
Abstract
Hybridization and introgression have played important roles in the history of various species, including lineage diversification and the evolution of adaptive traits. Hybridization can accelerate the development of reproductive isolation between diverging species, and thus valuable insight into the evolution of reproductive barrier formation may be gained by studying secondary contact zones. Hedgehogs of the genus Erinaceus, which are insectivores sensitive to changes in climate, are a pioneer model in Pleistocene phylogeography. The present study provides the first genome-wide SNP data regarding the Erinaceus hedgehogs species complex, offering a unique comparison of two secondary contact zones between Erinaceus europaeus and E. roumanicus. Results confirmed diversification of the genus during the Pleistocene period, and detected a new refugial lineage of E. roumanicus outside the Mediterranean basin, most likely in the Ponto-Caspian region. In the Central European zone, the level of hybridization was low, whereas in the Russian-Baltic zone, both species hybridise extensively. Asymmetrical gene flow from E. europaeus to E. roumanicus suggests that reproductive isolation varies according to the direction of the crosses in the hybrid zones. However, no loci with significantly different patterns of introgression were detected. Markedly different pre- and post-zygotic barriers, and thus diverse modes of species boundary maintenance in the two contact zones, likely exist. This pattern is probably a consequence of the different age and thus of the different stage of evolution of reproductive isolating mechanisms in each hybrid zone.
Similar content being viewed by others
Introduction
Allopatric divergence has been considered the most common mode of speciation; however, the application of modern genomic tools has highlighted that gene flow during species divergence (speciation with gene flow) is more widespread than previously thought (Smadja and Butlin 2011). Prior to our contemporary ability to detect introgression, it was estimated that at least 25% of plant species and 10% of animal species were involved in hybridization (Mallet 2005). Increasing genomic evidence suggests that this is an underestimation, when considering the complexity of the species´ history (Payseur and Rieseberg 2016). Even modern humans experienced substantial introgression from their sister species (Green et al. 2010; Reich et al. 2010), and similar patterns were revealed in many domesticated animals (e.g., Wu et al. 2018; Ai et al. 2015), as well as wildlife species (e.g., Li et al. 2016, Andrade et al. 2019; Kumar et al. 2017). In the light of research focused on whole genomes, it appears that species boundaries are often semipermeable, with permeability being a function of the genomic region (Harrison and Larson 2014; Taylor and Larson 2019).
Vast information regarding the role of introgressive hybridization in speciation is likely to be obtained from contact zones, located at the parapatric borders of the species’ ranges (Hewitt 1988). The formation of these contact zones is often related to the Pleistocene climate and range oscillations, which enabled divergent lineages at various stages of evolution to meet and interact. Thus, important evolutionary insights could be achieved through comparative phylogeographic studies of multispecies contact (suture) zones (Remington 1968; Johannesson et al. 2020). For example, Central Europe is traversed by a suture zone which originated from the secondary contact between the Apennine and Balkan glacial refugial populations (Hewitt 1999). Within this zone, one of the most well-recognized contacting species pair is the European hedgehog (Erinaceus europaeus) and the Northern White-breasted hedgehog (E. roumanicus). Together with the brown bear (Taberlet and Bouvet 1994) and grasshopper (Cooper et al. 1995), these model species were used to formulate the basic paradigm of European phylogeography. Insight into the evolutionary history of the genus Erinaceus was achieved successively, together with appropriate methodological development, using morphology (Kratochvíl 1966; Corbet 1988), restriction enzymes (Bannikova et al. 1995), allozymes (Filippucci and Lapini 1988), and mitochondrial DNA (Santucci et al.1998) (Fig. 1). However, a more comprehensive characterization of diversification within this genus using more comprehensive genomic information is still lacking.

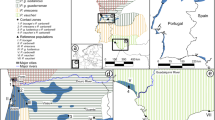
Each pie chart represents one individual and the proportion of each colour represents assignments to each lineage identified with Admixture. Non-admixed individual with foreign mitochondrial DNA is marked with white star. Species and lineage colours are indicated as following: the two shades of blue represent the Iberian and Apennine lineages of E. europaeus, red and brown for the Asian and Balkan lineages of the E. roumanicus, respectively, green indicates E. concolor, yellow indicates E. amurensis, grey indicates Hemiechinus auritus and white, Atelerix albiventris. The two contact zones in Central Europe and Russian-Baltic zones are highlighted in pink. Inset of the figure is based on an IUCN distribution map of species (Amori 2016; Amori et al. 2016a, 2016b; Cassola 2016).
The Central European (CE) hybrid zone between the Apennine lineage of E. europaeus and the Balkan lineage of E. roumanicus (Hewitt 1999; Seddon et al. 2001) spans across Poland, Germany, Czech Republic, Austria, and Italy. The establishment of this contact zone dates back to the period of Neolithic deforestation (10,000–12,000 years ago), following the end of the Last Glacial Maximum (20,000 years ago) (Hewitt 1999; Bolfíková and Hulva 2012). The zone of sympatry is relatively broad (Kratochvíl 1966, 1975). Given the differences in spatial distribution of the two species which may reflect differences in ecology (Bolfíková and Hulva 2012), the CE zone is compatible with a mosaic hybrid zone. Using observation data, the area of sympatry was reported to be expanding within the Czech Republic (Kratochvíl 1966, 1975). However, genetic data detected an expansion only in E. roumanicus (Bolfíková and Hulva 2012). Erinaceus europaeus is estimated to be the first species in the area of Czechia (Kratochvíl 1966), which is compatible with the stable population sizes, and higher abundance compared to E. roumanicus in the region (Bolfíková and Hulva 2012). Although both species show a syntopic distribution in some localities within the CE contact zone, recent hybridization is reported to be rare (Bolfíková and Hulva 2012, Černá Bolfíková et al. 2017; Curto et al. 2019).
The E. europaeus - E. roumanicus species pair is particularly interesting, as a second geographically distant contact zone occurs across Latvia, Estonia, and Russia (Fig. 1). This Russian-Baltic (RB) zone was reported to be younger, compared to the CE zone, and it is likely that its establishment correlates with Sub-Boreal (5000–2000 years ago) climatic changes (Hewitt 1999; Seddon et al. 2001; Sommer 2007). Seddon et al. (2001) presumed that the RB zone is occupied by lineages of the same refugial origin as those within the CE zone; however, detailed biogeographical analysis of the RB zone has not yet been performed. Furthermore, evidence of the distribution of the hedgehog species in central Russia is missing. A high rate of hybridization in the RB contact zone has been recently reported using classical genetic markers (Bogdanov et al. 2009; Zolotareva et al. 2021), but the mechanisms maintaining species isolation remain unclear. In contrast to the CE zone, the more abundant species in the RB is E. roumanicus, while signal of expansion was only detected in E. europaeus (Zolotareva et al. 2021).
Furthermore, the status of a potential contact zone between E. roumanicus and the Southern White-breasted hedgehog (E. concolor) from Asia Minor is unknown, while the Amur hedgehog (E. amurensis), which inhabits the Amur basin and Korea, has a disjunct range relative to other members of the genus (Fig. 1). The whole Erinaceus genus was reported to be monophyletic, based on classical genetic and morphological data (He et al. 2012; Bannikova et al. 2014); however, a well-defined hypothesis regarding the evolutionary history of the hedgehog species is still lacking. To understand the role of hybridization in speciation, it is important to infer the phylogenetic relationships between the target species.
The aim of the study was thus to compare two distant contact zones between the ranges of Erinaceus europaeus and E. roumanicus using SNP data generated via the RAD-Seq approach to gain a much better resolution of admixture patterns compared to markers used previously (microsatellites). Our main objectives were to assess the species evolutionary history and describe the patterns of hybridization and introgression within the contact zones, including the amount and direction, using state-of-the-art population genomics tools. We hypothesize that the rate of hybridization is lower in the CE zone compared to the RB contact zone because the former was established earlier, and the reproductive isolating mechanisms started to evolve before than in the RB zone.
Materials and methods
Samples
The sample list included 33 specimens of Erinaceus europaeus, 48 specimens of E. roumanicus, five E. concolor, two E. amurensis, and one specimen for Hemiechinus auritus and Atelerix albiventris, each. One of the sequenced individuals was a known hybrid between the species E. europaeus and E. roumanicus from the Central European contact zone, according to the microsatellite and mtDNA analyses performed by Černá Bolfíková et al. (2017). All tissue samples were obtained from roadkill animals, or from mortalities in rescue centres, and thus no animals were sacrificed for the purpose of this study. The samples from Russia were imported to the Czech Republic during 2019, following stipulated veterinary conditions for import, and under the reference number: SVS/2019/061603-G. The remainder of the samples were collected before 2013. The tissue samples were fixed in 96% ethanol and stored at −20 °C in a laboratory approved (No CZ 11712934) for the storage and use of animal material, according to § 48(1)(i) of Act No 166/1999s, pursuant to Article 17(1) of the Regulation of the European Parliament and the Council (EC) No 169/2009, and Article 27(1) of the Commission Regulation (EU) No 142/2011. The sample names, their collection coordinates, species classification according to D loop and the samples used in each dataset are listed in Table S1.
DNA extractions and Restriction site associated DNA sequencing
DNA was extracted using spin-column protocol (Qiagen DNeasy Blood and Tissue kit, Qiagen). A combination of microsatellites and mitochondrial control regions was used for species determination prior to sequencing, according to the protocols described by Bolfíková and Hulva (2012), for individuals from the CE zone. Only mitochondrial control region sequences were used for species determination of the samples from the RB contact zone. Species classification was also later re-evaluated. Restriction site associated DNA (RAD) libraries were prepared for 50 samples, using the restriction enzyme Sbfl1 (performed at the European Molecular Biology Laboratory in Heidelberg) according to modified protocol from Baird et al. (2008) (barcodes and primers used are listed in Table S2). For DNA tagmentation, adapter ligation, purification, and fragment size segregation, the NEBNext® Ultra™ DNA Library Prep Kit for Illumina® (Illumina Inc.) protocol was followed. Two paired-end libraries were sequenced at the European Molecular Biology Laboratory (Heidelberg) using the Illumina HiSeq2000 sequencer, in two lanes, according to the manufacturer’s protocol (Illumina Inc.). A single-end library was subsequently prepared for 40 samples from the RB contact zone, using the same restriction enzyme. The library was then sequenced using Illumina NextSeq at the European Molecular Biology Laboratory (Heidelberg). The rationale for using the two sequencing strategies was to make coverage more homogenous across samples.
Data processing
Sequence demultiplexing, based on individual specific barcodes (Table S1), was performed using Sabre (https://github.com/najoshi/sabre), and a read quality assessment was performed in FastQC 0.11.5 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Further per individual quality assessment (the number of obtained reads and coverage) were performed in R 3.6.3 (R Core Team 2013) (Fig. S1). Reads were then mapped to the published contigs of the Erinaceus europaeus annotated reference genome (https://pre.ensembl.org/Erinaceus_europaeus/Info/Index) using Bowtie2 2.3.4 (Langmead et al. 2009) in a very sensitive mode. This mode describes the highest effort of the software to find the alignment for each read in terms of number of attempts which Bowtie dedicates to the extension of an alignment. Only end-to-end alignment was allowed, with the suppression of unpaired alignments for paired reads. Resultant bam files were further processed using Samtools 1.3.1 (Li et al. 2009). Reads mapping to mitochondrial DNA were discarded. Because the reference genome is not assembled into chromosomes, it was not possible to discard or analyse separately the variation specific to sex chromosomes. Freebayes 1.1 (Garrison and Marth 2012) variant caller was used to call genotypes, considering only sites having both a mapping and base quality higher than 20. Genotypes were then filtered with VCFTools 0.1.15 (Danecek et al. 2011) and only sites with a quality of over 50 in Phred scale were kept, which refers to the confidence that there is polymorphism at the loci through estimating base and mapping quality (QUAL column in vcf). Minimal and maximal per site coverage per individual were set to between 8 and 200 for Admixture, PCA, TreeMix, MrBayes, D and f statistics, and between 6 and 200 for Bgc. The upper limit was set to avoid the inclusion of SNPs from repetitive regions. Further filtering allowed a maximum of 15–25% of missing data per SNP site. The threshold of minimum allele frequency (MAF) was set to 0.0125, to exclude singletons which may be a result of sequencing errors, as recommended by Linck and Battey (2019). Using a greater MAF threshold could lead to inaccurate estimates of admixture (Linck and Battey 2019) and to the removal of the variance between E. amurensis and E. concolor, which are represented by only two samples. To avoid linkage disequilibrium between loci, only one SNP per stacks was kept. This was performed with VCFTools 0.1.15 (Danecek et al. 2011), keeping only SNPs that were at least 261 nucleotides apart to allow for some variability in the length of stacks resulting from reverse reads (the read lengths obtained from Illumina sequencing were 100–150 bp). Individuals displaying more than 50% of missing SNPs were removed from the individual-based downstream analysis, with the single exception of A. albiventris (61% of missing data) which was included in the MrBayes analysis to reconstruct the phylogenetic relationship between the studied taxa. This reduction resulted in a total of 67 individuals in Dataset 1 that were used for Admixture, PCA, TreeMix and MrBayes. Dataset 2, used to calculate D and f statistics, contained individuals regardless of the individual percentage of missing data, because both D and f statistics are based on per site allele frequencies comparisons among species. However, the same E. roumanicus and E. europaeus individuals as used for Admixture evaluation were used for D and f if they were identified as non-admixed, to be sure no hybrid was included. In Dataset 2, data from at least one individual from each Erinaceus species, and from one individual in the case of an outgroup were required to be present at a site, with the maximum missing data per site set to 50% (further filtering unchanged). Lastly, Dataset 3 was used to assess the patterns of introgression in Bgc, and contained only E. roumanicus and E. europaeus individuals plus their hybrids, with no missing genotypes allowed (further filtering unchanged). For a summary of the Datasets used in this study and the mean per site coverage of each dataset, see Table S3, for the coverage per individual before and after filtering see Fig. S1. The conversion of the obtained filtered vcf files into various input file formats was performed using softwares Populations (Catchen et al. 2013), PGDSpider (Lischer and Excoffier 2012), PLINK (Purcell et al. 2007), and shell scripts (Data archiving).
Phylogeny
Phylogenetic analyses were performed with Dataset 1 using variable SNPs only. Nucleotide substitution model was tested in Jmodeltest (Posada 2008) and the best fitting model was evaluated using Bayesian information criterion (BIC) (Schwarz 1978). A phylogenetic tree of all samples was generated through MrBayes (Ronquist and Huelsenbeck 2003), with a total chain length of 1,100,000 generations, sampled every 200 generations. Four independent runs, each with four chains–one cold and three heated–were applied. The burn-in period was set to 10% of the sampled trees. Convergence was checked in Tracer (Rambaut and Drummond 2007) using effective sample size (ESS) values (>200) and average standard deviation of split frequencies (<0.001). The time of divergence was estimated using Beast v2.6.2 (Bouckaert et al. 2014) without admixed individuals (detected by Admixture, as discussed in the next paragraph) with following parameters: strict molecular clocks, GTR substitution model with estimated base frequencies, gamma model for site heterogeneity and four gamma categories, Yule model of branching, and calibration date based on the results of Bannikova et al. (2014) –5.7–7.4 Myr for the most recent common ancestor of Erinaceinae. Twelve runs consisting of 10,000,000 iterations, sampled every 1000 generations, each were combined in LogCombiner, and checked in Tracer, to reach ESS > 200. The resulting tree was assembled in TreeAnnotator, with a burn-in percentage of 20%.
Population structure
Dataset 1 was used to perform maximum likelihood estimations of the individuals’ ancestry in Admixture 1.3.0 (Alexander et al. 2009; Alexander and Lange 2011), showing the assignments of each individual to a cluster. Unsupervised clustering under default settings were used to run Admixture for K 2–10. Convergence of the analyses was assessed using the quasi-Newton acceleration method. The optimal number of clusters was identified, based on the lowest value of the cross-validation error (CV error). Individuals displaying a proportion of foreign ancestry (higher than 0.00002), as identified by the Q coefficient of Admixture (Table S4), are hereon further referred to as “recently admixed”. This threshold was used because in the admixed individuals the proportion of estimated foreign ancestry was two orders of magnitude higher, therefore values between 0.00001 and 0.00002 are likely a result of either stochasticity in estimation, historical introgression or incomplete lineage sorting. A principal component analysis (PCA) was used to further assess the population structure in a model-free manner. The PCA was performed in R package Adegenet 2.0.1 (Jombart and Ahmed 2011) using Dataset 1, for a) E. roumanicus, E. europaeus, and admixed individuals, and b) for E. roumanicus only, to explore the population structure of this species in detail.
To assess the pairwise genetic differentiation between Asian and Balkan lineages of E. roumanicus and Apennine and Iberian lineage of E. europaeus, the Weir and Cockerham’s (Weir and Cockerham 1984) mean FST values were estimated with the exclusion of all admixed individuals using Dataset 1 and VCFtools (Danecek et al. 2011).
Detection of introgression
To compare the level of introgression between E. roumanicus and E. europaeus in each contact zone, the D statistic (Green at al. 2010) and corresponding f statistics were computed. The implementation of the D statistic is embodied within the ABBA BABA test, which calculates a genome-wide representation of two shared derived patterns, namely ABBA, and BABA. Using three ingroup populations/species of known phylogeny ((P1,P2),P3) plus an outgroup, the test counted sites displaying shared derived alleles, between either P1 and P3 (pattern BABA) or between P2 and P3 (pattern ABBA). Under the expectations of incomplete lineage sorting, no recurrent mutations, and a randomly mating ancestral population (Durand et al. 2011), both patterns should be equally distributed within the genome. The significant excess of one of the patterns indicates introgression between the affected lineages. When multiple individuals are tested from each species, the estimation of allele frequencies at each tested SNP site is more effective than counting the patterns side by side between all individuals. It is equivalent to counting each pattern using all possible sets of four haploid samples at each site (Martin et al. 2013).
The f statistic represents the fraction of the genome affected by gene flow. The three estimators of f computed were: fhom, fd (Martin et al. 2015) and fdM (Malinsky et al. 2015). The fhom estimator is the modification of the original f estimator used by Green et al. (2010). It is calculated based on the assumption that gene flow occurs only in the direction from P3 to P2, and may lead to spurious values when this assumption is violated (Martin et al. 2015). It further assumes complete homogenization of allele frequencies between hybridizing taxa, which would only be true if the gene flow is extreme. Therefore, two more estimators were used to avoid possible bias originating from violations of these assumptions. The fd represents a more dynamic f estimator, where the donor population may vary at each site, and the donor is always the population with the higher frequency (Martin et al. 2015). The fdM is suitable for counting the proportions of genome shared between species in both cases, when D is either positive or negative. The fdM is positive when introgression occurs between P2 and P3, and negative when introgression occurs between P1 and P3 (Malinsky et al. 2015).
In this study, E. concolor is always represented by P1, E. roumanicus by P2, and E. europaeus by P3. Atelerix albiventris was used as an outgroup in all comparisons. Only the sites at which the outgroup was homozygous were included to ensure the ancestral state of the allele. The D and three f estimators were calculated across the genome, and were used to test for differences in the rate of introgression across each hybrid zone. Therefore, the estimations of D and f contained only non-admixed individuals, as identified in the Admixture analysis (Q value ≥0.00002) of both species from each contact zone, separately. To assess the significance of D, specifically when the excess of shared ancestry was evenly distributed in the genome, a jackknife procedure was performed, with a block size length of 106 bp. The jackknife procedure allows for computing the variance of D, by calculating the standard deviation for pseudovalues of the mean genome-wide D. By excluding genomic blocks of 106 bp length, each pseudovalue was computed and compared with the mean genome-wide D, when the block is included. The chosen block size of 106 corresponded to a safe genomic distance at which linkage disequilibrium decays and no autocorrelation occurs. The D and f statistics were computed using a combination of scripts provided by Simon Martin (https://github.com/simonhmartin/genomics_general) and custom script (Data archiving).
To explore the possibility of an asymmetry in the gene flow, a TreeMix (Pickrell and Pritchard 2012) was performed using Dataset 1. Individuals were grouped into populations according to the topology assessed by MrBayes. The only excluded sample was the F1 hybrid identified in Admixture (confirmed by analyses in NewHybrids (Anderson and Thompson 2002) with a burn-in period of 10,000 steps followed by 100,000 steps) which displayed intermediate ancestry between both species. Several runs with different sub-sets of individuals were performed. Within the initial analysis, all individuals regardless of their contact zone origin were evaluated. Afterwards, individuals belonging to each contact zone were analysed separately, to test if the direction of introgression differs between the contact zones. The number of individuals was limited in these comparisons −35 from the CE contact zone and 37 from the RB zone, including individuals of other species. The final analyses performed tested the contribution of lineages on hybridization; individuals were grouped by the lineage origin (according to Q coefficient). The maximum likelihood tree was inferred, with up to six migration events over five replicates. The bootstrap support was inferred from 100 replicates, which were summarised using Dendropy package 4.5.1 (Sukumaran and Holder 2010) and plotted in FigTree 1.4.4 (Rambaut 2018). Using the linear method implemented in OptM package 0.1.3 (Fitak 2019), the optimum number of migration edges was inferred using nonlinear least squares and the simple exponential method.
Detection of outlier loci in Russian-Baltic contact zone
To identify loci under selection likely involved in reproductive isolation, Bgc 1.03 (Gompert and Buerkle 2012) was used with Dataset 3 as input. The individuals of hybrid origin were compared with both parental species in the RB contact zone, because the number of hybrid individuals was only sufficient to perform the test in this zone. The Bgc software is based on the Bayesian genomic clines model (Gompert and Buerkle 2011), which quantifies two cline parameters for each locus, named α and β, describing the introgression based on ancestry. Cline parameter α describes the excess of ancestry at the locus from either species one (positive value) or species two (negative value), whereas β describes the locus specific transition rate, when compared to the genome-wide ancestry (Gompert and Buerkle 2012). Bgc was set to run with 150,000 MCMC steps, a burn-in of 10,000, and a thinning interval of 100, using the sequencing error model in two independent chains to evaluate convergence.
Results
RAD-Seq data description
RAD-Seq libraries resulted in ~2–22.5 million reads per sample and all samples having less than 7.5 million reads were discarded (Fig. S1). The percentage of reads which aligned exactly one time ranged between 55–60% on average for the single-end library and between 80–85% on average for two paired-end libraries. A total of 70 individuals from the 90 sequenced passed the quality filtering and were involved in further analyses. The filtering strategy based on mapping and sequencing quality, MAF, minimum and maximum coverage, missing data per site, and pruning by keeping one SNP per stacks, resulted in 9854–49,291 SNPs with a mean coverage of between 11.52 and 12.59 (depending on the included individuals and percentage of missing data per site) in Dataset 1. For Dataset 2, 38,467 SNPs (a mean coverage of 9.79) and 31,523 SNPs (mean coverage of 9.41), were recovered for CE and RB zone, respectively. Dataset 3 yielded 18,786 SNPs with a mean coverage of 12.05.
Phylogeny
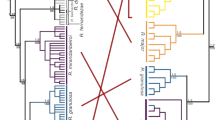
The phylogenetic reconstruction using MrBayes revealed 22 specimens within the Erinaceus europaeus clade, 39 specimens within E. roumanicus, two E. concolor and two E. amurensis, all separated from the outgroups Atelerix albiventris, and Hemiechinus auritus (Fig. 2). The most supported substitution model was GTR + I + G. The topology and branch support obtained from BEAST was congruent with the topology obtained from MrBayes when considering non-admixed individuals. The genus Erinaceus was supported as monophyletic with the most recent common ancestor (MRCA) dating to 1.0 Myrs ago (95% CI: 0.83–1.19). Within the genus Erinaceus, E. europaeus − E. amurensis (MRCA − 0.7 Myr, 95% CI: 0.57–0.83 Myr) and E. roumanicus − E. concolor (MRCA − 0.5 Myr, 95% CI: 0.44–0.63 Myr) formed sister clades. The genus Atelerix had a sister position to the whole Erinaceus group. The two refugial lineages of E. europaeus started diverging before 0.3 Myr (95%CI: 0.20–0.30 Myr), and the split between the two lineages of E. roumanicus was estimated at 0.1 Myr (95% CI: 0.07–0.11 Myr).

Each taxon (species or lineage) is coloured according to the sampling map (Fig. 1). Posterior probabilities from MrBayes indicating support of each node are reported before the forward slash symbol while the age of each node is reported after. CI represents 95% HPD intervals estimated using Beast 2.6.4. and the estimates are given in million of years.
Population structure
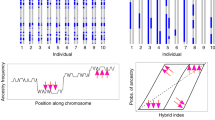
Using the lowest cross-validation (CV) error, the optimal number of clusters identified with Admixture reached K = 6. The values of the CV error increased sharply in K lower or greater than 6, and therefore, only K = 6 is reported (Fig. 3). This division into six clusters recognized all four Erinaceus species, where both E. roumanicus and E. europaeus comprised two differentiated lineages. The clusters observed in Erinaceus europaeus correspond to the previously known Iberian and Apennine lineages. Erinaceus roumanicus was differentiated into two previously unrecognized clusters of Balkan and Asian origin. Twelve individuals were identified as recently admixed, and in four of those admixed individuals (Table S1), the mitochondrial species determination did not match the majority of the estimated ancestry assessed by Admixture. All four samples had mtDNA from E. europaeus, but the majority of their nuclear genome was of E. roumanicus origin. One F1 hybrid presented the typical E. europaeus mtDNA. Moreover, one individual with the mtDNA from E. roumanicus was identified as pure E. europaeus. The majority of the identified hybrids (10/12) were from the RB contact zone, specifically from the Moscow region, and the majority of them (8/10) were identified as E. europaeus according to mtDNA. The proportion of shared ancestry varied from a minimum of 3.9% of E. europaeus ancestry to that of a F1. Two admixed specimens were from Central Europe −a priori known hybrid from Slovakia with ~25% of E. europaeus ancestry, and one from Austria, with 4.2% of E. europaeus ancestry. Both individuals carried mtDNA from E. roumanicus. The population structure was also explored with ANGSD (Korneliussen et al. 2014) as a tool alternative to Admixture and the same results were obtained (data not shown).

The individuals are shown by vertical bars. Asterisks labels samples where cytonuclear conflict was found. Hashtag labels the individual from the CE zone which was known to be hybrid prior to sequencing based on a combination of mtDNA and microsatellites. The hybrid individuals are marked with pie plots according to sampling map: red and blue pie plots represent hybrids from the RB contact zone, whereas two brown, blue and turquoise plots are from CE contact zone.
The results of the PCA supported the outcomes obtained from Admixture (Fig. 4). When exploring E. roumanicus and E. europaeus (Fig. 4a) the PC1 explained 65% of the variability in the data. The admixed individuals formed a transition between the two species on the PC1 axis. The two lineages of E. europaeus form clusters on PC2, accounting for 4.33% of variability. In the case of E. roumanicus (Fig. 4b), PC1 explained 18.3% of variability, differentiating individuals of Balkan and Asian origin, while PC2 explained 8.4% of variability, separating the Crete population from the Balkan lineage.

The PCA was performed for a) Erinaceus europaeus and E. roumanicus and their hybrids, and b) for E. roumanicus separately.
The interspecific pairwise FST values (Table S5) showed much higher differentiation between the Iberian and Apennine lineages of E. europaeus (0.2430) than between the Balkan and Asian lineages of E. roumanicus (0.0723).
Detection of introgression
An unequal distribution of the shared derived pattern of ABBA over BABA was detected in all comparisons, indicating introgression between E. roumanicus and E. europaeus (Table 1). In the CE contact zone, the level of historical introgression among recently non-admixed individuals was substantially lower (f between 0.005–0.009%) than in the RB contact zone (f between 0.02–0.03%). All obtained values of D and f were significant (p < 0.05); however, the Z score, which is calculated from the value of D statistic divided by its standard error, were only higher than 3 for the RB contact zone. By convention, only the values of the Z score higher than 3 indicate strong pattern of introgression, corresponding to values of p < 0.0013 (Green et al. 2010; Patterson et al. 2012; Zheng and Janke 2018).
The TreeMix analysis supported a single migration event as the optimal number of migration edges based on a simple exponential and nonlinear model in all comparisons. When analysing all individuals together, the TreeMix indicated gene flow from E. europaeus into E. roumanicus (Fig. S2), and the estimated weight of migration was low. When analysing each zone separately, gene flow from E. europaeus to E. roumanicus was inferred in three out of five replicate runs in the CE zone, and in five out five in the RB zone. In the case of lineages, gene flow was inferred from the Apennine lineage of E. europaeus to Asian lineage of E. roumanicus in four out of five runs (data not shown).
Testing for locus specific selection in the Russian-Baltic contact zone
Bgc did not detect any outlier loci that would differ from the average pattern of genome-wide introgression, in either of the two cline parameters α and β. When comparing the parental species from the RB contact zone with the admixed individuals from the RB contact zone (Fig. S3), all analysed SNPs included zero in their credible intervals (CI) in the estimates of both cline parameters.
Discussion
We used population genetics and phylogenetic analyses of genome-wide SNP data to study the evolutionary history of the Erinaceus genus and patterns of admixture across two hybrid zones of E. roumanicus and E. europaeus. By leveraging the power of genomic data, we provide a robust and improved hypothesis for the divergence of major hedgehog lineages, and we found evidence for different levels of hybridization across the CE and the RB contact zones. Below, we discuss the implications of our findings for an improved understanding of hedgehog phylogeography and the evolution of reproductive barriers between hybridizing species.
Phylogenetic and phylogeographic patterns
The results support the sister position of genus Atelerix and Erinaceus proposed by He et al. (2012) but later questioned by Bannikova et al. (2014). Previous studies provided low support for branching within Erinaceus (He et al. 2012) or for the grouping of Erinaceus with Atelerix (Bannikova et al. 2014). For the first time, genome-wide data has been used to reconstruct the phylogeny within the Erinaceidae family, providing better resolution and higher support compared to morphological traits and a limited number of genes, as used in previous studies. Classical phylogeographic studies based on only a few markers suggested speciation even before the Pleistocene period (Hewitt 1999; Seddon et al. 2001). However, the present results, based on the genome-wide SNP data, support divergence of all four species during the Pleistocene, and therefore climatic changes were probably the main factor driving the divergence between the species.
Both Iberian and Apennine lineages of E. europaeus, and the lineage of E. roumanicus from the Balkan proposed by Seddon et al. (2001) and Hewitt (1999), were detected in the present study. Additionally, a new Asian lineage of E. roumanicus was detected, even if the differentiation from the Balkan lineage was lower than the divergence between the two E. europaeus lineages. This substructure has not been previously revealed using classical genetic markers, which illustrates the power of genomic markers in the resolution of phylogeographic patterns. The Asian lineage of E. roumanicus is distributed within the north-eastern part of the range; this observed signal may have several explanations, including an isolated glacial refugium outside the Mediterranean region for this lineage. Seddon et al. (2001) suggested possible divergent routes of expansion from the Balkan area to the north and to the east; however, suitable habitats may have also been present in the Ponto-Caspian region during the Last Glacial Maximum (LGM) (Varga 2010; Allen et al. 2010), as the interglacial expansion of hedgehogs is typically correlated with the spread of deciduous woodlands. The Ponto-Caspian area is a system of basins with specific microclimatic conditions (Cordova et al. 2009), where patches of forest may have been preserved during the LGM.
Zolotareva et al. (2021) detected a signal of population growth in E. europaeus within the RB zone, and stated that this species colonized East Europe after E. roumanicus, whose population size is stable in the RB zone. This conclusion is compatible with our hypothesis regarding the existence of a previously unrecognized glacial refugia, situated outside of the Balkan peninsula. Phylogenetic analyses suggested that the split between the Asian and Balkan lineages of E. roumanicus started during the last glacial period, while in the case of E. europaeus, the Apennine and Iberian lineages differentiated much earlier. This so-called extra-Mediterranean refugia has been described in a variety of animals, such as brown bears (García-Vázquez et al. 2019), bank voles (Kotlík et al. 2006), moor frogs (Babik et al. 2004), and other species (Schmit and Varga 2012).
Hybridization of hedgehogs is far more extensive in the RB zone, and therefore a higher rate of introgression could have contributed to increase the divergence between of the Balkan and Asian lineages. Continuous sampling, and samples from the far eastern part of the range, are needed to improve our knowledge concerning the evolutionary history of E. roumanicus.
Comparison of the hybrid zones
In hybrid zone research, different transects (i.e. the allele frequency clines between different populations) are typically compared to address the variation in species boundaries maintenance. In the case of mosaic hybrid zones, different parts of the zone could have different attributes, as for example, the two sides of the Carpathian arch (Zieliński et al. 2019). The present study reports the contrasting genomic hallmarks of reproductive isolation in a unique model of two geographically distant secondary contact zones, between a pair of hybridizing mammal species.
The rate of hybridization is substantially higher in the RB zone than in the CE zone. This result agrees with previous detection of early stage hybrids based on microsatellite loci (Bolfíková and Hulva 2012; Černá Bolfíková et al. 2017; Curto et al. 2019; Zolotareva et al. 2021). Only three hybrids (two from Austria, one from Slovakia) out of the 280+ studied specimens in the CE zone were found (Bolfíková and Hulva 2012; Černá Bolfíková et al. 2017; Curto et al. 2019), while Zolotareva et al. (2021) confirmed 21 hybrids from the 140 specimens studied in the RB zone. Even though low numbers of microsatellites loci do not have the power to detect older generations of hybrids, the previous studies (based on 11 microsatellite loci) correctly assigned the a priori known admixed individual from Slovakia as a hybrid resulting from second backcrossing. The second hybrid from the CE zone (from Austria) was previously unrecognized as such by previous studies, probably resulting from an older hybridization event (4.2%, Table S4). All known hybrids from the CE zone originated from the easternmost edge of the E. europaeus distribution. The observed pattern suggests that contemporary hybridization in the CE zone is uncommon, probably occurring in contribution to the Allee effect (Stephens and Sutherland 1999), i.e. occurs when population density of conspecifics drops rapidly at the edge of the contact zone
Different rates of hybridization in each zone are compatible with the hypothesis that the CE contact zone was established earlier than the RB zone. To reach the current distribution in Russia, E. europaeus must have spread from Central Europe northward, through the Baltic states, and was later replaced by E. roumanicus in this area (Seddon et al. 2001). Therefore, based on phylogeographic and paleontological data (Seddon et al. 2001; Sommer 2007), the formation of the RB contact zone must have followed the formation of the CE contact. We propose that a prolonged period of contact in the CE zone led to the development of stronger reproductive barriers than in the RB contact zone. Within the coupling or trapping hypothesis, the centre of hybrid zones originating with contribution of intrinsic genetic incompatibilities is expected to coincide with environmental ecotones and density troughs (Barton and Hewitt 1985; Bierne et al. 2011; Johannesson et al. 2020). The CE zone could hypothetically coincide with the transition from oceanic to continental climate. Regular extensions of maritime weather from the North Atlantic to the Baltic could also explain the expansion of the E. europaeus range to the north-east, and formation of the RB zone. According to Zolotareva et al. (2021) the species ranges do not substantially overlap in the RB contact zone and it can be assumed that the RB contact zone is narrower than the CE zone. However, the exact location, width, and length of the RB zone are currently poorly described and further detailed studies with dense sampling are needed.
Asymmetrical gene flow
The Treemix and Admixture results confirmed that E. roumanicus received more introgression than E. europaeus in both contact zones. Asymmetrical gene flow often occurs among species pairs which are under different selection pressures, like polar bears and brown bears (Liu et al. 2014). In the case of hedgehogs, published studies have not found any adaptive differences in the ecology of the species, which could be responsible for the observed pattern (Kratochvíl 1975; Reeve 1994). However, in the future it is necessary to explore the ecology of eastern species, as the majority of information currently exists for the E. europaeus species only.
Experiments in captivity, conducted by Poduschka and Poduschka (1983) using individuals captured in Central Europe, provide further evidence for this asymmetry. In the aforementioned research, F1 and F2 generations of hybrids between E. roumanicus and E. europaeus were produced. Whilst the backcrossing of F1 hybrids with E. roumanicus produced offspring, backcrossing with E. europaeus was unsuccessful. In this case of experimental crossing, developed postzygotic barriers seems to be the most plausible reason for failure of backcrossing between F1 and E. europaeus. The gene flow asymmetry reported in the present study may be a result of postzygotic barriers, making the backcross mating of F1 hybrids with E. europaeus less successful.
Both zones differ regarding their dominant species, and in both zones, different species show signals of population growth or expansion. This suggests that density and the phenomena connected to range expansion are not primary factors explaining the different rates of hybridization or asymmetry in gene flow. Zolotareva et al. (2021) explained the contrasting rates of recent hybridization between the zones through unsuccessful backcrossing to E. europaeus together with unevenness in the population densities of each species within the zones. Zolotareva et al. (2021) stated that hybridization in the RB zone might be frequency-dependent, when males of the high-density population (E. roumanicus) hybridise preferentially with females from the low-density population (E. europaeus). If this would be true, it would mean that in the CE zone, females of E. roumanicus would have preferentially mated with males of E. europaeus, which are a more abundant species, and at the same time backcrossing to E. europaeus would be less successful. The only recent hybridization events found in the CE zone occurred at the very edge of distribution of E. europaeus, where the situation is more similar to that of the RB contact zone. In those specific localities where the hybrids were found, E. roumanicus has a higher density; however, in contrast with what is expected based on the theory of different densities, both hybrids contained mtDNA of E. roumanicus. Thus, even though differences in abundance together with less successful backcrossing to one of the species has the power to generate slightly different rates of hybridization across the zones, it cannot exclusively explain the direction of the hybridization towards E. roumanicus and the almost complete absence of recent hybridization in the CE zone.
Reproductive isolation
The evolution of reproductive barriers can be driven by selection against hybrids (Dobzhansky 1937; Coyne and Orr 2004). Genomic regions harbouring loci influenced by divergent selection remain distinct displaying high differentiation despite gene flow (Mallet 2005). In the present study, no loci that could be potentially influenced by selection causing reproductive isolation in the RB contact zone were identified.
One of the possible explanations for this is that introgression is prevented by selection against the early generation hybrids. When selection is strong and acting on a single gene with simple genomic architecture, as, for example, genes linked to colour pattern differences in Heliconius butterflies (Nadeau et al. 2014), the detection of divergent genomic regions is straightforward. However, when selection acts on multiple genes of small effect, or includes epistasis, the genomic scans will likely fail to locate differences in allele frequencies between species or populations (Coyne and Orr 2004; Rockman 2012).
The dataset included first to older generation hybrids crossed to various degrees with both parental forms, which eventually means that every hybrid individual possessed a different combination of alleles. This could complicate the detection of varying introgression patterns within the limited number of hybrids detected in this study. Also, RAD-seq data represents only a minor fraction of the genome, and therefore it is possible that the data were not dense enough to detect loci involved in reproductive isolation (Szarmach et al. 2021), although it has been proved capable of detecting genes under selection for reproductive isolation (Nadeau et al. 2014). Population data with a higher number of individuals from both contact zones, and more loci with a higher coverage, are necessary to search for regions involved in reproductive isolation and for the presence of postzygotic incompatibilities.
It can also be hypothesized that selection acts on prezygotic reproductive isolating mechanisms in hedgehogs, which evolve during the ontogenesis of individuals, and might be related to heterochrony, behavioural, olfactory, or bioacoustic signalling. Erinaceus europaeus is well-known for its complex courtship behaviour, which includes courtship dances and specific bioacoustics display (Reeve and Morris 1986), while the role of chemical signalling cannot be excluded in a macrosmatic mammal. The ritual may take place for more than an hour in these hedgehogs, with the majority of encounters ending unsuccessfully (Jackson 2006). To date, no study of the reproductive behaviour of E. roumanicus has been published, and therefore differences between the species are unknown.
Spatio-temporal components of mating should also be considered. Differences in the timing of hibernation, mating, and litter production between species have been observed in the CE contact zone (Kratochvíl 1975). The RB contact zone is situated farther north, and thus a lack of flexibility in the timing of reproduction is possible, thereby further accelerating hybridization.
Whether the differences in the rate of gene flow between both contact zones can be attributed to prezygotic barriers or postzygotic incompatibilities, the mechanism of maintaining species cohesion may be independent in each zone and may involve different genomic regions and context-dependent selective pressures, as has been shown when studying multiple contact zones of crows (sub)species (Vijay et al. 2016).
Conclusion
From a chorological point of view, the two zones could be considered as the result of the discontinuity of a single contact zone between two terrestrial ranges by water body (in the present case, the Baltic sea). The contrasting rates of gene flow in each zone negatively correlate with the age of the contact inferred from the biogeographic scenario previously proposed for these taxa. Whilst the current rate of hybridization is low within the presumably older Central European contact zone, both hedgehog species hybridize extensively in the presumably younger Russian-Baltic zone, formed with contribution of newly recognized Asian lineage of E. roumanicus. Thus, from an evolutionary point of view, different reproductive isolating mechanisms operate in two portions of a putatively continuous hybrid zone. From the range geometry point of view, it is possible to refer to two distant hybrid zones, or a single discontinuous hybrid zone. However, further data are necessary to reconstruct the evolutionary dynamics of the spatial topography of contact lines between the two ranges in particular interglacials. Asymmetrical gene flow from E. europaeus into E. roumanicus was reported in both zones. This is likely caused by pre- and/or post-mating reproductive barriers, which prevent hybridization and further introgression in the direction to E. europaeus. Future studies should address the mechanisms of maintaining the zones, using detailed mapping of genomic clines in selected transects, as well as to investigate the potential role of environmental factors in shaping both contact zones.
Data availability
The data that supports findings of this study are publically available in Dryad under reference: 10.5061/dryad.2547d7wnt. Scripts used to process genomic data, produced plots and graphs are publicly available on GitHub: https://github.com/eliasok/hedgehog_RAD and https://github.com/IgnasiLucas/hedgehog.
References
Ai H, Fang X, Yang B, Huang Z, Chen H, Mao L et al. (2015) Adaptation and possible ancient interspecies introgression in pigs identified by whole-genome sequencing. Nat Genet 47:217–225
Alexander DH, Lange K (2011) Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinforma 12:246
Alexander DH, Novembre J, Lange K (2009) Fast model-based estimation of ancestry in unrelated individuals. Genome Res 19:1655–1664
Allen JRM, Hickler T, Singarayer JS, Sykes MT, Valdes PJ, Huntley B (2010) Last glacial vegetation of Northern Eurasia. Quat Sci Rev 29:2604–2618
Amori G (2016) Erinaceus europaeus. The IUCN Red List of Threatened Species: e.T29650A2791303
Amori G, Hutterer R, Kryštufek B, Yigit N, Mitsain G & Palomo LJ (2016a) Erinaceus concolor (errata version published in 2017) The IUCN Red List of Threatened Species: e.T40605A115174497
Amori G, Hutterer R, Kryštufek B, Yigit, N, Mitsain G & Palomo LJ (2016b) Erinaceus roumanicus (errata version published in 2017) The IUCN Red List of Threatened Species: e.T136344A115206348.
Anderson EC, Thompson EA (2002) A model-based method for identifying species hybrids using multilocus genetic data. Genetics 160:1217–1229
Andrade P, Pinho C, i de Lanuza GP, Afonso S, Brejcha J, Rubin CJ et al. (2019) Regulatory changes in pterin and carotenoid genes underlie balanced color polymorphisms in the wall lizard. Proc Natl Acad Sci USA 116:5633–5642
Babik W, Branicki W, Sandera M, Litvinchuk S, Borkin LJ, Irwin JT et al. (2004) Mitochondrial phylogeography of the moor frog, Rana arvalis. Mol Ecol 13:1469–1480
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA et al. (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One 3:3376
Bannikova AA, Lebedev VS, Abramov AV, Rozhnov VV (2014) Contrasting evolutionary history of hedgehogs and gymnures (Mammalia: Erinaceomorpha) as inferred from a multigene study. Biol J Linn Soc 112:499–519
Bannikova AA, Fedorova LV, Fedorov AN, Troitskiĭ AV, Grechko VV, Dolgov VA et al. (1995) Comparison of repeat DNA sequences in the Erinaceidae mammal family by restriction analysis. Genetika 31:1498–1506
Barton NH, Hewitt GM (1985) Analysis of Hybrid Zones. Annu Rev Ecol Syst 16:113–148
Bierne N, Welch J, Loire E, Bonhomme F, David P (2011) The coupling hypothesis: why genome scans may fail to map local adaptation genes. Mol Ecol 20:2044–2072
Bogdanov AS, Bannikova AA, Pirusskii YM, Formozov NA(2009) The first genetic evidence of hybridization between West European and Northern white-breasted hedgehogs (Erinaceus europaeus and E. roumanicus) in Moscow region. Biol Bull 36:647–651
Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu CH, Xie D et al. (2014) BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLoS Comput Biol 10:e1003537
Cassola F (2016) Erinaceus amurensis (errata version published in 2017) The IUCN Red List of Threatened Species: e.T40604A115174360
Catchen J, Bassham S, Wilson T, Currey M, O’Brien C, Yeates Q et al. (2013) The population structure and recent colonization history of Oregon threespine stickleback determined using restriction-site associated DNA-sequencing. Mol Ecol 22:2864–2883
Černá Bolfíková B, Eliášová K, Loudová M, Kryštufek B, Lymberakis P, Sándor AD et al. (2017) Glacial allopatry vs. postglacial parapatry and peripatry: The case of hedgehogs. PeerJ 5:e3163
Cooper SJB, Ibrahim KM, Hewitt GM (1995) Postglacial expansion and genome subdivision in the European grasshopper Chorthippus parallelus. Mol Ecol 4:49–60
Corbet GB (1988) The family Erinaceidae: a synthesis of its taxonomy, phylogeny, ecology and zoogeography. Mamm Rev 18:117–172
Cordova CE, Harrison SP, Mudie PJ, Riehl S, Leroy SAG, Ortiz N (2009) Pollen, plant macrofossil and charcoal records for palaeovegetation reconstruction in the Mediterranean-Black Sea Corridor since the Last Glacial Maximum. Quat Int 197:12–26
Coyne J, Orr H (2004) Speciation. Sinauer Associates, Sunderland
Curto M, Winter S, Seiter A, Schmid L, Scheicher K, Barthel LMF et al. (2019) Application of a SSR-GBS marker system on investigation of European hedgehog species and their hybrid zone dynamics. Ecol Evol 9:2814–2832
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA et al. (2011) The variant call format and VCFtools. Bioinformatics 27:2156–2158
Dobzhansky T (1937) Genetics and the origin of species. Columbia University Press, New York
Durand EY, Patterson N, Reich D, Slatkin M (2011) Testing for ancient admixture between closely related populations. Mol Biol Evol 28:2239–2252
Filippucci MG, Lapini L (1988) First data on the genetic differentiation between Erinaceus europaeus Linnaeus, 1758 and Erinaceus concolor Martin, 1838 in North-eastern Italy (Mammalia, Insectivora, Erinaceidae). Gortania 9:227–236
Fitak RR (2019) optM: an R package to optimize the number of migration edges using threshold models. v0.1.3. J Hered. https://cran.r-project.org/web/packages/OptM/index.html
García-Vázquez A, Pinto Llona AC, Grandal-d’Anglade A (2019) Post-glacial colonization of Western Europe brown bears from a cryptic Atlantic refugium out of the Iberian Peninsula. Hist Biol 31:618–630
Garrison E, Marth G (2012) Haplotype-based variant detection from short-read sequencing. arXiv:1207.3907. http://arxiv.org/abs/1207.3907
Gompert Z, Buerkle CA (2011) Bayesian estimation of genomic clines. Mol Ecol 20:2111–2127
Gompert Z, Buerkle CA (2012) Bgc: Software for Bayesian estimation of genomic clines. Mol Ecol Resour 12:1168–1176
Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M et al. (2010) A draft sequence of the neandertal genome. Science 328:710–722
Harrison RG, Larson EL (2014) Hybridization, introgression, and the nature of species boundaries. J Hered 105:795–809
He K, Chen J-H, Gould GC, Yamaguchi N, Ai H-S, Wang Y-X et al. (2012) An estimation of erinaceidae phylogeny: a combined analysis approach. PLoS One 7:e39304
Hewitt GM (1988) Hybrid zones-natural laboratories for evolutionary studies. Trends Ecol Evol 3:158–167
Hewitt GM (1999) Post-glacial re-colonization of European biota. Biol J Linn Soc 68:87–112
Jackson DB (2006) The breeding biology of introduced hedgehogs (Erinaceus europaeus) on a Scottish Island: Lessons for population control and bird conservation. J Zool 268:303–314
Johannesson K, Le Moan A, Perini S, André C (2020) A Darwinian Laboratory of Multiple Contact Zones. Trends Ecol Evol 35:1021–1036
Jombart T, Ahmed I (2011) Adegenet 1.3-1: New tools for the analysis of genome-wide SNP data. Bioinformatics 27:3070–3071
Korneliussen TS, Albrechtsen A, Nielsen R (2014) ANGSD: analysis of next generation sequencing data. BMC Bioinforma 15:356. https://doi.org/10.1186/s12859-014-0356-4
Kotlík P, Deffontaine V, Mascheretti S, Zima J, Michaux JR, Searle JB (2006) A northern glacial refugium for bank voles (Clethrionomys glareolus). Proc Natl Acad Sci USA 103:14860–14864
Kratochvil J (1966) Zur Frage der Verbreitung des Igels (Erinaceus) in der ČSSR. Zool List 15:291–304
Kratochvíl J (1975) Zur Kenntnis der Igel der Gattung Erinaceus in der ČSSR (Insectivora, Mamm.). Zool List 24:297–312
Kumar V, Lammers F, Bidon T, Pfenninger M, Kolter L, Nilsson MA et al. (2017) The evolutionary history of bears is characterized by gene flow across species. Sci Rep. 7:46487
Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25
Li G, Davis BW, Eizirik E, Murphy WJ (2016) Phylogenomic evidence for ancient hybridization in the genomes of living cats (Felidae). Genome Res 26:1–11
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079
Linck E, Battey CJ (2019) Minor allele frequency thresholds strongly affect population structure inference with genomic data sets. Mol Ecol Resour 19:639–647
Lischer HEL, Excoffier L (2012) PGDSpider: an automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 28:298–299
Liu S, Lorenzen ED, Fumagalli M, Li B, Harris K, Xiong Z et al. (2014) Population genomics reveal recent speciation and rapid evolutionary adaptation in polar bears. Cell 157:785–794
Malinsky M, Challis RJ, Tyers AM, Schiffels S, Terai Y, Ngatunga BP et al. (2015) Genomic islands of speciation separate cichlid ecomorphs in an East African crater lake. Science 350:1493–1498
Mallet J (2005) Hybridization as an invasion of the genome. Trends Ecol Evol 20:229–237
Martin SH, Davey JW, Jiggins C (2015) Evaluating the Use of ABBA-BABA Statistics to Locate Introgressed Loci. Mol Biol Evol 32:244–257
Martin SH, Dasmahapatra KK, Nadeau NJ, Salazar C, Walters JR, Simpson F et al. (2013) Genome-wide evidence for speciation with gene flow in Heliconius butterflies. Genome Res 23:1817–1828
Nadeau NJ, Ruiz M, Salazar P, Counterman B, Medina JA, Ortiz-Zuazaga H et al. (2014) Population genomics of parallel hybrid zones in the mimetic butterflies, H. melpomene and H. erato. Genome Res 24:1316–1333
Patterson N, Moorjani P, Luo Y, Mallick S, Rohland N, Zhan Y et al. (2012) Ancient admixture in human history. Genetics 192:1065–1093
Payseur BA, Rieseberg LH (2016) A genomic perspective on hybridization and speciation. Mol Ecol 25:2337–2360
Pickrell JK, Pritchard JK (2012) Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet 8:e1002967
Poduschka W, Poduschka C (1983) Kreuzungsversuche an mitteleuropäischen Igeln. Säugetierkundliche Mitteilungen 31:1–12
Posada D (2008) jModelTest: phylogenetic model averaging. Mol Biol Evol 25:1253–1256
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D et al. (2007) PLINK: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575
R Core Team (2013) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/
Rambaut A (2018) FigTree v1.4.4. http://tree.bio.ed.ac.uk/software-/figtree/
Rambaut A, Drummond A (2007) Tracer v1.4. http://tree.bio.ed.ac.uk/software/tracer/
Reeve NJ, Morris PA (1986) Mating strategy in the hedgehog (Erinaceus europaeus). J Zool 210:613–614
Reeve N (1994) Hedgehogs. T. & A.D. Poyser: London
Reich D, Green RE, Kircher M, Krause J, Patterson N, Durand EY et al. (2010) Genetic history of an archaic hominin group from Denisova cave in Siberia. Nature 468:1053–1060
Remington CL (1968) Suture-Zones of Hybrid Interaction Between Recently Joined Biotas. In: Dobzhansky T, Hecht MK, Steere CW (eds) Evolutionary Biology. Plenum Press, New York, p 321–428
Rockman MV (2012) The QTN program and the alleles that matter for evolution: All that’s gold does not glitter. Evolution (NY) 66:1–17
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
roumanicus in Central Europe. Heredity 108:248–255
Santucci F, Emerson BC, Hewitt GM (1998) Mitochondrial DNA phylogeography of European hedgehogs. Mol Ecol 7:1163–1172
Schmitt T, Varga Z (2012) Extra-Mediterranean refugia: The rule and not the exception? Front Zool 9:22
Schwarz G (1978) Estimating the dimension of a model. Ann Stat 6:461–464
Seddon JM, Santucci F, Reeve NJ, Hewitt GM (2001) DNA footprints of European hedgehogs, Erinaceus europaeus and E. concolor: Pleistocene refugia, postglacial expansion and colonization routes. Mol Ecol 10:2187–2198
Smadja CM, Butlin RK (2011) A framework for comparing processes of speciation in the presence of gene flow. Mol Ecol 20:5123–5140
Sommer RS (2007) When east met west: the sub-fossil footprints of the west European hedgehog and the northern white-breasted hedgehog during the Late Quaternary in Europe. J Zool 273:82–89
Stephens PA, Sutherland WJ (1999) Consequences of the Allee effect for behaviour, ecology and conservation. Trends Ecol Evol 14:401–405
Sukumaran J, Holder MT (2010) DendroPy: A Python library for phylogenetic computing. Bioinformatics 26:1569–1571
Szarmach SJ, Brelsford A, Witt CC, Toews DPL (2021) Comparing divergence landscapes from reduced-representation and whole genome resequencing in the yellow-rumped warbler (Setophaga coronata) species complex. Mol Ecol 30:5994–6005
Taberlet P, Bouvet J (1994) Mitochondrial DNA polymorphism, phylogeography, and conservation genetics of the brown bear Ursus arctos in Europe. Proc R Soc Lond Ser B Biol Sci 255:195–200
Taylor SA, Larson EL (2019) Insights from genomes into the evolutionary importance and prevalence of hybridization in nature. Nat Ecol Evol 3:170–177
Varga Z (2010) Extra-mediterranean refugia, post-glacial vegetation history and area dynamics in Eastern Central Europe. In: Habel J, Assmann T (eds) Relict Species: Phylogeography and Conservation Biology. Springer-Verlag, Berlin, Heidelberg, p 57–87
Vijay N, Bossu CM, Poelstra JW, Weissensteiner MH, Suh A, Kryukov AP et al. (2016) Evolution of heterogeneous genome differentiation across multiple contact zones in a crow species complex. Nat Commun 7:13195–13205
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution (NY) 38:1358
Wu DD, Ding XD, Wang S, Wójcik JM, Zhang Y, Tokarska M et al. (2018) Pervasive introgression facilitated domestication and adaptation in the Bos species complex. Nat Ecol Evol 2:1139–1145
Zheng Y, Janke A (2018) Gene flow analysis method, the D-statistic, is robust in a wide parameter space. BMC Bioinforma 19:10
Zieliński P, Dudek K, Arntzen JW, Palomar G, Niedzicka M, Fijarczyk A et al. (2019) Differential introgression across newt hybrid zones: evidence from replicated transects. Mol Ecol 28:4811–4824
Zolotareva KI, Belokon MM, Belokon YS, Rutovskaya MV, Hlyap LA, Starykov VP et al. (2021) Genetic diversity and structure of the hedgehogs Erinaceus europaeus and Erinaceus roumanicus: evidence for ongoing hybridization in Eastern Europe. Biol J Linn Soc 132:174–195
Acknowledgements
We thank Lukáš Choleva and Bianka Baying for assisting us with the preparation of the Illumina libraries, and Jindřich Brejcha for creating the sampling map. For language proofreading, we thank Science Unleashed. The project was funded by the Grant Agency of Charles University in Prague, project no. 538218 (KE, ML, BČB), Czech University of Life Sciences CIGA 20185006 (BČB) and IGA 20223108 (BČB), Spanish Ministry of Economy and Competitivity RYC-2012-11872 (JILL) and Russian Foundation for Basic Research N20-04-00081a (AB, KZ).
Author information
Authors and Affiliations
Contributions
BČB and PH conceived the study; BČB, PH, KE and JILL designed the study; ML, PH and KE performed the laboratory work; KE, JILL, JHG and BČB analysed the data; BČB, KE, PH, JILL, ML, VB, AB, KZ contributed to samples/reagents/materials; KE, BČB and PH drafted the manuscript, and all authors contributed to the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Associate editor: Rui Faria.
Supplementary information
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Eliášová, K., Lucas Lledó, J.I., Grau, J.H. et al. Contrasting levels of hybridization across the two contact zones between two hedgehog species revealed by genome-wide SNP data. Heredity 129, 305–315 (2022). https://doi.org/10.1038/s41437-022-00567-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-022-00567-5
