo-geminal-Br-substituted amido-PCP 1f was examined under the standard KR conditions, which readily provided the C-H amination product 3f in 46% yield with 97% ee, as well as the recovered (Sp)-1f in 51% yield with 89% ee, corresponding to an s factor of 198. The absolute configure of recovered 1f was assigned as Sp, which was unambiguously determined by X-ray crystallography analysis, and the configurations of the other amido-PCPs were assigned by analogy to this structure. Additionally, a series of amido-PCPs bearing aryl, alkenyl, alkynyl and alkyl groups at the pseudo-geminal-position were all amenable to this method, which provided good to high KR performances (1g–1k). Notably, the pseudo-geminal OAc-substituted amido-PCP 1l was also a feasible variant under the KR conditions, which was a valuable chiral synthon for the development of chiral catalysts and ligands11. A wide range of substitutions at the pseudo-ortho-position of the amido-PCPs were also investigated, which were all well compatible with this KR method, giving s factors up to 180 (1m–1v). It is worth mentioning that diverse functional groups were readily feasible at this position, including Br (1m), OAc (1s), CHO (1t), CH2OH (1u) and CO2Me (1v), which would facilitate the further derivatizations of these chiral PCP building blocks. The pseudo-meta- and pseudo-para-substituted amido-PCPs bearing bromo, aryl, alkenyl and alkyl groups were studied with the KR method as well, which could give both the recovered SM (Rp)-1 and the C-H amination products 3 up to >95% ee, albeit slighted modified conditions were employed for these substrates (1w–1z for pseudo-meta-substituted amido-PCPs and 1aa–1ad for pseudo-para-substituted amido-PCPs). Furthermore, the enantioselective desymmetrization of the achiral pseudo-para-substituted diamido-PCP 4a was studied using this method by modifying the reaction conditions, which gave access to the expected C-H amination product 5a in 40% yield with 99% ee, with the unreacted 4a in 23% yield and achiral di-amination product as the major byproducts.

Reactions were run with 1 (0.2 mmol), 2a (0.14 mmol) and the CPA (R)-A5 (0.02 mmol, 10 mol%) in CCl4 (2 ml) with 4 Å molecular sieves (200 mg) at −10 °C. Yields refer to isolated yields. The ee values were determined by HPLC analysis on a chiral stationary phase. Conversion (C) = ees/(ees + eep). s = ln[(1 − C)(1 − ees)]/ln[(1 − C)(1 + ees)]. Note: (a) Reactions were performed in toluene at −20 °C. (b) Reactions were performed in toluene at −30 °C. (c) Reactions were performed at 0 °C. (d) Reactions were performed using 2a (0.65 equiv) at 0 °C. (e) Reactions were performed using 2a (0.6 equiv) at 0 °C.
Mechanistic studies
To shed light on the detailed mechanism for these reactions, a series of control experiments were performed (Fig. 3). The treatment of the N-Boc-N-Me-substituted amido-PCP 6a with the standard KR conditions failed to provide the desired C-H amination product, which suggested the N-H moiety of the substrate played a crucial role in this reaction (Fig. 3a). Surprisingly, the 2,5-dimethyl-substituted N-Boc aniline 6b, a simplified noncyclophane-type analogue of 1a, was also found to be unreactive to the standard KR conditions, suggesting the indispensable role of the [2.2]paracyclophane scaffold in the reactivity of this reaction (Fig. 3b). Additionally, other N-substitution groups apart from the N-alkoxycarbonyl groups in the amido-PCP substrates were also examined in the KR reaction (Fig. 3c). Notably, the amido-PCPs bearing various N-acyl substitutions resulted in dramatically diminished KR performances (6c–6e, with s of 3.8 ~ 9.5), while the N-Ts substituted PCP barely exhibited any KR effect (with both recovered 6f and the C-H amination product 7f obtained in racemic form). These results suggested that the N-substitutions of the amido-PCPs play a critical role for the high stereoselectivity of these KR reactions, which may potentially affect the interaction between the CPA catalyst and the amido group. The N-unsubstituted 4-amino[2.2]paracyclophane 6g was also subjected to examination under the standard conditions, which surprisingly did not yield the expected C-H amination product 7g after 24 h. Instead, the triazane product 8g was afforded via the direct addition of the amino group to the azodicarboxylate44 (Fig. 3d). However, both the triazane product 8g and the recovered 6g were obtained with low enantioselectivities. Interestingly, raising the temperature to 20 °C and prolonging the reaction time to 48 h resulted in a mixture of 8g and the expected C-H amination product 7g, with poor KR performance. Moreover, during the screening of the substrate scope, a significant amount of triazane product 8 was also generated with high enantioselectivity in the KR of pseudo-meta-substituted amido-PCPs when performing the reaction at −10 °C (compared to 0 °C in the substrate scope). This type of intermediate was also observed for the pseudo-para-substituted amido-PCP 1ac, when analyzing the reaction mixture at the early stage of the reaction. For instance, the KR reaction of pseudo-meta-Ph-substituted amido-PCP 1x under the standard conditions (−10 °C, 40 h) provided the recovered (Rp)-1x in 51% yield with 92% ee and the C-H amination product 3x in 37% yield with 94%, as well as the triazane 8x in 8% yield with 95% ee (Fig. 3e). Careful monitoring this reaction by chiral HPLC analysis indicated that the triazane 8x was actually formed first, which was then slowly converted into the C-H amination product 3x. To study the transformation between 8x and 3x, several control experiments were conducted. In the absence of CPA catalyst, no conversion between 8x and 3x was observed, while the enantioenriched 8x could be readily converted into chiral 3x with retained enantiopurity in the presence of a racemic phosphoric acid catalyst. Treatment of racemic triazane 8x with CPA (R)-A5 resulted in the formation of 3x with 25% ee and the recovered 8x with 63% ee, corresponding to an s factor of 2.9 (Fig. 3f). These results suggested that the CPA-catalyzed rearrangement from 8x to 3x was not the primary stereoselective step in the KR reaction. To further elucidate this hydrazine-shift reaction, a cross-over experiment was performed. A 1:1 mixture of racemic triazane 8x and amido-PCP 1a was subjected to the standard conditions, which afforded the C-H amination product 3x in 43% yield with 32% ee and the recovered 8x in 43% yield with 34% ee (s = 2.6, Fig. 3g). Notably, no C-H amination product 3a derived from 1a was detected, which implied that the hydrazine-shift reaction proceeded through an intramolecular manner. Based on these results, a plausible reaction mechanism for this KR reaction was proposed (Fig. 3h). The CPA-catalyzed direct addition of the amido group to azodicarboxylate (formation of triazanes 8) was believed to be the stereodetermining step, in which the (Sp)-amido-PCP 1 was the kinetically favored enantiomer due to the dual-hydrogen bonding activation between CPA catalyst with substrates and azodicarboxylate. The acid-promoted hydrazine-shift from the amino group to the para-position was proposed to proceed through a para-semidine-type rearrangement, involving either a stepwise double N[1,3]-sigmatropic shift53,54 or a one-step N[1,5]-sigmatropic shift process55,56. In most cases, the rearrangement step is faster than the triazane formation step, and the corresponding triazane intermediate cannot be detected. However, due to the steric hindrance around the para-position of aniline for pseudo-meta- and pseudo-para-substituted amido-PCPs, the rearrangement step is slower than the initial triazane formation step, leading to the detection or isolation of the triazanes 8.

a Control experiment of N-Boc-N-methylated amido-PCP. b Control experiment of N-Boc aniline 6b. c KR of amido-PCPs bearing various N-acyl groups. d Control experiment of the N-unsubstituted amino-PCP 6g. e Monitoring the reaction of pseudo-meta-substituted amido-PCP 1x at −10 °C. f Reaction of triazane 8x in the presence of CPA catalysts. g Cross-over experiment of triazane 8x and amido-PCP 1a. h Proposed reaction mechanism.
Derivatizations of chiral products
To demonstrate the practicability of this method, a large-scale KR of racemic 1a (2.0 mmol) was performed using the standard conditions, which readily gave access to the C-H amination product 3a in 51% yield with 94% ee and the recovered (Rp)-1a in 47% yield with 99% ee, corresponding to an s factor of 170 (Fig. 4a). The derivatizations of these planar chiral products were then investigated to showcase the utilities of this method. Firstly, the derivatizations of the C-H amination products were examined. The treatment of 3a with strong basic conditions at 70 °C led to the facile removal of the hydrazine moiety, and provided the (Sp)-1a in 94% yield with 94% ee41 (Fig. 4b). Therefore, both enantiomers of the amido-PCPs could be readily afforded without the need for two KR reactions using opposite enantiomer of the CPA catalyst. Moreover, the substituted hydrazine group in 3a was readily converted into a NH2 group (9a) via facile catalytic hydrogenation, and this type of planar chiral diamine scaffold may find diverse applications in organic synthesis and material science. For instance, the coupling of 9a with isothiocyanate afforded the primary amine-thiourea-type derivative 10a, which could be further modified into a planar chiral bis-thiourea derivative 11a. Notably, both of these two products have the potential to serve as chiral organocatalysts in asymmetric catalysis. In addition, treatment of 9a with NaNO2 and KI afforded the para-iodinated amido-PCPs 12a, which is a versatile planar chiral building block for further derivatizations as well. Next, the derivatizations of the recovered chiral amido-PCPs were also studied (Fig. 4c). After switching the N-protecting group of the pseudo-geminal-Br-substituted amido-PCP (Sp)-1f from Boc to Piv group, the NHPiv group directed sp2-C–H borylation57 yielded a functional group-rich PCP derivative 14f in 80% yield with 91% ee, which bear one amido, one bromide and one boronate group. In addition, after removal of the N-Boc group of (Sp)-1f, the Combes quinoline synthesis was performed with acetyl acetone, yielding the corresponding quinoline-containing PCP derivative 15f. In addition, the Pd-catalyzed para-iodination of (Rp)-1a with NIS afforded 16a in 72% yield with 97% ee, which was also the enantiomer of 12a. Furthermore, after release of the NH2 group, the four-component reaction of the aniline moiety with glyoxal dimethyl acetal, formic acid, and benzyl isocyanide gave the indole-containing PCP derivative 17a58. The utilization of these planar chiral amido-PCP skeletons in the development of chiral organocatalysts was explored as well. Sequential coupling of the amino-PCP segment and (1R,2R)- or (1S,2S)-N,N-dimethylcyclohexane-1,2-diamine 18 with thiophosgene (SCCl2) afforded the diastereomeric tertiary amine-thiourea bifunctional catalysts 19a and 20a, respectively. These two products, along with the commercially available chiral tertiary amine-thiourea catalysts (24a and 24b), were screened as chiral bifunctional catalysts in the asymmetric [4 + 2] cyclizations of 2-benzothiazolimine 21a with homophthalic anhydride 22a59 (Fig. 4d). Notably, the reaction catalyzed by (1R,1S,Rp)-19a (15 mol%) yielded the cyclization product 23a in 87% yield with 10:1 dr and 89% ee, whereas the catalysts with the achiral aniline moiety (24a and 24b) produced the product with erosive yield, diastereoselectivity, and enantioselectivity. A significant match-mismatch effect was observed since the reaction enabled by (1S,2S,Rp)-20a generated the opposite enantiomer of product with both diminished dr and ee values.

a Large scale KR of amido-PCP 1a. b Derivatizations of the C-H amination product 3a. c Derivatizations of the recovered amido-PCPs. d Applications of chiral amido-PCP in the development of chiral organocatalysts.
In conclusion, we have developed an efficient kinetic resolution method for amido-PCPs through the CPA-catalyzed asymmetric C-H amination using commercially available azodicarboxylates. This KR method features a remarkably broad substrate scope, enabling the successful resolution of various pseudo-geminal-, pseudo-ortho-, pseudo-meta- and pseudo-para-substituted amido-PCPs with excellent KR performances (with s factors up to 371). Moreover, this reaction allows for the enantioselective desymmetrization of an achiral diamido-PCP substrate. Detailed experimental studies unraveled a new reaction mechanism for the electrophilic aromatic C-H amination, involving the sequential triazane formation and N[1,5]-rearrangement process, with the CPA-catalyzed direct addition of the amido group to azodicarboxylate as the stereodetermining step. The feasibility of large-scale KR reaction, along with the potential for diverse derivatizations of both the recovered chiral starting materials and the C-H amination products, particularly the utilization of the planar chiral amino-PCP scaffold in chiral organocatalysts development, further underscore the value of this methodology.
Methods
Representative procedure
To a solution of racemic 1 (0.2 mmol, 1.0 equiv.), (R)-A5 (0.02 mmol, 0.1 equiv.) and activated 4 Å MS (200 mg) in CCl4 or toluene (1 ml) was added a solution of 2a (0.14 mmol, 0.7 equiv.) in dry CCl4 or toluene (1 ml) at designed temperature under N2 atmosphere. After achieving appropriate conversion as indicated by HPLC analysis of the reaction mixture, the reaction mixture was quenched with Et3N (20 μl) and concentrated under vacuum to give a residue, which was purified by column chromatography to afford the recovered product 1 and product 3.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and Supplementary Information file, or from the corresponding author upon request. The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers CCDC 2248134 (for (Sp)-1f). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
References
Brown, C. J. & Farthing, A. C. Preparation and structure of Di-p-Xylylene. Nature 164, 915–916 (1949).
Cram, D. J. & Steinberg, H. Macro rings. I. Preparation and spectra of the paracyclophanes. J. Am. Chem. Soc. 73, 5691–5704 (1951).
Morisaki, Y. & Chujo, Y. Through-space conjugated polymers based on cyclophanes. Angew. Chem. Int. Ed. 45, 6430–6437 (2006).
David, O. R. P. Syntheses and applications of disubstituted [2.2]paracyclophanes. Tetrahedron 68, 8977–8993 (2012).
Hassan, Z., Spuling, E., Knoll, D. M., Lahann, J. & Bräse, S. Planar chiral [2.2]paracyclophanes: from synthetic curiosity to applications in asymmetric synthesis and materials. Chem. Soc. Rev. 47, 6947–6963 (2018).
Hassan, Z., Spuling, E., Knoll, D. M. & Bräse, S. Regioselective functionalization of [2.2]paracyclophanes: recent synthetic progress and perspectives. Angew. Chem. Int. Ed. 59, 2156–2170 (2020).
Felder, S., Wu, S., Brom, J., Micouin, L. & Benedetti, E. Enantiopure planar chiral [2.2]paracyclophanes: synthesis and applications in asymmetric organocatalysis. Chirality 33, 506–527 (2021).
Kitagaki, S., Ueda, T. & Mukai, C. Planar chiral [2.2]paracyclophane-based bis(thiourea) catalyst: application to asymmetric Henry reaction. Chem. Commun. 49, 4030–4032 (2013).
Ma, Y. et al. Asymmetric addition of aryl boron reagents to enones with rhodium dicyclophane imidazolium carbene catalysis. Angew. Chem. Int. Ed. 42, 5871–5874 (2003).
Sharma, N. et al. Turn on of sky-blue thermally activated delayed fluorescence and circularly polarized luminescence (CPL) via increased torsion by a bulky carbazolophane donor. Chem. Sci. 10, 6689–6696 (2019).
Schneider, J. F., Falk, F. C., Fröhlich, R. & Paradies, J. Planar-chiral thioureas as hydrogen-bond catalysts. Eur. J. Org. Chem. 2010, 2265–2269 (2010).
Liu, X. et al. Synthesis of cyclophanes with planar and helical chirality. J. Org. Chem. 76, 1953–1956 (2011).
Duan, W. et al. Synthesis of new alkoxy/sulfonate-substituted carbene precursors derived from [2.2]paracyclophane and their application in the asymmetric arylation of aldehydes. Tetrahedron Asymmetry 23, 1369–1375 (2012).
Zhao, L. et al. Asymmetric β-boration of α,β-unsaturated N-acyloxazolidinones by [2.2]paracyclophane-based bifunctional catalyst. Org. Lett. 14, 5780–5783 (2012).
Chen, J. et al. [2.2]Paracyclophane-based carbene–copper catalyst tuned by transannular electronic effects for asymmetric boration. RSC Adv. 6, 75144–75151 (2016).
Chen, C.-H. & Zheng, W.-H. Planar chiral B–N heteroarenes based on [2.2]paracyclophane as circularly polarized luminescence emitters. Org. Lett. 23, 5554–5558 (2021).
Xu, D. & Zheng, W.-H. Synthesis and chiroptical properties of planar chiral azahelicenes based on [2.2]paracyclophane. Org. Lett. 23, 8612–8616 (2021).
Yuan, S., Liao, C. & Zheng, W.-H. [2.2]Paracyclophane-based isothiourea-catalyzed highly enantioselective α-fluorination of carboxylic acids. Org. Lett. 23, 4142–4146 (2021).
Zhang, Z. et al. Molecular design and synthesis of dicarbazolophane-based centrosymmetric through-space donors for solution-processed thermally activated delayed fluorescence OLEDs. Org. Lett. 23, 6697–6702 (2021).
Braun, C., Bräse, S. & Schafer, L. L. Planar-chiral [2.2]paracyclophane-based amides as proligands for titanium- and zirconium-catalyzed hydroamination. Eur. J. Org. Chem. 2017, 1760–1764 (2017).
Rowlands, G. J. The synthesis of enantiomerically pure [2.2]paracyclophane derivatives. Org. Biomol. Chem. 6, 1527–1534 (2008).
Cipiciani, A. et al. Synthesis of chiral (R)−4-hydroxy- and (R)−4-halogeno[2.2]paracyclophanes and group polarizability. Optical rotation relationship. J. Org. Chem. 62, 3744–3747 (1997).
Friedmann, C. J., Ay, S. & Bräse, S. Improved synthesis of enantiopure 4-hydroxy[2.2]paracyclophane. J. Org. Chem. 75, 4612–4614 (2010).
Morisaki, Y., Gon, M., Sasamori, T., Tokitoh, N. & Chujo, Y. Planar chiral tetrasubstituted [2.2]paracyclophane: optical resolution and functionalization. J. Am. Chem. Soc. 136, 3350–3353 (2014).
Dominguez, B., Zanotti-Gerosa, A. & Hems, W. Electrophilic substitution of dibromoparacyclophane: a route to novel paracyclophane phosphine ligands. Org. Lett. 6, 1927–1930 (2004).
Vedejs, E. & Jure, M. Efficiency in nonenzymatic kinetic resolution. Angew. Chem. Int. Ed. 44, 3974–4001 (2005).
Pellissier, H. Catalytic non-enzymatic kinetic resolution. Adv. Synth. Catal. 353, 1613–1666 (2011).
Liu, W. & Yang, X. Recent advances in (dynamic) kinetic resolution and desymmetrization catalyzed by chiral phosphoric acids. Asian J. Org. Chem. 10, 692–710 (2021).
Liu, W., Wang, D., Zhang, D. & Yang, X. Catalytic kinetic resolution and desymmetrization of amines. Synlett 33, 1788–1812 (2022).
Rossen, K., Pye, P. J., Maliakal, A. & Volante, R. P. Kinetic resolution of rac-4,12-Dibromo[2.2]paracyclophane in a palladium [2.2]PHANEPHOS catalyzed amination. J. Org. Chem. 62, 6462–6463 (1997).
Braddock, D. C., MacGilp, I. D. & Perry, B. G. Improved synthesis of (±)−4,12-Dihydroxy[2.2]paracyclophane and its enantiomeric resolution by enzymatic methods: planar chiral (R)- and (S)-phanol. J. Org. Chem. 67, 8679–8681 (2002).
Mori, K., Kishi, H. & Akiyama, T. Highly efficient kinetic resolution of PHANOL by chiral phosphoric acid catalyzed asymmetric acylation. Synthesis 49, 365–370 (2017).
Dorizon, P., Martin, C., Daran, J.-C., Fiaud, J.-C. & Kagan, H. B. A practical kinetic resolution of 4-acetyl[2.2]paracyclophane. Tetrahedron Asymmetry 12, 2625–2630 (2001).
Delcourt, M.-L., Turcaud, S., Benedetti, E. & Micouin, L. Efficient and scalable kinetic resolution of racemic 4-Formyl[2.2]paracyclophane via asymmetric transfer hydrogenation. Adv. Synth. Catal. 358, 1213–1218 (2016).
Delcourt, M.-L. et al. Highly enantioselective asymmetric transfer hydrogenation: a practical and scalable method to efficiently access planar chiral [2.2]paracyclophanes. J. Org. Chem. 84, 5369–5382 (2019).
Zippel, C., Hassan, Z., Parsa, A. Q., Hohmann, J. & Bräse, S. Multigram-scale kinetic resolution of 4-acetyl[2.2]paracyclophane via Ru-catalyzed enantioselective hydrogenation: accessing [2.2]paracyclophanes with planar and central chirality. Adv. Synth. Catal. 363, 2861–2865 (2021).
Delcourt, M.-L., Felder, S., Benedetti, E. & Micouin, L. Highly enantioselective desymmetrization of centrosymmetric pseudo-para-Diformyl[2.2]paracyclophane via asymmetric transfer hydrogenation. ACS Catal. 8, 6612–6616 (2018).
Zhao, Y., Wang, H., Wu, B. & Zhou, Y.-G. Synthesis of paracyclophanes with planar and central chirality: kinetic resolution of [2.2]paracyclophane aldimines via palladium-catalyzed addition of arylboronic acids. Org. Chem. Front. 6, 3956–3960 (2019).
Zhao, Y., Ding, Y.-X., Wu, B. & Zhou, Y.-G. Nickel-catalyzed asymmetric hydrogenation for kinetic resolution of [2.2]paracyclophane-derived cyclic N-sulfonylimines. J. Org. Chem. 86, 10788–10798 (2021).
Zhao, Y., Wang, X.-Q., Yu, Y.-J. & Zhou, Y.-G. Kinetic resolution of [2.2]paracyclophane-derived cyclic N-sulfonylimines via palladium-catalyzed addition of arylboronic acids. J. Org. Chem. 86, 1262–1272 (2021).
Chen, Y., Zhu, C., Guo, Z., Liu, W. & Yang, X. Asymmetric synthesis of hydroquinolines with α,α-disubstitution through organocatalyzed kinetic resolution. Angew. Chem. Int. Ed. 60, 5268–5272 (2021).
Pan, Y. et al. Kinetic resolution of α-tertiary propargylic amines through asymmetric remote aminations of anilines. ACS Catal. 11, 8443–8448 (2021).
Xie, J. et al. Kinetic resolution of 1,2-diamines via organocatalyzed asymmetric electrophilic aminations of anilines. Chin. J. Chem. 40, 1674–1680 (2022).
Liu, W., Jiang, Q. & Yang, X. A versatile method for kinetic resolution of protecting-group-free BINAMs and NOBINs through chiral phosphoric acid catalyzed triazane formation. Angew. Chem. Int. Ed. 59, 23598–23602 (2020).
Wang, D., Liu, W., Tang, M., Yu, N. & Yang, X. Atroposelective synthesis of biaryl diamines and amino alcohols via chiral phosphoric acid catalyzed para-aminations of anilines and phenols. iScience 22, 195–205 (2019).
Uraguchi, D. & Terada, M. Chiral brønsted acid-catalyzed direct Mannich reactions via electrophilic activation. J. Am. Chem. Soc. 126, 5356–5357 (2004).
Akiyama, T., Itoh, J., Yokota, K. & Fuchibe, K. Enantioselective Mannich-type reaction catalyzed by a chiral brønsted acid. Angew. Chem. Int. Ed. 43, 1566–1568 (2004).
Parmar, D., Sugiono, E., Raja, S. & Rueping, M. Complete field guide to asymmetric BINOL-phosphate derived brønsted acid and metal catalysis: history and classification by mode of activation; brønsted acidity, hydrogen bonding, ion pairing, and metal phosphates. Chem. Rev. 114, 9047–9153 (2014).
Rahman, A. & Lin, X. Development and application of chiral spirocyclic phosphoric acids in asymmetric catalysis. Org. Biomol. Chem. 16, 4753–4777 (2018).
Wang, D., Shao, Y.-B., Chen, Y., Xue, X.-S. & Yang, X. Enantioselective synthesis of planar-chiral macrocycles through asymmetric electrophilic aromatic amination. Angew. Chem. Int. Ed. 61, e202201064 (2022).
Bao, H., Chen, Y. & Yang, X. Catalytic asymmetric synthesis of axially chiral diaryl ethers through enantioselective desymmetrization. Angew. Chem. Int. Ed. 62, e202300481 (2023).
Kagna, H. B. & Fiaud, J. C. in Kinetic Resolution, Vol. 18 (eds Eliel, E. L. & Wilen, S. H.) 249–330 (Wiley, 1988).
Hou, S., Li, X. & Xu, J. N[1,3]-sigmatropic shift in the benzidine rearrangement: experimental and theoretical investigation. Org. Biomol. Chem. 12, 4952–4963 (2014).
Yang, Z. et al. Regioselectivity of the ortho- and para-semidine, and diphenyline rearrangements. Tetrahedron 72, 2186–2195 (2016).
Heesing, A. & Schinke, U. Zum Mechanismus der Semidin-Umlagerung unter Zwei-Protonen-Katalyse. Chem. Ber. 110, 3319–3323 (1977).
Shine, H. J., Zmuda, H., Kwart, H., Horgan, A. G. & Brechbiel, M. Benzidine rearrangements. 17. The concerted nature of the one-proton p-semidine rearrangement of 4-methoxyhydrazobenzene. J. Am. Chem. Soc. 104, 5181–5184 (1982).
Lv, J. et al. Metal-free directed sp2-C–H borylation. Nature 575, 336–340 (2019).
Lei, X., Angeli, G. K., Neochoritis, C. G. & Dömling, A. Sustainable multicomponent indole synthesis with broad scope. Green. Chem. 24, 6168–6171 (2022).
Liu, S.-J. et al. Rational design of axially chiral styrene-based organocatalysts and their application in catalytic asymmetric (2+4) cyclizations. Angew. Chem. Int. Ed. 61, e202112226 (2022).
Acknowledgements
The authors gratefully acknowledge NSFC (grant nos. 22171186, 22222107), Double First-Class Initiative Fund of ShanghaiTech University, and ShanghaiTech University start-up funding for financial support. The authors thank the support from Analytical Instrumentation Center (# SPST-AIC10112914), SPST, ShanghaiTech University and Mr. Huanchao Gu for the X-ray crystallographic analysis.
Author information
Authors and Affiliations
Contributions
S.Y., H.B. and D.Z. performed the experiments. X.Y. directed the project and wrote the paper with the feedback from other authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, S., Bao, H., Zhang, D. et al. Kinetic resolution of substituted amido[2.2]paracyclophanes via asymmetric electrophilic amination. Nat Commun 14, 5239 (2023). https://doi.org/10.1038/s41467-023-40718-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-40718-8
This article is cited by
-
Organocatalytic desymmetrization provides access to planar chiral [2.2]paracyclophanes
Nature Communications (2024)
-
Asymmetric synthesis of P-stereogenic phosphindane oxides via kinetic resolution and their biological activity
Nature Communications (2024)
-
(Parallel) kinetic resolution of 3,3-disubstituted indolines via organocatalyzed reactions with azodicarboxylates
Science China Chemistry (2024)
-
Catalytic asymmetric synthesis of sulfur-containing atropisomers by C-S bond formations
Science China Chemistry (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
