
Abstract
Knowledge on the genetic composition of Quercus petraea in south-eastern Europe is limited despite the species’ significant role in the re-colonisation of Europe during the Holocene, and the diverse climate and physical geography of the region. Therefore, it is imperative to conduct research on adaptation in sessile oak to better understand its ecological significance in the region. While large sets of SNPs have been developed for the species, there is a continued need for smaller sets of SNPs that are highly informative about the possible adaptation to this varied landscape. By using double digest restriction site associated DNA sequencing data from our previous study, we mapped RAD-seq loci to the Quercus robur reference genome and identified a set of SNPs putatively related to drought stress-response. A total of 179 individuals from eighteen natural populations at sites covering heterogeneous climatic conditions in the southeastern natural distribution range of Q. petraea were genotyped. The detected highly polymorphic variant sites revealed three genetic clusters with a generally low level of genetic differentiation and balanced diversity among them but showed a north–southeast gradient. Selection tests showed nine outlier SNPs positioned in different functional regions. Genotype-environment association analysis of these markers yielded a total of 53 significant associations, explaining 2.4–16.6% of the total genetic variation. Our work exemplifies that adaptation to drought may be under natural selection in the examined Q. petraea populations.
Similar content being viewed by others

Introduction
Forest ecosystems are undergoing unprecedented changes in environmental conditions due to global change impacts (González de Andrés 2019; Pouresmaeily 2022; Pörtner et al. 2022). These changes involve the simultaneous and rapid alteration of several key environmental parameters that control the dynamics of forests (Thom and Seidl 2022). As a result, modifications in tree species’ dominance and distribution, productivity, and nutrient cycles are expected (Gea‐Izquierdo and Sánchez‐González 2022; Boonman et al. 2022; Elsen et al. 2022). It is important to determine how the life processes of trees will be affected by new specific competitive and climatic conditions, since large-scale, consistent monitoring of forest ecosystems plays a key role in preparing for the impact of extreme events (Kijowska-Oberc et al. 2020).
Economic evaluation based on the output of the large-scale scenario model EFISCEN grouped major European tree species according to their economic performance (https://efi.int/knowledge/models/efiscen). Among seven groups of tree species, Quercus petraea (Matt.) Liebl. was ranked fourth in terms of economic importance, being of considerable productivity (Hanewinkel et al. 2013). In addition to its economic importance, the species is highly valued in nature conservation terms (Mölder et al. 2019). In terms of adaptation, on one hand, sessile oak is widely regarded as a forest species with high adaptiveness (Kohler et al. 2020) since its drought tolerance and storm resistance are superior to those of other common trees (Kunz et al. 2018). On the other hand, the climate is shifting rapidly (Loarie et al. 2009) and due to the long lifespan of oaks, shifts in the genetic composition of populations are slow, opportunities for adaptation are limited, making the predictions about its response to climate change contradictory (Sáenz-Romero et al. 2017). Considering oaks adaptiveness, important works (Temunović et al. 2020; Lang et al. 2021; Guichoux et al. 2013) analyse potential candidate genes for stress responses. Available resources on functional drought candidate genes are considered the works of Homolka et al. (2013), Lepoittevin et al. (2015) and Rellstab et al. (2016). However, as future adaptedness requires beneficial alleles from habitats currently matching future climatic conditions, the importance of collecting more information about the Balkans in terms of adaptation of sessile oak has high priority in genetic research.
The genetic diversity of Q. petraea has been evaluated using different markers at different scales by numerous authors, including but not limited to the morphological research of Streiff et al. (1998), Bruschi et al. (2003), isoenzyme research of Zanetto and Kremer (1995) and Kremer and Zanetto (1997); microsatellite research of Muir et al. (2004); research using chloroplast genetic markers of Petit et al. (1993), Bordács et al. (2002), and Mátyás and Sperisen (2001); markers developed from EST databases (Lang et al 2021) or by using next generation sequencing of the whole genome (Leroy et al. 2020).
Reduced representation sequencing (Miller et al. 2007; Lewis et al. 2007; Baird et al. 2008) is a simple, cost-effective approach for generating large amounts of SNP data and is gaining popularity in species conservation (Fuentes‐Pardo and Ruzzante 2017) and phylogenetic studies (Hipp et al. 2018). Such genome-wide studies can also provide valuable information on adaptive gene flow, selection and speciation (Hipp et al. 2014, 2018; Cavender-Bares et al. 2015; Eaton et al. 2015; Fitz-Gibbon et al. 2017; Pham et al. 2017; Deng et al. 2018; Kim et al. 2018; Ortego et al. 2018; Jiang et al. 2019; Blanc-Jolivet et al. 2020; Degen et al. 2021).
As we mentioned in our previous work (Tóth et al. 2021), while sessile oak populations are well studied in Europe, the Central-Eastern European region, including the Balkan Peninsula, with the exception of some important works (Zanetto and Kremer 1995; Gömöry et al. 2001; Bordács et al. 2002; Slade et al. 2008; Neophytou et al. 2010) has been less investigated with high-resolution and genome-wide genetic markers. The genetic diversity in the Balkan region is generally high due to many refugia in this area harbouring differentiated genetic lineages and high environmental heterogeneity (Birks and Willis 2008; Gömöry et al. 2020). For this reason, the area is considered an important source of genetic material not only for forestry (Tzedakis 2004; Feliner 2011; Fassou et al. 2020) but also for research on the genomic architecture of adaptation.
Local adaptation occurs when individuals from a population have higher average fitness due to genetic changes in their local environment than individuals from other populations of the same species (Savolainen et al. 2013). Exploring this phenomenon, to identify the environmental factors responsible for genetic variation and gene variants that drive adaptation to the environment, are the most important aims. While some models predict that specific genes with large effects may be more important than other loci, other theoretical works emphasize the importance of polygenic traits in mediating local adaptation (Kawecki and Ebert 2004). However, to apply any of these approaches to elucidate the genetic variation underlying local adaptation, generation of large numbers of SNPs is required.
To address the need for optimal marker resolution, RAD sequencing (Peterson et al. 2012) fulfils the requirements for determining individual sequence genotypes that can be tuned to sample a wide range of randomly distributed regions genome-wide. Nevertheless, while the approach clearly permits genotyping of multiple individuals, it has its limit, namely, the ability to genotype for only the number and type of markers needed for the experiment. To overcome this deficiency, RAD sequencing data can be coupled with a reference genome or a study-specific annotated sequence database, which is advantageous for several reasons: improving the reliability of genotype calls (Torkamaneh et al. 2016), reducing the required coverage for accurate genotyping (Davey et al. 2011), providing a greater number of SNPs, improving downstream population genetic inferences (Shafer et al. 2017) and allowing SNP annotation with gene information (Gurgul et al. 2019). SNP annotation can identify the target genes of the analysis and to separate the functional vs. non-functional diversity of the genome, with high conservation implications (Johnson et al. 2018).
One of the major interests of current quantitative genetics is to explore the exact number, distribution and interaction of loci affecting the variations in adaptively important traits. The first step in this exploration process is SNP discovery. The second is to distinguish the molecular variation of the SNPs that are neutral from those that are subject to selection since selection is the predominant driver of differentiation in phenotypic traits (Darwin 1859; Rieseberg et al. 2002; Matesanz et al. 2010; Kremer and Hipp 2020). However, species have different sensitivity in terms of growth, survival and reproduction. Changes in environment are not always reflective on the selection signature and moreover, one SNP could have multiple associations with different environmental variables (Ahrens et al. 2018). Still, searching for loci under selection with important considerations regarding the underlying neutral genetic structure may provide valuable information on the adaptation to local conditions, as adaptation can cause subtle changes in allele frequencies (Rellstab et al. 2015). For this purpose, various specific strategies have been developed. These approaches include DNA-based, mRNA-DNA, protein-DNA, DNA-environment, and phenotype-based methods (Vasemägi and Primmer 2005). The FST-based method is a DNA-based approach that uses multiple-population tests to estimate outliers (Beaumont 2005). During the identification of outlier loci by this method, it is necessary to use different approaches to minimize the false-positive rate since the validation of the detected outliers is highly important (Tsumura et al. 2012). However, even by different approaches, outlier tests make the assumption that selection pressures differ among populations and also do not link specific selection pressures that underlie adaptation (Rellstab et al. 2015). Additionally, identifying outlier loci is complicated by the occurrence of asymmetric introgression between oak species, which may be attributed to differences in colonization history and result in outliers retaining signatures of past introgression events (Bierne et al. 2011; Guichoux et al. 2013). For this reason, another way is needed to identify loci under selection, to see which of them are correlated with environmental gradients using allele distribution models (Joost et al. 2007; Holderegger et al. 2008; Manel and Segelbacher 2009). The basic hypothesis of allele distribution models is that natural selection due to heterogeneity generates changes in allele frequencies at loci linked to selected genes (Endler 1986; Hirao and Kudo 2004; Schmidt et al. 2008). The highly differentiated markers assessed by these methods become candidate genes for adaptation to the environmental factors in question, tracing the patterns of fundamental evolutionary processes: differential survival or reproduction of genetically based phenotypes in response to environmental challenges. However, environmental association analyses also have important limitations. Their main limit is that they might result in high rates of false positives (Frichot et al. 2015, Rellstab et al. 2015), requiring preliminary assessments of population structure to avoid false positive associations (Ahrens et al. 2018, Capblancq and Forester 2021). To avoid false positives, rather than contrasting association analyses and outlier detection methods, a more effective approach may be to combine them and link the positively identified SNPs to gene function using gene ontology analyses (Rellstab et al. 2015).
In this study, we identified and investigated the genetic variation at several stress-response loci in natural sessile oak populations from the Central-Eastern European region. The objectives of the study were (i) to reveal the genetic structure, diversity and differentiation of the sampled populations; (ii) to identify stress-response loci in our RAD sequencing dataset after mapping paired-end reads to the Q. robur reference genome; (iii) to explore the number and distribution of loci affecting the variations in the adaptively important traits by FST outlier detection and associations of SNP allele frequencies with environmental variation; and (iv) by annotating these loci, to describe the biological process, molecular function and cellular component, assuming the same function in both the resulted, already annotated model species, and Q. petraea.
Materials and methods
Plant materials
Sampling was designed to cover heterogeneous climatic conditions in the southeastern natural distribution range (CE-Europe) of Q. petraea, extending to Bulgaria, Hungary, Romania, Serbia, Bosnia and Herzegovina, Kosovo and Albania (Fig. S1). Altogether, 180 individuals from 18 natural populations were sampled in the framework of a former project, aiming to genotype a large number of samples and single-nucleotide polymorphisms (SNPs) from key geographic regions by RAD-seq (restriction-site associated DNA sequencing) (Table S1). Sampling was carried out using the classification system and taxonomic descriptors of Schwarz (1936). Detailed information on the sampling strategy can be found in Tóth et al. (2021).
Genotyping
A variation of RAD sequencing, that is suitable for high-confidence genome-wide SNP loci discovery, double-digest RAD-seq (ddRAD-seq), was used to genotype all individuals (Peterson et al. 2012). The details of the wet-laboratory procedure, sequencing and raw sequence editing are described in Tóth et al. (2021). Key wet-laboratory steps are provided in the Supplementary Materials. Raw data have been deposited in the NCBI Sequence Read Archive (SRA); BioProject ID: PRJNA699096. The dataset can also be accessed at https://doi.org/10.5281/zenodo.3908963.
Sequence editing and genome mapping
Bioinformatics processing was carried out on a Silicon Computers (SGI) Unix-based HPC server, allocating 40 cores (80 threads) and 38 GB of RAM. Key steps of the bioinformatics pipeline are shown in Fig. 1.

The main bioinformatic steps are visualised, including data processing, assembly, mapping, filtering, variant calling, reference mapping, diversity-differentiation analyses, outlier SNP detection and association.
Short read sequences (~77 M) were demultiplexed and adaptor-trimmed by using MiSeq Control Software (Illumina, San Diego, CA, USA). The resulting sequences (69,993,001) were 3′ and 5′ end-trimmed using the FastQ Toolkit. Reads with a mean quality score <30 and shorter than 200 bp were filtered. Computational processing of short-read data was carried out with Stacks 2.0 (Catchen et al. 2013; Rochette et al. 2019). Whole reads were quality filtered using a sliding-window method (15% of read length) implemented with ‘process_radtags’ (Rochette et al. 2019). Reads having a quality score below 90% (raw Phred score of 10) were discarded (Catchen et al. 2011). Using ‘process_radtags’, reads were truncated to 200 bp as a prerequisite for further processing and to avoid the lower-quality bases present at the ends of the reads (Catchen et al. 2011). During this filtering step, 42,273 sequences were discarded. In addition, one individual was removed from the dataset (BU2-10) since an insufficient number of high-quality reads remained after filtering.
Paired-end reads were mapped to the Q. robur reference genome (PM1N [haploid version]; http://www.oakgenome.fr; Plomion et al. 2018) using BWA-MEM v0.7.17 (Li 2013), which is designed for mapping reads (70 bp to 1 Mbp) against large reference genomes and has already been successfully used in oak studies (Fitz-Gibbon et al. 2017; Konar et al. 2017; Ramos et al. 2018). During mapping, parameters were set to default (Li 2013), and the unassigned scaffolds of the reference genome were excluded. Sequences of individual samples in SAM format were sorted with SAMtools v1.10 software (Li et al. 2009) and then converted to a BAM file. Reads with a mapping quality less than five were removed (MAPQ > = 5). The result of mapping was evaluated with the SAMtools ‘flagstat’ option and used for calculating the individual- and population-level summary statistics presented in Table S2, Table S3 and Fig. S2. The successfully mapped sequences (~64 M), later termed RAD loci, were kept for further downstream processing.
In Stacks, the ‘gstacks’ programme reconstructs loci and creates a SNP catalogue by incorporating paired-end reads that have been aligned to the reference genome using a sliding window algorithm (Catchen et al. 2013; Rochette et al. 2019). Unpaired reads were removed to avoid reads supporting a variant aligned to only one strand (strand-bias error). The ‘populations’ programme was used to call SNPs across the whole sample set. In this step, SNP markers with a minor allele frequency <0.05, a missing individual rate >0.8, and significant deviation from Hardy–Weinberg equilibrium (HWE, p < 1 × 10−5) were filtered out (Xiong et al. 2009; Marees et al. 2018). In addition, only a single SNP per locus was kept to have independent loci for later model-based statistical approaches. The ‘minimum number of populations’ parameter was set to 18 to identify loci that were present in all populations.
Genetic structure and diversity
Population groups were estimated using three different methods, namely the Bayesian model-based clustering algorithm of fastStructure v 1.0 (Raj et al. 2014), fineRADstructure (Malinsky et al. 2018), and Principal Component Analysis (PCA).
fastStructure was run with default settings and 100-fold cross-validation on the 179 samples, testing for the best number of groups of populations (K) ranging from K = 2 to 9. The Python script ‘ChooseK’, included with the fastStructure package, was used to choose the number of groups that maximize the log-marginal likelihood lower bound (LLBO; Beal 2003) according to Raj et al. (2014). The mean population membership probability was manually calculated in MS Excel based on the Q-matrix values produced by fastStructure. fineRADstructure infers population structure via shared ancestry based on the autosomal loci, and focuses on the most recent coalescent events providing information on relatedness (estimates co-ancestry), which is informative in situations of contemporary gene flow. Individuals were assigned to populations using 1,000,000 iterations sampled every 1000 steps with a burn-in of 100,000. Estimated co-ancestry values were sorted according to populations and plotted as a heatmap. Principal component analysis (PCA) was performed using the ‘factoextra’ (Kassambara and Mundt 2017) and ‘FactoMineR‘ (Lê et al. 2008) packages in R (R Core Team 2013) to assess genetic differentiation among populations. The results were visualized using the ‘pophelper’ (Francis 2017) and ‘ggplot2’ (Wickham et al. 2016) packages and also plotted on a topographic map using ESRI ArcGIS (ArcMap 10.2.2, Redlands, CA, USA).
Expected heterozygosity (He), observed heterozygosity (Ho), and the inbreeding coefficient (FIS) were calculated for each population and for each genetic cluster in R using the ‘adegenet’ package (Jombart and Ahmed 2011). The significance of FIS values was calculated separately with the ‘hierfstat’ package (Goudet 2005) using 1000 permutations. Allelic richness (Ar) was calculated using the ‘popgenreport’ package (Adamack and Gruber 2014). Private alleles (PAs) were counted in each population using the ‘poppr’ package (Kamvar et al. 2014). The diversity statistics are presented in Table S4. Genetic differentiation between populations and clusters was measured using the fixation index (FST) (Nei 1973) by creating a pairwise distance matrix using the ‘hierfstat’ package and visualized as a heatmap (Fig. S3). In addition, an unweighted pair group method with arithmetic mean (UPGMA) dendrogram was created, with 1000 bootstrap resampling, based on the FST distances using the ‘ggdendro’ package in R (de Vries and Ripley 2013).
F ST outlier detection
Since the application of multiple selection tests (distinct approaches; e.g., FST frequency or Bayesian based) can outperform single tests in the detection of loci under directional selection, we applied distinct approaches to see if they converged and detected the same outliers (Tsumura et al. 2012; De La Torre et al. 2019). Altogether, two tests were performed: the PCA-based procedure implemented in PCAdapt 4.3.3 (Privé et al. 2020) and the FST frequency-based approach used by Outflank (Whitlock and Lotterhos 2015).
PCAdapt performs a PCA of a scaled genotype matrix and regresses all SNPs against the PCs to obtain a matrix of Z scores. Then, the robust Mahalanobis distances of these Z scores are computed to integrate all PCA dimensions into one multivariate distance for each SNP. Distances approximately follow a chi-squared distribution, thus enabling the calculation of a p value for each SNP (Privé et al. 2020). PCAdapt was found to be more powerful than former genome scans (Luu et al. 2017; Privé et al. 2020). Outflank identifies FST outliers, loci with atypical values of FST, by inferring a distribution of neutral FST using likelihood on a trimmed distribution of FST values. In this analysis, the ‘number_of_samples’ parameter was set to 18 (a number equal to the populations sampled), the ‘LeftTrimFraction’ was set to 0.08, the ‘RightTrimFraction’ to 0.30, and the Hmin parameter was left at the default setting (0.1). The false discovery rate threshold for calculating q-values first was 0.05, as set by default (Whitlock and Lotterhos 2015), however in our final analysis a more stringent value of 0.01 was used.
The loci containing the significant SNPs was extracted from the initial dataset and annotated using the NCBI’s BLASTN and BLASTX services (https://www.ncbi.nlm.nih.gov/), as an example presented in Table S5. Finally, the type of nucleotide change, substitution type (synonymous or non-synonymous) and product change were determined by DnaSP (Rozas et al. 2017). The sequence of each locus can be accessed at https://doi.org/10.5281/zenodo.7763329.
Outlier association
To test for associations of loci (SNPs) with environmental variables (GEA; genotype–environment associations), five different regression approaches were applied.
First, an environmental dataset of 84 monthly, seasonal, and annual variables was created by extracting climate data from WorldClim 1.4 (current: 1960–1990 based on De La Torre et al. 2019) in 30 arcsec-resolution (≤1 km) layers (Hijmans et al. 2005) and from ENVIREM 1.0 (current: 1960–1990) in 30 arcsec-resolution (≤1 km) layers (Title and Bemmels 2018) using the ‘raster’ (Hijmans and van Etten 2015), ‘rgeos’ (Bivand et al. 2017), and ‘rgdal’ (Bivand et al. 2015) R packages. The environmental variables were filtered for co-correlation between the environmental variables using Pearson’s correlation with the ‘caret’ package in R (Kuhn 2009), and by a step-by-step VIF (variance inflation factor) calculation, using a VIF threshold of 10, as implemented in the ‘usdm’ (Naimi 2017) and ‘fmsb’ (Nakazawa 2022) packages in R. Of the 84 variables, 14 variables were co-correlated with a r2 value below 0.75 and with a VIF < 10, thus were subsequently used for the association analyses. Selected environmental variables are detailed in Table S6.
Then, a matrix regression model (GDM) implemented in the ‘gdm’ package (Fitzpatrick et al. 2021), a latent factor mixed model (LFMM) implemented in the ‘LEA’ package in R (Frichot et al. 2013), a single-factor analysis of variance (SFA), a general linear model (GLM) (controlling for Q) and a mixed linear model (MLM) (controlling for Q + k) using TASSEL 5.2.73 (Bradbury et al. 2007) were performed.
GDM is a non-linear distance-based approach that models GEAs and adapts to a variable rate of change in allele frequencies along environmental gradients (Fitzpatrick and Keller 2015; Varas-Myrik et al. 2022). The method uses flexible splines (three as default) for fitting nonlinearity relationships between population dissimilarity and environmental variables as predictors (Ferrier et al. 2007). To investigate each outlier SNPs separately, we applied the protocol published in Fitzpatrick and Keller (2015) using the ‘gdm’ R package (Fitzpatrick et al. 2021). To measure population dissimilarities for each outlier SNP, locus-specific FST between all pairs of populations were calculated, as suggested by Fitzpatrick and Keller (2015), and using the ‘hierefstat’ R package. Best-fitted predictor’s spline present significant curvilinear relationships and indicates the total magnitude of change as a function and how the rate of change varies (Fitzpatrick and Keller 2015).
The LFMM approach uses a hierarchical Bayesian mixed model that accounts for covariation of alleles and the environment and for hidden population structure (via the K-value in the algorithm) while maintaining a relatively low false detection rate (de Villemereuil et al. 2014). For the analysis, our fastStructure-detected genetic clusters were chosen for the demographic background model. One hundred independent LFMM runs for each value of K with 10,000 iterations of the Gibbs sampling algorithm and a burn-in period of 5000 cycles were performed. |z | -scores for all loci were combined using the Fisher-Stouffer method (Brown 1975), and the resulting p values were adjusted using the genomic inflation factor (λ) (Devlin and Roeder 1999). A false discovery rate (FDR) correction of 1% was further used in p value adjustment using the ‘q-value’ package in R (Storey et al. 2021).
SFA, which does not consider population structure, was performed using each marker as the independent variable. Mean performance was compared between allelic classes using the general linear model (GLM), similarly to Kwon et al. (2013). The GLM (controlling for Q) and MLM (controlling for Q + k) analyses, for correcting population structure and relatedness, were performed using the Q-matrix (Q), which was obtained from the former fastStructure analysis, and a kinship (k) matrix which was calculated by the tool ‘Kinship’ with the Scald_IBS method built in TASSEL (Bradbury et al. 2007). We applied 1000 permutations for each test, and the p values of associated markers were tested against non-adjusted, Bonferroni-adjusted and FDR-adjusted significance thresholds at 0.05 (conservative), 0.01 and 0.001 (stringent) significance levels. Corrections of p values were carried out using the ‘dplyr’ (Mailund 2019) package in R.
Results
Bioinformatics data processing
Our pipeline yielded 64,378,333 short read sequences after filtering that were mapped onto the Q. robur reference genome with an average success rate of 92.43% (Table S2, Table S3 and Fig. S2). After processing, we reconstructed 5370 loci from an initial set of 410,445 loci, of which none had passed the filtering protocol (missing rate of sample/population and below the minor allele frequency (MAF) threshold). The final dataset consisted of 2,615,792 sites, in which we identified 2627 highly polymorphic variant sites (SNPs). The mean genotyped sites per locus was 474.24 base pairs (S. E. = 1.77). The Variant Call Format (VCF) file can be accessed at https://doi.org/10.5281/zenodo.7763329.
Population structure and diversity
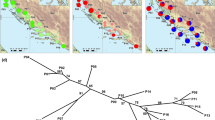
The Bayesian clustering algorithm of fastStructure resulted in K = 4 as the number of groups that maximized the log-marginal likelihood lower bound (LLBO) for the SNP data. At this LLBO, we identified three main genetic clusters (Clusters 1, 2 and 3) and few introduced individuals as a separate group (Fig. 2; within SE1 and SE3 populations). While the populations of Cluster 1 (AL, RO, SE, KO, and BU1–3) and Cluster 3 (BU4) were clearly separated, the individuals of Cluster 2 (populations of HU, BH1 and BH2) were highly admixed (Fig. 2). This pattern was also evident on the co-ancestry heat map of fineRADstructure. BU4 represented close familial relationships as indicated by an island of high co-ancestry values. In addition, high values were detected at the admixed populations of HU, BH1 and BH2. PCA, by explaining 27.56% of the variance at PC1 vs. PC2 and 19.87% of the variance at PC2 vs. PC3, separated Cluster 1 and 2, as well as Cluster 3 (BU4) in both ordinations (Fig. 2). The geographic extent of detected clusters indicated a north–southeast gradient, along which the southernmost Mediterranean Cluster 3 (red) towards the west, Cluster 1 (yellow) became dominant in continental regions and Cluster 2 (blue) increased towards Central Europe (Fig. 2).

a Bayesian estimation of population structure in 18 populations of Quercus petraea using reference-mapped loci at K 2-4, as determined by fastStructure (Raj et al. 2014). The most likely number of clusters is defined by the ‘ChooseK’ method that maximizes the log-marginal likelihood lower bound (LLBO) according to Raj et al. (2014). b Estimated co-ancestry heatmap of fineRADstructure (Malinksy et al. 2018), individuals are ordered according to the fastStructure output. c Principal component analysis (PCA) of populations (PC1-PC2 and PC2-PC3 axes). d Geographic map of estimated population structure at K = 4 (mean population probabilities). Population abbreviations are as explained in Table S1.
Genetic differentiation was generally low among populations (FST ranging from 0.003–0.119) and among the genetic clusters (FST ranging from 0.022–0.083). Cluster 3 showed the highest differentiation compared to Clusters 1 and 2 (FST = 0.065 and 0.083), while differentiation between Clusters 1 and 2 was much lower (FST = 0.022) (Fig. S3). The UPGMA dendrogram of Nei’s genetic distance showed the closest relationship (shortest genetic distance) between Cluster 1 and Cluster 2, while Cluster 3 was positioned on a separate branch with a much greater genetic distance (Fig. S3).
Moderate levels of genetic diversity were observed, and the values were balanced at the population level (He = 0.181–0.223; Ho = 0.166–0.210), as detailed in Table S4. Allelic richness values were also balanced (Ar = 1.570–1.746). Interestingly, BU4 showed the lowest diversity values (He = 0.181; Ho = 0.166; Ar = 1.570). Significant inbreeding depression was not detected (p < 0.05). Private alleles were absent, and only HU3 had a unique allele. Diversity among genetic clusters was similar; only Cluster 3, which contained only the BU4 population, showed slightly lower diversity values (Table S4).
F ST outlier detection
Outlier detection methods revealed different numbers of SNPs as being under selection with different levels of significance. PCAdapt identified 34 SNPs, while Outflank identified 38 SNPs. In both approaches, thresholds were highly stringent with q-values < 0.01 and p ≤ 0.001, respectively. However, we considered robust outliers to be the SNPs detected jointly with the two different approaches. In this way, nine SNPs (0.34%) located at different loci, and at six different chromosomes (Chr. 2: 50083_64, 96506_153, 96534_88; Chr. 5: 180975_104; Chr. 6: 227780_278, 238771_249; Chr. 7: 277051_56; Chr. 7: 277051_56; Chr. 9: 361438_297; Chr. 12: 437832_228) were considered robust outliers (Table 1, Fig. 3). Those SNPs that were presented in NCBI databases resided in four different functional regions. Two SNPs found in titin homologues, one in bHLH162 transcription factor, one in hydroxymethylglutaryl-CoA synthase coding region, and one in a hypothetical protein coding region (possible double-strand damage repair) (Table 2). Functional regions with known CDS parts (96506_153, 96534_88, 277051_56) consisted three non-synonymous substitution (Table 2).

Loci marked with yellow are significant for the specific approach, and loci with red are common among different approaches, as indicated in Table 1, and thus considered to be robust outliers. a The PCA-based method implemented in PCAdapt by Luu et al. (2017), and b the FST frequency-based method by Outflank (Whitlock and Lotterhos 2015).
Genotype–environment associations
GEAs were determined by five different methods, including GDM, LFMM, SFA, GLM where population membership (Q) served as a covariate, and last MLM where the average relationship was estimated by kinship (Q + k). Table 3 presents the significance levels at p ≤ 0.05, p ≤ 0.01 and p ≤ 0.001 for all SNPs for each analysis.
By using the nine robust SNPs, a total of 102 significant marker-environment associations were detected, 21 with GDM, 48 with the LFMM, 9 with SFA and 14–10 with the GLM and MLM. However, similar to outlier analysis, we considered only those marker-environment associations that were detected with at least two different methods. Among the 53 jointly detected associations, the significance levels were different and ranged from 0.05 to 0.001.
The GDM method found relationships only for four SNPs (180975_104, 227780_278, 277051_56, 361438_297). In these relationships, PETCQ, prec6_16, bio3_16 were the most frequent, and presented the highest relative importance (‘highest magnitude of biological change’; Fitzpatrick and Keller 2015) (Fig. 4). The rate of change in allele frequencies (shape of function) were different in each environmental variable. For all SNPs, in decreasing importance, PETCQ showed rapid change in allele frequencies at the beginning of the spline, while no change elsewhere. On the other hand, prec_6_16 showed rapid turnover at the end of the gradient. In case of bio3_16, the frequency of change was gradually increasing (Fig. 4).

a Heatmap of the relative importance of each environmental predictor (darker colours indicate higher importance) and the relationship of ecological and genetic distances (left: relationship between predicted ecological distance and observed compositional dissimilarity; right: predicted versus observed genetic distance). b–e The three most important environmental predictors for each of the four outlier SNPs (180975_104, 227780_278, 277051_56, 361438_297), where associations were detected. Variable abbreviations are as explained in Table S6.
The LFMM revealed associations for all nine outlier SNPs, in these |z | -score values ranged from 2.232 to 6.414 (S.D.: 4.182). The highest |z | -score values (i.e. the highest significance of an environmental effect) were found for three SNPs, namely for 227780_278, 277051_56 and 361438_297, in association with PETCQ (6.036 and 6.289), prec6_16 (6.022) and aridity (6.414). The single-factor analysis (SFA) was similar to LFMM, where the LFMM |z | -scores values were high, the SFA adjusted R2 values were also high. Thus, the highest R2 values were detected for 227780_278, 277051_56 and 361438_297, in association with PETCQ (0.166 and 0.219 R2), prec6_16 (0.167 R2) and aridity (0.189 R2), and explaining 16.6–21.9% of variance (Table 3).
The GLM and MLM detected much less numbers of associations (14 and 10), also the significance thresholds were largely different. The GLM detected associations with significant FDR and Bonferroni-adjusted thresholds (p values ranging from 0.05 to 0.001), however, the same associations detected by MLM, were not significant with FDR and Bonferroni-adjusted significance thresholds. The highest R2 values were detected for 50083_64 and 238771_249 in association with mCBTemp10 (both 0.150 R2). MLM, similarly to LFMM and SFA, revealed the highest significant association between 277051_56 and PETCQ (0.045 R2).
Altogether, the environmental variables explained 2.4–16.6% of the genetic variation (Table 3; average adjusted R2). SNP 50083_64, 180975_104 and 227780_278 was explained chiefly by PETCQ (avg. R2adj = 10.7, 10.1 and 16.6%), 96506_153, 96534_88, 361438_297 and 437832_228 by aridity (avg. R2adj = 10.8, 11.6, 10.9 and 14.4%), 238771_249 by mCBTemp10 (avg. R2adj = 8.2%), 277051_56 by bio8_18 (avg. R2adj = 11.2%). By summarizing the environmental predictors, in decreasing order, aridity (81.7%), PETCQ (79.9%), bio8_16 (51.9%) and prec6_16 (49.7%) explained the highest amount of the genetic variation, thus considered to be the most important (Tables 3, S7).
Discussion
Previous studies on Q. petraea suggest on the one side generally high genetic diversity in the Balkan region due to the large number of refugia in this area and the high environmental heterogeneity (Zanetto and Kremer 1995; Gömöry et al. 2001; Tzedakis 2004), on the other side differential population responses to climate change (Sáenz-Romero et al. 2017). Although populations possess characteristics that can facilitate adaptation (prolific seed production and masting, high levels of genetic diversity, extensive gene flow) (Kremer 2016), they are becoming detached from the local climatic conditions to which they have previously adapted (Cheaib et al. 2012).The results of our study may provide information on how, despite continuous gene flow, populations from this area continued to differentiate as they successfully adapted to their new distinct environments. This may also suggest that adaptation to the climate might have occurred via many changes in the frequency of alleles for genes related to moisture deficit, temperature, and precipitation.
One important step of our method consisted of mapping our short reads to the Q. robur reference genome. Restriction-site-associated DNA sequencing or RAD-seq is increasingly used to identify large numbers of single-nucleotide polymorphisms (SNPs) (Allendorf et al. 2010; Davey and Blaxter 2010). Certainly, for the identification of genes under selection, the functional annotation of these genes and their location in the species’ genome are possible only through sequencing of the reference genome of a given, or a closely related species. Lacking a Q. petraea genome, we mapped our paired-end reads to the haploid version of the Q. robur genome (PM1N; http://www.oakgenome.fr; Plomion et al. 2018). After filtering, the short-read sequences were mapped onto the genomic reference with an average success rate of 92.43%. This percentage was relatively high considering that López de Heredia et al. (2020) managed to align their Quercus suber L. filtered reads to only 67.8% of the available Q. suber genome assembly. However, since PstI/MspI were selected as restriction enzymes in both studies, the mapping difference can be attributed to the different types of sequencing [single-end in the work of López de Heredia et al., paired-end in the work of Tóth et al. (2021)]. Another reason for the difference may be attributed to the different lengths and quality scores of the reads [mean quality score of at least 30 and length of at least 200 bp in our previous work, 20 and at least 20 bp in the work of López de Heredia et al. (2020)]. Finally, we must not forget the fact that the reference genomes assemblies were also different. Mapping our reads to the Q. robur genome allowed precise location of many of the loci in specific genes of known function.
Population structure and diversity
Three main clusters were identified based on our results. The geographic extent of all clusters indicated a north–southeast gradient. The first cluster (Cluster 1) (yellow cluster in Fig. 2a) is dominant in continental regions, while the second cluster (Cluster 2) (blue) was found mainly in Central Europe (Fig. 2d). The southernmost Mediterranean group proved to be the third cluster (Cluster 3) (red) positioned at the border of Black Sea region. This geographical distribution corresponds with results from the work of both Zanetto and Kremer (1995) and Petit et al. (1993), in which longitudinal appeared to be more pronounced than latitudinal gradients. Regarding current knowledge of potential glacial refugia, in case of oaks, the western Balkans constituted a major refugium during the glacial periods (Bennett et al. 1991; Bordács et al. 2002; Petit et al. 2002). Recolonisation pathways probably followed a north-western postglacial migration route from the Balkans and Black Sea refugia. (Zanetto and Kremer 1995). These recolonization patterns may explain not only the north–southeast gradient but also the tendency to increase in probability towards the west of Cluster 3. The highly admixed nature of Cluster 2 (populations of HU and BH) could be a consequence of a transition zone, shown not only by Hewitt (1993) but also by Zanetto and Kremer (1995). The fact that Cluster 3 not only showed the highest differentiation but was also positioned on a separate branch with a much greater genetic distance from Clusters 1 and 2 also supports this assumption. We observed moderate levels of genetic diversity and allelic richness, with balanced values at the population level and no private alleles. As distribution of genetic diversity within and among populations is a function of the rate of gene flow (Bruschi et al. 2003), in our opinion this result can be attributed to different processes. First, woody species with large geographic ranges, outcrossing breeding systems, and wind-facilitated seed dispersal show less variation among populations than woody species with other combinations of traits (Hamrick et al. 1992; Hamrick and Godt 1996). Another possible interpretation of the observed differentiation pattern involves consequences of rapid migration. Oak species that expanded rapidly over large distances usually exhibit widely distributed haplotypes (Pham et al. 2017; Sork et al. 2016). Introgression of genes from Q. pubescens may be another reason the lack of differentiation (Bruschi et al. 2003). Rare alleles might have been lost because of selection pressures or during population bottlenecks (Allendorf 1986). Being associated with decreased fitness, selection against rare alleles occurred where viability selection was strongest at optimal growing sites (Bush and Smouse 1992). Of all populations, BU4 showed the lowest diversity values. Our sampling in Bulgaria was designed to cover the western Balkan, Rila, Rhodope and Strandzha Mts., geographic regions corresponding to the major genetic clusters. The BU4 population originates from the Strandzha Mts., as can be traced from the EUFORGEN distribution map (http://www.euforgen.org/species/quercus-petraea/), and this population is located at the eastern edge of the Black Sea refugium and in the eastern part of the current European distribution range of the species. Additionally, individuals of this region are considered to be a distinct subspecies in the Euro+Med Plantbase (http://ww2.bgbm.org/EuroPlusMed/query.asp) and on The Plant List (http://www.theplantlist.org), Q. petraea subsp. iberica (Steven ex M. Bieb.) Krassiln., and Schwarz (1993) considered it a separate species: Q. polycarpa Schur. (Schwarz 1993). The low level of intrapopulation diversity of the BU4 population may be explained by genetic drift (Newman and Pilson 1997), anthropogenic activities or by the successive bottlenecks that occurred at the head of the migration front (Dumolin-Lapegue et al. 1997), which may have reduced diversity during the eastward expansion from this refugium. It should also be noted that the lower genetic diversity observed could be the consequence of the stringent filtering strategy that is commonly applied for SNP filtering and quality control in GEA studies (DeSilva and Dodd 2020).
Biological functions of outliers under selection
Local adaptation in natural populations may arise from differential selection pressures across heterogeneous environments (De La Torre et al. 2019). Therefore, different combinations of alleles might be favoured in these different environments and maintained as stable polymorphisms or experience partial sweeps due to selection acting on already standing variation (Hermisson and Pennings 2005). Our sampled populations experience different environmental conditions with different adaptations and selection pressures specific to their local habitats. Thus, we presumed that these populations evolve traits that provide an advantage in their local environment, and we performed different outlier detection methods, as these analyses can screen numerous markers in the genome to identify candidate genes for further investigation (Narum and Hess 2011). During our survey, as robust outliers, we detected nine SNPs located at different loci at six different chromosomes, and the FST values computed suggested positive (diversifying) selection. Those SNPs that were presented in NCBI databases resided in four different functional regions. Two SNPs found in a gene encoding the uncharacterized protein 115976593 (NCBI ID), one in bHLH162 transcription factor, one in hydroxymethylglutaryl-CoA synthase coding region, and one in a hypothetical protein coding region (possible double-strand damage repair). No biological function can be associated to uncharacterized proteins, however, bHLH transcription factor is related to NPF genes (Zhao et al. 2021), which have functions in stomatal opening and contributes to drought susceptibility in Arabidopsis sp. (Guo et al. 2003). Hydroxymethylglutaryl-CoA synthase has role in the biosynthesis of secondary metabolites under drought stress (Haider et al. 2017), by regulating the synthesis of mevalonic acid, the precursor of isoprenoid compounds (Bach 1986), that plays a role in various physiological processes, including cell membrane fluidity, plant defence under stress caused by environmental factors such as drought or high temperatures (Dani et al. 2014; Tattini et al. 2015).
Precipitation as the strongest possible determinant of adaptation in Quercus petraea
Drought stress and temperature variation impose limitations on the survival, growth, and productivity of many forest tree species, and the survival of sessile oak is also affected by these two climatic factors. Evidenced by high level of genetic differentiation observed in common garden experiments, oak populations have responded to climatic selection during historical global warming after the last glaciation (Kremer et al. 2014; Torres-Ruiz et al. 2019). Previous studies have also suggested differential responses to temperature and moisture across geographically distant populations of the species (Bruschi et al. 2003; Müller and Gailing 2019; Mátyás 2021), with higher population differentiation, for example, compared to that of Quercus robur L. (Kremer and Petit 1993).
In our study, nine SNPs (0.34%) showed signatures of selection according to differentiation outlier analyses. The observed proportion was lower than obtained in other studies on temperate broadleaf forest trees (Derory et al. 2010; Alberto et al. 2013; Csilléry et al. 2014; De Kort et al. 2014; Sork et al. 2016; Temunović et al. 2020).
Caution must be exercised when interpreting our results, taking into account the potential for false positive selection signatures (Excoffier et al. 2009; De Villemereuil et al. 2014; Whitlock and Lotterhos 2015; Hoban et al. 2016) and past introgression events (Bierne et al. 2011; Guichoux et al. 2013). Should be also noted that the proportion we obtained from SNPs detected by two different approaches, including our restriction site-associated DNA sequencing method, was based on only a few SNPs per gene region (Luikart et al. 2003). Our results revealed 53 associations between markers and environment, with significance levels ranging from p ≤ 0.05 to 0.001. The variations seen in the analyses are due to differences in the methods used to account for demographic history or neutral genetic structure, which results in different statistical strengths for different sampling schemes or complicated demographic situations (De Mita et al. 2013; Forester et al. 2018; Aguirre-Liguori 2021). Altogether, the environmental variables explained 2.4–16.6% of the genetic variation. SNP 50083_64 (hypothetical protein), 180975_104 (transcription factor bHLH162), 227780_278 (F-box protein At-B) and 437832_228 (hydroxymethylglutaryl-CoA synthase) associated with aridity, 238771_249 (uncharacterized protein) with mCBTemp10. By summarizing the environmental predictors, in decreasing order, aridity (81.7%), PETCQ (79.9%), bio8_16 (51.9%) and prec6_16 (49.7%) explained the highest amount of the genetic variation. In general, our analyses discovered stronger connections with environmental changes than prior studies. Although it is challenging to compare with our study owing to the use of distinct markers, analytical techniques, and climate variables utilized, Homolka et al. (2013) found significant population differentiation for three genes with strong correlations with the local temperature–precipitation regime, Neophytou et al. (2015) did not detect any significant relationships between the considered environmental variables and neutral genetic variation, only Rellstab et al. (2016) found 521 associations in 224 SNPs. The different signal power we noticed may be somewhat attributable to methodological factors, given that our analysis employed a different dataset and statistical methods than Homolka et al. (2013) and Neophytou et al. (2015). Additionally, our selection of SNP loci was more narrowly focused on climate factors than Rellstab et al. (2016), since we aimed for genes likely implicated in traits that are susceptible to climate shifts, especially drought-related ones. Our result also provide evidence for adaptation to drought detected in the provenance study of Bert et al. (2020), who observed provenances originating from sites with wet summers displaying the strongest responses to summer drought, particularly in the driest common garden.
The above list of significant marker-environment associations clearly shows, that most variables are linked to aridity. To validate functional mechanisms underlying selection signatures, it is generally recommended to use annotations, which can provide additional support beyond relying solely on FST outlier and environmental association analyses, which are only correlative measures (Ahrens et al. 2018). As we previously mentioned when annotating the genes with significant environmental associations, according to literature bHLH transcription factor (marker 180975_104) is related to functions in stomatal opening and contributes to drought susceptibility (Babitha et al. 2015; Qian et al. 2021), marker 227780_278 resides in a region encoding F-box protein At-B, whose transcription is induced by abscisic acid, which regulates the regulation of abiotic stress responses (Sah et al. 2016; Park and Kim 2021) and marker 437832_228 is in a region encoding hydroxymethylglutaryl-CoA synthase with role in the biosynthesis of secondary metabolites under drought stress (Ghasemi et al. 2019; Rogowska and Szakiel 2020). By matching gene functions with relevant climatic factors we can provide biological significance to the genetic basis of local adaptation and the impact of climate on divergent selection among our studied sessile oak populations. Nonetheless, assigning functions to specific candidates in nonmodel species should be approached with caution, and further association genetics and functional studies are needed to validate their role in adaptive traits, as recommended by Pavlidis et al. (2012).
Conclusion
While sessile oak populations are well studied in Europe, in the Central-Eastern European region, including the Balkan Peninsula it is seldom tested with genome-wide genetic markers. The identification of genetic variants within populations of this region may hold the key to facilitating the sessile oak’s adaptation to the deleterious effects of modern climate change, thus underscoring the crucial significance of further investigation in this field. Through our examination, we found molecular evidence indicating that important climate-related factors, such as drought, may have influenced the adaptive divergence of sessile oak populations in the Central-Eastern European region. Moreover, our results suggest that these populations may contain beneficial genetic variants that could aid the species in responding to the rapidly changing climate. In light of these findings, it is vital that European forest tree species conservation and management programmes incorporate the Central-Eastern European sessile oak populations, including facilitated gene flow, into their strategies to combat the significant threat that climate change poses to forest tree populations.
Data availability
The genomic dataset analysed during the current study is available in Tóth et al. (2021). The dataset can also be accessed at https://doi.org/10.5281/zenodo.3908963. The sequences of each outlier loci are available at https://doi.org/10.5281/zenodo.7763329.
References
Adamack AT, Gruber B (2014) PopGenReport: simplifying basic population genetic analyses in R. Methods Ecol Evol 5:384–387. https://doi.org/10.1111/2041-210x.12158
Aguirre-Liguori JA, Ramírez-Barahona S, Gaut BS (2021) The evolutionary genomics of species’ responses to climate change. Nat Ecol Evol 5:1350–60. https://doi.org/10.1038/s41559-021-01526-9
Ahrens CW, Rymer PD, Stow A, Bragg J, Dillon S, Umbers KD et al. (2018) The search for loci under selection: trends, biases and progress. Mol Ecol 27:1342–56. https://doi.org/10.1111/mec.14549
Alberto FJ, Derory J, Boury C, Frigerio JM, Zimmermann NE, Kremer A (2013) Imprints of natural selection along environmental gradients in phenology-related genes of Quercus petraea. Genetics 195:495–512. https://doi.org/10.1534/genetics.113.153783
Allendorf FW (1986) Genetic drift and the loss of alleles versus heterozygosity. Zoo Biol 5:181–190. https://doi.org/10.1002/zoo.1430050212
Allendorf FW, Hohenlohe PA, Luikart G (2010) Genomics and the future of conservation genetics. Nat Rev Genet 11:697–709. https://doi.org/10.1038/nrg2844
Babitha KC, Vemanna RS, Nataraja KN, Udayakumar M (2015) Overexpression of EcbHLH57 transcription factor from Eleusine coracana L. in tobacco confers tolerance to salt, oxidative and drought stress. PLoS One 10(9):e0137098. https://doi.org/10.1371/journal.pone.0137098
Bach TJ (1986) Hydroxymethylglutaryl-CoA reductase, a key enzyme in phytosterol synthesis. Lipids 21(1):82–88. https://doi.org/10.1007/BF02534307
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA et al. (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 3:e3376. https://doi.org/10.1371/journal.pone.0003376
Beal MJ (2003) Variational algorithms for approximate Bayesian inference. University of London, University College London, United Kingdom
Beaumont MA (2005) Adaptation and speciation: what can Fst tell us? Trends Ecol Evol 20:435–440. https://doi.org/10.1016/j.tree.2005.05.017
Bennett KD, Tzedakis PC, Willis KJ (1991) Quaternary refugia of north European trees. J Biogeogr 103-115. https://doi.org/10.2307/2845248
Bert D, Lebourgeois F, Ponton S, Musch B, Ducousso A (2020) Which oak provenances for the 22nd century in Western Europe? Dendroclimatology in common gardens. PLoS One 15:e0234583. https://doi.org/10.1371/journal.pone.0234583
Bierne N, Welch J, Loire E, Bonhomme F, David P (2011) The coupling hypothesis: why genome scans may fail to map local adaptation genes. Mol Ecol 20:2044–72. https://doi.org/10.1111/j.1365-294X.2011.05080.x
Birks HJ, Willis KJ (2008) Alpines, trees, and refugia in Europe. Plant Ecol Divers 1:147–60. https://doi.org/10.1080/17550870802349146
Bivand R, Keitt T, Rowlingson B, Pebesma E, Sumner M, Hijmans R et al. (2015) Package ‘rgdal’.: Bindings for the Geospatial Data Abstraction Library. R package. https://cran.r-project.org/web/packages/rgdal/index.html (Accessed: July 20, 2021)
Bivand R, Rundel C, Pebesma E, Stuetz R, Hufthammer KO, Bivand MR (2017) Package ‘rgeos’.: Interface to Geometry Engine - Open Source (‘GEOS’). R package. https://cran.r-project.org/web/packages/rgeos/index.html (Accessed: July 20, 2021)The Comprehensive R Archive Network (CRAN). https://doi.org/10.1007/978-1-4614-7618-4
Blanc-Jolivet C, Bakhtina S, Yanbaev R, Yanbaev Y, Mader M, Guichoux E et al. (2020) Development of new SNPs loci on Quercus robur and Quercus petraea for genetic studies covering the whole species’ distribution range. Conserv Genet Resour 12:597–600. https://doi.org/10.1007/s12686-020-01141-z
Boonman CC, Huijbregts MA, Benítez‐López A, Schipper AM, Thuiller W, Santini L (2022) Trait‐based projections of climate change effects on global biome distributions. Diversity Distrib 28:25–37. https://doi.org/10.1111/ddi.13431
Bordács S, Popescu F, Slade D, Csaikl UM, Lesur I, Borovics A et al. (2002) Chloroplast DNA variation of white oaks in northern Balkans and in the Carpathian Basin. Ecol Manag 156:197–209. https://doi.org/10.1016/s0378-1127(01)00643-0
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635. https://doi.org/10.1093/bioinformatics/btm308
Brown MB (1975) Method for combining non-independent, one-sided tests of significance. Biometrics 31:987–992. https://doi.org/10.2307/2529826
Bruschi P, Vendramin GG, Bussotti F, Grossoni P (2003) Morphological and molecular diversity among Italian populations of Quercus petraea (Fagaceae). Ann Bot 91:707–716. https://doi.org/10.1093/aob/mcg075
Bush RM, Smouse PE (1992) Evidence for the adaptive significance of allozymes in forest trees. New 6:179–196. https://doi.org/10.1007/bf00120644
Capblancq T, Forester BR (2021) Redundancy analysis: A Swiss Army Knife for landscape genomics. Methods Ecol Evol 12:2298–2309. https://doi.org/10.1111/2041-210X.13722
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA (2013) Stacks: an analysis tool set for population genomics. Mol Ecol 22:3124–3140. https://doi.org/10.1111/mec.12354
Catchen JM, Amores A, Hohenlohe P, Cresko W, Postlethwait JH (2011) Stacks: building and genotyping loci de novo from short-read sequences. G3: Genes Genom Genet 1:171–182. https://doi.org/10.1534/g3.111.000240
Cavender-Bares J, Gonzalez-Rodriguez A, Eaton DAR, Hipp AL, Beulke A, Manos PS (2015) Phylogeny and biogeography of the American live oaks (Quercus subsection Virentes): a genomic and population genetics approach. Mol Ec 24:3668–3687. https://doi.org/10.1111/mec.13269
Cheaib A, Badeau V, Boe J, Chuine I, Delire C, Dufrêne E et al. (2012) Climate change impacts on tree ranges: model inter-comparison facilitates understanding and quantification of uncertainty. Ecol Lett 15:533–544. https://doi.org/10.1111/j.1461-0248.2012.01764.x
Csilléry K, Lalagüe H, Vendramin GG, González‐Martínez SC, Fady B, Oddou‐Muratorio S (2014) Detecting short spatial scale local adaptation and epistatic selection in climate‐related candidate genes in European beech (Fagus sylvatica) populations. Mol Ecol 23:4696–708.v. https://doi.org/10.1111/mec.12902
Dani KS, Jamie IM, Prentice IC, Atwell BJ (2014) Evolution of isoprene emission capacity in plants. Trends Plant Sci 19:439–46. https://doi.org/10.1016/j.tplants.2014.01.009
Darwin C (1859) On the Origin of Species by Means of Natural Selection. J. Murray, London
Davey JW, Blaxter ML (2010) RADSeq: next-generation population genetics. Brief Funct Genomics 9:416–423. https://doi.org/10.1093/bfgp/elq031
Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML (2011) Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat Rev Genet 12:499–510. https://doi.org/10.1038/nrg3012
Degen B, Blanc-Jolivet C, Bakhtina S, Ianbaev R, Yanbaev Y, Mader M et al. (2021) Applying targeted genotyping by sequencing with a new set of nuclear and plastid SNP and indel loci for Quercus robur and Quercus petraea. Conserv Genet Resour 1-3. https://doi.org/10.1007/s12686-021-01207-6
De Kort H, Vandepitte K, Bruun HH, Closset‐Kopp D, Honnay O, Mergeay J (2014) Landscape genomics and a common garden trial reveal adaptive differentiation to temperature across Europe in the tree species Alnus glutinosa Mol Ecol 23:4709–21. https://doi.org/10.1111/mec.12813
De La Torre AR, Wilhite B, Neale DB (2019) Environmental genome-wide association reveals climate adaptation is shaped by subtle to moderate allele frequency shifts in loblolly pine Genome Biol Evol 11:2976–2989. https://doi.org/10.1093/gbe/evz220
De Mita S, Thuillet AC, Gay L, Ahmadi N, Manel S, Ronfort J et al. (2013) Detecting selection along environmental gradients: analysis of eight methods and their effectiveness for outbreeding and selfing populations Mol Ecol 22:1383–99. https://doi.org/10.1111/mec.12182
Deng M, Jiang X-L, Hipp AL, Manos PS, Hahn M (2018) Phylogeny and biogeography of East Asian evergreen oaks (Quercus section Cyclobalanopsis; Fagaceae): insights into the Cenozoic history of evergreen broad-leaved forests in subtropical Asia. MPE 119:170–181. https://doi.org/10.1016/j.ympev.2017.11.003
Derory J, Scotti-Saintagne C, Bertocchi E, Le Dantec L, Graignic N, Jauffres A et al. (2010) Contrasting relationships between the diversity of candidate genes and variation of bud burst in natural and segregating populations of European oaks. Heredity 104:438–48. https://doi.org/10.1038/hdy.2009.134
DeSilva R, Dodd RS (2020) Association of genetic and climatic variability in giant sequoia, Sequoiadendron giganteum, reveals signatures of local adaptation along moisture‐related gradients. Ecol Evolution 10:10619–10632. https://doi.org/10.1002/ece3.6716
de Villemereuil P, Frichot É, Bazin É, François O, Gaggiotti OE (2014) Genome scan methods against more complex models: when and how much should we trust them? Mol Ecol 23:2006–2019. https://doi.org/10.1111/mec.12705
de Vries A, Ripley BD (2013) Ggdendro: tools for extracting dendrogram and tree diagram plot data for use with ggplot. R package version 0.1–12. https://cran.r-project.org/web/packages/ggdendro/vignettes/ggdendro.html (Accessed: July 20, 2021)
Devlin B, Roeder K (1999) Genomic control for association studies. Biometrics 55:997–1004. https://doi.org/10.1111/j.0006-341x.1999.00997.x
Dumolin-Lapegue S, Demesure B, Fineschi S, Le Come V, Petit RJ (1997) Phylogeographic structure of white oaks throughout the European continent. Genetics 146:1475–1487. https://doi.org/10.1093/genetics/146.4.1475
Eaton DAR, Hipp AL, González-Rodríguez A, Cavender-Bares J (2015) Historical introgression among the American live oaks and the comparative nature of tests for introgression. Evolution 69:2587–2601. https://doi.org/10.1111/evo.12758
Elsen PR, Saxon EC, Simmons BA, Ward M, Williams BA, Grantham HS et al. (2022) Accelerated shifts in terrestrial life zones under rapid climate change. Glob 28:918–935. https://doi.org/10.1111/gcb.15962
Endler JA (1986) Natural selection in the wild. Monographs in population biology. no. 21. Princeton Univ. Press, Princeton, NJ
Excoffier L, Hofer T, Foll M (2009) Detecting loci under selection in a hierarchically structured population. Heredity 103:285–98. https://doi.org/10.1038/hdy.2009.74
Fassou G, Kougioumoutzis K, Iatrou G, Trigas P, Papasotiropoulos V (2020) Genetic diversity and range dynamics of Helleborus odorus subsp. cyclophyllus under different climate change scenarios. Forests 11:620. https://doi.org/10.3390/f11060620
Feliner GN (2011) Southern European glacial refugia: a tale of tales. Taxon 60:365–372. https://doi.org/10.1002/tax.602007
Ferrier S, Manion G, Elith J, Richardson K (2007) Using generalized dissimilarity modelling to analyse and predict patterns of beta diversity in regional biodiversity assessment. Divers Distrib 13:252–264. https://doi.org/10.1111/j.1472-4642.2007.00341.x
Fitz-Gibbon S, Hipp AL, Pham KK, Manos PS, Sork VL (2017) Phylogenomic inferences from reference-mapped and de novo assembled short-read sequence data using RADseq sequencing of California white oaks (Quercus section Quercus). Genome 60:743–755. https://doi.org/10.1139/gen-2016-0202
Fitzpatrick MC, Keller SR (2015) Ecological genomics meets community‐level modelling of biodiversity: Mapping the genomic landscape of current and future environmental adaptation. Ecol Lett 18:1–16. https://doi.org/10.1111/ele.12376
Fitzpatrick MC, Mokany K, Manion G, Nieto-Lugilde D, Ferrier S (2021) gdm: Generalized dissimilarity modeling. R package. https://cran.r-project.org/web/packages/gdm/index.html (Accessed 20 July 2021)
Forester BR, Lasky JR, Wagner HH, Urban DL (2018) Comparing methods for detecting multilocus adaptation with multivariate genotype–environment associations. Mol Ecol 27:2215–33. https://doi.org/10.1111/mec.14584
Francis RM (2017) pophelper: an R package and web app to analyse and visualize population structure. Mol Ecol Resour 17:27–32. https://doi.org/10.1111/1755-0998.12509
Frichot E, Schoville SD, Bouchard G, François O (2013) Testing for associations between loci and environmental gradients using latent factor mixed models. Mol Biol Evol 30:1687–1699. https://doi.org/10.1093/molbev/mst063
Frichot E, Schoville SD, de Villemereuil P, Gaggiotti OE, Francois O (2015) Detecting adaptive evolution based on association with ecological gradients: orientation matters. Heredity 115:22–28. https://doi.org/10.1038/hdy.2015.7
Fuentes‐Pardo AP, Ruzzante DE (2017) Whole‐genome sequencing approaches for conservation biology: Advantages, limitations and practical recommendations. Mol Ecol 26:5369–5406. https://doi.org/10.1111/mec.14264
Gea‐Izquierdo G, Sánchez‐González M (2022) Forest disturbances and climate constrain carbon allocation dynamics in trees. Glob 28:4342–4358. https://doi.org/10.1111/gcb.16172
Ghasemi S, Kumleh HH, Kordrostami M (2019) Changes in the expression of some genes involved in the biosynthesis of secondary metabolites in Cuminum cyminum L. under UV stress. Protoplasma 256:279–90. https://doi.org/10.1007/s00709-018-1297-y
Gömöry D, Zhelev P, Brus R (2020) The Balkans: a genetic hotspot but not a universal colonization source for trees. Plant Syst Evol 306:1–9. https://doi.org/10.1007/s00606-020-01647-x
Gömöry D, Yakovlev I, Zhelev P, Jedináková J, Paule L (2001) Genetic differentiation of oak populations within the Quercus robur/Quercus petraea complex in Central and Eastern Europe. Heredity 86:557–563. https://doi.org/10.1046/j.1365-2540.2001.00874.x
González de Andrés E (2019) Interactions between climate and nutrient cycles on forest response to global change: The role of mixed forests. Forests 10:609. https://doi.org/10.3390/f10080609
Goudet J (2005) Hierfstat, a package for R to compute and test hierarchical F‐statistics. Mol Ecol Notes 5:184–186. https://doi.org/10.1111/j.1471-8286.2004.00828.x
Guichoux E, Garnier‐Géré P, Lagache L, Lang T, Boury C, Petit RJ (2013) Outlier loci highlight the direction of introgression in oaks. Mol Ecol 22:450–462. https://doi.org/10.1111/mec.12125
Guo FQ, Young J, Crawford NM (2003) The nitrate transporter AtNRT1. 1 (CHL1) functions in stomatal opening and contributes to drought susceptibility in Arabidopsis. Plant Cell 15:107–17. https://doi.org/10.1105/tpc.006312
Gurgul A, Miksza-Cybulska A, Szmatoła T, Jasielczuk I, Piestrzyńska-Kajtoch A, Fornal A et al. (2019) Genotyping-by-sequencing performance in selected livestock species. Genomics 111:186–195. https://doi.org/10.1016/j.ygeno.2018.02.002
Haider MS, Zhang C, Kurjogi MM, Pervaiz T, Zheng T, Zhang C et al. (2017) Insights into grapevine defense response against drought as revealed by biochemical, physiological and RNA-Seq analysis. Sci Rep. 7:1–5. s41598-017-13464-3
Hamrick JL, Godt MW (1996) Effects of life history traits on genetic diversity in plant species. Philos Trans R Soc Lond B Biol Sci 351:1291–1298. https://doi.org/10.1098/rstb.1996.0112
Hamrick JL, Godt MJW, Sherman-Broyles SL (1992) Factors influencing levels of genetic diversity in woody plant species. In Population genetics of forest trees. Springer, Dordrecht. pp. 95-124.
Hanewinkel M, Cullmann DA, Schelhaas MJ, Nabuurs GJ, Zimmermann NE (2013) Climate change may cause severe loss in the economic value of European forest land. Nat Clim Chang 3:203–207. https://doi.org/10.1038/nclimate1687
Hermisson J, Pennings PS (2005) Soft sweeps: molecular population genetics of adaptation from standing genetic variation. Genetics 169:2335–2352. https://doi.org/10.1534/genetics.104.036947
Hewitt GM (1993) Postglacial distribution and species substructure: lessons from pollen, insects and hybrid zones. Evolut patterns Process 14:97–123
Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A (2005) Very high resolution interpolated climate surfaces for global land areas. Int J Climatol 25:1965–1978. https://doi.org/10.1002/joc.1276
Hijmans RJ, van Etten J (2015) Raster: geographic data analysis and modeling. R package. https://cran.r-project.org/web/packages/raster/index.html (Accessed: July 20, 2021) http://CRAN.R-project.org/package=raster
Hipp AL, Eaton DAR, Cavender-Bares J, Fitzek E, Nipper R, Manos PS (2014) A framework phylogeny of the American oak clade based on sequenced RAD data. PLoS ONE 9:e93975. https://doi.org/10.1371/journal.pone.0093975
Hipp AL, Manos PS, González-Rodríguez A, Hahn M, Kaproth M, McVay JD et al. (2018) Sympatric parallel diversification of major oak clades in the Americas and the origins of Mexican species diversity. N. Phytologist 217:439–452. https://doi.org/10.1111/nph.14773
Hirao AS, Kudo G (2004) Landscape genetics of alpine-snowbed plants: comparisons along geographic and snowmelt gradients. Heredity 93:290–298. https://doi.org/10.1038/sj.hdy.6800503
Hoban S, Kelley JL, Lotterhos KE, Antolin MF, Bradburd G, Lowry DB et al. (2016) Finding the genomic basis of local adaptation: pitfalls, practical solutions, and future directions. Am Nat 188:379–97. https://doi.org/10.1086/688018
Holderegger R, Herrmann D, Poncet B, Gugerli F, Thuiller W, Taberlet P et al. (2008) Land ahead: using genome scans to identify molecular markers of adaptive relevance. Plant Ecol Divers 1:273–283. https://doi.org/10.1080/17550870802338420
Homolka A, Schueler S, Burg K, Fluch S, Kremer A (2013) Insights into drought adaptation of two European oak species revealed by nucleotide diversity of candidate genes. Tree Genet 9:1179–1192. https://doi.org/10.1007/s11295-013-0627-7
Jiang X-L, Hipp AL, Deng M, Su T, Zho Z-K, Yan M-X (2019) East Asian origins of European holly oaks via the Tibet-Himalayas. J Biogeogr 46:2203–2214. https://doi.org/10.1111/jbi.13654
Johnson RN, O’Meally D, Chen Z, Etherington GJ, Ho SY, Nash WJ et al. (2018) Adaptation and conservation insights from the koala genome. Nat Genet 50:1102–1111. https://doi.org/10.1038/s41576-018-0039-5
Jombart T, Ahmed I (2011) adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics 27:3070–3071. https://doi.org/10.1093/bioinformatics/btr521
Joost S, Bonin A, Bruford MW, Després L, Conord C, Erhardt G et al. (2007) A spatial analysis method (SAM) to detect candidate loci for selection: towards a landscape genomics approach to adaptation. Mol Ecol 16:3955–3969. https://doi.org/10.1111/j.1365-294x.2007.03442.x
Kamvar ZN, Tabima JF, Grünwald NJ (2014) Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2:e281. https://doi.org/10.7717/peerj.281
Kassambara A, Mundt F (2017) Package “factoextra” for R: Extract and Visualize the Results of Multivariate Data Analyses. R Package. https://cran.r-project.org/web/packages/factoextra/index.html (Accessed: Nov 22, 2022)
Kawecki TJ, Ebert D (2004) Conceptual issues in local adaptation. Ecol Lett 7:1225–1241. https://doi.org/10.1111/j.1461-0248.2004.00684.x
Kijowska-Oberc J, Staszak AM, Kamiński J, Ratajczak E (2020) Adaptation of forest trees to rapidly changing climate. Forests 11:123. https://doi.org/10.3390/f11020123
Kim BY, Wei X, Fitz-Gibbon S, Lohmueller KE, Ortego J, Gugger PF et al. (2018) RADseq data reveal ancient, but not pervasive, introgression between Californian tree and scrub oak species (Quercus sect. Quercus: Fagaceae). Mol Ecol 27:4556–4571. https://doi.org/10.1111/mec.14869
Kohler M, Pyttel P, Kuehne C, Modrow T, Bauhus J (2020) On the knowns and unknowns of natural regeneration of silviculturally managed sessile oak (Quercus petraea (Matt.) Liebl.) forests—a literature review. Ann Sci 77:1–19. https://doi.org/10.1007/s13595-020-00998-2
Konar A, Choudhury O, Bullis R, Fiedler L, Kruser JM, Stephens MT et al. (2017) High-quality genetic mapping with ddRADseq in the non-model tree Quercus rubra. BMC Genomics 18:1–12. 10.1186/s12864-017-3765
Kremer A (2016) Microevolution of European temperate oaks in response to environmental changes. C R Biol 339:263–267. https://doi.org/10.1016/j.crvi.2016.04.014
Kremer A, Zanetto A (1997) Geographical structure of gene diversity in Quercus petraea (Matt.) Liebl. II: Multilocus patterns of variation. Heredity 78:476–489. https://doi.org/10.1038/hdy.1997.76
Kremer A, Hipp AL (2020) Oaks: an evolutionary success story. N. Phytol 226:987–1011. https://doi.org/10.1111/nph.16274
Kremer A, Potts BM, Delzon S (2014) Genetic divergence in forest trees: understanding the consequences of climate change. Funct 28:22–36. https://doi.org/10.1111/1365-2435.12169
Kremer A, Petit RJ (1993) Gene diversity in natural populations of oak species. In: Annales des sciences forestières (50, No. Supplement, pp. 186s-202s). EDP Sciences. https://doi.org/10.1051/forest:19930717
Kuhn M (2009) The caret package. J Stat Softw 28:1–26. https://doi.org/10.18637/jss.v028.i05
Kunz J, Löffler G, Bauhus J (2018) Minor European broadleaved tree species are more drought-tolerant than Fagus sylvatica but not more tolerant than Quercus petraea. Ecol Manag 414:15–27. https://doi.org/10.1016/j.foreco.2018.02.016
Kwon S, Simko I, Hellier B, Mou B, Hu J (2013) Genome-wide association of 10 horticultural traits with expressed sequence tag-derived SNP markers in a collection of lettuce lines. Crop J 1:25–33. https://doi.org/10.1016/j.cj.2013.07.014
Lang T, Abadie P, Léger V, Decourcelle T, Frigerio JM, Burban C et al. (2021) High-quality SNPs from genic regions highlight introgression patterns among European white oaks (Quercus petraea and Q. robur). bioRxiv, 388447. https://doi.org/10.1101/388447
Lê S, Josse J, Husson F (2008) FactoMineR: an R package for multivariate analysis. J Stat Softw 25(1):1–18. https://doi.org/10.18637/jss.v025.i01
Lepoittevin C, Bodénès C, Chancerel E, Villate L, Lang T, Lesur I et al. (2015) Single‐nucleotide polymorphism discovery and validation in high‐density SNP array for genetic analysis in European white oaks. Mol Ecol Res 15:1446–1459. https://doi.org/10.1111/1755-0998.12407
Leroy T, Louvet JM, Lalanne C, Le Provost G, Labadie K, Aury JM et al. (2020) Adaptive introgression as a driver of local adaptation to climate in European white oaks. N. Phytol 226:1171–1182. https://doi.org/10.1111/nph.16095
Lewis ZA, Shiver AL, Stiffler N, Miller MR, Johnson EA, Selker EU (2007) High-density detection of restriction-site-associated DNA markers for rapid mapping of mutated loci in Neurospora. Genetics 177:1163–1171. https://doi.org/10.1534/genetics.107.078147
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al. (2009) The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics 25:2078–2079. https://doi.org/10.1093/bioinformatics/btp352
Li H (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.3997
Loarie SR, Duffy PB, Hamilton H, Asner GP, Field CB, Ackerly DD (2009) The velocity of climate change. Nature 462:1052–5. https://doi.org/10.1038/nature08649
López de Heredia U, Mora-Márquez F, Goicoechea PG, Guillardín-Calvo L, Simeone MC, Soto Á (2020) ddRAD sequencing-based identification of genomic boundaries and permeability in Quercus ilex and Q. suber hybrids. Front Plant Sci 11:1330. https://doi.org/10.3389/fpls.2020.564414
Luikart G, England PR, Tallmon D, Jordan S, Taberlet P (2003) The power and promise of population genomics: from genotyping to genome typing. Nat Rev Genet 4:981–94. https://doi.org/10.1038/nrg1226
Luu K, Bazin E, Blum MG (2017) pcadapt: an R package to perform genome scans for selection based on principal component analysis. Mol Ecol Resour 17:67–77. https://doi.org/10.1111/1755-0998.12592
Mailund T (2019) Manipulating data frames: dplyr. In: R Data Science Quick Reference. Apress, Berkeley, CA. pp. 109-160
Malinsky M, Trucchi E, Lawson DJ, Falush D (2018) RADpainter and fineRADstructure: population inference from RADseq data. Mol Biol Evol 35:1284–1290. https://doi.org/10.1093/molbev/msy023
Manel S, Segelbacher G (2009) Perspectives and challenges in landscape genetics. 18(9), 1821-1822. https://doi.org/10.1111/j.1365-294x.2009.04151.x
Marees AT, de Kluiver H, Stringer S, Vorspan F, Curis E, Marie‐Claire C et al. (2018) A tutorial on conducting genome‐wide association studies: Quality control and statistical analysis. Int J Methods Psychiatr Res 27:e1608. https://doi.org/10.1002/mpr.1608
Matesanz S, Gianoli E, Valladares F (2010) Global change and the evolution of phenotypic plasticity in plants. Ann NY Acad Sci 1206:35–55. https://doi.org/10.1111/j.1749-6632.2010.05704.x
Mátyás C (2021) Adaptive pattern of phenotypic plasticity and inherent growth reveal the potential for assisted transfer in sessile oak (Quercus petraea L.). Ecol Manag 482:118832. https://doi.org/10.1016/j.foreco.2020.118832
Mátyás G, Sperisen C (2001) Chloroplast DNA polymorphisms provide evidence for postglacial re-colonisation of oaks (Quercus spp.) across the Swiss Alps. Theor Appl Genet 102:12–20. https://doi.org/10.1007/s001220051613
Miller MR, Atwood T, Eames BF, Eberhart J, Yan Y-L, Postlethwait J et al. (2007) RAD marker microarrays enable rapid mapping of zebrafish mutations. Genome Biol 8:R105. https://doi.org/10.1186/gb-2007-8-6-r105
Mölder A, Meyer P, Nagel RV(2019) Integrative management to sustain biodiversity and ecological continuity in Central European temperate oak (Quercus robur. Q. petraea) forests: an overview For Ecol Manage 437:324–339
Muir G, Lowe AJ, Fleming CC, Vogl C (2004) High nuclear genetic diversity, high levels of outcrossing and low differentiation among remnant populations of Quercus petraea at the margin of its range in Ireland. Ann Bot 93:691–697. https://doi.org/10.1093/aob/mch096
Müller M, Gailing O (2019) Abiotic genetic adaptation in the Fagaceae. Plant Biol 21:783–795. https://doi.org/10.1111/plb.13008
Naimi B (2017) Package ‘usdm’: uncertainty analysis for species distribution models. R package. https://cran.r-project.org/web/packages/usdm/index.html (Accessed: July 20, 2021)
Nakazawa M (2022) Package ‘fmsb’: Functions for Medical Statistics Book with some Demographic Data. R package. https://cran.r-project.org/web/packages/fmsb/index.html (Accessed: July 20, 2021)
Narum SR, Hess JE (2011) Comparison of FST outlier tests for SNP loci under selection. Mol Ecol Resour 11:184–194. https://doi.org/10.1111/j.1755-0998.2011.02987.x
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci USA 70:3321–3. https://doi.org/10.1073/pnas.70.12.3321
Neophytou C, Aravanopoulos FA, Fink S, Dounavi A (2010) Detecting interspecific and geographic differentiation patterns in two interfertile oak species (Quercus petraea (Matt.) Liebl. and Q. robur L.) using small sets of microsatellite markers. For Ecol Manag 259:2026–35. https://doi.org/10.1016/j.foreco.2010.02.013
Neophytou C, Gärtner SM, Vargas-Gaete R, Michiels HG (2015) Genetic variation of Central European oaks: shaped by evolutionary factors and human intervention? Tree Genet 11:1–5. https://doi.org/10.1007/s11295-015-0905-7
Newman D, Pilson D (1997) Increased probability of extinction due to decreased genetic effective population size: experimental populations of Clarkia pulchella. Evolution 51:354–62. https://doi.org/10.1111/j.1558-5646.1997.tb02422.x
Ortego J, Gugger PF, Sork VL (2018) Genomic data reveal cryptic lineage diversification and introgression in Californian golden cup oaks (section Protobalanus). N. Phytol 218:804–818. https://doi.org/10.1111/nph.14951
Park EJ, Kim TH (2021) Arabidopsis OSMOTIN 34 functions in the ABA signaling pathway and is regulated by proteolysis. Int J Mol Sci 22:7915. https://doi.org/10.3390/ijms22157915
Pavlidis P, Jensen JD, Stephan W, Stamatakis A (2012) A critical assessment of storytelling: gene ontology categories and the importance of validating genomic scans. Mol Biol Evol 29:3237–48. https://doi.org/10.1093/molbev/mss136
Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE (2012) Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PloS one 7(5):e37135. https://doi.org/10.1371/journal.pone.0037135
Petit RJ, Kremer A, Wagner DB (1993) Geographic structure of chloroplast DNA polymorphisms in European oaks. Theor Appl Genet 87:122–128. https://doi.org/10.1007/bf00223755
Petit RJ, Brewer S, Bordács S, Burg K, Cheddadi R, Coart E et al. (2002) Identification of refugia and post-glacial colonisation routes of European white oaks based on chloroplast DNA and fossil pollen evidence. Ecol Manag 156:49–74. https://doi.org/10.1016/S0378-1127(01)00634-X
Pham KK, Hipp AL, Manos PS, Cronn RC (2017) A time and a place for everything: phylogenetic history and geography as joint predictors of oak plastome phylogeny. Genome 60:720–732. https://doi.org/10.1139/gen-2016-019
Plomion C, Aury JM, Amselem J, Leroy T, Murat F, Duplessis S et al. (2018) Oak genome reveals facets of long lifespan. Nat Plants 4:440–452. https://doi.org/10.1111/1755-0998.12425
Pörtner HO, Roberts DC, Adams H, Adler C, Aldunce P, Ali E et al. (2022) Climate change 2022: Impacts, adaptation and vulnerability. IPCC Sixth Assessment Report. Cambridge University Press, Cambridge, UK and New York, NY, USA, p 3056. 10.1017/9781009325844
Pouresmaeily M (2022) Ecological Responses to Climate Change. In: Bandh SA (eds) Climate Change. Springer, Cham, 10.1007/978-3-030-86290-9_9
Privé F, Luu K, Vilhjálmsson BJ, Blum MG (2020) Performing highly efficient genome scans for local adaptation with R package pcadapt version 4. Mol Biol Evol 37:2153–2154. https://doi.org/10.1093/molbev/msaa053
Qian Y, Zhang T, Yu Y, Gou L, Yang J, Xu J et al. (2021) Regulatory mechanisms of bHLH transcription factors in plant adaptive responses to various abiotic stresses. Front 12:677611. https://doi.org/10.3389/fpls.2021.677611
R Core Team (2013) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria
Raj A, Stephens M, Pritchard JK (2014) fastSTRUCTURE: variational inference of population structure in large SNP data sets. Genetics 197:573–589. https://doi.org/10.1093/genetics/197.2.NP
Ramos AM, Usié A, Barbosa P, Barros PM, Capote T, Chaves I et al. (2018) The draft genome sequence of cork oak. Sci Data 5:180069. https://doi.org/10.1038/sdata.2018.69
Rellstab C, Gugerli F, Eckert AJ, Hancock AM, Holderegger R (2015) A practical guide to environmental association analysis in landscape genomics. Mol Ecol 24:4348–4370. https://doi.org/10.1111/mec.13322
Rellstab C, Zoller S, Walthert L, Lesur I, Pluess AR, Graf R et al. (2016) Signatures of local adaptation in candidate genes of oaks (Quercus spp.) with respect to present and future climatic conditions. Mol Ecol 25:5907–5924. https://doi.org/10.1111/mec.13889
Rieseberg LH, Widmer A, Arntz AM, Burke JM (2002) Directional selection is the primary cause of phenotypic diversification. Proc Natl Acad Sci USA 99:12242–12245. https://doi.org/10.1073/pnas.19236089
Rochette NC, Rivera‐Colón AG, Catchen JM (2019) Stacks 2: Analytical methods for paired‐end sequencing improve RADseq‐based population genomics. Mol Ecol 28:4737–4754. https://doi.org/10.1111/mec.15253
Rogowska A, Szakiel A (2020) The role of sterols in plant response to abiotic stress. Phytochem 19:1525–38. https://doi.org/10.1007/s11101-020-09708-2
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE et al. (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol 34:3299–3302. https://doi.org/10.1093/molbev/msx248
Sáenz‐Romero C, Lamy JB, Ducousso A, Musch B, Ehrenmann F, Delzon S et al. (2017) Adaptive and plastic responses of Quercus petraea populations to climate across Europe. Glob 23:2831–2847. https://doi.org/10.1111/gcb.13576
Sah SK, Reddy KR, Li J (2016) Abscisic acid and abiotic stress tolerance in crop plants. Front 7:571. https://doi.org/10.3389/fpls.2016.00571
Savolainen O, Lascoux M, Merilä J (2013) Ecological genomics of local adaptation. Nat Rev Genet 14:807–820. https://doi.org/10.1038/nrg3522
Schmidt PS, SerrÃo EA, Pearson GA, Riginos C, Rawson PD, Hilbish TJ et al. (2008) Ecological genetics in the North Atlantic: environmental gradients and adaptation at specific loci. Ecology 89:S91–S107. https://doi.org/10.1890/07-1162.1
Schwarz O (1936) Entwurf zu einem natürlichen System der Cupuliferen und der Gattung Quercus L. Notizbl Bot Gart Berl-Dahl 13:1–22. https://doi.org/10.2307/3994908
Schwarz O (1993) Quercus L. In: Tutin TG, Heywood VH, Burges NA, Valentine DH, Walters SM, Webb DA (Eds) Flora Europaea, vol. 1: Lycopodiaceae to Platanaceae, 2nd Ed. Cambridge University Press, Cambridge, p 72–76
Shafer AB, Peart CR, Tusso S, Maayan I, Brelsford A, Wheat CW et al. (2017) Bioinformatic processing of RAD‐seq data dramatically impacts downstream population genetic inference. Methods Ecol Evol 8:907–917. https://doi.org/10.1111/2041-210x.12700
Slade D, Škvorc Ž, Ballian D, Gračan J, Papes D (2008) The chloroplast DNA polymorphisms of White Oaks of section Quercus in the Central Balkans. Silvae Genet 57:227–34. https://doi.org/10.1515/sg-2008-0035
Sork VL, Squire K, Gugger PF, Steele SE, Levy ED, Eckert AJ (2016) Landscape genomic analysis of candidate genes for climate adaptation in a California endemic oak, Quercus lobata. Am J Bot 103:33–46. https://doi.org/10.3732/ajb.1500162
Storey JD, Bass AJ, Dabney A, Robinson D (2021) qvalue: Q-value estimation for false discovery rate control. R package version 2.24.0. http://www.bioconductor.org/packages/release/bioc/html/qvalue.html (Accessed: July 20, 2021)
Streiff R, Labbe T, Bacilieri R, Steinkellner H, Glössl J, Kremer A (1998) Within‐population genetic structure in Quercus robur L. and Quercus petraea (Matt.) Liebl. assessed with isozymes and microsatellites. Mol Ecol 7:317–328. https://doi.org/10.1046/j.1365-294x.1998.00360.x
Tattini M, Loreto F, Fini A, Guidi L, Brunetti C, Velikova V et al. (2015) Isoprenoids and phenylpropanoids are part of the antioxidant defense orchestrated daily by drought‐stressed P latanus× acerifolia plants during Mediterranean summers. N. Phytol 207:613–26. https://doi.org/10.1111/nph.13380
Temunović M, Garnier‐Géré P, Morić M, Franjić J, Ivanković M, Bogdan S et al. (2020) Candidate gene SNP variation in floodplain populations of pedunculate oak (Quercus robur L.) near the species’ southern range margin: Weak differentiation yet distinct associations with water availability. Mol Ecol 29:2359–2378. https://doi.org/10.1111/mec.15492
Thom D, Seidl R (2022) Accelerating mountain forest dynamics in the Alps. Ecosystems 25:603–17. https://doi.org/10.1007/s10021-021-00674-0
Title PO, Bemmels JB (2018) ENVIREM: an expanded set of bioclimatic and topographic variables increases flexibility and improves performance of ecological niche modeling. Ecography 41:291–307. https://doi.org/10.1111/ecog.02880
Torkamaneh D, Laroche J, Belzile F (2016) Genome-wide SNP calling from genotyping by sequencing (GBS) data: a comparison of seven pipelines and two sequencing technologies. PLoS One 11:e0161333. https://doi.org/10.1371/journal.pone.0161333
Torres-Ruiz JM, Kremer A, Carins Murphy MR, Brodribb T, Lamarque LJ, Truffaut L et al. (2019) Genetic differentiation in functional traits among European sessile oak populations. Tree physiol 39:1736–1749. https://doi.org/10.1093/treephys/tpz090
Tóth EG, Köbölkuti ZA, Cseke K, Kámpel JD, Takács R, Tomov VT et al. (2021) A genomic dataset of single‐nucleotide polymorphisms generated by ddRAD tag sequencing in Q. petraea (Matt.) Liebl. populations from Central-Eastern Europe and Balkan Peninsula. Ann Sci 78:1–13. https://doi.org/10.1007/s13595-021-01051-6
Tsumura Y, Uchiyama K, Moriguchi Y, Ueno S, Ihara-Ujino T (2012) Genome scanning for detecting adaptive genes along environmental gradients in the Japanese conifer, Cryptomeria japonica. Heredity 109:349–360. https://doi.org/10.1038/hdy.2012.50
Tzedakis PC (2004) The Balkans as Prime Glacial Refugial Territory of European Temperate Trees. In: Griffiths HI, Kryštufek B, Reed JM (Eds.) Balkan Biodiversity - Pattern and Process in the European Hotspot. Springer, Netherlands, Dordrecht, p 49–68. https://doi.org/10.1007/978-1-4020-2854-0
Varas-Myrik A, Sepúlveda-Espinoza F, Fajardo A, Alarcón D, Toro-Núñez Ó, Castro-Nallar E et al. (2022) Predicting climate change-related genetic offset for the endangered southern South American conifer Araucaria araucana. Ecol Manag 504:119856. https://doi.org/10.1016/j.foreco.2021.119856
Vasemägi A, Primmer CR (2005) Challenges for identifying functionally important genetic variation: the promise of combining complementary research strategies. Mol Ecol 14:3623–3642. https://doi.org/10.1111/j.1365-294X.2005.02690.x
Whitlock MC, Lotterhos KE (2015) Reliable detection of loci responsible for local adaptation: Inference of a null model through trimming the distribution of FST. Am Nat 186:S24–S36. https://doi.org/10.1086/682949
Wickham H, Chang W, Wickham MH (2016) Package ‘ggplot2’. Creat Elegant Data Vis Using Gramm Graph 2:1–189
Xiong S, Hao Y, Rao S, Huang W, Hu B, Wang Y (2009) Effects of cutoff thresholds for minor allele frequencies on HapMap resolution: A real dataset-based evaluation of the Chinese Han and Tibetan populations. Chin Sci Bull 54:2069–2075. https://doi.org/10.1007/s11434-009-0302-4
Zanetto A, Kremer A (1995) Geographical structure of gene diversity in Quercus petraea (Matt.) Liebl. I. Monolocus patterns of variation. Heredity 75:506–517. https://doi.org/10.1038/hdy.1995.167
Zhao W, Liu Y, Li L, Meng H, Yang Y, Dong Z et al. (2021) Genome-wide identification and characterization of bHLH transcription factors related to anthocyanin biosynthesis in red walnut (Juglans regia L.). Front Genet 12:632509. https://doi.org/10.3389/fgene.2021.632509
Acknowledgements
The authors are grateful to Ivan Iliev (University of Forestry, Bulgaria); Péter Ábrán (Romanian National Environmental Protection Agency Mures, Romania); Srdjan Stojnic, Sasa Orlovic and Erna Vastag (University of Novi Sad, Serbia); Milan Mataruga and Vanja Daničić (University of Banja Luka, Bosnia and Herzegovina); and Egzon Tahirukaj (Connecting Natural Values and People Foundation, Kosovo) for their help in sample collection and valuable discussions in the course of which they shared their insights.
Funding
This study was supported by the Ministry of Agriculture of Hungary (Kaán Károly Project, nos.: EVgF/549/2018, EGF/178/2019) and by the National Science Fund of Bulgaria (research grant no.: KP-06-H26/4). Project no. TKP2021-NKTA-43 has been implemented with support provided by the Ministry of Innovation and Technology of Hungary from the National Research, Development and Innovation Fund, financed under the TKP2021-NKTA funding scheme. Open access funding provided by University of Sopron.
Author information
Authors and Affiliations
Contributions
Contributions of the co-authors: EGT and ZAK conceived the study. EGT, KC, ABe, VTT, PZ, JDK, ABo, and ZAK contributed to the sampling of plant materials. EGT, ZAK and VT processed genomic data and performed statistical analyses and simulations. EGT and ZAK wrote the manuscript. EGT, KC, ABe, BBL, VTT, PZ, JDK, ABo, and ZAK provided feedback on the manuscript draughts.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Associate editor: Pår Ingvarsson.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tóth, E.G., Cseke, K., Benke, A. et al. Key triggers of adaptive genetic variability of sessile oak [Q. petraea (Matt.) Liebl.] from the Balkan refugia: outlier detection and association of SNP loci from ddRAD-seq data. Heredity 131, 130–144 (2023). https://doi.org/10.1038/s41437-023-00629-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-023-00629-2
This article is cited by
-
ddRAD-seq generated genomic SNP dataset of Central and Southeast European Turkey oak (Quercus cerris L.) populations
Genetic Resources and Crop Evolution (2024)
