
Abstract
Fluorination using chiral catalytic methods could result in a direct access to asymmetric fluorine chemistry. However, challenges in catalytic asymmetric fluorinations, especially the longstanding stereochemical challenges existed in BF3·Et2O-based fluorinations, have not yet been addressed. Here we report the catalytic asymmetric nucleophilic fluorination using BF3·Et2O as the fluorine reagent in the presence of chiral iodine catalyst. Various chiral fluorinated oxazine products were obtained with good to excellent enantioselectivities (up to >99% ee) and diastereoselectivities (up to >20:1 dr). Control experiments (the desired fluoro-oxazines could not be obtained when Py·HF or Et3N·3HF were employed as the fluorine source) indicated that BF3·Et2O acted not only as a fluorine reagent but also as the activating reagent for activation of iodosylbenzene.
Similar content being viewed by others
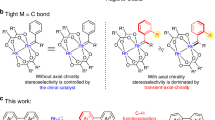

Introduction
Being called as “a small atom with a big ego”, fluorine acts as a significant and increasingly important role in the fields of organic chemistry, pharmaceuticals, agrochemicals and material chemistry1,2,3,4. The fluorinated molecules often display higher thermal and metabolic stabilities, lower polarity, and weaker intermolecular interactions due to the strong C−F bond and unique properties of F atom5. Therefore, these unique properties of fluorine-contained compounds make the development of efficient strategies, especially of catalytic asymmetric reaction for fluorination of molecules as one of the hottest areas in organic synthesis2,3. Nevertheless, the asymmetric fluorine organic chemistry still represents a considerable challenge to date6. In the wake of the emergence of the first electrophilic enantioselective fluorination of enolates using chiral N-fluoro camphorsultam reagent reported by the group of Lang7, significant progress for enantioselective fluorination studies have been presented4,8,9 because of contributing to the development of catalytic asymmetric methodologies for electrophilic fluorine reagents (F+ reagents), such as N-fluorobenzenesulfonimide (NFSI)10,11,12, N-fluoropyridinium salts3, and Selectfluor (Fig. 1a)13,14,15,16,17,18. These reagents exhibited efficient transfer of fluorine atom under the asymmetric fluorination, however, their industrial applications were significantly restricted by the poor atom economy in fluorination, expensive synthesis and other inherent characteristics of electrophilic reagent. Alternatively, nucleophilic fluorine reagents (F− reagents) have been attracting considerable interest recently since the relative stability and low-cost. Considerable advances have recently been achieved in this field involving catalytic asymmetric fluorination of keto esters19,20 and alkenes21,22,23,24,25 employing pyridine·HF as a fluorine reagent, catalytic asymmetric fluorination of allylic trichloroacetimidates using a combination of Et3N·HF with Iridium complex26, asymmetric ring-opening fluorinations of meso-epoxides (aziranes) using PhCOF, HF-reagents or AgF as the fluorine source in metal-catalyzed system27,28,29, and other asymmetric transformations in the presence of metal fluorides (KF, CsF or AgF)30,31,32 (Fig. 1b). Despite these elegant works, several practical disadvantages still discouraged their further large-scale utilization in the area: the high toxicity and biohazard for HF-bases, and metal fluorides poor solubility in organic solvents coupled with limited strategies to control reactivity.

a Asymmetric fluorinations using electrophilic fluorine (F+) reagents. b Asymmetric fluorinations using nucleophilic fluorine (F−) reagents. c The first example of using BF3·Et2O as F− Reagent for asymmetric fluorinations (this work). [F+], electrophilic fluorine reagents; [F−], nucleophilic fluorine reagents; PTC, phase-transfer catalyst; [M], metal catalyst; L, ligand; HBPTC, hydrogen bonding phase-transfer catalysis; [O], oxidant; m-CPBA, Meta-Chloroperoxybenzoic acid.
Ideally, one low-toxic, stable and commercially cheap available nucleophilic fluorine reagent would drastically promote enantioselective fluorine synthetic innovation and industrial development. As a versatile Lewis acid, commercially available BF3·Et2O is easy to prepare and is widely being used in various organic transformations. As early as 1960, it was discovered that as a nucleophilic fluorine reagent could be applied in the fluorination of ring opening of mesoepoxides33. The development of BF3·Et2O mediated reactions in half a century reveals that the BF3·Et2O can also be applied to fluorinations of alkenes34,35,36, alkynes37, arenes38,39 and other fluorine organic chemistry40,41,42,43. Although these efficient achiral methodologies have been well-established, to date, the longstanding stereochemical challenges of the BF3·Et2O-based fluorination have not yet been addressed, probably impeded by several hurdles: intense competition for the role of BF3 between a nucleophilic fluorine reagent and a Lewis acid, the difficulty in achieving stereocontrol of fluorine atom, the competition from the uncatalyzed background reaction and other side reactions. Undoubtedly, the advent of enantioselective approach is long overdue that would be welcome.
In terms of the operational and environmental advantages associated with organocatalysis, we speculated that a metal-free mild reaction system with a chiral iodine catalyst (CIC) might meet the aforementioned challenges21,44,45. Its activated oxidants forms could form the iodine (III) catalyst with a typical structure type of trigonal bipyramidal geometry, thus this type of robust organocatalyst has been commonly used for asymmetric nucleophilic addition reactions44,45. The unique stereoscopic configuration of iodine (III) and well-defined steric hindrance of the iodine (III) catalyst bearing chiral ligand can be readily applied, leading to complete stereo-control in fluorination of olefins using BF3·Et2O as a nucleophilic reagent. Rapid cyclization and its simultaneous BF3·Et2O nucleophilic fluorination are viable with a CIC to suppress the pure intramolecular cycloaddition and other side reactions.
Herein, we introduce a catalytic asymmetric fluorination strategy that involves BF3·Et2O as nucleophilic fluorine source with chiral iodine catalysts. Various F-contained products were obtained with good to excellent diastereoselectivities (>20:1 dr) and enantioselectivities (up to >99% ee) (Fig. 1c).
Results and discussion
Catalyst optimization
After several initial trials (Supplementary Table 1), we set out to optimize the model catalytic asymmetric aminofluorination of N-cinnamylbenzamide (1a) in the presence of 15 mol% of ligand loading using BF3·Et2O as the fluorine reagent in DCE at −25 °C with m-CPBA as an oxidate (Fig. 2). The significant difference in stereoselectivity was observed under these reaction conditions, whereas good yields were afforded with the addition of structure diverse chiral iodine catalyst CICs. These results suggested that the substituents of the catalysts have a strong influence on the stereochemistry of the reaction. In general, compared to spiro-CICs, the linear CICs could give the higher stereoselectivity. The CIC1 was proved to be the best catalyst, providing the desired chiral fluorinated oxazine 1b with excellent diastereoselectivity (>20:1 dr) and enantioselectivity (86% ee) in 70% yield.

Notations for CICs: the reaction of 1a (0.1 mmol), BF3·Et2O (1.0 mmol), m-CPBA (0.12 mmol) and catalyst (15% mol) was carried out in 4.0 ml of 1,2-dichloroethane (DCE) at −25 °C for 48 h; isolated yields are reported. The ee values were determined by chiral HPLC analysis and the dr values were determined by 19F NMR.
Asymmetric aminofluorination of N-cinnamylbenzamides using BF3·Et2O
Results of experiments under the optimized conditions that probe the scope of the reaction are summarized in Fig. 3. Substrate scope of the reaction was investigated with a variety of substituted N-cinnamylbenzamides under the optimal reaction conditions. As shown in Fig. 3a, variation of the electronic properties of substituents at either Ar1 or Ar2 of N-cinnamylbenzamides with different steric parameters were tolerated, affording the desired products with good to excellent diastereoselectivities (2:1–>20:1 dr) and enantioselectivities (80–>99% ee) in good yields (45–75%). Gratifyingly, the fluorinated oxazine products bearing Ar2 with high steric hindrance were still obtained in good yields and excellent stereoselectivities (85–>99% ee, 15b–24b). The naphthyl-substituted N-cinnamylbenzamides could also be tolerated, and gave the corresponding fluorinated products (26b, 27b and 31b) in excellent diastereoselectivities (up to >20:1dr) and enantioselectivities (86–87% ee) with good yields. Furthermore, as expected, the catalytic system also proved to be efficient for the N-cinnamylbenzamides with heterocycles or “complex substituents” on Ar2 ring (Fig. 3b, 35b–42b), again leading to good yields (45–72%) in high to excellent diastereoselectivities (10:1–>20:1dr) and enantioselectivities (80-99% ee). It is worth noting that the catalytic asymmetric aminofluorination of complex natural product structures could also be achieved efficiently in high to excellent diastereoselectivities and enantioselectivities.

Reactions were conducted on a 0.2 mmol scale with 10.0 equivalents of BF3·Et2O in DCE (8.0 mL) at −25 °C for 48 h. The absolute configuration of 4b was assigned by X-ray crystallography (structure shown), and the configuration of all other products was assigned by analogy. a Substrate scope of N-cinnamylbenzamides bearing various substituents on Ar1 and Ar2 ring. b Substrate scope of N-cinnamylbenzamides with “complex substituents” on Ar2 ring and Ar2 = heterocyclic groups. c Gram-scale reaction with 4a, CIC1, m-CPBA and BF3·Et2O. Isolated yields are reported. *The ee values of 30b and 31b could not be detected by HPLC. †CIC2 was employed as the catalyst for 42b. The ee values were determined by chiral HPLC analysis and the dr values were determined by 19F NMR. t-Bu, tert-butyl; Ph, phenyl; Me, methyl; Ac, acetyl.
Additionally, the gram-scale experiment was conducted to evaluate the applicability of our asymmetric fluorination method by using 4a (Fig. 3c), the desired product was obtained with excellent diastereoselectivity (>20:1 dr) and enantioselectivity (92% ee). The result suggested that our protocol was promising in future industrial applications. It was interesting that compared to the PTC catalyzed process46, different fluorinated products could be obtained from the same substrates using current process (indicating a different catalytic progress). The relative and absolute configurations of the products were determined by X-ray crystal structure analysis of 4b (see the Supplementary Information).
Asymmetric aminofluorination of N-(2-(prop-1-en-2-yl)phenyl)benzamides
To further understanding of the scope of this catalytic system, substrates 43a–54a were employed to undergo the fluorination process. To our delight, various substituted N-(2-(prop-1-en-2-yl)phenyl)benzamides including either electron-donating substituents or steric hindrance substituents at different positions on the Ar4 ring, as well as 3,5-ditrifluoromethyl substituents could be tolerated, affording the corresponding fluorinated products (43b–54b) with high enantioselectivities (80–85% ee) and isolated yields (80–88% yield) (Fig. 4). These substrate scope expanding experiments showed the hypothesis to introduce the combination of BF3·Et2O and CIC into direct, catalytic asymmetric fluorinations can be achieved. This method expanded the structures of the fluorinated products and provided a benign access to asymmetric nucleophilic fluorinations.

Reactions were conducted on a 0.2 mmol scale with 10.0 equivalents of BF3·Et2O in C6H5F (8.0 mL) at −42 °C for 20 h; isolated yields are reported. *CIC2 was applied as the catalyst for the formation of 53b and 54b. The ee values were determined by chiral HPLC analysis. Me, methyl; t-Bu, tert-butyl; Et, ethyl.
Mechanism studies
On the basis of the experimental results described above, we have proposed a possible mechanism to explain the stereochemistry of the catalytic asymmetric nucleophilic fluorinations (Fig. 5). To gain a better understanding of the process of this catalytic fluorination system, we also conducted control experiments (Fig. 5a) and density functional theory (DFT) calculations (Fig. 6). It is worth noting that we could not obtain the desired fluorinated products when we used or Py·HF or Et3N·3HF instead of BF3·Et2O (Fig. 5a, equation 1). When PhIF247 was applied as the hypervalent iodine reagent and fluorine source, we didn’t obtain the 1b as well (Fig. 5a, equation 1). Thus, these results indicate that BF3·Et2O acted not only as the nucleophilic fluorine source, but also as the activating reagents for activation of iodosylbenzene (Int1). which is distinct from previously catalytic nucleophilic fluorination process reported by Jacobsen’s group21. Based on previous work29,35,48,49, control experiments and DFT calculations, the plausible catalytic cycle was shown in Fig. 5b, and associated free energy profile was shown in Fig. 6.

a Control experiments. b Plausible catalytic cycle.

a DFT-computed reaction profile for the catalytic asymmetric fluorination of 1a and BF3·Et2O in the presence of CIC1 and m-CPBA. b DFT-optimized stereo-determining transition state (TS1) structures and their relative energies for Int2.
At first, the aryl iodine catalyst is oxidized by m-CPBA to form Ar−I=O (Int1), and Int1 is found to be 2.8 kcal/mol lower in energy than ArI. (Fig. 6). Then Int2 is formed through the activation of Ar−I=O by BF3·Et2O44, the energy of this intermediate is calculated to be 23.8 kcal/mol lower than Int1 (Fig. 6). The electrophilic addition of iodine (III) to the double bond of la forms Int334, during which the energy barrier is found to be 3.6 kcal/mol. Then with the assistance of BF3, Int3 releases anionic [BF4]- to form Int3+. The nucleophilic attack of [BF4]− (generated in previous step) on Int3+ at C1 position afford the TS143, the energy barrier of this step is 13.1 kcal/mol. There were intramolecular n−σ* interactions between the electron-deficient iodine (III) center and the carbonyl groups50,51. For the impacts of steric hindrance, the nucleophilic attack of F− on the Si face was favored (Fig. 5b), and this is in consistence with the experimental results and DFT calculations (Fig. 6b). As seen from the energy profile, the enantioselectivity is determined by the addition of F- with [BF4]- as fluorine source to the cationic Int3+ (Fig. 6a). The energy of TS1 versus ent-TS1, the transition state to the minor product, is compared. Surprisingly the two TSs exhibit huge energy difference of 14.9 kcal/mol (Fig. 6b). A closer observation on the geometry shows that ent-TS1 is much later, and bears a significantly longer I-O distance. In both TSs, the hypervalent I(III) atom is stabilized by interaction with the amide oxygen atom in the alkene. It could be proposed that the relative direction of alkene and catalyst in ent-TS1 disabled the feasible I-O interaction that provides essential stabilization to the iodane, leading to both a later transition state and much higher energy. The formation of Int4 is achieved through the interaction between TS1 and BF3, and Int4 is found to be 23.7 kcal/mol lower in energy than TS1. Then Int4 releases anionic [BF2OBF3]− to form Int4+, and Int4+ is found to be 5.5 kcal/mol lower in energy than Int4. Dearomatization of Ar1 ring of Int4+ by intramolecularly nucleophilic attack of the Ar1 on Si face at C2 position (Fig. 5b) furnish the cyclopropyl compound TS2 (Fig. 5b)49, which was calculated to be +11.1 kcal/mol in energy relative to Int4+. And then Int5 is formed with 1.7 kcal/mol of energy barrier relative to TS2. The hypervalent iodine (III) Int5 underwent reductive elimination to afford Int6 with 6.8 kcal/mol of energy barrier. TS3 was formed by the intramolecularly nucleophilic attack of the amide oxygen on C2 position with 4.5 kcal/mol of energy barrier relative to Int6. Here the nucleophilic attack of the amide oxygen takes place regioselectively at the higher substituted carbon atom of the cyclopropane unit49. Ring opening of the spirocyclopropyl ring TS3 takes place intramolecularly via a cyclization with simultaneous ring expansion to the six-membered cationic Int7 (Fig. 6)49. The calculated energy barrier for this step is −38.9 kcal/mol relative to TS5. Control experiment (Fig. 5a, equation 2) demonstrated the necessity of Ar1 ring. Moreover, the aromatic ring (Ar2) is essential to stabilize cationic Int7 (Fig. 5a, equation 3). With the assistance of [BF2OBF3]- anion, Int7 can be deprotonated to generate final product 1b (Fig. 6a). Besides, lengthening of carbon chain could not result in a desired fluoro-product according to the control experiment (Fig. 5a, equation 4). In a word, the formation of fluorinated oxazines follows a fluorination/1, 2-aryl migration/cyclization cascade49.
We have disclosed an efficient asymmetric fluorinations process that has enabled the development of the first highly enantioselective fluorination reaction (up to 99% ee and >20:1 dr) using BF3·Et2O as the fluorine source and dual-activating reagent. Moreover, the substrate expanding experiments further demonstrated the wide applicability of current method. This process provides not only a direct access to fluoro‐oxazine/benzoxazepine skeletons, but also a foundation for further development of new types of asymmetric nucleophilic fluorinations in future applications. The studies of the applicability of this asymmetric fluorination methodology using other substrates are going on in our group.
Methods
General procedure for synthesis of 1b–42b
The substrate (0.2 mmol) and catalyst (15 mol%) were mixed into the reaction tube, and then DCE (8.0 ml) was added. The mixture was cooled to −25 °C, after stirring for 10 min at this temperature, m-CPBA (1.2 equiv.) was added in one portion, followed by addition of BF3·Et2O (10.0 equiv.) dropwise. The reaction was run at −25 °C for 48 h. The reaction mixture was poured into saturated NaHCO3 (aq), the organic layer was collected and washed with brine, dried over Na2SO4, and concentrated under reduced pressure in the presence of basic Al2O3, Column chromatography (basic Al2O3, 200–300 mesh, EtOAc-hexane (0.5% Et3N) elution: hexane/EtOAc (V/V) = 50:1 ~ 5:1) gave the corresponding fluorinated products.
General procedure for synthesis of 43b–54b
The substrate (0.2 mmol) and catalyst (20 mol%) were mixed into the reaction tube, and then C6H5F (8.0 ml) was added. The mixture was cooled to −42 °C, after stirring for 5 min at this temperature, m-CPBA (1.2 equiv) was added in one portion, followed by addition of BF3·Et2O (10.0 equiv) dropwise. The reaction was run at −42 °C for 20 h. The reaction mixture was poured into saturated NaHCO3 (aq) sulotion, the organic layer was collected and washed with brine, dried over Na2SO4, and concentrated under reduced pressure in the presence of basic Al2O3, Column chromatography (basic Al2O3, 200–300 mesh, EtOAc-Hexanes (0.5% Et3N) elution: hexanes/EtOAc (V/V) = 100:1 ~ 25:1) gave the corresponding fluorinated products.
DFT calculations
All calculations were carried out with the Gaussian 09 software52. The B3LYP functional53 was adopted for all calculations in combination with the D3BJ dispersion correction54. For geometry optimization and frequency calculations, the SDD ECP and basis set55 was used for I and 6-31 G(d) for others56,57. The singlet point energy calculations were performed with a larger basis set combination, in which the def2-TZVP basis set58 was used for I, and 6–311 + G(d,p)59,60 for others. The SMD implicit solvation model61 was used to account for the solvation effect of DCE when performing single point energy calculations.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All relevant data are available from the corresponding author upon reasonable request. All the data supporting the findings of this study are available within this article, and supplementary information files. The authors declare that all data generated or analyzed during this study are included in this Article (and its Supplementary Information). The X-ray crystallographic coordinates for the structure of 4b are available free of charge from the Cambridge Crystallographic Data Centre under deposition number CCDC 1960281.
References
Amii, H. & Uneyama, K. C−F bond activation in organic synthesis. Chem. Rev. 109, 2119–2183 (2009).
Gouverneur, V. & Seppelt, K. Introduction: fluorine chemistry. Chem. Rev. 115, 563–565 (2015).
Champagne, P. A. et al. Monofluorination of organic compounds: 10 years of innovation. Chem. Rev. 115, 9073–9174 (2015).
Zhu, Y. et al. Modern approaches for asymmetric construction of carbon−fluorine quaternary stereogenic centers: synthetic challenges and pharmaceutical needs. Chem. Rev. 118, 3887–3964 (2018).
Petrone, D. A., Ye, J. & Lautens, M. Modern transition-metal-catalyzed carbon–halogen bond formation. Chem. Rev. 116, 8003–8104 (2016).
Ma, J. A. & Cahard, D. Asymmetric fluorination, trifluoromethylation, and perfluoroalkylation reactions. Chem. Rev. 104, 6119–6146 (2004).
Differding, E. & Lang, R. W. New fluorinating reagents - I. The first enantioselective fluorination reaction. Tetrahedron Lett. 29, 6087–6090 (1988).
Yang, X., Wu, T., Phipps, R. J. & Toste, F. D. Advances in catalytic enantioselective fluorination, mono-, di-, and trifluoromethylation, and trifluoromethylthiolation reactions. Chem. Rev. 115, 826–870 (2015).
Kohlhepp, S. V. & Gulder, T. Hypervalent iodine(III) fluorinations of alkenes and diazo compounds: new opportunities in fluorination chemistry. Chem. Soc. Rev. 45, 6270–6288 (2016).
Ishimaru, T. et al. Cinchona alkaloid catalyzed enantioselective fluorination of allyl silanes, silyl enol ethers, and oxindoles. Angew. Chem. Int. Ed. 47, 4157–4161 (2008).
Miró, J., del Pozo, C., Toste, F. D. & Fustero, S. Enantioselective palladium‐catalyzed oxidative β,β‐fluoroarylation of α,β‐unsaturated carbonyl derivatives. Angew. Chem. Int. Ed. 55, 9045–9049 (2016).
Buckingham, F. et al. Organomediated enantioselective 18F fluorination for PET applications. Angew. Chem. Int. Ed. 54, 13366–13369 (2015).
Rauniyar, V., Lackner, A. D., Hamilton, G. L. & Toste, F. D. Asymmetric electrophilic fluorination using an anionic chiral phase-transfer catalyst. Science 334, 1681 (2011).
Wang, L. M. et al. Enantio‐ and diastereoselective hydrofluorination of enals by N‐heterocyclic carbene catalysis. Angew. Chem. Int. Ed. 58, 7410–7414 (2019).
Egami, H. et al. Asymmetric fluorolactonization with a bifunctional hydroxyl carboxylate catalyst. J. Am. Chem. Soc. 137, 10132–10135 (2015).
He, Y., Yang, Z. Y., Thornbury, R. T. & Toste, F. D. Palladium-catalyzed enantioselective 1,1-fluoroarylation of aminoalkenes. J. Am. Chem. Soc. 137, 12207–12210 (2015).
Sandford, C., Rasappan, R. & Aggarwal, V. K. Synthesis of enantioenriched alkylfluorides by the fluorination of boronate complexes. J. Am. Chem. Soc. 137, 10100–10103 (2015).
Hiramatsu, K., Honjo, T., Rauniyar, V. & Toste, F. D. Enantioselective synthesis of fluoro-dihydroquinazolones and -benzooxazinones by fluorination-initiated asymmetric cyclization reactions. ACS Catal. 6, 151–154 (2016).
Suzuki, S. et al. Iodoarene-catalyzed fluorination and aminofluorination by an Ar-I/HF·pyridine/mCPBA system. Chem. Sci. 5, 2754–2760 (2014).
Pluta, R. et al. Metal-free catalytic asymmetric fluorination of keto esters using a combination of hydrogen fluoride (HF) and oxidant: experiment and computation. ACS Catal. 8, 2582–2588 (2018).
Banik, S. M., Medley, J. W. & Jacobsen, E. N. Catalytic, asymmetric difluorination of alkenes to generate difluoromethylated stereocenters. Science 353, 51–54 (2016).
Woerly, E. M., Banik, S. M. & Jacobsen, E. N. Enantioselective, catalytic fluorolactonization reactions with a nucleophilic fluoride source. J. Am. Chem. Soc. 138, 13858–13861 (2016).
Mennie, K. M., Banik, S. M., Reichert, E. C. & Jacobsen, E. N. Catalytic diastereo- and enantioselective fluoroamination of alkenes. J. Am. Chem. Soc. 140, 4797–4802 (2018).
Scheidt, F. et al. Enantioselective, catalytic vicinal difluorination of alkenes. Angew. Chem. Int. Ed. 57, 16431–16435 (2018).
Levin, M. D. et al. Catalytic enantioselective synthesis of difluorinated alkyl bromides. J. Am. Chem. Soc. 142, 14831–14837 (2020).
Zhang, Q. et al. Iridium-catalyzed enantioselective fluorination of racemic, secondary allylic trichloroacetimidates. J. Am. Chem. Soc. 137, 11912–11915 (2015).
Bruns, S. & Haufe, G. Enantioselective introduction of fluoride into organic compounds: First asymmetric ring opening of epoxides by hydrofluorinating reagents. J. Fluor. Chem. 104, 247–254 (2000).
Haufe, G. & Bruns, S. Salen chromium complex mediated asymmetric ring opening of meso‐ and racemic epoxides with different fluoride sources. Adv. Synth. Catal. 344, 165–171 (2002).
Kalow, J. A. & Doyle, A. G. Enantioselective ring opening of epoxides by fluoride anion promoted by a cooperative dual-catalyst system. J. Am. Chem. Soc. 132, 3268–3269 (2010).
Zhu, J. T., Tsui, G. C. & Lautens, M. Rhodium‐catalyzed enantioselective nucleophilic fluorination: ring opening of oxabicyclic alkenes. Angew. Chem. Int. Ed. 51, 12353–12356 (2012).
Pupo, G. et al. Asymmetric nucleophilic fluorination under hydrogen bonding phase-transfer catalysis. Science 360, 638–642 (2018).
Katcher, M. H., Sha, A. & Doyle, A. G. Palladium-catalyzed regio- and enantioselective fluorination of acyclic allylic halides. J. Am. Chem. Soc. 133, 15902–15905 (2011).
Edwards, J. A., Ringold, H. J. & Djerassi, C. Steroids. CXXXVI.1 Synthesis of a new class of potent cortical hormones. 6α-fluoro- and 6α, 9α-difluoro-16α-methylprednisolone and related steroids2. J. Am. Chem. Soc. 82, 2318–2322 (1960).
Wei, Z. Y., Wang, D., Li, J. S. & Chan, T. H. Lewis acid-promoted condensation of allylalkoxysilanes with carbonyl compounds. Synthesis of tetrahydropyrans. J. Org. Chem. 54, 5768–5774 (1989).
Cui, J. et al. Boron trifluoride etherate functioning as a fluorine source in an iodosobenzene-mediated intramolecular aminofluorination of homoallylic amines. Org. Lett. 16, 1442–1445 (2014).
Launay, G. G., Slawin, A. M. Z. & O’Hagan, D. Prins fluorination cyclisations: preparation of 4-fluoro-pyran and -piperidine heterocycles. Beilstein J. Org. Chem. 6, 41 (2010).
Yeh, M. C. P. et al. Transition-metal-free carbofluorination of TBS-protected nitrogen-containing cyclic enynols: synthesis of fluorinated azabicycles. J. Org. Chem. 78, 5521–5529 (2013).
Demeio, G. V. & Pinhey, J. T. Aryl fluorides from the reaction of boron trifluoride with aryl-lead (IV) triacetates, which may be generated in situ from aryltrimethylsilanes, triarylboroxines, and arenes. J. Chem. Soc. Chem. Commun. 15, 1065–1066 (1990).
Meio, G. D., Morgan, J. & Pinhey, J. T. Aryl fluoride syntheses involving reaction of aryllead triacetates with boron trifluoride-diethyl ether complex. Tetrahedron 49, 8129–8138 (1993).
Xu, Z. F., Dai, H. C., Shan, L. H. & Li, C. Y. Metal-free synthesis of (E)-monofluoroenamine from 1-sulfonyl-1,2,3-triazole and Et2O·BF3 via stereospecific fluorination of α-diazoimine. Org. Lett. 20, 1054–1057 (2018).
Cresswell, A. J. et al. Pinacolatoboron fluoride (pinBF) is an efficient fluoride transfer agent for diastereoselective synthesis of benzylic fluorides. Tetrahedron Lett. 56, 3373–3377 (2015).
Ding, C. H., Dai, L. X. & Hou, X. L. An efficient and highly regioselective fluorination of zziridines using BF3·OEt2 as fluorine source. Synlett 2004, 2218–2220 (2004).
Cresswell, A. J., Davies, S. D., Roberts, P. M. & Thomson, J. E. Beyond the Balz−Schiemann reaction: the utility of tetrafluoroborates and boron trifluoride as nucleophilic fluoride sources. Chem. Rev. 115, 566–611 (2015).
Yoshimura, A. & Zhdankin, V. V. Advances in synthetic applications of hypervalent iodine compounds. Chem. Rev. 116, 3328–3435 (2016).
Wirth, T. Hypervalent iodine chemistry in synthesis: scope and new directions. Angew. Chem. Int. Ed. 44, 3656–3665 (2005).
Egami, H. et al. Dianionic phase-transfer catalyst for asymmetric aluoro-cyclization. J. Am. Chem. Soc. 140, 2785–2788 (2018).
Masanori, S., Shinichi, A. & Shoji, H. A practical synthetic method for iodoarene difluorides without fluorine gas and mercury salts. Synthesis 2002, 1802–1803 (2002).
Scheidt, F. et al. Enantioselective, catalytic vicinal difluorination of alkenes. Angew. Chem. Int. Ed. 130, 16669–16673 (2018).
Ulmer, A. et al. A fluorination/aryl migration/cyclization cascade for the metal‐free synthesis of fluoro‐benzoxazepines. Chem. Eur. J. 22, 3660–3664 (2016).
Uyanik, M., Yasui, T. & Ishihara, K. Hypervalent compounds. Angew. Chem. Int. Ed. 49, 2175–2177 (2010).
Haubenreisser, S. et al. Structurally defined molecular hypervalent iodine catalysts for intermolecular enantioselective reactions. Angew. Chem. Int. Ed. 55, 413–417 (2016).
Gaussian 09, rev. D.01 (Gaussian Inc., 2013).
Stephens, P. J., Devlin, F. J., Chabalowski, C. F. & Frisch, M. J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 98, 11623–11627 (1994).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Bergner, A. et al. Ab initio energy-adjusted pseudopotentials for elements of groups 13-17. Mol. Phys. 80, 1431–1441 (1993).
Hariharan, P. C. & Pople, J. A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 28, 213–222 (1973).
Gnan, N. et al. Dynamical properties of different models of elastic polymer rings: Confirming the link between deformation and fragility. J. Chem. Phys. 56, 2257–2261 (1972).
Weigend, F. & Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297 (2005).
Clark, T., Chandrasekhar, J., Spitznagel, G. W. & Schleyer, P. V. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li-F. J. Comput. Chem. 4, 294–301 (1983).
Krishnan, R., Binkley, J. S., Seeger, R. & Pople, J. A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 72, 650–654 (1980).
Marenich, A. V., Cramer, C. J. & Truhlar, D. G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 113, 6378–6396 (2009).
Acknowledgements
We appreciate the financial support from the National Natural Science Foundation of China (No. 91853106), the Program for Guangdong Introducing Innovative and Enterpre-neurial Teams (No. 2016ZT06Y337), Guangdong Provincial Key Laboratory of Construction Foundation (No. 2019B030301005), Shenzhen Science and Technology Program (JSGG20200225153121723), and the Fundamental Research Funds for the Central Universities (No. 19ykzd25). We also thank Dr. Chaoxian Yan for his help in DFT calculations.
Author information
Authors and Affiliations
Contributions
X.X.J. and W.W.Z. conceived of and directed the project; W.W.Z. developed the catalytic asymmetric fluorinations; W.W.Z., X.Z. and J.K.A. conducted most of the experiments; J.Y.W., Y.P.C. and W.W.Z. conducted the DFT calculations and provided mechanism analysis; X.Y.M., J.K.L, Y.J.Q, H.Z. and J.J.X. helped the conduction of the project; and X.X.J. and W.W.Z. co-wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional information
Experimental procedures, characterization of new compounds, and all other data supporting the findings are available in the supplementary materials.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhu, W., Zhen, X., Wu, J. et al. Catalytic asymmetric nucleophilic fluorination using BF3·Et2O as fluorine source and activating reagent. Nat Commun 12, 3957 (2021). https://doi.org/10.1038/s41467-021-24278-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-24278-3
This article is cited by
-
Lateral dipole moments induced by all-cis-pentafluorocyclohexyl groups cause unanticipated effects in self-assembled monolayers
Nano Research (2023)
-
Electrophilic Halogen Reagents-mediated Halogenation: Synthesis of Halogenated Dihydro-1,3-oxazine Derivatives
Chemical Research in Chinese Universities (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
