
Abstract
Lung malignant tumors are a type of cancer with high incidence and mortality rates worldwide. Non-small cell lung cancer (NSCLC) accounts for over 80% of all lung malignant tumors, and most patients are diagnosed at advanced stages, leading to poor prognosis. Over the past decades, various oncogenic driver alterations associated with lung cancer have been identified, each of which can potentially serve as a therapeutic target. Rat sarcoma (RAS) genes are the most commonly mutated oncogenes in human cancers, with Kirsten rat sarcoma (KRAS) being the most common subtype. The role of KRAS oncogene in NSCLC is still not fully understood, and its impact on prognosis remains controversial. Despite the significant advancements in targeted therapy and immune checkpoint inhibitors (ICI) that have transformed the treatment landscape of advanced NSCLC in recent years, targeting KRAS (both directly and indirectly) remains challenging and is still under intensive research. In recent years, significant progress has been made in the development of targeted drugs targeting the NSCLC KRASG12C mutant subtype. However, research progress on target drugs for the more common KRASG12D subtype has been slow, and currently, no specific drugs have been approved for clinical use, and many questions remain to be answered, such as the mechanisms of resistance in this subtype of NSCLC, how to better utilize combination strategies with multiple treatment modalities, and whether KRASG12D inhibitors offer substantial efficacy in the treatment of advanced NSCLC patients.
Similar content being viewed by others
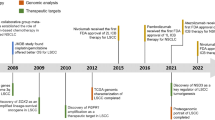


Introduction
Lung cancer is the most common and deadly cancer worldwide [1]. Non-small cell lung cancer (NSCLC) accounts for ~80% of all lung cancers, with lung adenocarcinoma (LUAD) being the most common histological subtype, often accompanied by oncogenic driver mutations [2]. With the development of molecular targeted therapy, some LUAD patients can benefit from specific drugs targeting multiple gene loci, such as EGFR, ALK, ROS1, etc. The RAS proteins (H-RAS, K-RAS, and N-RAS) are GTPases that regulate various cellular activities, including proliferation, survival, growth, migration, differentiation, and cytoskeletal dynamics, through signal transduction pathways. When mutated, RAS proteins remain activated, leading to excessive activation of downstream signaling pathways and ultimately triggering tumor formation. Among them, KRAS gene mutations are found in ~30% of lung adenocarcinomas (LUAD). Unlike lung cancers with mutated EGFR and ALK expressions, KRAS has long been considered a challenging therapeutic target, even regarded as “undruggable”. It is generally believed that the difficulty in developing direct KRAS inhibitors is due in part to the pico-molar affinity of GTP and GDP for KRAS (which have much higher cellular concentrations) and to the lack of suitable deep pockets for conformational regulation [3]. Therefore, the development of targeted drugs for KRAS has been difficult for many years. This dilemma was broken in 2013 when American chemical biologist Kevan Shokat discovered that when KRASG12C protein is in the inactive GDP-bound state, the switch II domain (SIIP) of its mutated cysteine residue exhibits a pocket structure that can covalently bind to small molecule drugs [4]. These targeted drugs are now collectively known as KRASG12C inhibitors, including Sotorasib (AMG 510) from Amgen and Adagrasib (MRTX 849) from Mirati, used to treat non-small cell lung cancer patients with KRASG12C mutations [5]. Although KRASG12C inhibitors need further research on drug resistance and toxic side effects, their emergence has been a significant breakthrough in the field of KRAS-targeted therapy [6]. KRASG12D, as the most common mutation (33%) in KRAS-mutant tumors, has its unique molecular mechanism and clinical features, and targeted therapies against it are still under continuous research. Despite significant clinical advancements in targeted treatments, meeting the clinical demands for KRASG12D mutation remains challenging to date.
Characteristics of KRAS mutations in NSCLC
Molecular background
Kirsten-rat-sarcoma viral oncogene homolog (KRAS) is the most common mutation driving factor, accounting for 15–30% of all human malignant tumors, and KRAS mutations are particularly common in pancreatic ductal adenocarcinoma, colorectal cancer, and NSCLC [7]. Its gene product was initially discovered as p21 GTPase. The conformation of KRAS protein cycles between two different conformational states. When KRAS protein binds to GTP, it is in an active state, and when it binds to GDP, it is in an inactive state. KRAS, in its active state, binds to GTP and has an intrinsic enzymatic activity to cleave the terminal phosphate of nucleotides, converting it to GDP. Its conversion rate is usually slow but can be significantly increased with the assistance of Guanosine triphosphatase-activating protein (GAP). Meanwhile, KRAS can bind to guanine nucleotide exchange factors (GEFs) (such as SOS), causing the bound nucleotide (GDP) to be released and binding of KRAS to GTP [8]. In normal mammalian cells, endogenous KRAS protein mainly exists in an inactive state. However, the oncogenic mutation of KRAS protein interferes with GTP hydrolysis, causing the protein to remain in an active GTP state and continuously transmit signals to the downstream pathway to recruit and activate the proteins required for growth factor and other receptors (such as RAF and PI3K) signal transduction [8].
Furthermore, with the deepening research in recent years, the focus of gene studies has shifted toward non-coding RNAs that play regulatory roles. Among them, long non-coding RNAs (lncRNAs) are a class of non-coding transcripts with a length exceeding 200 nucleotides (nt), and involved in many physiological and pathological processes. Yang et al. reported on HIF1A-As2, a KRAS-responsive long non-coding RNA (lncRNA), confirming its positive correlation with KRAS through RT-qPCR. Further experiments revealed that HIF1A-As2 guides a key member of the DExD/H-box helicase family, DHX9, to the promoter region of the oncogenic transcription factor gene MYC, thereby enhancing MYC signaling transduction. Activated MYC further promotes cell proliferation and migration in KRAS-driven NSCLC. Simultaneously, KRAS, through MYC, promotes HIF1A-As2, forming a positive feedback loop [9]. MicroRNAs (miRNAs) are another important class of non-coding RNAs that play a role in gene regulation by degrading mRNA or inhibiting translation. Shi et al. used NanoString technology and Real-time PCR to identify the most upregulated microRNAs (miR-30c and miR-21) in cells overexpressing KRASWT and KRASG12D. Through experiments, they demonstrated that miR-30c downregulates BID, NF1, RASA1, and RASSF8 at the transcriptional level, while miR-21 inhibits the protein expression of RASA1 and RASSF8 to contributes to tumorigenesis [10].
Incidence
Statistics on RAS mutations in 17,993 cancer patients showed that the total mutation frequency of RAS genes was 22.58%, with KRAS mutations being the most frequent and most commonly observed in pancreatic cancer (685/842, 81.35%), colorectal cancer (85/175, 48.57%), and colon cancer (1609/3329, 48.33%) [11]. KRAS mutations are concentrated in codons 12 and 13 of exon 2 and codon 61 of exon 3. The most common mutation subtypes in KRAS-mutant lung adenocarcinoma are codon 12 substitution mutations (purine is replaced by pyrimidine or vice versa) G12C (39%) and G12V (18–21%), followed by transition mutations (purine is replaced by purine or pyrimidine is replaced by pyrimidine) G12D (14–18%) and G12A (10–11%). G12C is the most common mutation (45%) in former/current smokers, and G12D is the most common mutation (46%) in never-smokers [12]. In addition, KRAS also shows a different frequency depending on the patient’s race (more common in white populations than Asians). A multicenter retrospective cohort study of 216 Asian KRAS-mutant NSCLC patients analyzed by Lee et al. found that most patients were male (70.8%), Eastern Cooperative Oncology Group (ECOG) physical status scores were mostly 0–1 (92.1%), histological subtypes included adenocarcinoma (89.8%), squamous cell carcinoma (4.2%), and others (6.0%), and KRASG12D was the most common subtype (25.5%), mostly in never-smokers, suggesting that KRAS-mutant lung cancer in Asian patients may be driven by factors other than smoking [13]. Cooper et al. studied the clinical characteristics of KRASG12D-mutant NSCLC and found that among 107 KRASG12D-mutant NSCLC patients, most had a history of smoking (80 cases, 74.8%), and the histological subtype of the tumor was mostly adenocarcinoma (93 cases, 86.9%). Coexisting mutations were more common in KRASG12D-mutant NSCLC, including STK11 (17/107, 15.9%), TP53 (36/107, 33.6%), and KEAP1 (10/107, 9.4%). The co-occurrence of STK11 and KEAP1 mutations was associated with a poor clinical outcome, while the co-occurrence of TP53 did not affect survival [14]. Chen et al. analyzed 18,224 KRAS-mutant NSCLC patients to investigate the clinical characteristics of KRAS-mutant NSCLC in China. Among them, G12C (29.6%) was the most common subtype, followed by G12D (18.1%) and G12V (17.5%). The highest incidence of co-mutation was with TP53 (33.6%), followed by EGFR (11.6%), STK11 (10.4%), KEAP1 (6.2%), and CDKN2A (6.0%) [15].
The prognostic
The impact of KRAS mutations remains controversial, with inconsistent findings in various studies [16]. Wahl et al. investigated the correlation between KRAS status (KRAS wt vs. KRAS mut), KRASG12 status (KRAS wt vs. KRASG12C vs. KRAS non-G12C mutations), and KRAS mutation type (G12C, G12V, G12D, and G12A) and survival in multivariate analysis, entire cohorts, curative resection patients, or advanced patients, and none of the control groups showed any correlation with survival [12] (Table 1). However, Cai et al. revealed that KRASG12D mutation was associated with the shortest progression-free survival (PFS) and overall survival (OS) when compared with KRASG12C and KRASG12V, and the KRAS G > T group had better PFS and OS than the KRAS G > C and KRAS G > A groups at the amino acid substitution level [1]. Similarly, Johnson et al. found that KRAS mutation was an independent factor associated with shorter survival because of inherent biological differences in KRAS-driven lung cancer rather than differences in receiving prolonging life treatments like platinum-based chemotherapy and bevacizumab [17]. Aredo et al. explored the impact of KRAS mutation and co-mutations in the prognosis of NSCLC patients and found that KRASG12D mutation was significantly associated with OS in multivariate analysis, and STK11 co-mutation was also significantly associated with OS [18]. Arbour et al. identified different biological subtypes of KRAS-mutant lung adenocarcinoma that could be associated with co-mutations and investigated the effect of co-mutations on patient prognosis and treatment response. For the 330 patients with advanced KRAS-mutant lung cancer screened, the most common co-occurring genomic alterations were TP53 (42%), STK11 (29%), and KEAP1/NFE2L2 (27%), and KEAP1/NFE2L2 co-mutation was an independent prognostic factor that predicted shorter survival [19]. Shepherd et al. summarized the prognostic and predictive roles of KRAS mutation status and subtype in early-stage resected NSCLC in four adjuvant chemotherapy trials and found that KRAS mutation status was not a significant independent prognostic factor, especially for amino acid substitution mutations in the 12th codon [20]. Yu et al. retrospected 677 patients with metastatic or recurrent KRAS-mutant lung cancer and evaluated the relationship between KRAS mutation type, clinical factors, and overall survival. The data showed that the median overall survival for all patients with KRAS-mutant advanced lung cancer was 1.2 years, and the median overall survival ranged from 0.7 years (G13C) to 1.5 years (G12F) for specific KRAS point mutations, and no significant differences in survival were observed when comparing different KRAS point mutations [21]. Chen et al. treated 497 KRAS-mutant patients with different combinations of chemotherapy drugs and with or without immune checkpoint inhibitors (ICIs) and found no significant differences in PFS among the most common three subtypes of G12C, G12D, and G12V, with median PFS of 5.7, 6.6, and 6.6 months, respectively. The data also indicated that the patients who received ICI plus chemotherapy had significantly longer survival than those who received monotherapy [15].
Sensitivity to treatment
Currently, immunotherapy alone or in combination with platinum-based chemotherapy is the standard first-line treatment for KRAS-mutant NSCLC, but its efficacy is influenced by multiple factors. Fancelli et al. conducted a retrospective analysis showing that single-agent chemotherapy had limited efficacy in the KRAS-mutant NSCLC population, while single-agent ICI or ICI plus chemotherapy could benefit this population, which was not related to PD-L1 overexpression [22]. Sun et al. demonstrated that the clinical outcomes of KRAS-mutant NSCLC patients varied in response to ICI-based first-line treatment to some extent with different KRAS mutation subtypes and co-mutations. However, patients with KRAS/SMAD 4 co-mutation had a poor prognosis and were considered a new genotype that was insensitive to treatment [23]. Ghimessy et al. analyzed the effect of the KRAS mutation subtype on bevacizumab (vascular endothelial growth factor inhibitor) and found that BEV had lower activity in KRAS-mutant tumors, especially in KRASG12D-mutant stage III–IV LUAD patients than in non-KRAS-mutant LUAD patients [24]. Passiglia et al. investigated the efficacy and safety of nivolumab (immune checkpoint inhibitor) in 530 pretreated patients with advanced NSCLC and KRAS mutations. The data showed that KRAS status did not affect the efficacy of nivolumab in NSCLC, and TP53 co-mutated NSCLC patients also showed significant and durable clinical benefits from anti-PD-1 therapy, while patients with LKB1/STK11 tumor suppressor gene co-mutations were associated with shorter PFS and OS, possibly suggesting that LKB1 deficiency is a major driver of immune escape and a genomic biomarker of innate resistance to ICIs [25].
Activation of KRASG12D mutation-related signaling pathways
Currently, the KRAS-related signaling pathways have been extensively studied, as shown in Fig. 1. However, the abnormal activation of other proteins in the pathway carried by KRAS-mutated NSCLC will affect the treatment and prognosis of these patients. The ERBB family of receptor tyrosine kinases (RTKs), which includes 4 subtypes, can form dimers and bind to a range of ligands, to jointly activate a signaling network driven by ERBB. While KRAS mutations have traditionally been considered independent of upstream regulation, this perspective has been challenged by the work of Kruspig et al. Their research shows that multiple ERBB receptors are expressed and activated from the earliest stages of KRAS-driven lung tumor development. Additionally, lung tumors driven by KRASG12D mutation express multiple ERBB ligands, and active ERBB enhances signal transduction through the core RAS → ERK pathway, promoting cancer cell proliferation and tumor progression [26]. The RAS-specific guanine nucleotide exchange factors Son of sevenless (SOS) activates RAS by catalyzing the exchange of GDP for GTP, facilitating the activation of RAS. Mutant KRAS reduces its own GTPase-activating protein (GAP) activity, allowing SOS1 to serve as a central hub in regulating KRAS. SOS1 catalyzes the formation of KRAS-GTP complex to activate multiple downstream signaling pathways that trigger cancer cell proliferation. Fernando et al. discovered that the loss of SOS1 impairs the development and progression of KRASG12D-driven LUAD lung tumors. Their experiments demonstrated that SOS1 deficiency specifically inhibits the rates of lung tumor cell proliferation (Ki67) and ERK activation (pERK), while also significantly affecting the pro-tumor activities of various cell subpopulations within the LUAD microenvironment [27]. Ihle et al. studied the different effects of KRAS mutations on drug sensitivity and the impact on different signaling pathways. They found that NSCLC cell lines carrying the KRASG12D mutation have activated phosphatidylinositol 3-kinase (PI3K) and mitogen-activated protein kinase/extracellular signal-regulated kinase (MEK) signaling pathways. The PI3K/AKT signaling pathway is constitutively activated by mutant KRASG12D and is not inhibited by mTOR. However, the bulky Asp of KRASG12D interferes with the formation of KRAS homodimers and the binding of RALGDS, and downstream effector RAL is not activated. Therefore, the substitution of different amino acids induces heterogeneity in the behavior of KRAS protein, resulting in different signal outputs. This has profound implications for identifying and treating KRAS-driven tumors, and different combinations of downstream signaling pathway inhibitors may be required when treating KRAS mutation lung cancer with different amino acid substitutions [28]. Hung et al. studied the different mechanisms of RhoA/Wnt-induced NSCLC metastasis in different KRAS mutation subtypes. Immunoblotting results showed that KRASG12D mutation activates RhoA and further eliminates the activation of Wnt/β-catenin protein signaling, reducing the metastatic activity of KRASG12D-mutated NSCLC. This study confirms the relevance of the KRAS/RhoA/Wnt/β-catenin signaling pathway in NSCLC metastasis [29].

Upon activation by receptor tyrosine kinases, the receptors bind with the adapter protein RAS. RAS also activates phosphatidylinositol 3-kinases (PI3Ks) through protein growth factor receptor-binding protein 2 (GRB2). Then, GRB2 binds to participate in the activation of multiple downstream effectors, including SOS1 or 2 (guanine nucleotide exchange factors [GEFs] that cause phosphoinositide-dependent kinase-1 (PDK1) and protein kinase B (PKB, also known as AKT) to be activated by nucleotide exchange and replacement of GDP with GTP. AKT plays an important role in inhibiting NF-KB and multiple RAS functions. Once activated, RAS interacts with multiple effector transcription factors and activates the mammalian target of rapamycin (mTOR). PI3K also activates the monomeric GTPase RAC involved in mitogen-activated protein kinase/extracellular signal-regulated kinase (MEK1 and 2) and catalyzes cell migration. Finally, RAS also activates the RALGDS protein, which is a guanine nucleotide exchange factor for MAPK and RAL and phospholipase C activation, leading to the transcription of multiple transcription factors, including ELK1 and activation protein 1 (AP1). It also activates protein kinase C (PKC) and mobilizes intracellular calcium. RAF protein is essential for the proliferative action of PKA signaling protein kinase A. The mutant KRAS maintains a low level of GTPase activity, resulting in a weak response to upstream signals and a constitutively active state. G12D continuously activates downstream pathways, mainly through the RAF/MEK/ERK signaling pathway, leading to abnormal cell proliferation.
Targeted therapy for KRASG12D mutation
For decades, mutated KRAS has been recognized as an attractive drug target for treating multiple types of cancer, but the development of targeted drugs has not been as successful as anticipated. The difficulties in targeting KRAS are due to several factors: (1) the broad scope of KRAS’s activity, including its essential role in many normal cellular functions, meaning drugs that directly inhibit KRAS may have significant toxicity and strong side effects; (2) KRAS’s primary functional domain involves a pocket that binds to GDP or GTP. Unlike protein kinases, which have a weak affinity for ATP, KRAS’s binding to GTP or GDP is extremely strong, with an affinity coefficient on the picomolar (10–12) level, while the concentration of GDP and GTP in normal cells is on the micromolar (10–6) level. This means that finding a small molecule compound with a binding ability to KRAS that is equivalent to GDP or GTP is extremely challenging; (3) designing a drug that selectively inhibits the activity of mutated KRAS protein while minimizing the impact on normal KRAS activity requires a compound with good selectivity for mutated KRAS, which is another difficult challenge in drug design; and (4) indirect strategies for targeting KRAS are also fraught with challenges, including the fact that KRAS signaling pathway is a necessary pathway for normal cell growth and survival, and targeting essential pathways is often associated with significant toxicity that reduces the therapeutic window to the point where it may be absent, compensatory escape mechanisms, and feedback and redundancy resulting from strict regulation.
Direct targeting drugs
KRASG12D inhibitors
Wang et al. developed MRTX1133 (Fig. 2), a potent, selective, non-covalent KRASG12D inhibitor with picomolar binding affinity, through a series of structurally-based optimizations on the KRASG12C inhibitor adagrasib. Firstly, the compound has a pyrido[4,3-d]pyrimidine scaffold, and three substituents were searched to interact widely with KRASG12D protein. The C4 position is a [3.2.1]bicyclic diamino substituent to achieve optimal interaction with Asp12 and Gly60 in the mutant. The C2 position is modified with the pyrrolizidine with a 2-fluoro substituent which forms a strong ionic interaction with the negatively charged Glu62 carboxylate salt. Finally, the C7 substituent is an optimized 7-fluoro and 8-ethynyl group that lies well within the hydrophobic pocket of KRASG12D protein and forms a well-organized hydrogen-bonding network. The interaction of hydrogen bonds allows the terminal ethynyl group to effectively bridge the lipophilic and polar regions of the KRASG12D protein switch II pocket. Experimental validation demonstrated that MRTX1133 inhibits KRASG12D signal transduction in cells and in vivo, and its anti-tumor effects have been confirmed in a mouse model, showing robust in vivo efficacy and potential for targeted therapy against this “undruggable” target [30].

The chemical structure of the KRASG12D inhibitor MRTX1133.
Mao et al. used a strategy based on strong interactions (salt bridges) between the alkylamine moiety and Asp 12 on the inhibitor to design a series of potent inhibitors (TH-Z816, TH-Z827, and TH-Z835) that can form salt bridges with the Asp 12 residue of KRASG12D, using the G12C inhibitor MRTX 22 as a scaffold and characterized their in vitro and in vivo activity. ITC experiments showed that these salt bridge-forming inhibitors bound to both GDP and GTP-bound KRASG12D and effectively disrupted KRAS-CRAF interaction, but did not bind to wild-type or G12C mutant KRAS. These molecules also disrupted the activation of MAPK and PI3K/mTOR signaling in different cancer cells and displayed anti-proliferative and anti-tumor effects. This study demonstrated proof-of-concept for the strategy of targeting KRASG12D by inducing adaptation pockets via salt bridge formation [3].
Meanwhile, Zhou et al. discovered an effective, selective, biologically stable, and cell-permeable peptide drug, NKTP-3, which targets NRP1 and KRASG12D. NKTP-3 first binds to NRP1 on the cancer cell membrane and is then delivered into the cell. Once inside the cell, it binds to KRASG12D and significantly inhibits downstream signaling, including AKT and ERK phosphorylation, leading to anti-tumor effects. Strong anti-tumor activity of the NRP1/KRASG12D dual-targeting cyclic peptide NKTP-3 was demonstrated in xenografts derived from A427 cells and primary lung cancer model driven by KRASG12D, with no obvious toxicity. These findings suggest that NKTP-3 may be a potential drug for treating KRASG12D-driven lung cancer [31].
In 2017, a synthetic cyclic peptide, KRpep-2d, was discovered as the first selective inhibitor of KRASG12D. The two Cys residues in the peptide are essential for its cyclic structure and control of its binding and inhibitory activity, but the bond is cleaved under intracellular reducing conditions, limiting application. Sakamoto et al. generated KS-58, a KRpep-2d derivative identified as a bicyclic peptide with a non-proteinogenic amino acid structure. KS-58 enters cells and exerts anti-cancer effects by blocking two pathways: RASGDP-SOS1 interaction (i.e., GDP-GTP exchange on RAS) and RASGTP-BRAF interaction. KS-58 was shown to selectively bind to KRASG12D and inhibit the in vitro proliferation of both the A427 human lung cancer cell line and the PANC-1 human pancreatic cancer cell line that expresses KRASG12D. However, the pharmacokinetic properties and high doses required for the treatment of this drug still need improvement. Nevertheless, KS-58 is an attractive lead molecule for developing new cancer drugs that target KRASG12D [32].
Pan-KRAS inhibitors
The broad-spectrum KRAS inhibitor is defined as a non-covalent inhibitor that exhibits high affinity for the inactive state of KRAS and can block nucleotide exchange to prevent the activation of wild-type KRAS and a wide range of KRAS mutants [33]. Using the selective KRASG12C inhibitor BI-0474 as a starting point, Kim et al. designed a broad-spectrum KRAS inhibitor, BI-2865, with potent non-covalent inhibitory activity. Experimental evidence showed that this inhibitor demonstrated similar efficacy to BI-0474 against KRASG12C mutant cells while also significantly inhibiting cell proliferation in G12D or G12V mutant cells. The inhibitor functions by preferentially targeting the inactive state of KRAS to prevent its reactivation through nucleotide exchange. Additionally, the research group conducted experiments with BI-2865 to inhibit NRAS and HRAS mutant cells, revealing that the inhibitor’s ability to inhibit nucleotide exchange in HRAS or NRAS is several orders of magnitude lower than that in KRAS, and this difference is attributed to direct and/or indirect constraints imposed by three residues in the G domain. Consequently, in cells with wild-type KRAS, the use of this inhibitor leads to increased activation of other RAS homologs, thereby limiting its antiproliferative effects [33].
SOS1 is a key guanine nucleotide exchange factor (GEF) for KRAS, which binds to KRAS protein at its catalytic binding site and promotes the exchange of GDP for GTP, thereby activating the KRAS protein. In addition to its catalytic site, SOS1 can also bind to GTP-bound KRAS at an allosteric site, forming a positive feedback regulation mechanism [34]. Hofmann et al. reported the discovery of a highly efficient, selective, and orally bioavailable small molecule SOS1 inhibitor BI-3406, which binds to the catalytic domain of SOS1, thereby preventing its interaction with KRAS. Experimental evidence suggests that BI-3406 reduces the formation of GTP-loaded KRAS and restricts the growth of most tumor cells driven by KRAS variants at positions G12 and G13. Furthermore, BI-3406 can weaken the feedback reactivation induced by MEK inhibitors, thereby enhancing the sensitivity of KRAS-dependent cancers to MEK inhibition. Thus, the development of clinical SOS1 compounds in combination with MEK inhibitors and potentially other RTK/MAPK pathway inhibitors holds promise for significant clinical benefits [35]. Hillig et al. designed a pan-KRAS inhibitor, BAY-293, using a dual-screening approach and structure-guided design. The study demonstrated that this inhibitor binds to a surface pocket on SOS1, preventing the formation of the KRAS-SOS1 complex. This pocket is located adjacent to the KRAS binding site and thus blocks the reloading of KRAS with GTP, leading to anti-proliferative activity. BAY-293 also exhibits synergistic effects with covalent inhibitors of KRASG12D, highlighting the potential of combined therapy targeting both KRAS and SOS1 [36].
Indirectly targeted drugs
MEK inhibitor
Due to the difficulty of directly targeting KRASG12D drugs, targeting the KRAS signaling pathway has always focused on downstream targets, one of which is MEK. Drugs targeting MEK downstream in the MAPK cascade via inhibition of signal transduction pathways are less effective in treating KRAS-mutant NSCLC in multiple experiments. For example, Pasi et al. found that the addition of the MEK inhibitor trametinib to docetaxel did not improve progression-free survival in advanced KRAS-mutant non-small cell lung cancer patients compared to docetaxel alone [37]. The main reason is that although targeting MEK blocks MAPK cascade signaling, other downstream pathways of KRAS (such as PI3K-AKT, RAL, etc.) are strengthened. Lee et al. demonstrated synergistic effects of combination therapy with MEK inhibitor cobimetinib and immunotherapy for the treatment of KRAS-mutant NSCLC, showing anti-tumor effects and improved survival in a mouse model [38].
GRP78 inhibitor
Studies have shown that newly synthesized KRAS is cytoplasmic and inactive and undergoes a series of translation and post-translational modifications on the cytoplasmic surface of the endoplasmic reticulum (ER), which are mediated by enzymes that act as transmembrane ER proteins. Therefore, ER is the main site of KRAS maturation, and perturbation of ER homeostasis and protein quality control may be detrimental to KRAS-driven LUAD. The 78-kDa glucose-regulated protein GRP 78/BiP is a critical chaperone protein in the ER and a major pro-survival effector in the unfolded protein response (UPR). The loss of GRP78 induces UPR and apoptotic markers, which are associated with the loss of cell viability in lung cancer cell lines carrying the same KRAS mutation [39]. Ha et al. targeted GRP78 with small molecule inhibitors (such as HA15 and YUW70) with anti-cancer activity, which consistently reduced the levels of oncogenic KRAS protein in the tested cell lines. They also found that GRP78 deficiency can inhibit PI3K, AKT, TGF-β, and CD44 signaling pathways, as well as many other signaling pathways. Combined with ER stress-induced cell apoptosis and autophagy, this will provide a strong defense against the development of cancer cell resistance before cancer cells are eliminated [40].
NFkB activating kinase inhibitor
Preclinical studies have provided evidence that both classical and non-classical NF-κB pathways are co-activated in LUAD-carrying KRAS mutation. The specificity of IKK (NFkB activation kinase) synergistically induces tumorigenesis with mutant KRAS in an autocrine manner, providing survival advantages for mutant cells in vitro and in vivo. The NCT01833143 phase II single-center clinical trial of bortezomib subcutaneously administered to patients with advanced NSCLC carrying KRASG12D mutations or without a previous smoking history at Memorial Sloan Kettering Cancer Center showed some anti-tumor activity, especially in a unique subtype of lung adenocarcinoma, invasive mucinous adenocarcinoma (IMA), while boron-tazoxime is inactive in most patients with advanced KRASG12D mutant lung adenocarcinomas. Therefore, novel inhibitors of the NF-κB pathway need to be explored [41].
HSP inhibitor
Inhibiting heat shock proteins has been identified as another potential therapeutic strategy for KRAS-mutant NSCLC. Molecular chaperone Hsp 90 is essential for protein stability and maturation and prevents protein degradation by the proteasome. Vreka et al. found that IKKα is a partner of KRAS non-oncogene addiction, and specifically synergizes with mutant KRAS to induce tumorigenesis, providing survival advantages for mutant cells in vitro and in vivo. The Hsp 90 inhibitor 17-DMAG can block IKK function and have better efficacy against KRASG12D-mutant lung adenocarcinoma, opening up a new way to prevent/treat KRAS-mutant LUAD [42].
ERBB inhibitors
Previous research has shown that EGFR mutations and KRAS mutations rarely occur together, and the use of EGFR-targeted drugs alone to treat KRAS-mutant lung adenocarcinoma has not shown significant clinical benefits. However, recent experimental results suggest that the independence of mutated KRAS from upstream signaling pathways may not be absolute. Kruspig et al. demonstrated through experiments that the initiation and progression of KRAS-driven lung tumors require the involvement of ERBB family receptor tyrosine kinases (RTKs), and inhibition of the ERBB network weakens the activation of a series of downstream signaling proteins (such as pERK, STAT3, etc.), while transient pharmacological inhibition of the ERBB network enhances the therapeutic benefits of MEK inhibitors in the autologous tumor environment. Multiple ERBB inhibitors almost completely inhibit the formation of KRASG12D-driven lung tumors and enhance the benefits of MEK inhibition in tumor therapy [26].
SHP2 inhibitors
SHP2, encoded by the PTPN11 gene, plays an important role in signal transduction downstream of growth factor receptors by mainly regulating cell survival and proliferation through activation of the RAS-ERK signaling pathway. Ruess et al. found that PTPN11 gene deletion significantly inhibited tumor development in KRAS-driven pancreatic ductal adenocarcinoma and non-small cell lung cancer mouse models, providing evidence for the critical dependence of mutant KRAS on SHP2 in the process of carcinogenesis [43]. Chen et al. found that SHP2 is activated by peptides and proteins containing appropriately spaced phosphotyrosine residues, which bind the N-terminal and C-terminal SH2 domains in a bidentate manner, releasing it from the self-inhibitory interface, and make the active site available for substrate recognition and turnover. They used this natural regulatory mechanism to screen out the SHP2 inhibitor SHP099, which locks SHP2 in an autoinhibitory conformation and directly targets the inhibition of MAPK signaling and proliferation in RTK-dependent cells. This provides a feasible strategy for targeting RTK for cancer treatment [44]. Nichols et al. found through the treatment with another SHP2 inhibitor, RMC-4550 (a small molecule allosteric inhibitor), that it reduces oncogenic RAS-RAF-MEK-ERK signaling and cancer growth by disrupting SOS1-mediated RAS-GTP loading, highlighting SHP2 inhibition as a promising molecular therapeutic strategy for nucleotide cycling oncogenic KRAS in cancer [45].
Immune therapies
The inhibition and rewiring of the immune system play a crucial role in the onset and development of tumors. Immune therapies aim to reactivate anti-tumor immune cells and overcome the tumor’s immune escape mechanisms. Tumor immunotherapy, represented by immune checkpoint blockade and adoptive cell transfer, has made enormous clinical successes by inducing long-term remission of some tumors that are difficult to treat with all other therapies. Among them, immune checkpoint blockade therapy represented by PD-1/PD-L1 inhibitors (nivolumab) and CTLA-4 inhibitors (ipilimumab) has shown encouraging therapeutic effects in the treatment of various malignancies, such as non-small cell lung cancer (NSCLC) and melanoma, etc. [46].
Immune checkpoint inhibitors (ICIs), mainly represented by PD-1/PD-L1 inhibitors (nivolumab), have been widely used in the treatment of non-small cell lung cancer (NSCLC). ICIs are currently used as single-agent therapy or in combination with other treatments for first-line and subsequent therapy of metastatic NSCLC. In addition, ICI in the neoadjuvant and adjuvant treatment setting has shown efficacy for resectable disease patients, highlighting the potential of ICIs to improve outcomes for this patient group [47]. However, research has shown that most tumors have immune inhibitory mechanisms that limit the effectiveness of immunotherapy, including PD-1 expression on tumor-infiltrating T cells and the accumulation of inhibitory T cells, such as CD4+Foxp3+ regulatory T (Treg) cells, in the tumor microenvironment, which hinders anti-tumor immune responses. Studies have shown that KRASG12D mutation correlates with reduced TMB, and KRASG12D/TP53 co-mutation has a significant effect on reducing TMB and PD-L1 expression and reducing immune cell infiltration. KRASG12D mutation, especially in combination with TP53 co-mutation, maybe a negative predictive biomarker for PD-1/PD-L1 immune checkpoint inhibitors in NSCLC patients [48]. Eliminating these inhibitory mechanisms in tumors can pave the way for more effective anti-tumor responses.
Martinez-Usatorre et al. improved the efficacy of PD-1/PD-L1 inhibitors by modulating the tumor microenvironment in a KRASG12D/+;TP53−/− genetically engineered mouse model. They induced vascular normalization and facilitated T cell trafficking through inhibition of angiogenic factors VEGFA and ANGPT2 with A2 V, which improved maturation and antigen presentation of tumor-associated macrophages (TAMs). They also used CSF1R inhibitor 2G2 and platinum-based chemotherapy to deplete TAMs and reduce Treg cell numbers, respectively, to enhance the response of KRAS tumors to A2 V and anti-PD-1 dual therapy. The combination of these three agents induced an immune-infiltrated tumor microenvironment with increased CD4+ and CD8+ T cells, and decreased TAMs and Treg cells, leading to improvement in the response of KP tumors to checkpoint inhibitors [49]. Adeegbe et al. improved the response of KRAS-mutant NSCLC to immune therapy by combining JQl (a BET family inhibitor containing a bromodomain) with anti-PD-1 treatment. Bromodomain proteins are epigenetic regulators that cause growth inhibition and/or cell cytotoxicity in tumor cells. Treatment with JQl alone induced a decrease in Treg cell numbers, while combination with anti-PD-1 enhanced activation of infiltrated T cells in the tumor bed and improved effector function, leading to increased expression of Th1 cytokine profile, which is consistent with the persistent antitumor response observed with this novel treatment combination [48]. In addition, Lee et al. improved the prognosis of KRAS-driven lung cancer by combining MEK inhibitors with immunomodulatory anti-PD-1 and anti-PD-L1 antibodies. Low-dose MEKi (trametinib) and anti-PD-L1 combined therapy in KRAS-mutant NSCLC enhanced tumor microenvironment and T cell infiltration, reduced Ly6Ghigh PMN-MDSCs (myeloid-derived suppressor cells, a type of immune inhibitory cells that mainly suppress natural killer cells and effector CD8+ T cells) in tumor tissue, and inhibited tumor cell proliferation, leading to tumor cell apoptosis. MEKi acted as a sensitizer for KRAS-mutant tumors that were previously unresponsive to immune therapy [38].
Conclusion
In the context of advanced NSCLC, targeted therapy against driver oncogenic mutations has already changed the treatment paradigm. Given the high incidence of KRAS mutations in NSCLC patients, this is a promising treatment target. The 2023 NCCN guidelines for the first time recommended KRASG12C targeted drug Adagrasib for the treatment of KRASG12C mutant NSCLC, which is a significant breakthrough for KRAS treatment targets. However, other coexisting mutations and changes in the immune microenvironment may be critical for its function and biological impact. Therefore, combination therapy with chemotherapy, immunotherapy, targeted therapy, and other treatment modalities may further improve the poor prognosis of KRAS-mutant NSCLC. Moreover, as KRAS mutation involves multiple downstream signaling pathways, targeting multiple different targets with combination targeted therapy may improve the current inadequate therapeutic efficacy of targeted drugs. Shortly, new molecules or treatment strategies may radically alter the outcome of patients with KRAS-driven NSCLC. Further research is required to better understand the pathways involved in KRAS mutation and to develop more targeted and effective drugs.
References
Cai D, Hu C, Li L, Deng S, Yang J, Han-Zhang H, et al. The prevalence and prognostic value of KRAS co-mutation subtypes in Chinese advanced non-small cell lung cancer patients. Cancer Med. 2020;9:84–93. https://doi.org/10.1002/cam4.2682.
Zhou X, Padanad MS, Evers BM, Smith B, Novaresi N, Suresh S, et al. Modulation of mutant KRASG12D -driven lung tumorigenesis in vivo by gain or loss of PCDH7 function. Mol Cancer Res. 2019;17:594–603. https://doi.org/10.1158/1541-7786.MCR-18-0739.
Mao Z, Xiao H, Shen P, Yang Y, Xue J, Yang Y, et al. KRAS(G12D) can be targeted by potent inhibitors via formation of salt bridge. Cell Discov. 2022;8:5. https://doi.org/10.1038/s41421-021-00368-w.
Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–51. https://doi.org/10.1038/nature12796.
Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, et al. Sotorasib for lung cancers with KRAS p.G12C mutation. N Engl J Med. 2021;384:2371–81. https://doi.org/10.1056/NEJMoa2103695.
Tang D, Kang R. Glimmers of hope for targeting oncogenic KRAS-G12D. Cancer Gene Ther. 2023;30:391–3. https://doi.org/10.1038/s41417-022-00561-3.
Ha DP, Huang B, Wang H, Rangel DF, Van Krieken R, Liu Z, et al. Targeting GRP78 suppresses oncogenic KRAS protein expression and reduces the viability of cancer cells bearing various KRAS mutations. Neoplasia. 2022;33:100837. https://doi.org/10.1016/j.neo.2022.100837.
Xu K, Park D, Magis AT, Zhang J, Zhou W, Sica GL, et al. Small molecule KRAS agonist for mutant KRAS cancer therapy. Mol Cancer. 2019;18:85. https://doi.org/10.1186/s12943-019-1012-4.
Yang K, Zhang W, Zhong L, Xiao Y, Sahoo S, Fassan M, et al. Long non-coding RNA HIF1A-As2 and MYC form a double-positive feedback loop to promote cell proliferation and metastasis in KRAS-driven non-small cell lung cancer. Cell Death Differ. 2023;30:1533–49. https://doi.org/10.1038/s41418-023-01160-x.
Shi L, Middleton J, Jeon YJ, Magee P, Veneziano D, Laganà A, et al. KRAS induces lung tumorigenesis through microRNAs modulation. Cell Death Dis. 2018;9:219. https://doi.org/10.1038/s41419-017-0243-9.
Zhou S, Zhang D, Li J, Zhao J, Wang G, Zhang Y, et al. Landscape of RAS variations in 17,993 pan-cancer patients identified by next-generation sequencing. Pathol Oncol Res. 2020;26:2835–7. https://doi.org/10.1007/s12253-020-00845-9.
Wahl SGF, Dai HY, Emdal EF, Berg T, Halvorsen TO, Ottestad AL, et al. The prognostic effect of KRAS mutations in non-small cell lung carcinoma revisited: a Norwegian multicentre study. Cancers. 2021;13:4294. https://doi.org/10.3390/cancers13174294.
Lee J, Tan AC, Zhou S, Yoon S, Liu S, Masuda K, et al. Clinical characteristics and outcomes in advanced KRAS-mutated NSCLC: a multicenter collaboration in Asia (ATORG-005). JTO Clin Res Rep. 2021;3:100261. https://doi.org/10.1016/j.jtocrr.2021.100261.
Cooper AJ, Muzikansky A, Lennerz J, Narinesingh F, Mino-Kenudson M, Hung YP, et al. Clinicopathologic characteristics and outcomes for patients with KRAS G12D-mutant NSCLC. JTO Clin Res Rep. 2022;3:100390. https://doi.org/10.1016/j.jtocrr.2022.100390.
Chen H, Huang D, Lin G, Yang X, Zhuo M, Chi Y, et al. The prevalence and real-world therapeutic analysis of Chinese patients with KRAS-mutant non-small cell lung cancer. Cancer Med. 2022;11:3581–92. https://doi.org/10.1002/cam4.4739.
Martin P, Leighl NB, Tsao MS, Shepherd FA. KRAS mutations as prognostic and predictive markers in non-small cell lung cancer. J Thorac Oncol. 2013;8:530–42. https://doi.org/10.1097/JTO.0b013e318283d958.
Johnson ML, Sima CS, Chaft J, Paik PK, Pao W, Kris MG, et al. Association of KRAS and EGFR mutations with survival in patients with advanced lung adenocarcinomas. Cancer. 2013;119:356–62. https://doi.org/10.1002/cncr.27730.
Aredo JV, Padda SK, Kunder CA, Han SS, Neal JW, Shrager JB, et al. Impact of KRAS mutation subtype and concurrent pathogenic mutations on non-small cell lung cancer outcomes. Lung Cancer. 2019;133:144–50. https://doi.org/10.1016/j.lungcan.2019.05.015.
Arbour KC, Jordan E, Kim HR, Dienstag J, Yu HA, Sanchez-Vega F, et al. Effects of co-occurring genomic alterations on outcomes in patients with KRAS-mutant non-small cell lung cancer. Clin Cancer Res. 2018;24:334–40. https://doi.org/10.1158/1078-0432.CCR-17-1841.
Shepherd FA, Domerg C, Hainaut P, Jänne PA, Pignon JP, Graziano S, et al. Pooled analysis of the prognostic and predictive effects of KRAS mutation status and KRAS mutation subtype in early-stage resected non-small-cell lung cancer in four trials of adjuvant chemotherapy. J Clin Oncol. 2013;31:2173–81. https://doi.org/10.1200/JCO.2012.48.1390.
Yu HA, Sima CS, Shen R, Kass S, Gainor J, Shaw A, et al. Prognostic impact of KRAS mutation subtypes in 677 patients with metastatic lung adenocarcinomas. J Thorac Oncol. 2015;10:431–7. https://doi.org/10.1097/JTO.0000000000000432.
Fancelli S, Caliman E, Mazzoni F, Paglialunga L, Gatta Michelet MR, Lavacchi D, et al. KRAS G12 isoforms exert influence over up-front treatments: a retrospective, multicenter, Italian analysis of the impact of first-line immune checkpoint inhibitors in an NSCLC real-life population. Front Oncol. 2022;12:968064. https://doi.org/10.3389/fonc.2022.968064.
Sun Y, Li Z, Jian H, Xia L, Lu S. Impact of KRAS mutation subtypes and co-occurring mutations on response and outcome in advanced NSCLC patients following first-line treatment. J Clin Med. 2022;11:4003. https://doi.org/10.3390/jcm11144003.
Ghimessy AK, Gellert A, Schlegl E, Hegedus B, Raso E, Barbai T, et al. KRAS mutations predict response and outcome in advanced lung adenocarcinoma patients receiving first-line bevacizumab and platinum-based chemotherapy. Cancers. 2019;11:1514. https://doi.org/10.3390/cancers11101514.
Passiglia F, Cappuzzo F, Alabiso O, Bettini AC, Bidoli P, Chiari R, et al. Efficacy of nivolumab in pre-treated non-small-cell lung cancer patients harbouring KRAS mutations. Br J Cancer. 2019;120:57–62. https://doi.org/10.1038/s41416-018-0234-3.
Kruspig B, Monteverde T, Neidler S, Hock A, Kerr E, Nixon C, et al. The ERBB network facilitates KRAS-driven lung tumorigenesis. Sci Transl Med. 2018;10:eaao2565. https://doi.org/10.1126/scitranslmed.aao2565.
Baltanás FC, García-Navas R, Rodríguez-Ramos P, Calzada N, Cuesta C, Borrajo J, et al. Critical requirement of SOS1 for tumor development and microenvironment modulation in KRASG12D-driven lung adenocarcinoma. Nat Commun. 2023;14:5856. https://doi.org/10.1038/s41467-023-41583-1.
Ihle NT, Byers LA, Kim ES, Saintigny P, Lee JJ, Blumenschein GR, et al. Effect of KRAS oncogene substitutions on protein behavior: implications for signaling and clinical outcome. J Natl Cancer Inst. 2012;104:228–39. https://doi.org/10.1093/jnci/djr523.
Hung PS, Huang MH, Kuo YY, Yang JC. The inhibition of Wnt restrain KRASG12V-driven metastasis in non-small-cell lung cancer. Cancers. 2020;12:837. https://doi.org/10.3390/cancers12040837.
Wang X, Allen S, Blake JF, Bowcut V, Briere DM, Calinisan A, et al. Identification of MRTX1133, a noncovalent, potent, and selective KRASG12D inhibitor. J Med Chem. 2022;65:3123–33. https://doi.org/10.1021/acs.jmedchem.1c01688.
Zhou Y, Zou Y, Yang M, Mei S, Liu X, Han H, et al. Highly potent, selective, biostable, and cell-permeable cyclic d-peptide for dual-targeting therapy of lung cancer. J Am Chem Soc. 2022;144:7117–28. https://doi.org/10.1021/jacs.1c12075.
Sakamoto K, Masutani T, Hirokawa T. Generation of KS-58 as the first K-Ras(G12D)-inhibitory peptide presenting anti-cancer activity in vivo. Sci Rep. 2020;10:21671. https://doi.org/10.1038/s41598-020-78712-5.
Kim D, Herdeis L, Rudolph D, Zhao Y, Böttcher J, Vides A, et al. Pan-KRAS inhibitor disables oncogenic signalling and tumour growth. Nature. 2023;619:160–6. https://doi.org/10.1038/s41586-023-06123-3.
Freedman TS, Sondermann H, Friedland GD, Kortemme T, Bar-Sagi D, Marqusee S, et al. A Ras-induced conformational switch in the Ras activator Son of sevenless. Proc Natl Acad Sci USA. 2006;103:16692–7. https://doi.org/10.1073/pnas.0608127103.
Hofmann MH, Gmachl M, Ramharter J, Savarese F, Gerlach D, Marszalek JR, et al. BI-3406, a potent and selective SOS1-KRAS interaction inhibitor, is effective in KRAS-driven cancers through combined MEK inhibition. Cancer Discov. 2021;11:142–57. https://doi.org/10.1158/2159-8290.CD-20-0142.
Hillig RC, Sautier B, Schroeder J, Moosmayer D, Hilpmann A, Stegmann CM, et al. Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS-SOS1 interaction. Proc Natl Acad Sci USA. 2019;116:2551–60. https://doi.org/10.1073/pnas.1812963116.
Jänne PA, van den Heuvel MM, Barlesi F, Cobo M, Mazieres J, Crinò L, et al. Selumetinib plus docetaxel compared with docetaxel alone and progression-free survival in patients with KRAS-mutant advanced non-small cell lung cancer: the SELECT-1 randomized clinical trial. JAMA. 2017;317:1844–53. https://doi.org/10.1001/jama.2017.3438.
Lee JW, Zhang Y, Eoh KJ, Sharma R, Sanmamed MF, Wu J, et al. The combination of MEK inhibitor with immunomodulatory antibodies targeting programmed death 1 and programmed death ligand 1 results in prolonged survival in KRAS/p53-driven lung cancer. J Thorac Oncol. 2019;14:1046–60. https://doi.org/10.1016/j.jtho.2019.02.004.
Rangel DF, Dubeau L, Park R, Chan P, Ha DP, Pulido MA, et al. Endoplasmic reticulum chaperone GRP78/BiP is critical for mutant KRAS-driven lung tumorigenesis. Oncogene. 2021;40:3624–32. https://doi.org/10.1038/s41388-021-01791-9.
Ha DP, Huang B, Wang H, Rangel DF, Van Krieken R, Liu Z, et al. Targeting GRP78 suppresses oncogenic KRAS protein expression and reduces viability of cancer cells bearing various KRAS mutations. Neoplasia. 2022;33:100837. https://doi.org/10.1016/j.neo.2022.100837.
Drilon A, Schoenfeld AJ, Arbour KC, Litvak A, Ni A, Montecalvo J, et al. Exceptional responders with invasive mucinous adenocarcinomas: a phase 2 trial of bortezomib in patients with KRAS G12D-mutant lung cancers. Cold Spring Harb Mol Case Stud. 2019;5:a003665. https://doi.org/10.1101/mcs.a003665.
Vreka M, Lilis I, Papageorgopoulou M, Giotopoulou GA, Lianou M, Giopanou I, et al. IκB kinase α is required for development and progression of KRAS-mutant lung adenocarcinoma. Cancer Res. 2018;78:2939–51. https://doi.org/10.1158/0008-5472.CAN-17-1944.
Ruess DA, Heynen GJ, Ciecielski KJ, Ai J, Berninger A, Kabacaoglu D, et al. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat Med. 2018;24:954–60. https://doi.org/10.1038/s41591-018-0024-8.
Chen YN, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG, et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature. 2016;535:148–52. https://doi.org/10.1038/nature18621.
Nichols RJ, Haderk F, Stahlhut C, Schulze CJ, Hemmati G, Wildes D, et al. RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat Cell Biol. 2018;20:1064–73. https://doi.org/10.1038/s41556-018-0169-1.
Wang DR, Wu XL, Sun YL. Therapeutic targets and biomarkers of tumor immunotherapy: response versus non-response. Signal Transduct Target Ther. 2022;7:331. https://doi.org/10.1038/s41392-022-01136-2.
Zhao D, Li H, Mambetsariev I, Mirzapoiazova T, Chen C, Fricke J, et al. Clinical and molecular features of KRAS-mutated lung cancer patients treated with immune checkpoint inhibitors. Cancers. 2022;14:4933. https://doi.org/10.3390/cancers14194933.
Adeegbe DO, Liu S, Hattersley MM, Bowden M, Zhou CW, Li S, et al. BET bromodomain inhibition cooperates with PD-1 blockade to facilitate antitumor response in KRAS-mutant non-small cell lung cancer. Cancer Immunol Res. 2018;6:1234–45. https://doi.org/10.1158/2326-6066.CIR-18-0077.
Martinez-Usatorre A, Kadioglu E, Boivin G, Cianciaruso C, Guichard A, Torchia B, et al. Overcoming microenvironmental resistance to PD-1 blockade in genetically engineered lung cancer models. Sci Transl Med. 2021;13:eabd1616. https://doi.org/10.1126/scitranslmed.abd1616.
Author information
Authors and Affiliations
Contributions
Tang Yining’s contribution included data curation, writing—the original draft, resources, and validation. Pu Xi and Yuan Xiao’s contribution included methodology and formal analysis. Pang Zhonghao’s contributions included conceptualization and investigation. Li Feng and Wang Xu’s contribution included project administration, writing—review & and editing. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tang, Y., Pu, X., Yuan, X. et al. Targeting KRASG12D mutation in non-small cell lung cancer: molecular mechanisms and therapeutic potential. Cancer Gene Ther (2024). https://doi.org/10.1038/s41417-024-00778-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41417-024-00778-4
