
Abstract
Mitochondria, with their intricate networks of functions and information processing, are pivotal in both health regulation and disease progression. Particularly, mitochondrial dysfunctions are identified in many common pathologies, including cardiovascular diseases, neurodegeneration, metabolic syndrome, and cancer. However, the multifaceted nature and elusive phenotypic threshold of mitochondrial dysfunction complicate our understanding of their contributions to diseases. Nonetheless, these complexities do not prevent mitochondria from being among the most important therapeutic targets. In recent years, strategies targeting mitochondrial dysfunction have continuously emerged and transitioned to clinical trials. Advanced intervention such as using healthy mitochondria to replenish or replace damaged mitochondria, has shown promise in preclinical trials of various diseases. Mitochondrial components, including mtDNA, mitochondria-located microRNA, and associated proteins can be potential therapeutic agents to augment mitochondrial function in immunometabolic diseases and tissue injuries. Here, we review current knowledge of mitochondrial pathophysiology in concrete examples of common diseases. We also summarize current strategies to treat mitochondrial dysfunction from the perspective of dietary supplements and targeted therapies, as well as the clinical translational situation of related pharmacology agents. Finally, this review discusses the innovations and potential applications of mitochondrial transplantation as an advanced and promising treatment.
Similar content being viewed by others
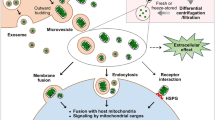
Introduction
Over the past three decades, mitochondria have emerged as promising therapeutic targets for common diseases, driven by substantial advancements in both mitochondrial biology and clinical research.1,2,3 (See Fig. 1) Evolving from their bacterial ancestor, mitochondria have retained a limited yet unique genomic material known as the mitochondrial DNA (mtDNA), capable of self-replication and encoding indispensable constituents of respiratory complexes located on the inner mitochondrial membrane.4,5,6 Thus, as essential endosymbionts within eukaryotic cells, mitochondria are recognized as the powerhouses orchestrating cellular activities.7,8 This central function is well-established on the electrochemical gradient generated by the respiratory chain.6,9,10 Mitochondria also play critical roles in mediating lipid metabolism, Ca2+ homeostasis, and apoptosis.2,11 The concept of mitochondrial dysfunction originates from bioenergetics terminology, which is now extended to a broader relationship corresponding to their cellular environment.12 Inherited mitochondrial dysfunction resulting from deficiencies of mtDNA maintenance, and translation, stand as determinants in defining primary mitochondrial diseases (PMD), such as Leigh syndrome, mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS), and chronic progressive external ophthalmoplegia (CPEO).13,14,15 The coexistence of mutant and wild-type mtDNA, referred to as mtDNA heterogeneity, accumulates during aging and age-associated pathogenesis, and is associated with worsened clinical performance.16 On the other hand, many common pathologies are convergent to mitochondria, causing secondary mitochondrial dysfunction (SMD), such as heart failure, neurodegeneration, and metabolic syndrome.17,18,19 Abnormal dynamics and quality control, dysfunctional proteostasis, inhibited ATP production, calcium dyshomeostasis, and metabolic reprogramming often simultaneously occur and interplay within pathological conditions, thereby impacting mitochondria in their capacity as biosynthetic and signaling hubs.1,20 Consequently, it is unsurprising that mitochondrial dysfunction is frequently implicated in the pathophysiology of various diseases and the aging process.21,22,23,24,25

The number of growing published articles or studies from 1980 to Aug 2023, based on mitochondrial medicine (pink), therapies (blue), and clinical trials (green). The publications of therapeutic targets increase following the pathfinding of mitochondria-related molecular mechanisms in many pathologies, including cardiac ischemia/reperfusion (IR) injury, stroke, and nonalcoholic steatohepatitis (NASH). Interventional clinical trials investigating the therapeutic potential of targeting mitochondrial dysfunction are also witnessing a yearly increment discernible in the volume. Data for this figure was extracted from PubMed by searching the term “mitochondri*” in combination with either “medicine”, “transplantation”, “transfer”, “administration”, “delivery”, “restore”, “rescue”, “treatment”, or “therap*”. Data of active clinical trials (recruiting, not yet recruiting, active, not recruiting, completed, enrolling by invitation, unknown status) were acquired from ClinicalTrials.gov
In response to these challenges, advances in mitochondrial biology have spurred the development of mitochondria-targeted therapeutic strategies. There are abundant preclinical studies targeting mitochondrial defects, but clinical studies in humans are still scarce, showing great potential. These range from dietary interventions aimed at counteracting nutritional deficiencies to pharmacological approaches designed to directly modulate mitochondrial dynamics, enhance biogenesis, and mitigate oxidative stress. Notable interventions include: exercise protocols to promote the expression of peroxisome proliferator-activated receptor-gamma coactivator-1 alpha (PGC-1α), dietary supplements to target primary nutrient deficiency, nicotinamide riboside (NR) to augment nicotinamide adenine dinucleotide (NAD) biosynthesis26,27, MitoQ for neutralizing mitochondria-derived reactive oxygen species (ROS)28,29, the global antioxidant Coenzyme Q10 (CoQ10)30, N-acetyl cysteine (NAC)31, and the mitochondrial inhibitor ME-344 (known for its anti-tumor properties).32. These small molecular drugs can either exert their effects directly on the mitochondria or influence the organelle indirectly by binding to their cytosolic or nuclear regulators.1 The strategies of gene replacement therapies and gene editing technologies have also provided hope for addressing the severe prognosis of genetic mitochondrial defect, although the unique mitochondrial genome still entails a more comprehensive mechanistic understanding.33
Molecular components from mitochondria, owing to the similarity to their bacterial origins, have long been recognized as potent extracellular signals that elicit immune responses.34,35,36 Recent research into intercellular communication indicated that mitochondria could shuttle across cells horizontally via various transfer mechanisms, a process independent of the vertical inheritance seen during cell division.37 Importantly, this paradigm has revealed that mitochondrial components from healthy cells can precipitate a protective response to reconstitute mitochondrial bioenergetics in recipient cells that are metabolically compromised.38,39,40 Additionally, studies also demonstrated that damaged mitochondria were outsourced, as a new form of quality control, to tissue-resided macrophages for clearance in response to oxidative stresses and thus regulating tissue homeostasis.41,42 These findings have sparked interest in leveraging transplantation of functionally intact mitochondria and mitochondrial components as therapies for common diseases. Endeavors across various animal models and, even pediatric patients who received central extracorporeal membrane oxygenation (ECMO) support have substantiated the beneficial outcomes of these interventions.43,44,45,46 However, the clinical translation is still limited, including unstable mitochondrial vitality, inefficient cellular internalization, and transient therapeutic effects, which collectively hinder the medical practice.47
Here, as the therapeutic landscape for mitochondrial dysfunction continues to evolve, this review aims to provide a comprehensive view of mitochondrial signaling that controls cell fate, as well as the pathophysiology of mitochondrial dysfunction in common diseases. We also highlight the innovative approaches being pursued to improve clinical management of mitochondrial dysfunction through a critical examination of dietary supplements, pharmacological agents, and mitochondrial transplantation. A deeper understanding of mitochondrial contributions to common pathologies will further elucidate their roles in disease and may reveal co-dependent therapeutic targets.
Overview of mitochondrial functions and behaviors
Mitochondria, the cell’s proverbial powerhouses, present a multifaceted biological landscape in biosynthesis and intra-/inter-cellular signaling pathways. Within these pivotal roles lie a harmonized regulation of static integrity and dynamic processes, such as membrane potential homeostasis and mitophagy, mtDNA maintenance and protein synthesis, inter-organelle contacts and Ca2+ regulation, enzymatic activity, and metabolic signaling. Here, we summarize the mitochondrial overview of cell-dependent behaviors, structure basis, and fuel metabolism (See Fig. 2).

Schematic overview of mitochondrial activities. Mitochondria play a critical role in maintaining cell homeostasis by quality control, energy production, and metabolic regulation. Mitochondrial dynamics, including the processes of fission and fusion, are crucial for shaping mitochondrial structure, ensuring proper distribution across the cell, and facilitating selective clearance of damaged or dysfunctional mitochondria via degradative or secretory pathways, which plays a critical role in mitochondrial quality control. The core of mitochondrial energy production lies in the respiratory chain, fueled by the Krebs cycle and electron transport. The metabolism of fatty acids and glutamine into acetyl-CoA and alpha-ketoglutarate (α-kG), respectively, feeds into the Krebs cycle, culminating in ATP synthesis, illustrating the mitochondria’s central role in cellular energy and metabolic regulation
In cellular architecture, the mitochondrial network is influenced by quality control systems (biogenesis, dynamics, mitophagy, and proteolysis).48,49 The transcriptional activation of mitochondrial biogenesis is governed by synergistic interactions with nucleus. The upregulation of the proliferator-activated receptor-γ co-activator 1α (PGC1α) initiates the transcription cascade, activating transcription factors like nuclear respiratory factors (NRF-1, NRF-2) to the final expression of mitochondrial transcription factor A (TFAM), which promotes mtDNA transcription and replication.50,51 These regulatory proteins are often subject to regulation via post-translational modifications (PTMs), exemplified by the action of AMP-activated kinase (AMPK). As an energy sensor, AMPK is allosterically activated by fluctuations in the ATP:ADP or ATP:AMP ratios, which downregulates energy-intensive anabolic pathways while concurrently upregulating energy-generating catabolic processes to restore energetic homeostasis.52 In addition, 99% of mitochondrial proteins are encoded by nuclear genes and imported into the mitochondrial compartments through different sorting and import machineries, which are under surveillance by cytosolic ribosomes, the ubiquitin–proteasome system, chaperones, and intramitochondrial proteases.48,53 Once synthesized in the cytosol, these precursor proteins, bearing distinctive amino-terminal cleavable targeting motifs, are recognized and translocated by the translocase of the outer/inner membrane (TOM/TIM) complex, such as TOM20, TOM22, TOM40, and TIM23.54,55,56,57 These channels and transporters are modulated by various PTMs, including phosphorylation, oxidative modifications, ions, and metabolites binding, glycosylation, acetylation, and others. On the other hand, mitochondrial fission and fusion define mitochondrial morphology, distribution, and turnover, which are regulated by highly conserved proteins and organelles to ensure optimal mitochondrial function. Dynamin-related protein 1 (Drp1) facilitates mitochondrial fission by translocating from the cytosol to the outer membrane of mitochondria, where it binds to the adaptors (including MID49, MID51, mitochondrial fission factor) and oligomerizes in a GTPase-dependent fashion to form the fission foci and rings.58,59,60 The depletion of Drp1 and the adaptor proteins leads to the elongation of mitochondria.61,62 Preceding the recruitment and assembly of Drp1, the endoplasmic reticulum-mitochondria membrane contact sites were marked in close proximity to future fission sites, contributing to pre-constriction, nucleoid replication, and deep regulation of fission fates.61,63,64,65 Mitophagy is a specialized form of autophagy, dedicated to the selective clearance of aged or damaged mitochondria. This process is tightly regulated by a host of signaling pathways and proteins, most notably the PINK1-Parkin pathway. Upon mitochondrial damage or stress, PINK1 accumulates on the mitochondrial outer membrane, recruiting the E3 ubiquitin ligase Parkin.66,67 This subsequently triggers the ubiquitination of mitochondrial proteins, effectively marking these damaged components for removal. These ubiquitinated mitochondria are subsequently engulfed by autophagosomes, which coalesce with lysosomes, facilitating the systematic degradation and recycling of mitochondrial contents. In addition, the mitochondria-derived vesicle represented another form of selective degradation, which might be directed to lysosomes, peroxisomes, or extracellular milieus.68,69,70,71
Within mitochondria, the folded mitochondrial inner membrane (IMM) provides an extensive surface area of protein import and effective oxidative phosphorylation.72,73 Embedded in the IMM, the respiratory chain consists of a series of protein complexes (I to IV) and ATP synthase, collectively forming the core components of mitochondrial energy production. These complexes function sequentially to transfer electrons derived from NADH and FADH2 to form an electrochemical gradient. This electron transport culminates in the reduction of molecular oxygen to water. The reduction potential difference driving electron movement through complexes I, III, and IV is used to pump protons, which builds up a protonmotive force (Δp) across the IMM. The resulting electrochemical gradient, or proton motive force, drives ATP synthesis as protons flow back into the mitochondrial matrix through ATP synthase, thereby coupling electron transport with oxidative phosphorylation.74 Integral to this process are key metabolic substrates: pyruvate, fatty acid, and glutamine are metabolized to form acetyl-CoA and alpha-ketoglutarate (α-kG), respectively.75 These feed into the tricarboxylic acid (TCA) cycle, further fueling electron transport and ATP production.
Mitochondrial as signaling hubs in defining cell fate
Mitochondria epitomize an example of endosymbiotic integration, coordinating the cellular and organelle signal transduction critical for adaptive responses and evolutionary processes. Central to this coordination is the Mitochondrial Information Processing System (MIPS), a sophisticated mechanism that decodes complex amalgamations of ions, proteins, nutrients, and energetic states into targeted genetic programs.20 These programs, in turn, initiate a strategic reorganization of metabolic pathways, thereby catalyzing cell proliferation, differentiation, contractility, secretion, and apoptosis, foundational for organismal survival and evolutionary fitness. However, the mechanism by which these pathways intersect within mitochondria to sustain homeostasis is yet to be elucidated. Subsequent discussions will delve into the convergent roles of mitochondria in calcium homeostasis, the intricate networks of energy and nutrient sensing orchestrated by AMPK, mTOR, and Sirtuins, as well as the influence of endogenous mtDNA on innate immune pathways through the cyclic GMP-AMP synthase (cGAS) and stimulator of interferon genes (STING), inflammasomes, and membrane-bound Toll-like receptors (TLRs) signaling. (See Fig. 3) These topics collectively underscore the central role of mitochondria as arbiters of cellular function and integrity, highlighting their potential as therapeutic targets for modulating health and disease states.

Convergence of cell signaling pathways to mitochondria. This figure delineates the integration of diverse cell signaling pathways converging on mitochondria, illustrating their central role in cellular homeostasis and stress responses. Highlighted pathways include calcium homeostasis, crucial for mitochondrial function and energy production; energy and nutrient sensing through AMPK and mTOR signaling; the innate immune response mediated by cGAS-STING, inflammasomes, and TLR9 endosomal pathways; apoptosis regulation; the unfolded protein response (UPR) as a key element of mitochondrial stress response; and the induction of cellular senescence. Together, these pathways underscore the mitochondria’s pivotal role in orchestrating cellular adaptation and survival mechanisms
Calcium homeostasis
Calcium ions (Ca2+) are critical intracellular second messengers that orchestrate a wide array of cellular functions. Central to this role are mitochondria, which modulate Ca2+ signals through sophisticated mechanisms of uptake, storage, and efflux, particularly in response to changes in energy demand, metabolic shifts, and apoptotic signals.76,77 This intricate management of calcium allows mitochondria to control the activation of Ca2+-sensitive dehydrogenases within the Krebs cycle, subsequently enhancing ATP production to meet cellular energy demands.78 Increased mitochondrial Ca2+ is beneficial up to a point; however, chronic mitochondrial Ca2+ overload triggered Bcl-2-mediated regulated cell death, which is primarily mediated through the induction of the mitochondrial permeability transition pore (mPTP).79 In addition, although mitochondrial Ca2+ plays a minor role in reducing cytosolic Ca2+ concentrations in cardiomyocytes, their function is essential in buffering intracellular calcium levels in non-muscle cells with limited SERCA (sarcoplasmic/endoplasmic reticulum Ca2+-ATPase) and NCX (Na+/Ca2+ exchanger) activity.80 In some spatially constrained domains, such as the proximal ER-mitochondria contact sites (also termed ‘mitochondria-associated membranes, MAMs’), calcium release is concentrated, and regulated by PTMs.81,82 The structural basis of the calcium release-associated MAMs is a series of tethering complexes that contain both subdomains from ER and mitochondria, such as vesicle-associated membrane protein-associated protein B (VAPB) on the ER and protein tyrosine phosphatase interacting protein 51 (PTPIP51) on the mitochondria (i.e., VAPB-PTPIP51); the inositol triphosphate receptor (IP3R)/glucose-regulated protein 75 (Grp75)/voltage-dependent anion (VDAC1) and sporadic PD-related protein DJ1 complex; mitofusin-2 (Mfn2) on the ER with Mfn1/Mfn2 on the OMM; Rho GTPases Miro1 and Miro2; and others.83,84,85,86,87 In aged endothelial cells, the enhancement of MAMs is associated with increased mitochondrial Ca2+ uptake but also cell death.88
The OMM is highly permeable to Ca2+ ions, primarily due to the expression of VDACs which facilitate the direct and indirect calcium flow from the ER, SR, and lysosome.89,90 VDACs are differentiated into three isoforms—VDAC1, VDAC2, and VDAC3—each displaying tissue-specific distributions and capable of adopting various voltage-dependent conformations.91 Importantly, while VDACs enable Ca2+ flux across their open and closed states, the permeability for Ca2+ is significantly enhanced in the closed state, where there is concurrently reduced permeability to metabolites.92 Furthermore, advanced single cell imaging data indicate that Ca2+ may facilitate the VDAC-mediated cation and ATP transport across the OMM.93 Additionally, mitochondrial Ca2+ uptake is driven by the membrane potential difference (ΔΨ) generated by the activity of the respiratory chain, which is critical for driving the ingress of positively charged ions into the mitochondrial matrix.94 To reach the mitochondrial matrix, cytosolic Ca2+ has to cross the IMM, mainly through the mitochondrial Ca2+ uniporter (MCU) complex. The MCU complex is composed of the channel-forming MCU, the integral scaffolding protein, the essential mitochondrial response element (EMRE), and the mitochondrial calcium uptake (MICU) that senses matrix Ca2+ to inhibit or sensitize MCU-mediated Ca2+ uptake.95,96,97,98 Patch-clamp electrophysiology indicated that this inwardly rectifying channel bound Ca2+ with extremely high affinity (dissociation constant <2 nM), despite the relatively low cytoplasmic Ca2+ concentrations.99 However, Ca2+ does not remain inside mitochondria but instead is rapidly extruded into the cytoplasm through a complex system of Ca2+ antiporters, restoring the basal state. The Na+/Ca2+ exchanger NCLX localized to the IMM is an established NCX family protein mediating the efflux of matrix mitochondrial calcium.100 Deletion of Slc8b1 (encoding NCLX) in adult mouse hearts causes heart failure and sudden death, correlated with Ca2+ overload induced by mPTP.101 In addition, the H+/Ca2+ exchanger, such as leucine zipper, EF hand-containing transmembrane protein 1 (LETM1), leads to mitochondrial Ca2+ release in mammalian cells.102 The Letm1-deficient mice showed reduced Ca2+ mitochondrial uptake at a low cytosolic Ca2+ level, with impaired mitochondrial ATP generation capacity, disrupted early embryonic development, altered glucose metabolism, and increased susceptibility to seizure.103
Energy and nutrient sensing
AMPK
AMPK is considered the primary sensor of decreased cellular AMP to ATP and ADP to ATP and subsequently acts to increase catabolic reactions and decrease anabolic reactions. Thus, AMPK activity is tightly intertwining its function with mitochondrial health, including biogenesis and dynamics. AMPK relies on PGC-1α to significantly influence GLUT4 and mitochondrial gene expression in skeletal muscle, and it activates PGC-1α through direct phosphorylation at threonine-177 and serine-538.104 In addition, AMPK subunit β1 and β2 isoforms knock out mice are physically inactive and have a drastically impaired capacity for treadmill running that is associated with reductions in skeletal muscle mitochondrial content.105 Treatment with the AMPK agonist AICAR led to partial correction of COX deficiency in the defective cytochrome c-oxidase (COX) mouse models, and, importantly, significant motor improvement up to normal in knockout/knockin mice for Sco2.106 Disruptions in adiponectin and adiponectin receptor 1-induced AMPK-PGC-1α pathway contributed to mitochondrial dysfunction and insulin resistance observed in diabetes.107 The underlying mechanism by which AMPK promotes mitochondrial biogenesis is through activation of nuclear respiratory factor-1 (NRF-1).108 In subarachnoid hemorrhage (SAH)-induced early brain injury, AMPK and subsequent PGC-1α were activated by exogenous hsp22, thereby maintaining neurological function via the regulation of NRF1/TFAM-dependent mitochondrial biogenesis and Drp1-mediated.109 Additionally, mitochondrial quality control is also under the control of AMPK. Direct pharmacological activation of AMPK was sufficient to rapidly promote mitochondrial fission through the mitochondrial fission factor (MFF) phosphorylation, even in the absence of mitochondrial stress.110 The AMPK signaling pathway could also activate OPA1‐related mitochondrial fusion and mitophagy, thus attenuating cardiac I/R injury and relieving mitochondrial stress.111 Acute exercise-induced mitochondrial oxidative stress initiates AMPK activation, leading to phosphorylated unc-51 like autophagy activating kinase 1 (Ulk1)-dependent mitophagy in skeletal muscle.112 This was possibly due to the phosphorylation event of Parkin by ULK1 at Ser108 in its nine amino acid (“ACT”) domain at the early phase of AMPK activation.113 Recently, under energy stress conditions, AMPK could translocate to MAMs and directly interact with MFN2 to initiate mitochondrial fission and autophagy.114 In addition, in response to the low energy status in mitosis, the energy sensor AMPK phosphorylates and activates MCU, allowing Ca2+ entry into mitochondria to boost mitochondrial respiration.115 Thus, AMPK emerges as a central regulator of cellular energy balance, directly influencing mitochondrial function and dynamics, with implications for treating metabolic disorders and enhancing mitochondrial resilience against stress and disease.
mTOR
Mammalian TOR (target of rapamycin) is a serine/threonine protein kinase in the PI3K-related kinase (PIKK) family that coordinates eukaryotic cell growth, metabolism, and autophagy with environmental inputs, including nutrients and growth factors.116 Transcriptionally, mTOR controls the expression of mitochondrial genes (such as Cytochrome c, Atp5g, and Cox5a) by regulating the coactivation of yin-yang 1 (YY1) and PGC-1α.117 In addition, mTORC1 controls mitochondrial activity and biogenesis by selectively promoting translation of nucleus-encoded mitochondria-related mRNAs via inhibition of the eukaryotic translation initiation factor 4E (eIF4E)-binding proteins (4EBPs).118 On the other hand, mTOR activity is closely related to mitochondrial metabolism.119 Metabolomics profiling showed that mTOR independently regulated mitochondrial metabolism in regulatory T cells.120 The inhibition of rapamycin-sensitive mTOR complex 1 (mTORC1) also leads to a metabolic shift of mitochondrial respiration to aerobic glycolysis in leukemic cells, possibly in a Bcl-xl-VDAC1-dependent manner.121 A recent study reported that FOXK1 suppressed mitochondrial fatty acid oxidation in an mTORC1-dependent manner during the pathogenesis of nonalcoholic fatty liver disease (NAFLD) to non-alcoholic steatohepatitis (NASH).122 Additionally, mTORC1 is a regulator of mitochondrial dynamics and cell survival. A study revealed that the nutrient-sensing mTORC1 pathway facilitates DRP1’s pro-fission activity of mitochondria and apoptosis via promoting the translation of mitochondrial fission process 1 (MTFP).123 Inhibition of active mTOR sites leads to mitochondrial hyperfusion by reducing MTFP1 translation through the eIF4E-binding protein pathway, showcasing MTFP1’s pivotal role in mTORC1’s modulation of mitochondrial dynamics. In addition to mTORC1, mTORC2 could translocate to MAM and control mitochondrial metabolism via Akt-mediated phosphorylation of the IP3R, Hexokinase 2, and phosphofurin acidic cluster sorting protein 2 (PACS-2).124 Conclusively, mTOR orchestrates a complex regulatory network influencing cell growth, metabolism, and autophagy through its impact on mitochondrial gene expression, biogenesis, metabolism, and dynamics.
Sirtuin
Sirtuins serve as crucial sensors of the NAD+ to NADH ratio, closely linking them to cellular metabolism, stress response, and longevity. Mitochondrial sirtuins (SIRT3–5) are part of the sirtuin family of NAD+-dependent deacylases and ADP-ribosyl transferases, containing an N-terminal mitochondrial signal sequence that dictates the mitochondrial localization.125,126 SIRT3 interacts with ATP synthase and is indispensable in mitochondrial membrane potential restoration under mitochondrial homeostasis.127 SIRT3 knockout caused decreased levels of superoxide dismutase, catalase, and mitochondrial complex enzymes I/II/III/IV, which exacerbated mitochondrial damage in acute kidney injuries.128 Cisplatin-induced decline of SIRT3 also contributed to DRP1-dependent mitochondrial fission in cultured human tubular cells.129 However, under mitochondrial stress during cardiac pathology, SIRT3 promoted mitochondrial fusion through the direct deacetylation of OPA1 at the Lys926 and Lys931 residues and thus replenishing the GTPase activity of OPA1 in a NAD+-dependent manner.130 In addition, an adiponectin receptor agonist promoted AMPK/SIRT3 and increased the protein levels of Mfn2 and OPA1 with no effect on the expression of Drp1, thereby promoting mitochondrial fusion and memory deficits in P301S mice.131 SIRT4 could specifically hydrolyze lipoamide cofactors from the DLAT E2 component of the pyruvate dehydrogenase (PDH) complex, thereby inhibiting PDH activity regulation.132 SIRT5 supports NADPH homeostasis and antioxidant defense via promoting the desuccinylation and deglutarylation of IDH2 and G6PD, respectively.133 Collectively, these insights highlight the intricate roles of mitochondrial sirtuins in modulating mitochondrial function and integrity.
Innate immunity
Innate immune cells express distinct germ-line-encoded pattern recognition receptors (PRRs) that detect conserved pathogen-associated molecular patterns (PAMPs) unique to microbes.134 Alternatively, PRRs could also be activated by the endogenous molecules as dangerous signals called damage-associated molecular patterns (DAMPs) in inflammatory diseases.135,136 These molecules include mtDNA, mitochondrial ROS, adenosine triphosphate (ATP), TFAM, cardiolipin, cytochrome c, mitochondrial Ca2+, and iron.137 Exemplified as mtDNA, mtDNA itself can be recognized by three important PRRs of the innate immune system, cGAS-STING, cytosolic inflammasomes, and TLR9, which triggers type 1 interferon (IFN) responses and/or NF-κB and other signaling pathways to induce proinflammatory cytokines.135 Thus, these DAMP-sensing receptors may be valuable targets for therapeutic approaches.
cGAS-STING
cGAS (also known as male abnormal 21 domain containing 1 (MB21D1)) is a cytosolic DNA sensor mediating innate immunity. It catalyzes the synthesis of a noncanonical cyclic dinucleotide, 2’,5’ cGAMP, that binds to STING.138 Subsequently, cGAS mediates the activation of TANK-binding kinase 1 (TBK1) in Golgi and phosphorylates the transcription factor interferon response factor 3 (IRF3) which then translocates to the nucleus and initiates the transcription of the IFN and ISG.139 These have been comprehensively reviewed elsewhere.140,141,142
Compelling evidence underscored the critical role of mtDNA in activating cGAS-STING signaling. Moderate mitochondrial stress, precipitated by TFAM insufficiency, facilitated the translocation of mtDNA into the cytosol, subsequently activating the cGAS-STING-IRF3 signaling cascade.143 Such activation is instrumental in engendering a comprehensive antiviral defense mechanism via enhanced expression of a subset of interferon-stimulated genes (ISG). In addition, during the pathogenesis of dengue virus infection, IL-1R-mediated signaling is implicated in the inducement of endogenous mtDNA release, thereby initiating the transcription of IFNB1 and IFNL1 in a paradigm that is unequivocally dependent on the functional integrity of STING and IRF3 pathways.144 On the other hand, mtDNA-release-mediated cGAS–STING signaling is involved in the inflammatory activation. STING-deficient mice showed improved inflammation and acute kidney injury (AKI) progression, evidenced by the deactivated mtDNA-cGAS-STING axis and the decreased phosphorylated TBK1 and p65.145 Aged microglia affirmed prominent cytosolic accumulation of mtDNA abundance and nucleoids adjacent to the mitochondria outer membrane, which triggered VDAC-dependent cGAS and several type I IFN and proinflammatory genes.146 To date, the mechanism of mtDNA release in these settings is not clear. Mechanistically, several studies have reported MOMP is regulated by several pore-forming proteins, such as BAK and BAX, and VDAC. The BAK/BAX is able to facilitate the macropore formation, resulting in MOMP and mtDNA release. This process, however, was directly evidenced by IMM herniation and mitochondrial inner membrane permeabilization (MIMP) rather than mPTP, as the pharmacological inhibitor cyclosporin A (CsA) failed to block the mtDNA and nucleoid release.147,148 However, despite the mPTP typically only allows passage of components smaller than 1.5 KDa, mtDNA fragments still can pass through the VDAC oligomer pores and mPTP to activate cGAS-STING.149,150 Indeed, the inhibition of mPTP opening with CsA reduced the cytosolic pools of mtDNA by 30%–40%, and VDAC oligomerization inhibition with VBIT-4 reduced cytosolic mtDNA by 50–60%.151 Alternatively, the MOMP that mediates mtDNA release and type I IFN response may depend on the level of mitochondrial stress. In moderately stressed mitochondria, mtDNA fragments interacted with the positively charged residues in the N-terminal domain of VDAC1, promoting VDAC1 oligomerization to form pores on the OMM and release mtDNA.149 During this procedure, immune recognition of cGAS-STING to mtDNA may be also enhanced by ROS exposure due to the decreased susceptibility to TREX1-mediated degradation.152 In addition, a recent study reported that urolithin A-induced mitophagy attenuated cGAS/STING activation by free cytosolic mtDNA, and ameliorated deterioration of neurological function, although the mechanism is unclear.153
Inflammasome
NLRP3 (Nucleotide-binding domain, Leucine-Rich Repeat, and Pyrin domain-containing protein 3) serves as a multifaceted intracellular sensor, adept at recognizing an extensive spectrum of microbial epitopes, endogenous danger signals, and environmental provocateurs. This recognition precipitates the assembly and activation of a pro-inflammatory signaling platform known as inflammasome. The NLRP3 inflammasome includes an adaptor protein characterized by a caspase activation and recruitment domain (ASC) that harbors two protein interaction domains: an amino-terminal pyrin domain (PYD) and a carboxy-terminal caspase recruitment domain (CARD).154 This assembly facilitates the proteolytic cleavage and caspase-1-dependent mobilization of pro-inflammatory cytokines (pro-IL-1β and pro-IL-18), as well as gasdermin D-mediated pyroptotic cellular demise.154 Mitochondria are implicated as critical regulators within the NLRP3 inflammasome activation pathway. As an important intracellular signal, ROS mediated the activation of caspase-1 through NLRP3, and thus coactivating the translocation of mtDNA and cytochrome c into the cytosol subject to LPS and ATP stimuli.155 Besides, inhibited mitophagy/autophagy leads to mitochondrial ROS generation, prompting the translocation of NLRP3 and its adaptor ASC to mitochondria.156 Upon activation by NLRP3 activators, TLR leads to the production of oxidized mtDNA fragments through the IRF1-dependent transcription of CMPK2, a key enzyme in mtDNA synthesis.157 This mechanism establishes a feedforward loop that augments inflammation by intertwining NLRP3 activation, ROS generation, and mtDNA release.135 In addition to ROS-induced activation, recent metabolomics profiling has shown that the NLRP3 inflammasome activation benefited from ETC integrity through phosphocreatine (PCr)-dependent generation of ATP, despite the existence of unidentified targets within this pathway.158 Intriguingly, mitochondrial cardiolipin could bind to NLRP3 and activate inflammasome in a ROS-independent manner.159 Additionally, The interrelation of inflammatory pathways with mitochondrial functionality suggests a layered complexity; for example, mtDNA release induced by degenerative retinal pigmented epithelium activated caspase-4- (caspase-11 in mice) and caspase-1-dependent inflammasome, which required cGAS-dependent IFNβ production and gasdermin D–dependent IL-18 secretion.160 In a counter-regulatory manner, the antiapoptotic protein Bcl-2 is demonstrated to inversely modulate mitochondrial dysfunction and NLRP3 inflammasome activation, highlighting the intricate balance of mitochondrial engagement in the regulation of inflammatory responses.161
TLR9
The TLRs constitute a class of type I integral membrane receptors characterized by a conserved structural architecture that encompasses an N-terminal ligand-binding domain, a singular transmembrane helix, and a C-terminal intracellular signaling domain.162 These receptors are distinguished by their extracellular domains’ horseshoe-shaped conformation, pivotal for the ligand-induced dimerization that initiates innate immune responses.163 TLRs are synthesized within the ER, proceeding through the Golgi apparatus before their distribution to intracellular loci, where they are poised to recognize unmethylated CpG-DNA motifs, notably including mtDNA. The release of mitochondrial DAMPs into the circulation upon cellular injuries activates human polymorphonuclear neutrophils (PMNs) via TLR9, precipitating a sepsis-like systemic inflammatory response syndrome (SIRS).34 Additionally, the engagement of the circulating free mtDNA (cf-mtDNA)-TLR9 axis is implicated in the inflammatory progression of NASH, myositis, septic AKI, and chronic restraint stress.164,165,166,167 Interestingly, mtDNA could shuttle to recipient cells in a PINK1-dependent EV packaging mechanism to promote endosomal trafficking and invasiveness.168 The downstream signaling of TLR9 involves the adaptor protein myeloid differentiation primary response protein 88 (MYD88), which activates mitogen-activated protein kinases (MAPKs) and nuclear factor-kappa B (NF-κB), orchestrating an inflammatory cascade.169,170 Moreover, mtDNA facilitates endogenous TLR9 activation by translocation into the lysosomal compartment. Intriguingly, mtDNA that escapes autophagy-mediated degradation in cardiomyocytes can initiate TLR9-mediated inflammatory pathways, resulting in myocarditis and dilated cardiomyopathy independent of extracellular mtDNA release.171 In the context of podocyte injury within glomerular diseases, TLR9 engagement with endogenously accumulated mtDNA within endolysosomes mediated apoptosis via the p38 MAPK and NFκB signaling pathways, elucidating a complex interplay between mitochondrial components and innate immune activation mechanisms.172
Apoptosis
Mitochondria are indispensable regulators of cell death signaling, playing a central role in intrinsic apoptosis through the dissemination of critical soluble proteins including cytochrome c, Htra2, and Smac after MOMP.173,174,175 The binding of cytochrome c with apoptotic peptidase activating factor 1 (APAF1) engenders the assembly of a pivotal multi-protein complex known as the apoptosome, heralding the initiation of the apoptotic cascade.176,177 This apoptotic induction is predicated upon the activation and subsequent oligomerization of pro-apoptotic proteins BAK and BAX, facilitating pore formation and cytochrome c release. Contrastingly, the extrinsic apoptotic pathway intersects with mitochondrial-mediated apoptosis through the caspase-8-facilitated cleavage of BID, a pro-apoptotic BH3-only constituent of the BCL-2 protein family. This enzymatic processing yields a truncated form of BID that robustly instigates MOMP, thereby irreversibly committing the cell to apoptosis. Regulation against MOMP, and consequently apoptosis, is chiefly exerted by the BCL-2 protein family’s anti-apoptotic members, including BCL-2, BCL-W, BCL-X_L, A1, and MCL1, which serve to preserve cell survival by inhibiting pro-apoptotic mechanisms.178,179,180
Intriguingly, it has been observed that the apoptotic machinery renders mitochondrial cGAS immunologically quiescent during apoptosis and subsequent apoptotic clearance.181,182 Nonetheless, this regulatory schema is not absolute. Recent empirical findings elucidate that a partial induction of MOMP, orchestrated by BAK/BAX under sublethal apoptotic stress in an APAF1-dependent manner, fails to completely abrogate cGAS–STING signaling.183 Instead, it permits the activation of the senescence-associated secretory phenotype (SASP) in senescent cells, indicating a refined interplay between apoptotic signaling pathways, mitochondrial integrity, and immune response modulation. This nuanced interaction underscores the intricate balance maintained by cellular mechanisms between death signaling and immune regulation, providing a fertile ground for further investigative endeavors into the implications of such mechanisms in cellular senescence and disease pathogenesis.
Mitochondrial dysfunction and related diseases
Milestone of research history for mitochondrial dysfunction and therapeutics
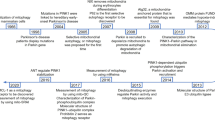
The historical narrative of mitochondrial exploration has roots extending to the 19th century, a period characterized by the biological paradigm of ‘bioblasts’ that described a cytoplasmic, bacteria-like structure of ubiquitous occurrence, functioning as ‘elementary organisms’. In 1898, Benda termed these structures as ‘mitochondrion’, which evolved from the Greek words, ‘mitos’ (thread) and ‘chondros’ (granules).184 Yet, the elucidation of the biochemistry of mitochondria remained silent, that must ‘find a way to make direct chemical analyses of mitochondria’.185 A significant breakthrough was achieved in 1934 through Bensley’s pioneering approach of employing differential centrifugation to isolate mitochondria from guinea pig liver.186 Subsequent decades were characterized by an elaborate delineation of the morphological and biochemical attributes of isolated mitochondria.187,188,189,190,191 An abundance of empirical evidence has demonstrated a compelling linkage between mitochondria and the process of electron transport and oxidative phosphorylation. Importantly, Kennedy’s seminal research revealed the mitochondrial localization of tricarboxylic acid (TCA) cycle intermediates, aerobic oxidation mechanisms of fatty acids, and enzymes implicated in oxidative phosphorylation.192 Nevertheless, it was not until 1961 that the elusive electrochemical nature of oxidative phosphorylation was expounded by Peter Mitchell’s ‘chemiosmotic coupling’ theory.193 After this revelation, mitochondria regained focus in the late 1990s due to the regulation of cellular apoptosis, particularly through the cytochrome c and Bcl-2 protein family.194
The early trajectory of mitochondrial medicine was anchored on the correlation of clinical symptoms with aberrant mitochondrial biochemical signatures and morphological anomalies, as demonstrated in the foundational reports on Luft and Leigh disease.195,196 The sequence of the mitochondrial genome in 1981 was a pivotal moment that authenticated the contribution of mtDNA mutations to human pathology.4 In the wake of this milestone, the first mitochondrial disorders were characterized, such as Leber’s Hereditary Optic Neuropathy (LHON) due to missense mutations, and Kearns-Sayre Syndrome (KSS) as a result of considerable mtDNA deletions.197,198 Subsequent investigations continued to uncover a strong correlation of mitochondrial genetic anomalies with degenerative diseases, aging, and cancer.199 This genetic complexity is fueled by diverse mtDNA mutations, intricate phenotypic manifestations, threshold effects, and widespread exceptions. Still, specific mtDNA mutations have been successfully correlated to their clinical phenotypes, including diseases like MELAS, CPEO, and maternally inherited diabetes and deafness (MIDD).200 Additionally, nuclear DNA (nDNA) has been elucidated to function directly on the maintenance, translation, and protein synthesis of mtDNA, further convoluting the inheritance patterns of mitochondria.201 In 2008, the introduction of a comprehensive mitochondrial proteome inventory - MitoCarta, marked another milestone, and it has now evolved to its 3.0 version.202,203
Pathophysiology of mitochondrial dysfunction in common diseases
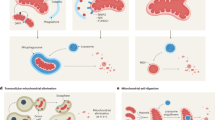
While the precise molecular mechanisms at play may differ depending on the disease context, common mitochondrial functions and behaviors include compromised bioenergetics, augmented oxidative stress, dysregulated calcium homeostasis, and modifications in mitochondrial dynamics along with quality control mechanisms. (See Fig. 4) In the following sections, we will elaborate on concrete disease examples.

Mechanisms of mitochondrial dysfunction in disease pathogenesis. This figure encapsulates the diverse mechanisms through which mitochondrial dysfunction contributes to common diseases. It illustrates six key dysfunctional processes: 1) Reverse Electron Transport: high Δp fuels the excessive ROS production during ischemia/reperfusion, leading to potential mtDNA damage; the accumulation of misfolded proteins and mistranslated respiratory complexes during AD and PD; imbalanced mitochondrial dynamics, mediated by ROS, Ca2+, and Aβ, affect mitochondrial morphology and function; various permeability transition pores lead to mitochondrial cargo release and initiate cell death; disrupted metabolism leading to energy imbalance and exacerbate insulin resistance; and mitochondrial genome instability resulting in altered gene expression and mitochondrial failure. Together, these mechanisms underscore the central role of mitochondria in cellular health and the etiology of various diseases, highlighting potential therapeutic targets for mitigating mitochondrial dysfunction
Ischemic/reperfusion injuries
Clinically, merely modulating energy balance by decreasing ventricular strain or heart rate might not rectify existing ventricular remodeling.22 Therefore, it’s imperative to target mitochondrial dysfunction, given its central role in cardio-cerebrovascular energy depletion and its influence on phenotype enhancement. Ischemia-reperfusion (IR) injury triggers a multifaceted series of metabolic and immune responses that ultimately lead to cell death and tissue remodeling. In the neural context, acute deprivation of oxygen and glucose swiftly impairs neuronal sodium (Na+) ion channels, precipitating cell swelling, degeneration, and necrosis.204 Concurrently, ischemic conditions activate the hypoxia-inducible factor (HIF), which engages in dual protective and damaging roles. By interacting with the von Hippel-Lindau (VHL) gene product, HIF inhibits proteases, while simultaneously initiating pro-inflammatory and apoptotic pathways via nuclear factor κB (NF-κB). This inflammatory response could exacerbate the hypoxic state by increasing metabolic demand.205 Moreover, hypoxia triggers HIF-1-dependent mitophagy, leading to a decrease in metabolic substrates, accumulation of metabolic waste, and dysregulation of calcium homeostasis.206 These changes collectively disrupt OXPHOS, diminishing ATP production and compromising mitochondrial permeability transition (MPT).17,207,208
Upon reperfusion, the abrupt restoration of blood flow introduces a surge of ROS, the key mediators of acute metabolic disturbance and the subsequent sterile inflammatory response.204,209 ROS contributes to cell and tissue impairment through lipid peroxidation within cell membranes and organelles, DNA oxidation, and the activation of matrix metalloproteinases and calpains.210,211 In addition, ROS could interact with nitric oxide (NO), fatty acids, or free iron, giving rise to highly reactive molecules such as peroxynitrite, peroxyl radicals, and hydroxyl radicals, which amplify cell death. Given the mitochondria’s role as the primary source of ROS in early reperfusion, their involvement is central to the pathophysiological mechanisms driving tissue injury in IR events, underscoring the critical role of mitochondrial dynamics in the etiology of reperfusion injuries.
Reverse electron transport (RET)
RET is a fundamental mechanism in ROS production during IR, involving the backward flow of electrons driven by an elevated proton motive force (Δp) and/or a reduced ubiquinone pool. This reverse flow leads to the incomplete reduction of molecular oxygen, resulting in the formation of reactive intermediates such as superoxide anion (O2−), hydrogen peroxide (H2O2), and the hydroxyl radical (HO•).212 In the mitochondrial electron transport chain, 11 potential sites have been delineated where electron leakage may occur, generating superoxide (O2-) and/or hydrogen peroxide (H2O2).213 Notably, the IQ site in Complex I and the IIIQo site in Complex III are significantly active, with Complex I identified as the predominant source of superoxide during the reperfusion phase.17,213 In the ischemic state, there is a reversal in the function of the succinate dehydrogenase complex (Complex II), which, by utilizing aspartate as a carbon source, increases the biosynthesis of succinate.17 This accumulated succinate is then swiftly oxidized within 5 min of reperfusion, sustaining a reduced ubiquinone pool and a high Δp. This scenario sets the stage for electrons to be driven back to Complex I in the reverse direction, thereby facilitating the reduction of NAD+ to NADH and thus promoting superoxide formation. Small molecules targeting suppressors of site IQ electron leak (S1QELs) during RET could reduce ROS production in primary astrocytes, caspase signaling in vivo, and IR injuries in mouse hearts.214 The complex I was thus established as the therapeutic target in the early phase of reperfusion, which provided insights to integrate the molecular mechanisms and pathways contributing to IR injury.215
Ca2+ overload and MPT
Under physiological conditions, mitochondria orchestrate calcium dynamics through the interaction with the sarcoplasmic/endoplasmic reticulum (SR/ER), which is crucial for maintaining cardiomyocyte bioenergetics and calcium homeostasis.216 During the onset of coronary artery occlusion, regional hypoxia switches mitochondrial OXPHOS to anaerobic metabolism, leading to rapid ATP exhaustion and cardiac contractile failure. This metabolic alteration, accompanied by the decreased function of SERCA and NCLX, perturbates mitochondrial Ca2+ homeostasis in the failing heart.217,218,219 When proceeding to the reperfusion phase, SR Ca2+ leak via type 2 ryanodine receptor (RyR2) channels triggers mitochondrial Ca2+ overload and exacerbates mitochondrial dysfunction and ROS production.220 Consequently, intracellular and mitochondrial Ca2+ overload activated the mitochondrial chaperone cyclophilin D (CypD), provoking cardiomyocyte death through mitochondrial permeability transition pore (MPTP) opening and excessive cardiac contraction.221 Overexpression of the mitochondrial Na+/Ca2+ exchanger (NCLX) could enhance Ca2+ clearance, reduce permeability transition, and prevent myocardial cell necrosis and heart failure due to ischemia. This approach was validated in vivo where ischemic preconditioning coupled with increased NCLX expression reduced infarct size by 43%, highlighting the therapeutic potential of modulating mitochondrial calcium for cardiac protection.101
Imbalanced mitochondrial dynamics
Mitochondrial fission is integrated by the translocation and binding of fission proteins, which are critical for stress resistance or apoptotic promotion. In adult cardiomyocytes, mitochondria exist as abundant and dispersed round monomers and exhibit low kinetics.222 However, their functional integrity is indispensable for cardiac activity, given the heart’s energy-intensive nature. Proper regulation of Drp1 is essential for cell health, illustrating the balance that exists between physiological resistance and apoptosis.223 For instance, while the physiological downregulation of Drp1 might be adaptive under specific conditions, its overexpression, especially in pathological states like glucose deprivation, can trigger apoptotic events in cardiomyocytes.224 In addition, genetic anomalies in mitochondrial fission and fusion processes can be detrimental during IR, leading to compromised mitophagy. This can, in turn, exacerbate cardiac dysfunction. Notably, there’s a noted association between dysfunctional mitochondrial dynamics and increased vulnerability of cardiomyocytes to IR.224 In studies utilizing the ex vivo Langendorff model and the in vivo LAD occlusion model of IR, inhibiting the interaction between Drp1 and Fis1, a key mediator of mitochondrial fission, yielded insightful results. These included decreased mitochondrial fragmentation and reduced release of cytochrome c (cyt c) - a marker of mitochondrial health and initiator of the intrinsic apoptotic pathway. Furthermore, a decline in ROS production was observed, which is crucial given the role of ROS in exacerbating IR injury. The pharmacological inhibition of Drp1-Fis1 interaction not only improved cardiac function but also effectively attenuated apoptosis and autophagy, as evidenced by decreased interactions with caspases and reduced LC3-II levels.225
Neurodegenerative diseases
Neurodegenerative diseases manifest region- and disease-specific alterations in brain energy metabolism that progress over time.226 These metabolic changes are characterized by decreased glucose uptake, impaired tricarboxylic acid (TCA) cycle function, deficiencies in oxidative phosphorylation (OXPHOS), and reduced energetic support provided to neurons by astrocytes and oligodendrocytes. Notably, these perturbations are parallel to mitochondrial dysfunctions, such as genetic defects, kinetics imbalcances, calcium dysregulation, and proteinopathies.1,227,228 These mitochondrial pathologies are prevalent across a range of neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD), and frontotemporal dementia (FTD).229 Due to the complex mitochondrial dysfunction and multifaced phenotypes of neurodegenerative diseases, it remains challenging to establish whether mitochondrial dysfunctions act as primary causes, consequences, or merely contributing factors in the progression of these diseases. Recent advancements in this field, however, are shedding light on potential underlying mechanisms.
Impaired mitochondrial proteostasis
Alzheimer’s Disease (AD), a prototypical neurodegenerative disorder characterized by initial synaptic failure and progressive loss of cholinergic neurons, manifests as memory and cognitive decline.230,231,232 As the hallmarks of AD’s pathology, amyloid-β (Aβ) accumulation and tau aggregation are associated with multifaceted mitochondrial disruption in respiration and dynamics.233,234 Aβ protein triggered mitochondrial fission, synaptic loss, and neuronal damage, at least partially via S-nitrosylation of Drp1 to activate GTPase activity.235 The binding of Aβ with mitochondria-localized Aβ-binding alcohol dehydrogenase underlies the neurotoxic mechanism, which induces mitochondrial ROS production, cytochrome c release, and ultimate cell death.236 On the other hand, the study revealed that Aβ peptides disrupted the turnover of peptides by inhibiting Cym1/PreP, a key mitochondrial metallopeptidase responsible for degrading mitochondrial targeting presequence peptides, resulting in enhanced protein degradation and widespread dysproteostasis in AD.237 Concurrently, amyloid precursor protein-derived C-terminal fragments (APP-CTFs) independently impaired mitochondrial morphology and were associated with a Parkin signature of mitophagy failure in human post-mortem AD brains.238
The mitochondrial cascade hypothesis, particularly relevant to sporadic and late-onset forms of AD, posits that accumulated oxidative stress reconfigures the cellular response to toxic proteins.239 This reconfiguration manifests as age-associated phenotypes driven by deficient cellular clearance mechanisms and disrupted cell cycle processes, highlighting the centrality of mitochondrial integrity in the progression of AD. Experimental models, including transgenic C. elegans models expressing human Aβ and tau proteins, have pinpointed defective mitophagy as a key pathological event in AD, suggesting it as a viable target for therapeutic interventions aimed at memory deficits.240 Furthermore, mitochondria may offer a protective response against Aβ-induced proteotoxic stress through the mitochondrial unfolded protein response (UPRmt) and enhanced mitophagy, slow the progression of AD, and improve lifespan by stabilizing mitochondrial proteostasis.241 Collectively, these insights not only illuminate the multifaceted roles of mitochondria in AD pathology but also suggest novel therapeutic strategies focused on restoring mitochondrial function and enhancing proteostasis.
Parkinson’s Disease (PD) is a progressive neurodegenerative disorder marked by the loss of specific neurons, including dopaminergic cells, where mitochondrial dysfunction frequently emerges as a contributing factor.242 This dysfunction is observed across genetic aberrations of the disease, including autosomal dominant cases with SNCA (alpha-synuclein) mutations and copy number variations, and autosomal recessive early-onset forms involving mutations in PARK2 and PARK6 (parkin) genes.243,244,245,246 Moreover, elevated mtDNA mutations linked to respiratory chain deficiencies are prevalent in neurons particularly vulnerable in PD and in the general process of brain aging, suggesting a possible etiological role of nucleotide pool imbalances and alterations in one-carbon metabolism pathways.228,247 The aggregation of alpha-synuclein (α-Syn), a defining feature of PD’s neuropathology through Lewy body formation, further compounds these mitochondrial challenges.248,249 Familial and sporadic PD cases often exhibit a dose-dependent mitochondrial accumulation of α-Syn aggregates, leading to the formation of toxic soluble oligomers.250,251 These oligomers provoke excessive ROS production and neuronal cell death, with the rate of these deleterious reactions accelerating due to increased oligomer formation and a breakdown in mitochondrial defensive mechanisms.252 Though this structural turnover is remarkedly slow due to the kinetic barrier, the reaction speed would sharply increase with enhanced aggregation, formation of β sheet oligomers, and dysregulation of compensatory mechanisms.252,253
At the molecular level, mitochondrial membrane constituents such as cardiolipin, ATP synthase, TOM20, and VDAC are known to interact with aggregated α-Syn and thus exacerbating mitochondrial dysfunction.254,255,256,257 This interaction notably impairs Complex I activity and leads to an overproduction of mitochondrial ROS, which in turn results in reduced ATP production and premature opening of MPTP, leading to neuronal apoptosis.257 This apoptotic cascade could potentially be attributed to a compromised protein import machinery caused by disrupted interaction with TOM22.256 However, the role of impaired Complex I function as a primary contributor to Parkinsonism is contentious. Conditional knockout of Ndufs4 (subunit of mitochondrial complex I) failed to recapitulate the loss of dopaminergic neurons or the motor deficits throughout the lifespan of mice, suggesting that Complex I dysfunction may not universally lead to parkinsonian symptoms.258 Conversely, recent findings from studies using Ndufs2-deficient mice indicate that Complex I dysfunction alone can precipitate a progressive, human-like parkinsonism phenotype, likely due to an early decline in axonal function driven by a shift in mitochondrial metabolic programming.259 This nuanced view underscores the complexity of mitochondrial involvement in PD and highlights the potential for therapeutic strategies targeting mitochondrial proteostasis to mitigate disease progression.
Aging
Aging is a complex universe that involves organismal ageing and cell senescence. The spectrum of mitochondrial-related aging signatures includes mtDNA mutation and enhanced methylation, genomic instability, decreased mitophagy, and compromised mitochondrial biogenesis.25,260 Both mtDNA point mutations and deletions in somatic cells have been shown to accumulate with age, thereby driving energy exhaustion and oxidative damage. In mice with mutated mtDNA polymerase gamma (PolgD257A/D257A), decreased life span, mitochondrial content, and activity of ETC complexes, as well as apoptotic activation were observed.261 Meanwhile, oxidants generated by mitochondria are considered the major source of the oxidative lesions that accumulate with age.262,263 In the past, the ‘free radical theory of ageing’ posits that biological aging results from the production of ROS.264 A case in point is the effective function of antioxidant enzymes in aging. Superoxide dismutase (SOD2) within the mitochondrial matrix is a critical antioxidant enzyme, mitigating oxidative stress by catalyzing the conversion of superoxide anions (O2-) into hydrogen peroxide (H2O2). In mice model of MnSOD+/− mice and ALDH-2−/− mice (aldehyde dehydrogenase), the assessment of acetylcholine-dependent vascular relaxation reveals a pronounced induction of vascular oxidative stress associated with aging, directly attributable to the compromised antioxidant defense due to partial SOD2 deficiency.265 Recent identification of five biomarkers in human plasma that were associated with the senescence-associated secretory phenotype (SASP) and an increased risk of clinical death, involving GDF15、RAGE、VEGFA, PARC, and MMP2.266 These hallmarks of biological and metabolic activity decline are often intertwined with mitochondrial dysfunction.267 Besides, ROS is also a crucial factor triggering senescence.268 In Sod2−/− mice, accumulated senescent cells and impaired complex II activity were observed.265 A reduction in mitochondrial ADP sensitivity during aging also increases mitochondrial H2O2 emission and contributes to age-associated redox stress.269 However, studies associated with age-dependent mitochondrial dysfunction engender controversial evidence in identifying the deleterious effect on longevity.270 Heterozygous Sod2+/− mice showed increased oxidative stress, as indicated by inactivation of ROS-sensitive enzymes, higher sensitivity to oxidative stress, impaired mitochondrial respiration, and higher levels of DNA oxidative damage in both the nucleus and mitochondria.271 Despite this, Sod2+/−mice appeared a wild-type lifespan. Recent advancement in understanding the aging process involves the role of apoptosis-induced changes in mitochondrial outer membrane permeability (MOMP), which have been found to facilitate the SASP through the release of pro-inflammatory mtDNA.183 This process is initiated by the activation of the pro-apoptotic proteins BAK and BAX, leading to IMM herniation and the subsequent release of mitochondrial matrix constituents, notably mtDNA and TFAM.147 Using the small-molecule BAX inhibitor BAI1 to inhibit MOMP could counteract age-associated sterile inflammation and improve lifespan.183 Furthermore, the relationship between mitochondrial dysfunction and lifespan is complex, possibly subject to a phenotype threshold or point of irreversibility. Intriguingly, increased oxidative stress resulting from the deletion of sod genes in C. elegans does not necessarily shorten lifespan; in some cases, it has been observed to extend it by altering mitochondrial function.272 This finding challenges the conventional understanding of oxidative stress and aging. Severe mitochondrial defects and reduced lifespan caused by the global deletion of ubiquinone (UQ) could also be reversed by restoring UQ levels.273 Together, these point to an uncoupled link between mitochondrial dysfunction and lifespan.
Metabolic syndrome
The indispensable involvement of mitochondria permeates numerous aspects of cellular metabolism, including the processing of diverse substrates like glucose, lipids, and amino acids.274 Metabolic syndrome refers to a cluster of metabolic disturbances including obesity, insulin resistance, dyslipidemia, and hypertension, and is associated with complications such as thrombosis, inflammation, cardiovascular disease, and NAFLD.275,276,277 The current evidence base has suggested that aberrations in mitochondrial biogenesis, lipid metabolism, and OXPHOS were involved in the pathogenesis of metabolic syndrome.277,278,279,280,281 Furthermore, alterations in mitochondrial dynamics and quality control mechanisms, as well as increased ROS production, contribute to a vicious cycle that exacerbates both mitochondrial dysfunction and metabolic syndrome.282,283 Mitochondrial impairments in key tissues such as adipose, skeletal muscle, and liver lead to systemic consequences, including insulin resistance and disrupted lipid homeostasis, which can manifest as components of metabolic syndrome.284,285,286 In light of these findings, targeting mitochondrial pathways may represent a promising therapeutic strategy to mitigate the pathological consequences of metabolic syndrome.
Insulin resistance
Insulin-resistant individuals exhibit impaired metabolism or tolerance to glucose challenge through obesity, sedentary lifestyle, high-fat diet, or genetic factors.276 Mitochondrial dysfunctions have been intricately linked to the development of insulin resistance, which is a key feature of metabolic syndrome. Metabolomic studies have provided detailed insights into the molecular mechanisms involved in this process. In skeletal muscle, decreased mitochondrial content and/or mitochondrial dysfunction led to a reduction in mitochondrial fatty acid oxidation, along with increased levels of intracellular diacylglycerol (DAG) and acyl CoA.287 These fatty acid derivatives activate a serine kinase cascade that augments the phosphorylation of Ser/Thr residues in insulin receptor substrate-1 (IRS-1), resulting in the inhibition of translocation of glucose transporter type 4 and glucose uptake.288,289,290 In the liver, mitochondrial dysfunction exacerbates hepatic insulin resistance and promotes the development of NAFLD by disrupting lipid metabolism, increasing mitochondrial uncoupling, and inducing pro-inflammatory cytokines.291 In skeletal muscle, mice lacking malonyl-CoA decarboxylase (MCD) showed reduced fat catabolism and resistance to diet-induced glucose intolerance, indicating a strong connection between skeletal muscle insulin resistance and lipid-induced mitochondrial stress.292 Accumulated intermediates in mitochondria from human muscle with insulin resistance, such as acylcarnitine and ceramide, also suggested the insulin resistance-associated stress pathway either by a deficiency in or an overload of mitochondrial oxidative capabilities.292,293 Acute and chronic consumption of a high-fat diet significantly enhanced the release of mitochondrial ROS, shifting to a more oxidized state and leading to insulin resistance within skeletal muscle.294 In addition, ROS signals could be secondary to respiratory alterations-induced insulin sensitivity when subjected to DAG accumulation, PKCθ activation, and impaired muscle insulin signaling.295 These mechanisms suggested that decreased mitochondrial function is regulated by damaging effects caused by nutrient excess and signal impairment.296 Moreover, it was also demonstrated that the impact of impaired mitochondrial dynamics and quality control mechanisms, such as mitophagy, on insulin resistance and metabolic syndrome.283 Increased Drp1 expression and mitochondrial fission were associated with impaired mitochondrial function, characterized by reduced ATP production and increased ROS generation, which contributed to impaired insulin signaling in skeletal muscle cells.297 Conversely, inhibiting Drp1 activity and reducing mitochondrial fission improved mitochondrial function and restored insulin signaling. Collectively, these findings underscore the complex interplay between mitochondrial dysfunction, lipid and amino acid metabolism, and insulin signaling, emphasizing the need for a multifaceted therapeutic approach to address the diverse aspects of metabolic syndrome.
Non-alcoholic fatty liver disease (NAFLD)
NAFLD is the most prevalent form of chronic liver disease globally and is closely associated with metabolic syndrome.298 This metabolic perturbation in NAFLD is characterized by the abnormal accumulation of carbohydrates and fats in the liver, often driven by processes such as the transformation of excessive fructose intake into triglycerides through de novo lipogenesis.299 Additionally, insulin resistance in skeletal muscle and liver also contributes to this metabolic shift towards lipogenesis.300,301 As NAFLD progresses to nonalcoholic steatohepatitis (NASH), mitochondria evidence a distinct shift to metabolic maladaptation, indicated by ultrastructural damage, compromised ETC activity, upregulated uncoupling protein content, and increased ROS production.302 The uncoupling of OXPHOS and increased ROS generation further compromise mitochondrial ETC function, thereby exacerbating ROS formation. In addition, the hepatocyte mitochondria’s capacity to sense overnutrition may precipitate a reverse cascade of proinflammatory signaling through the release of plasma mtDNA and subsequent activation of the intracellular Toll-like receptor 9 (TLR9) pathway.164 Interestingly, in obese rodent models, a decrease in mitochondrial functions, including carnitine palmitoyl-CoA transferase-1 (CPT1) activity, fatty acid oxidation, and cytochrome c protein content, were observed preceding the onset of insulin resistance and NAFLD.303 In contrast, in mice fed with an 8-week high-trans-fat high-fructose diet (TFD), changes in mitochondrial energetics and TCA flux lagged behind hepatic insulin resistance.304 High-resolution respirometry studies showed that obese human individuals, both with and without NAFLD, exhibit 4.3- to 5.0-fold higher maximal respiration rates of isolated mitochondria compared to lean individuals, yet these rates were 31–40% decreased in NASH individuals.291 Recent research also highlighted the role of autophagy-related gene 3 (ATG3) in steatosis during NAFLD progression, with studies indicating that knock-down of hepatic ATG3 could improve fatty acid metabolism and enhance mitochondrial complex activity, partly by reducing c-Jun N-terminal protein kinase 1 activity.305
Therapeutics for mitochondrial dysfunction
Dietary supplements and diet adjustments
Patients suffering from PMD confront an elevated risk of malnutrition, which is considered as a consequence of metabolic challenges.306,307 Similarly, several cellular dysfunctions in SMD associated with mitochondrial pathophysiology-such as escalated catabolic demands, bioenergetic depletion, calcium overload, and oxidative stresses-are known to result in deficiencies in both macro and micro-nutrients.308,309 Instances might lead to diminished active nutrient absorption where the disease pathology or therapeutic management involves gastrointestinal obstructions, thereby exacerbating the risk of malnutrition.310,311 It is well-established that mitochondrial activity relies heavily on a variety of essential nutrients, including cofactors and antioxidants that maintain bioenergetic production.308 Consequently, dietary supplementation strategies devised to counter mitochondrial dysfunction offer diverse nutritional options, including carnitine, CoQ, creatine, and vitamin B2. Carnitine is involved in the transport of long-chain fatty acids to mitochondria, and offers cellular protection by restricting the accretion of these fatty acids through binding with acyl-CoA.312,313 Ubiquinone or CoQ facilitates the electron transport from Complexes I and II to Complex III, while concurrently providing antioxidant protection to cell membranes and lipoproteins by suppressing lipid peroxidation.314 Creatine, by forming a complex with phosphoric acid, enables the replenishment of ATP, thus serving as an energy buffer reservoir in skeletal muscle.315 Riboflavin or Vitamin B2, acting as a cofactor for the electron transport chain, demonstrates the capacity to improve mitochondrial Complex I and II, and enzymatic activity upon supplementation.316
Human metabolism harnesses ketone bodies to provide alternative energy to various organs during periods of limited glucose availability.226,317,318 These ketone bodies - acetoacetate, β-hydroxybutyrate (βOHB), and acetone - are predominantly synthesized in the liver’s mitochondrial matrix through fatty acid oxidation. The ketogenic diet (KD), characterized by high fat and low carbohydrate content, simulates the metabolic state akin to prolonged fasting. Preclinical studies have highlighted the protective role of both ketogenic diets and exogenous supplementation of ketone body, showing benefits in conditions such as intractable childhood epilepsy, ischemic stroke, AD, PD, MELAS, advanced heart failure, and spinal cord injury.318,319,320,321,322,323,324,325 The potential mechanism involves increased ketolysis leading to oxidative stress in mitochondria, which triggers an adaptive cellular response.317 This hormetic response is characterized by the activation of key cell-defensive regulators like Nrf2, sirtuins 1 and 3, and AMPK, leading to upregulation of genes and pathways associated with antioxidative and anti-inflammatory responses, enhanced mitochondrial function and biogenesis, DNA repair, autophagy, and reduced anabolic energy expenditure. Notably, decanoic acid, a component of the ketogenic diet, acts as a peroxisome proliferator activator receptor γ (PPARγ) agonist, significantly enhancing mitochondrial citrate synthase and complex I activities in neuronal cell lines (SH‐SY5Y).326 However, extended exposure to KD has been linked to cardiac fibrosis in rats, a process involving the activation of Sirt7, which in turn inhibited the transcription of genes encoding mitochondrial ribosomes, subsequently impeding mitochondrial biogenesis.327
Despite the overall low risk, only a few studies exploring the use of dietary supplementation and structure adjustments have thus far adopted a randomized controlled trial (RCT) design, with most remaining as open-label studies. This is understandable considering the challenges associated with subject recruitment and financial costs due to the heterogeneity of PMD and SMD. Consensus on the clinical management, care, and refined administration of empirical interventions based on dietary supplements is accessible elsewhere.328
Targeted treatment using pharmacological agents
The transition of mitochondria from adaption to a dysfunctional state involves their dual roles: providing energy for cellular and organ function and acting as signal hubs to coordinate global cellular feedback. As the landscape of mitochondrial pathophysiology continues to evolve in common diseases, the spectrum of pharmacological targets for the treatment correspondingly expanded. Several intervention strategies are currently in focus: targeting the redox state of mitochondria, stimulating mitochondrial biogenesis, and modulating mitochondrial dynamics. Pharmacological agents corresponding to these targets are currently under investigation in active and randomized clinical trials (See Table 1). Regardless of the specificities of interventions in patients with PMD and SMD, current therapeutic approaches predominantly favor symptomatic treatment. Small molecule therapies have also progressed significantly, bringing these potential treatments closer to the bedside than ever before. Owing to the clinical heterogeneity of PMDs and SMDs, a panacea remains elusive; thus, the main objective of clinical care continues to be the reduction of morbidity and mortality.
Preventing mitochondrial stress
As highlighted previously, mitochondria constitute a principal source of ROS. Endogenous enzymatic sources, including transmembrane NADPH oxidases (NOXs) and the mitochondrial electron transport chain (ETC), principally responsible for the production of O2− and H2O2329,330. Accordingly, the levels of mitochondrial ROS serve as stress surveillance system for assessing mitochondrial dysfunction and cellular health.331 Meanwhile, mitochondria themselves are also targets of persistent oxidative stress, leading to lipid and mtDNA damage or protein posttranslational modifications.332,333,334 Oxidants are also involved in the bidirectional regulation of mitochondrial dynamics, such as mitochondrial cristae and network remodeling. High levels of oxidants can also lead to MPT, further triggering “ROS-induced ROS release (RIRR)”.335 Upon exposure to reactive oxygen or nitrogen species, [4Fe-4S]2+ clusters in mitochondrial respiratory proteins are oxidized and may result in reversible aconitase inactivation, which impairs the inhibition of ROS production though affecting mitochondrial metabolic flux.336 Antioxidants have long been thought to protect mitochondria from damage. Certain compounds specifically target the complex I site, including S1QELs and OP2113, inhibiting RET and ROS production without disturbing normal OXPHOS.214,337,338,339 Quinone-based antioxidants, encompassing CoQ analogs idebenone and EPI-743, alongside a water-soluble Vitamin E derivative sonlicromanol (also known as KH176), are employed as a frontline defense against mitochondrial oxidative damage. These antioxidants exert their protective roles by neutralizing reactive oxygen species and safeguarding the activity of glutathione, a crucial antioxidant within cells340,341,342 Some drugs target MPTP opening and thus reducing RIRR and cell death events, such as classic cyclophilin D (CypD) inhibitor CsA, which occlude the peptidyl-prolyl cis/trans isomerase (PPIase) active site of CypD.343 Despite their promising roles, the efficacy of these global antioxidants is constrained by their suboptimal localization within mitochondria.344 To overcome this therapeutic hurdle, researchers have turned to lipophilic cations, molecules that inherently possess a low transmembrane activation energy. This characteristic enables them to penetrate the phospholipid bilayer autonomously without necessitating an uptake mechanism, facilitating their accumulation within the mitochondria which boast a high membrane potential (−150 to −170 mV).345,346 The advent of this strategy has given rise to mitochondria-targeted antioxidants such as MitoQ and SKQ1.347,348 Szeto-Schiller (SS) peptide family (also known as SS-31, MTP-131 or Bendavia) also could directly target cardiolipin peroxidation, independent to mitochondria membrane potential, and inhibit cytochrome c peroxidase and prevent mitochondrial damage.349
Inducing mitochondrial biogenesis
Mitochondrial biogenesis represents a cellular mechanism of generating new mitochondria, which is instrumental in ensuring tissue homeostasis. Its pharmacological activation emerges as a potent strategy to mitigate diseases characterized by mitochondrial dysfunction.207 This multifaceted process comprises a network of therapeutic targets, including mitochondrial energy and nutrient sensors, and downstream effectors such as transcription factors, cofactors, and nuclear receptors (NRs).344 Among the molecular entities involved in mitochondrial biogenesis, the PGC-1α acts as a nodal regulator in this intricate network, integrating upstream signals to launch a downstream mitochondrial gene programs facilitating mitochondrial biogenesis.350 However, the transcriptional regulation and post-translational modifications of PGC-1α represent intricate molecular events, which introduces substantial challenges in considering PGC-1α as a direct target for pharmacological interventions.351
Upstream sensors respond to perturbations in cellular energy status and nutrient availability. For instance, AMPK, as an essential metabolic regulator of glucose and fatty acid, is activated under conditions of energy deprivation, characterized by a high AMP/ATP ratio. This activation triggers mitochondrial biogenesis through the promotion of PGC-1α activity and its translocation into the nucleus.352 The NAD+-dependent deacetylase sirtuin family, notably SIRT1, plays a significant role in steering mitochondrial biogenesis by deacetylating and activating PGC-1α, in addition to exerting a direct influence on mitochondrial gene expression.353 Conversely, the mTORC1, a sensor of nutrient and growth factor availability, exerts a suppressive effect on mitochondrial biogenesis under conditions rich in nutrients.
At the downstream spectrum, an assortment of transcription factors and nuclear receptors react to these upstream signals, orchestrating a concert of nuclear and mitochondrial gene expression to regulate mitochondrial biogenesis. Among these are nuclear respiratory factors 1 and 2 (NRF1 and NRF2), which dictate the transcription of a plethora of nuclear-encoded mitochondrial genes, and peroxisome proliferator-activated receptors (PPARs), governing lipid metabolism and mitochondrial functionality. The estrogen-related receptors (ERRs) also play a crucial role in the synchronization of gene expression necessary for oxidative phosphorylation. Other key players include the cAMP response element-binding protein 1 (CREB1), and forkhead box O (FOXO) transcription factors, which are known to induce PGC-1α transcription. NRs, serving as transcription factors, are also vital constituents in the signaling cascade governing mitochondrial biogenesis. They mediate gene expression in response to lipophilic hormones, vitamins, and dietary lipids, thus playing a role in shaping the mitochondrial biogenic response.
Several pharmacological agents have been identified to exert modulatory effects on these regulatory nodes, thereby enhancing mitochondrial biogenesis. For example, 5-Aminoimidazole-4-carboxamide ribonucleotide (AICAR) and PXL770 act as AMPK activators. AICAR, by mimicking the effects of AMP, stimulates AMPK which subsequently promotes the transcriptional activity of the coactivator PGC-1α, culminating in enhanced mitochondrial biogenesis.106,354 Similarly, PXL770 instigates mitochondrial biogenesis by acting as a direct stimulant of AMPK. In a preclinical investigation focusing on Autosomal dominant polycystic kidney disease (ADPKD), PXL770 treatment activates AMPK and restored mitochondrial DNA copy number and PGC-1α mRNA expression in mice with ADPKD.355 Another compound, Metformin, widely known for its role in diabetes management, also bolsters mitochondrial biogenesis by activating AMPK, leading to an upsurge in PGC-1α expression and activation.356 Furthermore, sirtuin-activating compounds (STACs), such as Resveratrol, enhance activity of the SIRT1-PGC-1α axis, thereby promoting muscle remodeling in Duchenne muscular dystrophy (DMD) mice.357 PPAR agonists, such as Bezafibrate and Thiazolidinediones, augment mitochondrial biogenesis by binding to and activating PPARs, which heterodimerize with Retinoid X Receptor (RXR) and bind to PPAR response elements in the DNA, enhancing the transcription of genes involved in mitochondrial biogenesis.358,359 Finally, REN001, a synthetic agonist of PPAR-δ, works by preferentially activating PPAR-δ, thereby promoting mitochondrial biogenesis and improving muscle function. These compounds, through their action on specific targets, highlight the potential for strategic pharmacological intervention to enhance mitochondrial biogenesis in the context of mitochondrial dysfunction.
Targeting mitochondrial dynamics
Impaired dynamics of mitochondrial fusion and fission have been implicated in numerous prevalent diseases, particularly those featuring neurodegeneration.360 The facilitation of mitochondrial fusion is orchestrated by GTPases, notably Mfn1, Mfn2, and Opa1, while mitochondrial fission is mediated by the DRP1.361 Mdivi-1, a derivative of quinazolinone, inhibits DRP1-dependent mitochondrial fragmentation and has been demonstrated to confer protection from cardiac IR injury and neurotoxicity.362,363 Recently, in vitro experimentation showed that Mdivi-1 partially ameliorated the aberrations in mitochondrial dynamics within human aortic smooth muscle cells (HAoSMCs) derived from bicuspid aortic valve (BAV) patients.364 Additionally, in vivo studies on mice with end-stage dilated cardiomyopathy (DCM) revealed that Mdivi-1 inhibited the activation of DRP1 signaling, partially mitigated autophagy inhibition and fatty acid metabolic disorder resultant from low-density lipoprotein receptor-related protein 6 (LRP6) deficiency, and ameliorated cardiac dysfunction.365
A critical facet of mitochondrial dynamics is the fragmentation and dynamic clearance of damaged mitochondria. Interestingly, recent findings have suggested that the physiological fragmentation of mitochondria under exercise conditions is an adaptation to energy demands.366 Physiological disruption mediated by DRP1 preserves mitochondrial membrane potential and attenuates the expression of factors related to mitophagy. It’s worth noting that inhibiting fission using P110 and Mdivi-1 significantly impairs exercise capacity. Furthermore, emerging evidence suggests that Mdivi-1 may exert effects that are independent of mediating fission, including the reversible inhibition of complex I and RET-mediated ROS production.367
Uncoupling mitochondrial respiration
OXPHOS is inherently inefficient due to proton leakage across the inner mitochondrial membrane, which facilitates the return of protons to the mitochondrial matrix, bypassing ATP synthase and ATP synthesis. This futile proton cycling, driven by specific respiratory chain complexes and uncontrolled proton permeability, is a crucial modulator of both ROS production and energy expenditure. Pertinently, such dynamics are accentuated in obesity, insulin resistance, and NAFLD. Intriguingly, mild mitochondrial uncoupling can yield therapeutic benefits. The mitochondrial uncoupler 2,4-dinitrophenol (DNP) was initially noted as having potential therapeutic benefits in obesity and metabolic diseases. However, it was ultimately discontinued in clinical practice due to neurological complications and fatalities resulting from overdose.368 This led to the development of safer alternatives with higher therapeutic indexes, such as DNP derivatives and controlled-release formulations like DNP-methyl ether (DNP-ME) and controlled-release mitochondrial protonophore (CRMP).369,370 The therapeutic mechanism involves reversing insulin resistance in the liver and muscle, associated with reductions in diacylglycerol content and decreased activity of protein kinase C epsilon (PKCε) and PKCθ. Recently, a phase 2a trial evaluated the safety and efficacy of HU6, a metabolic accelerator and mitochondrial uncoupler, in adults with NAFLD and high BMI.371 Participants received varying doses from 100 to 400 mg of HU6 or placebo. HU6 showed a significant reduction in liver fat content compared to placebo, with mild to moderate side effects like flushing, diarrhea, and palpitations. This trial highlights the potential of modulating mitochondrial uncoupling as a viable therapeutic approach in metabolic disorders.
Mitochondrial transplantation-a strategy of organelle replacement
Mitochondrial transplantation focusing on functional tissue repair has raised great interest in recent years. Mitochondrial transplantation refers to the artificial complementation or replacement of a dysfunctional mitochondrial network with viable respiratory-competent mitochondria. The basic process involves isolating mitochondrial preparations from healthy tissue and introducing them into the vicinity of the tissue area with mitochondrial dysfunction, either by regional or circulatory injection. Until now, the application list of mitochondrial transplantation has been broadened in various disease models, including IR injuries47,372,373,374,375,376,377, neurodegenerative diseases378,379,380,381,382,383, renal injury40, ARDS384,385, and cancer386,387 (See Table 2 and Fig. 5). Conducting mitochondrial component transplantation not only delineates molecular mechanisms of tissue revitalization but provides possible nodes of intervention in the future.

Therapeutic applications of mitochondrial and its component transplantation. This figure concludes therapeutic effects of mitochondria and associated components from different tissues and cells, to the mitochondria level (marked as grey arrows). The increased alterations in boxes are shown as up arrows while decreased alterations in boxes are shown as down arrows. Notably, though tissue level changes can vary, mitochondrial transplantation typically restores ATP production ability and reduces ROS production of damaged mitochondria. Besides, mitochondrial transplantation inhibited the intracellular try-IDO-kyn pathway, thereby improving the cognitive performance of psychiatric disorders and aging. In carbon tetrachloride (CCl4)-induced liver injury, transplanted mitochondria can increase a series of anti-oxidative enzymes to improve OXPHOS functions by triggering the UPRmt pathway. On the other hand, MSCs also repress TLR-signaling of macrophages via microRNA transfers to reduce inflammatory responses
Ischemia/reperfusion
Intervention targeting disrupted mitochondria at the early stage of reperfusion was proven to be effective in reducing subsequent cell necrosis.47,374,388 This was possibly due to prevention of the ROS burst derived from RET that induced reversible modification of Cys39 on the ND3 subunit of complex I.215,389 To reverse mitochondrial damage induced by ischemia, pioneering work of mitochondrial transplantation was conducted by McCully, J. D. et al. who directly applied autologous mitochondria into the infarction zone on hearts of New Zealand White rabbits instantly before reperfusion. Besides enhanced postischemic myocardial function and decreased ROS, data from proteomics also revealed that receiving exogenous mitochondria compensated for the impaired respiration and energy generation capability of myocytes.372 In addition, higher expression of non-mitochondrial cargos from donor induced pluripotent stem cell-derived cardiomyocytes (iCMs), such as PGC-1α and ERRγ mRNA, also facilitated mitochondrial biogenesis in cardiac IR injuries.390 The transplanted components upregulated mitochondrial ETC protein expression in a PGC-1α-dependent manner. In the rat livers and brains, mitochondrial transplantation also attenuated cell deaths and oxidative response caused by IR.375,376 Notably, it was evidenced that improvements in ventricular function could be achieved by mitochondrial transplantation in pediatric patients, though clinical trials in humans require further investigation.46
Neurodegenerative diseases and aging
Mitochondrial energetic disruption, oxidative injuries, and dynamic abnormalities are vital features of aging-related neurodegenerative diseases and traumatic injuries.18,226,391,392 Recently, mitochondrial-targeting therapy has revealed its potential in postponing the pathological course of PD. In a rat PD model induced by 6-hydroxydopamine (6-OHDA), both allogeneic and xenogeneic mitochondrial injection to the medial forebrain bundle (MFB) reversed the movement disorder and attenuated the deterioration of dopaminergic neurons.379 A similar strategy showed improvement of behavioral symptoms in PD mice after intravenous administration of HepG2 cell-derived mitochondria.380 Intranasal delivery of mitochondria to 6-OHDA-lesioned rats showed significant improvement in rotational and locomotor behavior, dopaminergic neuron survival, and oxidative damage, demonstrating potential for bypassing the blood-brain barrier and promoting neuronal recovery.393 In addition, hippocampal mitochondria showed neuroprotective and antidepressant effects in neuroinflammatory astrocytes and microglia.381 Transfer of active intact mitochondria into AD mice also significantly improved cognitive performance, decreased neuronal loss, ameliorated mitochondrial dysfunction in the brain and liver, without observed toxicity, suggesting a novel therapeutic approach for AD.394,395 In aging models, injecting mitochondria from young animals into aged ones has been shown to improve cognitive, motor, and depression-like behaviors, enhance mitochondrial function, and modulate metabolic pathways, presenting a promising approach for combating aging and age-related mood disorders.396,397
Psychiatric disorders
Multiple mechanisms of psychiatric disorders involve cerebral oxygen metabolic changes. Due to their important role in redox balance, it is possible for mitochondria to play a role in energy metabolism in psychiatric disorders.398,399 Attenuation of mitochondrial oxidative stress and structural damage were favorable in improving psychiatric symptoms, including depression, anxiety, and other cognitive deficits.381,397,400 In these models, ROS level and mitochondrial morphology were normalized via mitochondrial transplantation. In addition, BDNF signal transduction is vital to the regulation of depressive-like behaviors.401 Exogenous mitochondria promote BDNF-related neurogenesis in major depressive disorder (MDD), improving anti-depressant effects. Neuronal protection-related gene expression, such as that of Nfe2l1, was also upregulated after mitochondrial administration. Additionally, it was reported that dysregulated metabolites of tryptophan and activated indoleamine 2,3-dioxygenase (IDO) by inflammation were crucial parts of depression and aging regulated by the gut-brain axis.402 Supplementing mitochondria decreased the IDO activity and attenuated anxiety- and depression-like behaviors, highlighting the potential to treat psychiatric disorders.397
NAFLD and hepatic injuries
Failure of mitochondrial adaptation, particularly respiratory and metabolic deficiency, is one of the core features in the progression to insulin resistance and NAFLD.24,403,404 However, fat deposits gradually vanished after mitochondrial transplantation, along with decreased serum transaminase activity. Accordingly, it was observed that levels of ROS and malondialdehyde (MDA) were significantly reduced after mitochondrial transplantation, whereas Glutathione (GSH) content and superoxide dismutase (SOD) activity increased.405 These results suggested that mitochondrial transplantation could improve serum lipid metabolism and reduce hepatocyte injury from HFD-induced oxidation. Mitochondrial transplantation was also proven to have positive effects on hepatic injuries induced by toxic chemicals. In carbon tetrachloride (CCl4) induced hepatic injury, free radicals are formed and initiate lipid peroxidation to disrupt membrane integrity, resulting in mitochondrial damage and mass loss.406 However, the administration of liver mitochondria from healthy mice eliminated free radicals, upregulated GSH metabolism, and restored OXPHOS function in CCl4-induced chronic hepatic injury and fibrosis.407 Interestingly, the molecular mechanism of mitochondrial protection seemed to be correlated with the unfolded protein response (UPRmt) pathway, as it activated a series of anti-oxidative enzymes and increased mitophagy to rebuild mitochondrial homeostasis.407 Consistent results were observed in acetaminophen (APAP) induced liver injury where administration of exogenous mitochondria reversed mitochondrial structure disruption, GSH loss, and serum levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST).408 These provide novel therapeutic strategies to enhance resistance in response to hepatic injuries.
Acute respiratory distress syndrome/acute lung injury (ARDS/ALI)
With decades of foreshadowing of cell-based therapy, considerable potential has been shown to rescue the disrupted alveolar-capillary barrier in ARDS/ALI.409,410,411,412 As a protective mechanism of cell-based therapy, cell-based mitochondrial transfer has been reported to increase ATP content, promote an anti-inflammatory phenotype, and restore mitochondrial respiration in ARDS/ALI.413,414 Moreover, mitochondrial restoration which benefited from MSCs also protected epithelial cells from apoptosis in asthma.415,416 In cigarette smoke and LPS-induced chronic obstructive pulmonary disease (COPD), mitochondrial administration inhibited ROS production and thus decreased cholinergic sensitivity of the epithelium, resulting in altered inflammatory progression. However, direct protection of injured epithelial cells is not the only mechanism of mitochondrial transplantation against various tissue fates. It was reported that phagocytosis of macrophages was enhanced by mitochondria derived from MSCs, which partially involved the mechanism of antibacterial effects in an E. coli Pneumonia-induced ARDS model.417 In addition, an adoptive transfer model promoted M2 phenotype alteration and the phagocytic capacity of macrophages to resist LPS-induced ARDS. This was potentially achieved by increased OXPHOS capacity of macrophages via mitochondrial transfer induced by MSC-derived EVs.418 These studies highlighted cell-dependent mitochondrial transfer as an extension of cell-based therapy.
Spinal cord injury (SCI)
Mitochondrial dysfunction is increasingly recognized as a pivotal factor in the neuronal death cascades characteristic of central nervous system injuries.419 Empirical evidence has been garnered from a study using a Sprague-Dawley rat model, wherein local mitochondrial transplantation into the distal end of the injured nerve led to notable enhancements in neurobehaviors, nerve electrophysiology, and muscle activities.420 The administrated mitochondria mitigated oxidative stress and upregulated neurotrophic factor expression in both the injured nerves and denervated muscles, concurrently augmenting the pool of muscular progenitor cells, and increasing total muscle weight. In a parallel investigation, intraparenchymal infusion of allogeneic soleus muscles mitochondria into injured spinal cords resulted in the recovery of locomotor and sensory functions in rats with a reduction in SCI-induced cellular apoptosis and inflammation.421 Additionally, bone marrow stromal cells (BMSCs) have been observed to confer protection against SCI, attributed to their capacity to transfer mitochondria to injured neurons via intercellular gap junctions.422 Further advancements include the development of an engineered mitochondrial compound specifically designed to target macrophages within the SCI region.423 This approach demonstrated efficacy in attenuating pro-inflammatory profiles in macrophages, both in vitro and in vivo, thereby enhancing tissue regeneration and bolstering functional recovery in SCI models.
Other diseases
Studies on other diseases also illustrated the protective effect of mitochondrial transplantation, ranging from wound healing capacity to multiple sclerosis, tendinopathy, and osteoarthritis.424,425,426,427 Mitochondria from platelets were reported to stimulate the angiogenic potential of MSCs, thus promoting wound healing and tissue repair in chronic muscle injury.424 The directly administered mitochondria enhanced the TCA cycle and fatty acid synthesis, which was previously reported to favor epithelial cell proliferation for angiogenesis.428 In an autoimmune encephalomyelitis (EAE) model, intracerebroventricular induction of neural stem cells (NSCs) actively transferred mitochondria into mononuclear phagocytes and astrocytes, thereby ameliorating EAE disability in mice.425 In addition, mitochondria from L6 cells restored the protein levels of Tenascin C (TNC) in collagenase-damaged Achilles tendons, and were involved in the tendon protection process, along with the inhibition of apoptotic proteins (BID, Bax, and Bcl-2) and inflammatory cytokines (TNF-α, IL-1β, and IL-6).426 Similarly, the progression of osteoarthritis induced by monosodium iodoacetate was significantly inhibited by mitochondria from the skeletal (L6) myoblast cell line, which was also supported by improved pain phenotypes.427 Intravenous administration of mtDNA and TFAM mRNA-containing MSC-EVs was effective in reducing mitochondrial damage and inflammatory response in acute renal injury. This was possibly due to the protection of mitochondria in stressed HK-2 cells by TFAM-induced mtDNA maintenance.40 Additionally, it was discovered that mitochondria-located circular RNA (circRNA) and steatohepatitis-associated circRNA ATP5B regulator (circRNA SCAR) could reduce ROS production and avoid fibroblast activation via mitochondrial permeability transition during NAFLD.429 Through binding with ATP5B of the mPTP complex, the circRNA SCAR could block cyclophilin D-mPTP interactions, thereby inhibiting the opening of mPTP and reducing ROS production. Importantly, a decrease in ROS level and cytokine secretion was observed in circRNA SCAR-treated mice. The intravenous injection of mitochondria-targeting circRNA, which contained circRNA SCAR overexpression vectors, also evidenced the improvement of glucose and insulin resistance 2, along with weight loss and macrophage infiltration. These preclinical studies have broadened the applications of mitochondrial transplantation in non-ischemic diseases.
Conclusion and perspective
Mitochondrial medicine has unfolded a remarkable trajectory of advancement across the span of several decades. Historically, mitochondrial diseases were predominantly ascribed to genetic aberrations, however, the current understanding has broadened to incorporate many common pathologies, as critical contributors.1 This paradigm shift intensified the appeal of developing therapeutic strategies targeting mitochondria. This review underscores the significance of tailored dietary supplements and adjustments, particularly the administration of essential nutrients such as carnitine, CoQ, creatine, and vitamin B2, alongside the potential benefits of ketogenic diets in leveraging alternative energy sources through ketone bodies, aiming at mitigating the metabolic intricacies inherent in mitochondrial pathologies. In addition, conventional pharmacological targets - inclusive of those implicated in regulating mitochondrial stress responses, metabolite pools, biogenesis, and dynamics- are transitioning to rigorous evaluation in randomized clinical trials, which represent a strategic pivot towards the dual roles of mitochondria in energy production and cellular signaling. Concurrently, the necessity for the conception and implementation of highly precise therapies becomes increasingly evident. Small molecules that are intricately designed to target membrane potential and cardiolipin of mitochondria were included at the inception stages of drug design, with the objective of augmenting mitochondrial delivery.430 Nonetheless, the journey from bench to bedside is fraught with challenges, including the clinical heterogeneity of mitochondrial disorders and the potential side effects of long-term dietary and pharmacological interventions. For instance, the protective role of ketogenic diets, while beneficial in simulating a fasting-like metabolic state, raises concerns regarding nutritional balance and sustainability, not to mention the observed risk of cardiac fibrosis in animal models. Similarly, the clinical efficacy of antioxidants and compounds targeting mitochondrial dynamics necessitates a careful balance, avoiding interference with physiological ROS signaling mechanisms essential for cellular homeostasis.
Mitochondrial transplantation is another strategy burgeoning on the horizon of therapeutic innovation. This involves the isolation of active mitochondria from individuals devoid of mitochondrial defects, followed by their introduction into compromised cells. Recently, Hayashida, K. et al. conducted a systematic review of pertinent evidence from both animal and human studies concerning mitochondrial transplantation in IR injury, offering a practical assessment of the categorization, concentration, and accessibility of data within this field.431 Twenty animal and two human studies were included, with 14 studies (approximately 70%) focusing on IR models in heart and brain tissues with high energy demand and metabolism. The collective findings from animal studies endorsed the ability of mitochondrial transplantation to mitigate IR injury, although these results were inconsistent concerning specific biomarkers and pathological alterations.377,393,414,431 These exogenous mitochondria are postulated to integrate with the existing functional networks within the recipient cells.432 However, symbiosis was hardly observed between exogenous and endogenous mitochondria, which might explain why some studies consider exogenous mitochondrial function as a substitution for endogenous damaged mitochondria.376 While most studies reported favorable results after mitochondrial transplantation, elucidation of the underlying mechanisms pertinent to its clinical translation remains an open challenge, including risks such as susceptibility during mitochondrial isolation, immune adaptation, endocytosis, and lysosome escape.431,433 The potential for xenotransplantation, deliberation over the most suitable source of mitochondria-whether autologous, allogenic, or xenogeneic, is still governed by a matrix of factors encompassing immunological compatibility, ethical implications, and the nuanced demands of clinical application. Autologous mitochondria, procured from the patient’s own tissues, emerge as the frontrunner in terms of biocompatibility, significantly reducing the risk of immune rejection and sidestepping the ethical complexities tethered to xenogeneic sources. However, this approach is not without its limitations, particularly in the context of systemic or congenital mitochondrial disorders where the availability of functionally intact mitochondria is often compromised.434 Allogenic mitochondria, sourced from human donors, represent a pragmatic alternative, particularly in acute settings where the requirement for fresh mitochondria may be increased due to the limited intervention window of opportunity.388 Xenogeneic mitochondria, while presenting an abundant and diverse reservoir, are currently confined to the experimental realm, predominantly due to heightened immunogenic risks and profound ethical considerations. Recently, of great concern to researchers, a nanothylakoid unit (NTU) based on the controllable photosynthetic system was developed as cross-species ATP and NADPH providers to treat osteoarthritis.435 Plant-derived NTUs were extruded and encapsulated by the purified chondrocyte membrane (CM-NTUs) to avoid immune rejection. According to proteomic analysis, these chondrocyte membrane proteins were highly correlated with vesicle targeting and membrane fusion, which might result in the efficient uptake by chondrocytes rather than being eliminated by macrophages or intracellular lysosomes. In addition, proteomic profiling demonstrated that mitochondrial diversity reveals heterogeneity in morphology, state, function, and diseases, leaving a large mitochondrial pool to be discussed.436 For example, in brown adipocytes, PDMs are inactive in fusion-fission dynamics but favored with increased ATP synthesis and lipid expansion capacity, compared to cytoplasmic mitochondria.437 Stem cells might also be universal mitochondrial donors, as mitochondrial transfer partially participated in the therapeutic effect of MSCs.438 Viable transplanted mitochondria seemed to be indispensable to the postischemic restoration whereas deactivated mitochondria, such as frozen mitochondria, mitochondrial genomes, and ATP-containing vesicles, did not achieve the same results.43,372 To shorten the preparation time, McCully, J. D. et al. proposed a tissue pool to obtain fresh mitochondria, including pectoralis major muscle, rectus abdominis or even sternocleidomastoid muscle.439 Comprehensive mechanistic investigations and more extensive preclinical studies remain essential.
The mitochondrial transplantation process largely involves the internalization of transplanted mitochondria at the cell membrane level to mitigate the biological information in the cell. However, the molecular mechanism of how the mitochondria can be integrated into endogenous mitochondrial network remain heterogenous and even largely unknown. Several studies have suggested versatile mechanisms may be involved in mitochondrial horizontal transfer between cells. In vitro data showed that the autologous driving force of mitochondrial uptake included endocytosis of recipient cells, such as macropinocytosis, clathrin-mediated endocytosis, or actin polymerization, which vary among different mitochondrial recipients.440,441 Non-specific macropinocytosis was also involved in some cases, evidenced by the specific inhibitor of micropinocytosis, ethyl isopropyl amiloride (EIPA).85 Endocytosis is a cellular process whereby a vesicle engulfs extracellular material, and it was suggested that transplanted mitochondria can be internalized through this process.387,424,441 In addition, receptor, actin, and ATP-dependent phagocytosis was validated to participate in engulfment of extracellular mitochondria via membrane invagination.442 Gap junctions, which are intercellular channels that allow for the exchange of small molecules, have also been implicated in mitochondrial internalization.413,422,443 Tunneling nanotubes (TNT) are highly sensitive nanotube structures that allow for direct communication between cells and have long been implicated in mitochondrial transfer.417,444 Partial cell fusion is another mechanism that has been proposed, involving the direct fusion between cells and mitochondria-sharing after TNT was inhibited.445 Recent studies also suggested that fusogenic syncytins on mitochondria, along with their receptors, might facilitate the mitochondrial internalization process under cancerous circumstances.446 However, current evidence points out single forms of internalization, as transplantation efficiency appears to be hyperresponsive to single inhibitors.415,422,424,441 Additionally, the employment of mitochondrial dyes in assessing mitochondrial incorporation during transplantation, while a common practice in both in vitro and in vivo studies, is hindered by the propensity of these dyes to leak.447 This leakage can lead to non-specific staining and false-positive results, as these mitochondrial dyes may escape from both donor and recipient cells, inadvertently staining neighboring mitochondria. The optimal approach for studying mitochondrial transfer in both in vitro and in vivo settings involves the use of stable transgenes encoding mitochondrially localized tags or fluorescent proteins. This mitochondrial reporter system, adaptable for lineage-specific mitochondrial reporter mice or for broad cell labeling, offers stable and precise tracking of mitochondria in recipient cells. However, a major concern is to avoid misinterpretation due to unintended Cre activity in other cell types when establishing stringent cell-type-specific transgene expression based on Cre recombinase systems.447 Collectively, a better understanding of these internalization mechanisms will help optimize the efficacy and safety of mitochondrial transplantation for various clinical applications.
The future direction of mitochondrial dysfunction therapeutics likely resides in personalized medicine, wherein genetic and metabolic profiling could tailor interventions to individual patient needs, enhancing both efficacy and safety. As the therapeutic landscape for mitochondrial dysfunction continues to mature, the integration of dietary, pharmacological, and preventive strategies holds the promise of a more comprehensive and effective approach to managing these complex diseases. The endeavor to translate these advances into clinical practice underscores the need for a multidisciplinary effort, bridging the gap between molecular insights and therapeutic innovation to forge a path towards improved patient outcomes in the realm of mitochondrial pathology.
References
Murphy, M. P. & Hartley, R. C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 17, 865–886 (2018).
Nunnari, J. & Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 148, 1145–1159 (2012).
Picard, M., Wallace, D. C. & Burelle, Y. The rise of mitochondria in medicine. Mitochondrion 30, 105–116 (2016).
Anderson, S. et al. Sequence and organization of the human mitochondrial genome. Nature 290, 457–465 (1981).
Scarpulla, R. C. Transcriptional Paradigms in Mammalian Mitochondrial Biogenesis and Function. Physiol. Rev. 88, 611–638 (2008).
Friedman, J. R. & Nunnari, J. Mitochondrial form and function. Nature 505, 335–343 (2014).
Martijn, J., Vosseberg, J., Guy, L., Offre, P. & Ettema, T. J. G. Deep mitochondrial origin outside the sampled alphaproteobacteria. Nature 557, 101–105 (2018).
Ott, M., Gogvadze, V., Orrenius, S. & Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis 12, 913–922 (2007).
Hatefi, Y. The Mitochondrial Electron Transport and Oxidative Phosphorylation System. Annu Rev. Biochem. 54, 1015–1069 (1985).
Kummer, E. & Ban, N. Mechanisms and regulation of protein synthesis in mitochondria. Nat. Rev. Mol. Cell Bio. 22, 307–325 (2021).
Galluzzi, L., Kepp, O. & Kroemer, G. Mitochondria: master regulators of danger signalling. Nat. Rev. Mol. Cell Bio. 13, 780–788 (2012).
Mailloux, R. J., Treberg, J., Grayson, C., Agellon, L. B. & Sies, H. Mitochondrial function and phenotype are defined by bioenergetics. Nat. Metab. 5, 1641–1641 (2023).
Lake, N. J., Compton, A. G., Rahman, S. & Thorburn, D. R. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann. Neurol. 79, 190–203 (2016).
Goethem, G. V., Dermaut, B., Löfgren, A., Martin, J.-J. & Broeckhoven, C. V. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat. Genet. 28, 211–212 (2001).
Goto, Y., Nonaka, I. & Horai, S. A mutation in the tRNALeu(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 348, 651–653 (1990).
Wang, Y., Guo, X., Ye, K., Orth, M. & Gu, Z. Accelerated expansion of pathogenic mitochondrial DNA heteroplasmies in Huntington’s disease. Proc. Natl Acad. Sci. 118, e2014610118 (2021).
Chouchani, E. T. et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435 (2014).
Eldeeb, M. A., Thomas, R. A., Ragheb, M. A., Fallahi, A. & Fon, E. A. Mitochondrial quality control in health and in Parkinson’s disease. Physiol. Rev. 102, 1721–1755 (2022).
Prasun, P. Mitochondrial dysfunction in metabolic syndrome. Biochimica Et. Biophysica Acta Bba - Mol. Basis Dis. 1866, 165838 (2020).
Picard, M. & Shirihai, O. S. Mitochondrial signal transduction. Cell Metab. 34, 1620–1653 (2022).
Schon, E. A. & Przedborski, S. Mitochondria: The Next (Neurode)Generation. Neuron 70, 1033–1053 (2011).
Brown, D. A. et al. Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 14, 238–250 (2017).
Prakash, Y. S., Pabelick, C. M. & Sieck, G. C. Mitochondrial Dysfunction in Airway Disease. Chest 152, 618–626 (2017).
Begriche, K., Igoudjil, A., Pessayre, D. & Fromenty, B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion 6, 1–28 (2006).
Amorim, J. A. et al. Mitochondrial and metabolic dysfunction in ageing and age-related diseases. Nat. Rev. Endocrinol. 18, 243–258 (2022).
Brakedal, B. et al. The NADPARK study: A randomized phase I trial of nicotinamide riboside supplementation in Parkinson’s disease. Cell Metab. 34, 396–407.e6 (2022).
Zhou, B. et al. Boosting NAD Level Suppresses Inflammatory Activation of PBMC in Heart Failure. J. Clin. Invest. 130, 6054–6063 (2020).
Rossman, M. J. et al. Chronic Supplementation With a Mitochondrial Antioxidant (MitoQ) Improves Vascular Function in Healthy Older Adults. Hypertension 71, 1056–1063 (2018).
Snow, B. J. et al. A double‐blind, placebo‐controlled study to assess the mitochondria‐targeted antioxidant MitoQ as a disease‐modifying therapy in Parkinson’s disease. Mov. Disord. 25, 1670–1674 (2010).
Mortensen, S. A. et al. The Effect of Coenzyme Q10 on Morbidity and Mortality in Chronic Heart Failure Results From Q-SYMBIO: A Randomized Double-Blind Trial. Jacc Hear Fail 2, 641–649 (2014).
Berk, M. et al. A randomised controlled trial of a mitochondrial therapeutic target for bipolar depression: mitochondrial agents, N-acetylcysteine, and placebo. Bmc Med 17, 18 (2019).
Quintela-Fandino, M. et al. Randomized Phase 0/I Trial of the Mitochondrial Inhibitor ME-344 or Placebo Added to Bevacizumab in Early HER2-Negative Breast Cancer. Clin. Cancer Res 26, 35–45 (2020).
Slone, J. & Huang, T. The special considerations of gene therapy for mitochondrial diseases. npj Genom. Med. 5, 7 (2020).
Zhang, Q. et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464, 104–107 (2010).
Boudreau, L. H. et al. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood 124, 2173–2183 (2014).
Todkar, K. et al. Selective packaging of mitochondrial proteins into extracellular vesicles prevents the release of mitochondrial DAMPs. Nat. Commun. 12, 1971 (2021).
Borcherding, N. & Brestoff, J. R. The power and potential of mitochondria transfer. Nature 623, 283–291 (2023).
Tan, A. S. et al. Mitochondrial Genome Acquisition Restores Respiratory Function and Tumorigenic Potential of Cancer Cells without Mitochondrial DNA. Cell Metab. 21, 81–94 (2015).
Sansone, P. et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 114, E9066–E9075 (2017).
Zhao, M. et al. Mesenchymal Stem Cell-Derived Extracellular Vesicles Attenuate Mitochondrial Damage and Inflammation by Stabilizing Mitochondrial DNA. Acs Nano 15, 1519–1538 (2021).
Phinney, D. G. et al. Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs. Nat. Commun. 6, 8472 (2015).
Nicolás-Ávila, J. A. et al. A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell 183, 94–109.e23 (2020).
Hayakawa, K. et al. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 535, 551–555 (2016).
Kaza, A. K. et al. Myocardial rescue with autologous mitochondrial transplantation in a porcine model of ischemia/reperfusion. J. Thorac. Cardiovasc Surg. 153, 934–943 (2017).
Huang, T. et al. Efficient intervention for pulmonary fibrosis via mitochondrial transfer promoted by mitochondrial biogenesis. Nat. Commun. 14, 5781 (2023).
Emani, S. M., Piekarski, B. L., Harrild, D., Del Nido, P. J. & McCully, J. D. Autologous mitochondrial transplantation for dysfunction after ischemia-reperfusion injury. J. Thorac. Cardiovasc Surg. 154, 286–289 (2017).
McCully, J. D. et al. Injection of isolated mitochondria during early reperfusion for cardioprotection. Am. J. Physiol.-heart C. 296, H94–H105 (2009).
Pfanner, N., Warscheid, B. & Wiedemann, N. Mitochondrial proteins: from biogenesis to functional networks. Nat. Rev. Mol. Cell Bio 20, 267–284 (2019).
Youle, R. J. Mitochondria—Striking a balance between host and endosymbiont. Science 365, eaaw9855 (2019).
Virbasius, J. V. & Scarpulla, R. C. Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: a potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc. Natl Acad. Sci. 91, 1309–1313 (1994).
Wu, Z. et al. Mechanisms Controlling Mitochondrial Biogenesis and Respiration through the Thermogenic Coactivator PGC-1. Cell 98, 115–124 (1999).
Garcia, D. & Shaw, R. J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 66, 789–800 (2017).
Song, J., Herrmann, J. M. & Becker, T. Quality control of the mitochondrial proteome. Nat. Rev. Mol. Cell Biol. 22, 54–70 (2021).
Abe, Y. et al. Structural Basis of Presequence Recognition by the Mitochondrial Protein Import Receptor Tom20. Cell 100, 551–560 (2000).
van Wilpe, S. et al. Tom22 is a multifunctional organizer of the mitochondrial preprotein translocase. Nature 401, 485–489 (1999).
Melin, J. et al. Presequence Recognition by the Tom40 Channel Contributes to Precursor Translocation into the Mitochondrial Matrix. Mol. Cell. Biol. 34, 3473–3485 (2014).
Bauer, M. F., Sirrenberg, C., Neupert, W. & Brunner, M. Role of Tim23 as Voltage Sensor and Presequence Receptor in Protein Import into Mitochondria. Cell 87, 33–41 (1996).
Kalia, R. et al. Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature 558, 401–405 (2018).
Palmer, C. S. et al. MiD49 and MiD51, new components of the mitochondrial fission machinery. Embo Rep. 12, 565–573 (2011).
Otera, H. et al. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 191, 1141–1158 (2010).
Friedman, J. R. et al. ER Tubules Mark Sites of Mitochondrial Division. Science 334, 358–362 (2011).
Losón, O. C., Song, Z., Chen, H. & Chan, D. C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 24, 659–667 (2013).
Korobova, F., Ramabhadran, V. & Higgs, H. N. An Actin-Dependent Step in Mitochondrial Fission Mediated by the ER-Associated Formin INF2. Science 339, 464–467 (2013).
Lewis, S. C., Uchiyama, L. F. & Nunnari, J. ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 353, aaf5549 (2016).
Kleele, T. et al. Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature 593, 435–439 (2021).
Ge, P., Dawson, V. L. & Dawson, T. M. PINK1 and Parkin mitochondrial quality control: a source of regional vulnerability in Parkinson’s disease. Mol. Neurodegener. 15, 20 (2020).
Vives-Bauza, C. et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl Acad. Sci. 107, 378–383 (2010).
Soubannier, V. et al. A Vesicular Transport Pathway Shuttles Cargo from Mitochondria to Lysosomes. Curr. Biol. 22, 135–141 (2012).
Towers, C. G. et al. Mitochondrial-derived vesicles compensate for loss of LC3-mediated mitophagy. Dev. Cell 56, 2029–2042.e5 (2021).
Liang, W. et al. Mitochondria are secreted in extracellular vesicles when lysosomal function is impaired. Nat. Commun. 14, 5031 (2023).
Braschi, E. et al. Vps35 Mediates Vesicle Transport between the Mitochondria and Peroxisomes. Curr. Biol. 20, 1310–1315 (2010).
Hackenbrock, C. R. Ultrastructural Bases For Metabolically Linked Mechanical Activity In Mitochondria. J. Cell Biol. 37, 345–369 (1968).
Kühlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 13, 89 (2015).
Wallace, D. C., Fan, W. & Procaccio, V. Mitochondrial Energetics and Therapeutics. Annu. Rev. Pathol.: Mech. Dis. 5, 297–348 (2010).
Martínez-Reyes, I. & Chandel, N. S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 11, 102 (2020).
Boitier, E., Rea, R. & Duchen, M. R. Mitochondria Exert a Negative Feedback on the Propagation of Intracellular Ca2+ Waves in Rat Cortical Astrocytes. J. Cell Biol. 145, 795–808 (1999).
Pinton, P., Giorgi, C., Siviero, R., Zecchini, E. & Rizzuto, R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 27, 6407–6418 (2008).
McCormack, J. G., Halestrap, A. P. & Denton, R. M. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol. Rev. 70, 391–425 (1990).
Bonora, M. et al. Physiopathology of the Permeability Transition Pore: Molecular Mechanisms in Human Pathology. Biomol 10, 998 (2020).
Giorgi, C., Marchi, S. & Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 19, 713–730 (2018).
Vance, J. E. MAM (mitochondria-associated membranes) in mammalian cells: Lipids and beyond. Biochimica Et. Biophysica Acta Bba - Mol. Cell Biol. Lipids 1841, 595–609 (2014).
He, Q. et al. Control of mitochondria-associated endoplasmic reticulum membranes by protein S-palmitoylation: Novel therapeutic targets for neurodegenerative diseases. Ageing Res. Rev. 87, 101920 (2023).
Vos, K. J. D. et al. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 21, 1299–1311 (2012).
Szabadkai, G. et al. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 175, 901–911 (2006).
de Brito, O. M. & Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610 (2008).
Hirabayashi, Y. et al. ER-mitochondria tethering by PDZD8 regulates Ca2+ dynamics in mammalian neurons. Science 358, 623–630 (2017).
Bartok, A. et al. IP3 receptor isoforms differently regulate ER-mitochondrial contacts and local calcium transfer. Nat. Commun. 10, 3726 (2019).
Madreiter-Sokolowski, C. T. et al. Enhanced inter-compartmental Ca2+ flux modulates mitochondrial metabolism and apoptotic threshold during aging. Redox Biol. 20, 458–466 (2019).
Peng, W., Wong, Y. C. & Krainc, D. Mitochondria-lysosome contacts regulate mitochondrial Ca2+ dynamics via lysosomal TRPML1. Proc. Natl Acad. Sci. 117, 19266–19275 (2020).
Fernandez-Sanz, C. et al. Defective sarcoplasmic reticulum–mitochondria calcium exchange in aged mouse myocardium. Cell Death Dis. 5, e1573–e1573 (2014).
Rosencrans, W. M., Rajendran, M., Bezrukov, S. M. & Rostovtseva, T. K. VDAC regulation of mitochondrial calcium flux: From channel biophysics to disease. Cell Calcium 94, 102356 (2021).
Tan, W. & Colombini, M. VDAC closure increases calcium ion flux. Biochim. Biophys. Acta (BBA) - Biomembr. 1768, 2510–2515 (2007).
Báthori, G., Csordás, G., Garcia-Perez, C., Davies, E. & Hajnóczky, G. Ca2+−dependent Control of the Permeability Properties of the Mitochondrial Outer Membrane and Voltage-dependent Anion-selective Channel (VDAC). J. Biol. Chem. 281, 17347–17358 (2006).
Tilokani, L., Nagashima, S., Paupe, V. & Prudent, J. Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem. 62, 341–360 (2018).
Patron, M. et al. MICU1 and MICU2 Finely Tune the Mitochondrial Ca2+ Uniporter by Exerting Opposite Effects on MCU Activity. Mol. Cell 53, 726–737 (2014).
Plovanich, M. et al. MICU2, a Paralog of MICU1, Resides within the Mitochondrial Uniporter Complex to Regulate Calcium Handling. PLoS ONE 8, e55785 (2013).
Mallilankaraman, K. et al. MICU1 Is an Essential Gatekeeper for MCU-Mediated Mitochondrial Ca2+ Uptake that Regulates Cell Survival. Cell 151, 630–644 (2012).
Sancak, Y. et al. EMRE Is an Essential Component of the Mitochondrial Calcium Uniporter Complex. Science 342, 1379–1382 (2013).
Kirichok, Y., Krapivinsky, G. & Clapham, D. E. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427, 360–364 (2004).
Palty, R. et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. 107, 436–441 (2010).
Luongo, T. S. et al. The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 545, 93–97 (2017).
Jiang, D., Zhao, L. & Clapham, D. E. Genome-Wide RNAi Screen Identifies Letm1 as a Mitochondrial Ca2+/H+ Antiporter. Science 326, 144–147 (2009).
Jiang, D., Zhao, L., Clish, C. B. & Clapham, D. E. Letm1, the mitochondrial Ca2+/H+ antiporter, is essential for normal glucose metabolism and alters brain function in Wolf–Hirschhorn syndrome. Proc. Natl. Acad. Sci. 110, E2249–E2254 (2013).
Jäger, S., Handschin, C., St.-Pierre, J. & Spiegelman, B. M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc. Natl Acad. Sci. 104, 12017–12022 (2007).
O’Neill, H. M. et al. AMP-activated protein kinase (AMPK) β1β2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc. Natl Acad. Sci. 108, 16092–16097 (2011).
Viscomi, C. et al. In Vivo Correction of COX Deficiency by Activation of the AMPK/PGC-1α Axis. Cell Metab. 14, 80–90 (2011).
Iwabu, M. et al. Adiponectin and AdipoR1 regulate PGC-1α and mitochondria by Ca2+ and AMPK/SIRT1. Nature 464, 1313–1319 (2010).
Bergeron, R. et al. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am. J. Physiol. -Endocrinol. Metab. 281, E1340–E1346 (2001).
Fan, H. et al. Heat shock protein 22 modulates NRF1/TFAM-dependent mitochondrial biogenesis and DRP1-sparked mitochondrial apoptosis through AMPK-PGC1α signaling pathway to alleviate the early brain injury of subarachnoid hemorrhage in rats. Redox Biol. 40, 101856 (2021).
Toyama, E. Q. et al. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 351, 275–281 (2016).
Zhang, Y. et al. Melatonin attenuates myocardial ischemia‐reperfusion injury via improving mitochondrial fusion/mitophagy and activating the AMPK‐OPA1 signaling pathways. J. Pineal Res. 66, e12542 (2019).
Laker, R. C. et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 8, 548 (2017).
Hung, C.-M. et al. AMPK/ULK1-mediated phosphorylation of Parkin ACT domain mediates an early step in mitophagy. Sci. Adv. 7, eabg4544 (2021).
Hu, Y. et al. The AMPK-MFN2 axis regulates MAM dynamics and autophagy induced by energy stresses. Autophagy 17, 1142–1156 (2021).
Zhao, H. et al. AMPK-mediated activation of MCU stimulates mitochondrial Ca2+ entry to promote mitotic progression. Nat. Cell Biol. 21, 476–486 (2019).
Saxton, R. A. & Sabatini, D. M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 168, 960–976 (2017).
Cunningham, J. T. et al. mTOR controls mitochondrial oxidative function through a YY1–PGC-1α transcriptional complex. Nature 450, 736–740 (2007).
Morita, M. et al. mTORC1 Controls Mitochondrial Activity and Biogenesis through 4E-BP-Dependent Translational Regulation. Cell Metab. 18, 698–711 (2013).
Schieke, S. M. et al. The Mammalian Target of Rapamycin (mTOR) Pathway Regulates Mitochondrial Oxygen Consumption and Oxidative Capacity. J. Biol. Chem. 281, 27643–27652 (2006).
Chapman, N. M. et al. mTOR coordinates transcriptional programs and mitochondrial metabolism of activated Treg subsets to protect tissue homeostasis. Nat. Commun. 9, 2095 (2018).
Ramanathan, A. & Schreiber, S. L. Direct control of mitochondrial function by mTOR. Proc. Natl Acad. Sci. 106, 22229–22232 (2009).
Fujinuma, S. et al. FOXK1 promotes nonalcoholic fatty liver disease by mediating mTORC1-dependent inhibition of hepatic fatty acid oxidation. Cell Rep. 42, 112530 (2023).
Morita, M. et al. mTOR Controls Mitochondrial Dynamics and Cell Survival via MTFP1. Mol. Cell 67, 922–935.e5 (2017).
Betz, C. et al. mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc. Natl Acad. Sci. 110, 12526–12534 (2013).
Onyango, P., Celic, I., McCaffery, J. M., Boeke, J. D. & Feinberg, A. P. SIRT3, a human SIR2 homologue, is an NAD- dependent deacetylase localized to mitochondria. Proc. Natl Acad. Sci. 99, 13653–13658 (2002).
Michishita, E., Park, J. Y., Burneskis, J. M., Barrett, J. C. & Horikawa, I. Evolutionarily Conserved and Nonconserved Cellular Localizations and Functions of Human SIRT Proteins. Mol. Biol. Cell 16, 4623–4635 (2005).
Yang, W. et al. Mitochondrial Sirtuin Network Reveals Dynamic SIRT3-Dependent Deacetylation in Response to Membrane Depolarization. Cell 167, 985–1000.e21 (2016).
Fan, H., Le, J., Sun, M. & Zhu, J. Sirtuin 3 deficiency promotes acute kidney injury induced by sepsis via mitochondrial dysfunction and apoptosis. Iran. J. Basic Méd. Sci. 24, 675–681 (2021).
Morigi, M. et al. Sirtuin 3–dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Investig. 125, 715–726 (2015).
Samant, S. A. et al. SIRT3 Deacetylates and Activates OPA1 To Regulate Mitochondrial Dynamics during Stress. Mol. Cell. Biol. 34, 807–819 (2014).
Wang, C. et al. AdipoRon mitigates tau pathology and restores mitochondrial dynamics via AMPK-related pathway in a mouse model of Alzheimer’s disease. Exp. Neurol. 363, 114355 (2023).
Mathias, R. A. et al. Sirtuin 4 Is a Lipoamidase Regulating Pyruvate Dehydrogenase Complex Activity. Cell 159, 1615–1625 (2014).
Zhou, L. et al. SIRT5 promotes IDH2 desuccinylation and G6PD deglutarylation to enhance cellular antioxidant defense. EMBO Rep. 17, 811–822 (2016).
Janeway, C. A. Approaching the Asymptote? Evolution and Revolution in Immunology. Cold Spring Harb. Symp. Quant. Biol. 54, 1–13 (1989).
Marchi, S., Guilbaud, E., Tait, S. W. G., Yamazaki, T. & Galluzzi, L. Mitochondrial control of inflammation. Nat. Rev. Immunol. 23, 159–173 (2023).
Roh, J. S. & Sohn, D. H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw. 18, e27 (2018).
Lin, M., Liu, N., Qin, Z. & Wang, Y. Mitochondrial-derived damage-associated molecular patterns amplify neuroinflammation in neurodegenerative diseases. Acta Pharmacol. Sin. 43, 2439–2447 (2022).
Civril, F. et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature 498, 332–337 (2013).
Li, X. et al. Cyclic GMP-AMP Synthase Is Activated by Double-Stranded DNA-Induced Oligomerization. Immunity 39, 1019–1031 (2013).
Decout, A., Katz, J. D., Venkatraman, S. & Ablasser, A. The cGAS–STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 21, 548–569 (2021).
Gong, T., Liu, L., Jiang, W. & Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 20, 95–112 (2020).
Hopfner, K.-P. & Hornung, V. Molecular mechanisms and cellular functions of cGAS–STING signalling. Nat. Rev. Mol. Cell Biol. 21, 501–521 (2020).
West, A. P. et al. Mitochondrial DNA Stress Primes the Antiviral Innate Immune Response. Nature 520, 553–557 (2015).
Aarreberg, L. D. et al. Interleukin-1β Induces mtDNA Release to Activate Innate Immune Signaling via cGAS-STING. Mol. Cell 74, 801–815.e6 (2019).
Maekawa, H. et al. Mitochondrial Damage Causes Inflammation via cGAS-STING Signaling in Acute Kidney Injury. Cell Rep. 29, 1261–1273.e6 (2019).
Gulen, M. F. et al. cGAS–STING drives ageing-related inflammation and neurodegeneration. Nature 620, 374–380 (2023).
McArthur, K. et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 359, eaao6047 (2018).
Riley, J. S. et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. 37, e99238 (2018).
Kim, J. et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 366, 1531–1536 (2019).
Yu, C.-H. et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 183, 636–649.e18 (2020).
Xian, H. et al. Oxidized DNA fragments exit mitochondria via mPTP- and VDAC-dependent channels to activate NLRP3 inflammasome and interferon signaling. Immunity 55, 1370–1385.e8 (2022).
Gehrke, N. et al. Oxidative Damage of DNA Confers Resistance to Cytosolic Nuclease TREX1 Degradation and Potentiates STING-Dependent Immune Sensing. Immunity 39, 482–495 (2013).
Jiménez-Loygorri, J. I. et al. Mitophagy curtails cytosolic mtDNA-dependent activation of cGAS/STING inflammation during aging. Nat. Commun. 15, 830 (2024).
Swanson, K. V., Deng, M. & Ting, J. P.-Y. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 19, 477–489 (2019).
Nakahira, K. et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230 (2011).
Zhou, R., Yazdi, A. S., Menu, P. & Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 (2011).
Zhong, Z. et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560, 198–203 (2018).
Billingham, L. K. et al. Mitochondrial electron transport chain is necessary for NLRP3 inflammasome activation. Nat. Immunol. 23, 692–704 (2022).
Iyer, S. S. et al. Mitochondrial Cardiolipin Is Required for Nlrp3 Inflammasome Activation. Immunity 39, 311–323 (2013).
Kerur, N. et al. cGAS drives noncanonical-inflammasome activation in age-related macular degeneration. Nat. Med. 24, 50–61 (2018).
Shimada, K. et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 36, 401–414 (2012).
Bell, J. K. et al. The molecular structure of the Toll-like receptor 3 ligand-binding domain. Proc. Natl Acad. Sci. 102, 10976–10980 (2005).
Botos, I., Segal, D. M. & Davies, D. R. The Structural Biology of Toll-like Receptors. Structure 19, 447–459 (2011).
Garcia-Martinez, I. et al. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J. Clin. Invest 126, 859–864 (2016).
Rodríguez‐Nuevo, A. et al. Mitochondrial DNA and TLR9 drive muscle inflammation upon Opa1 deficiency. EMBO J. 37, e96553 (2018).
Tsuji, N. et al. Role of Mitochondrial DNA in Septic AKI via Toll-Like Receptor 9. J. Am. Soc. Nephrol. 27, 2009–2020 (2016).
Tripathi, A., Bartosh, A., Whitehead, C. & Pillai, A. Activation of cell-free mtDNA-TLR9 signaling mediates chronic stress-induced social behavior deficits. Mol. Psychiatry 28, 3806–3815 (2023).
Rabas, N. et al. PINK1 drives production of mtDNA-containing extracellular vesicles to promote invasiveness. J. Cell Biol. 220, e202006049 (2021).
West, A. P. & Shadel, G. S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 17, 363–375 (2017).
Ward, G. A. et al. Oxidized Mitochondrial DNA Engages TLR9 to Activate the NLRP3 Inflammasome in Myelodysplastic Syndromes. Int. J. Mol. Sci. 24, 3896 (2023).
Oka, T. et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485, 251–255 (2012).
Bao, W. et al. Toll-like Receptor 9 Can be Activated by Endogenous Mitochondrial DNA to Induce Podocyte Apoptosis. Sci. Rep. 6, 22579 (2016).
Hegde, R. et al. Identification of Omi/HtrA2 as a Mitochondrial Apoptotic Serine Protease That Disrupts Inhibitor of Apoptosis Protein-Caspase Interaction. J. Biol. Chem. 277, 432–438 (2002).
Du, C., Fang, M., Li, Y., Li, L. & Wang, X. Smac, a Mitochondrial Protein that Promotes Cytochrome c–Dependent Caspase Activation by Eliminating IAP Inhibition. Cell 102, 33–42 (2000).
Flores-Romero, H., Dadsena, S. & García-Sáez, A. J. Mitochondrial pores at the crossroad between cell death and inflammatory signaling. Mol. Cell 83, 843–856 (2023).
Yoshida, H. et al. Apaf1 Is Required for Mitochondrial Pathways of Apoptosis and Brain Development. Cell 94, 739–750 (1998).
Bock, F. J. & Tait, S. W. G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 21, 85–100 (2020).
Edlich, F. et al. Bcl-xL Retrotranslocates Bax from the Mitochondria into the Cytosol. Cell 145, 104–116 (2011).
Chen, H.-C. et al. An interconnected hierarchical model of cell death regulation by the BCL-2 family. Nat. Cell Biol. 17, 1270–1281 (2015).
Kazi, A. et al. The BH3 α-Helical Mimic BH3-M6 Disrupts Bcl-XL, Bcl-2, and MCL-1 Protein-Protein Interactions with Bax, Bak, Bad, or Bim and Induces Apoptosis in a Bax- and Bim-dependent Manner. J. Biol. Chem. 286, 9382–9392 (2011).
Rongvaux, A. et al. Apoptotic Caspases Prevent the Induction of Type I Interferons by Mitochondrial DNA. Cell 159, 1563–1577 (2014).
White, M. J. et al. Apoptotic Caspases Suppress mtDNA-Induced STING-Mediated Type I IFN Production. Cell 159, 1549–1562 (2014).
Victorelli, S. et al. Apoptotic stress causes mtDNA release during senescence and drives the SASP. Nature 622, 627–636 (2023).
Ernster, L. & Schatz, G. Mitochondria: a historical review. J. Cell Biol. 91, 227s–255s (1981).
Cowdry, E. V. & Bensley, R. R. Cytologist. Science 124, 972–973 (1956).
Bensley, R. R. & Hoerr, N. L. Studies on cell structure by the freezing‐drying method VI. The preparation and properties of mitochondria. Anat. Rec. 60, 449–455 (1934).
Claude, A. & Fullam, E. F. An Electron Microscope Study Of Isolated Mitochondria. J. Exp. Med. 81, 51–62 (1945).
Hogeboom, G. H., Schneider, W. C. & Pallade, G. E. Cytochemical studies of mammalian tissues i. Isolation of intact mitochondria from rat liver; some biochemical properties of mitochondria and submicroscopic particulate material. J. Biol. Chem. 172, 619–635 (1948).
Schneider, W. C. & Hogeboom, G. H. Intracellular distribution of enzymes v. Further studies on the distribution of cytochrome c in rat liver homogenates. J. Biol. Chem. 183, 123–128 (1950).
Hogeboom, G. H., Schneider, W. C. & Pallade, G. E. The Isolation of Morphologically Intact Mitochondria from Rat Liver. P Soc. Exp. Biol. Med. 65, 320–321 (1947).
Palade, G. E. An Electron Microscope Study Of The Mitochondrial Structure. J. Histochem. Cytochem. 1, 188–211 (1953).
Kennedy, E. P. & Lehninger, A. L. Oxidation Of Fatty Acids And Tricarboxylic Acid Cycle Intermediates By Isolated Rat Liver Mitochondria. J. Biol. Chem. 179, 957–972 (1949).
Mitchell, P. Coupling of Phosphorylation to Electron and Hydrogen Transfer by a Chemi-Osmotic type of Mechanism. Nature 191, 144–148 (1961).
Green, D. R. & Reed, J. C. Mitochondria and Apoptosis. Science 281, 1309–1312 (1998).
Luft, R., Ikkos, D., Palmieri, G., Ernster, L. & Afzelius, B. A Case Of Severe Hypermetabolism Of Nonthyroid Origin With A Defect In The Maintenance Of Mitochondrial Respiratory Control: A Correlated Clinical, Biochemical, And Morphological Study. J. Clin. Investig. 41, 1776–1804 (1962).
Willems, J. L. et al. Leigh’s encephalomyelopathy in a patient with cytochrome c oxidase deficiency in muscle tissue. Pediatrics 60, 850–857 (1977).
Holt, I. J., Harding, A. E. & Morgan-Hughes, J. A. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 331, 717–719 (1988).
Wallace, D. C. et al. Mitochondrial DNA Mutation Associated with Leber’s Hereditary Optic Neuropathy. Science 242, 1427–1430 (1988).
Wallace, D. C. Mitochondrial Diseases in Man and Mouse. Science 283, 1482–1488 (1999).
Nesbitt, V. et al. The UK MRC Mitochondrial Disease Patient Cohort Study: clinical phenotypes associated with the m.3243A>G mutation—implications for diagnosis and management. J. Neurol. Neurosurg. Psychiatry 84, 936 (2013).
Copeland, W. C. Inherited Mitochondrial Diseases of DNA Replication. Annu. Rev. Med. 59, 131–146 (2008).
Pagliarini, D. J. et al. A Mitochondrial Protein Compendium Elucidates Complex I Disease Biology. Cell 134, 112–123 (2008).
Rath, S. et al. MitoCarta3.0: an updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res 49, gkaa1011 (2020).
Iadecola, C. & Anrather, J. The immunology of stroke: from mechanisms to translation. Nat. Med 17, 796–808 (2011).
Schwartz, R. S., Eltzschig, H. K. & Carmeliet, P. Hypoxia and Inflammation. N. Engl. J. Med. 364, 656–665 (2011).
Zhang, H. et al. Mitochondrial Autophagy Is an HIF-1-dependent Adaptive Metabolic Response to Hypoxia. J. Biol. Chem. 283, 10892–10903 (2008).
Whitaker, R. M., Corum, D., Beeson, C. C. & Schnellmann, R. G. Mitochondrial Biogenesis as a Pharmacological Target: A New Approach to Acute and Chronic Diseases. Annu Rev. Pharm. 56, 1–21 (2015).
Lesnefsky, E. J., Chen, Q., Tandler, B. & Hoppel, C. L. Mitochondrial Dysfunction and Myocardial Ischemia-Reperfusion: Implications for Novel Therapies. Annu Rev. Pharm. 57, 535–565 (2015).
Eltzschig, H. K. & Eckle, T. Ischemia and reperfusion—from mechanism to translation. Nat. Med 17, 1391–1401 (2011).
Granger, D. N. & Kvietys, P. R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 6, 524–551 (2015).
Kalogeris, T., Bao, Y. & Korthuis, R. J. Mitochondrial reactive oxygen species: A double edged sword in ischemia/reperfusion vs preconditioning. Redox Biol. 2, 702–714 (2014).
D’Autréaux, B. & Toledano, M. B. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Bio 8, 813–824 (2007).
Brand, M. D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Bio Med 100, 14–31 (2016).
Brand, M. D. et al. Suppressors of Superoxide-H2O2 Production at Site IQ of Mitochondrial Complex I Protect against Stem Cell Hyperplasia and Ischemia-Reperfusion Injury. Cell Metab. 24, 582–592 (2016).
Chouchani, E. T. et al. A Unifying Mechanism for Mitochondrial Superoxide Production during Ischemia-Reperfusion Injury. Cell Metab. 23, 254–263 (2016).
Lopez-Crisosto, C. et al. Sarcoplasmic reticulum–mitochondria communication in cardiovascular pathophysiology. Nat. Rev. Cardiol. 14, 342–360 (2017).
Maack, C. et al. Elevated Cytosolic Na+ Decreases Mitochondrial Ca2+ Uptake During Excitation–Contraction Coupling and Impairs Energetic Adaptation in Cardiac Myocytes. Circ. Res. 99, 172–182 (2006).
Despa, S., Islam, M. A., Weber, C. R., Pogwizd, S. M. & Bers, D. M. Intracellular Na+ Concentration Is Elevated in Heart Failure But Na/K Pump Function Is Unchanged. Circulation 105, 2543–2548 (2002).
Shattock, M. J. et al. Na+/Ca2+ exchange and Na+/K+−ATPase in the heart. J. Physiol. 593, 1361–1382 (2015).
Santulli, G., Xie, W., Reiken, S. R. & Marks, A. R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl Acad. Sci. 112, 11389–11394 (2015).
Yellon, D. M. & Hausenloy, D. J. Myocardial Reperfusion Injury. N. Engl. J. Med. 357, 1121–1135 (2007).
Song, M. & Dorn, G. W. Mitoconfusion: Noncanonical Functioning of Dynamism Factors in Static Mitochondria of the Heart. Cell Metab. 21, 195–205 (2015).
Morales, P. E. et al. Emerging role of mitophagy in cardiovascular physiology and pathology. Mol. Asp. Med. 71, 100822 (2020).
Ikeda, Y. et al. Endogenous Drp1 Mediates Mitochondrial Autophagy and Protects the Heart Against Energy Stress. Circ. Res. 116, 264–278 (2015).
Disatnik, M. et al. Acute Inhibition of Excessive Mitochondrial Fission After Myocardial Infarction Prevents Long‐term Cardiac Dysfunction. J. Am. Hear. Assoc. 2, e000461 (2013).
Cunnane, S. C. et al. Brain energy rescue: an emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 19, 609–633 (2020).
Briston, T. & Hicks, A. R. Mitochondrial dysfunction and neurodegenerative proteinopathies: mechanisms and prospects for therapeutic intervention. Biochem Soc. T 46, 829–842 (2018).
Suomalainen, A. & Battersby, B. J. Mitochondrial diseases: the contribution of organelle stress responses to pathology. Nat. Rev. Mol. Cell Bio 19, 77–92 (2018).
Joardar, A., Manzo, E. & Zarnescu, D. C. Metabolic Dysregulation in Amyotrophic Lateral Sclerosis: Challenges and Opportunities. Curr. Genet. Med. Rep. 5, 108–114 (2017).
Querfurth, H. W. & LaFerla, F. M. Alzheimer’s Disease. N. Engl. J. Med. 362, 329–344 (2010).
Selkoe, D. J. Alzheimer’s Disease Is a Synaptic Failure. Science 298, 789–791 (2002).
Liu, P.-P., Xie, Y., Meng, X.-Y. & Kang, J.-S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target Ther. 4, 29 (2019).
Guo, T. et al. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 15, 40 (2020).
Wang, W., Zhao, F., Ma, X., Perry, G. & Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Mol. Neurodegener. 15, 30 (2020).
Cho, D.-H. et al. S-Nitrosylation of Drp1 Mediates β-Amyloid-Related Mitochondrial Fission and Neuronal Injury. Science 324, 102–105 (2009).
Lustbader, J. W. et al. ABAD Directly Links Aß to Mitochondrial Toxicity in Alzheimer’s Disease. Science 304, 448–452 (2004).
Mossmann, D. et al. Amyloid-β Peptide Induces Mitochondrial Dysfunction by Inhibition of Preprotein Maturation. Cell Metab. 20, 662–669 (2014).
Vaillant-Beuchot, L. et al. Accumulation of amyloid precursor protein C-terminal fragments triggers mitochondrial structure, function, and mitophagy defects in Alzheimer’s disease models and human brains. Acta Neuropathol. 141, 39–65 (2021).
Swerdlow, R. H. & Khan, S. M. A. “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses 63, 8–20 (2004).
Fang, E. F. et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 22, 401–412 (2019).
Sorrentino, V. et al. Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature 552, 187–193 (2017).
Schapira, A. H. V. et al. Mitochondrial Complex I Deficiency In Parkinson’s Disease. Lancet 333, 1269 (1989).
Polymeropoulos, M. H. et al. Mutation in the α-Synuclein Gene Identified in Families with Parkinson’s Disease. Science 276, 2045–2047 (1997).
Singleton, A. B. et al. α-Synuclein Locus Triplication Causes Parkinson’s Disease. Science 302, 841–841 (2003).
Valente, E. M. et al. Localization of a Novel Locus for Autosomal Recessive Early-Onset Parkinsonism, PARK6, on Human Chromosome 1p35-p36. Am. J. Hum. Genet. 68, 895–900 (2001).
Kitada, T. et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608 (1998).
Bender, A. et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet 38, 515–517 (2006).
Spillantini, M. G. et al. α-Synuclein in Lewy bodies. Nature 388, 839–840 (1997).
Burmann, B. M. et al. Regulation of α-synuclein by chaperones in mammalian cells. Nature 577, 127–132 (2020).
Chartier-Harlin, M.-C. et al. α-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364, 1167–1169 (2004).
Zambon, F. et al. Cellular α-synuclein pathology is associated with bioenergetic dysfunction in Parkinson’s iPSC-derived dopamine neurons. Hum. Mol. Genet. 28, ddz038 (2019).
Cremades, N. et al. Direct Observation of the Interconversion of Normal and Toxic Forms of α-Synuclein. Cell 149, 1048–1059 (2012).
Morimoto, R. I. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Gene Dev. 22, 1427–1438 (2008).
Choi, M. L. et al. Pathological structural conversion of α-synuclein at the mitochondria induces neuronal toxicity. Nat. Neurosci. 25, 1134–1148 (2022).
Jacobs, D. et al. Probing Membrane Association of α-Synuclein Domains with VDAC Nanopore Reveals Unexpected Binding Pattern. Sci. Rep.-UK 9, 4580 (2019).
Maio, R. D. et al. α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 8, 342ra78 (2016).
Ludtmann, M. H. R. et al. α-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat. Commun. 9, 2293 (2018).
Kim, H.-W. et al. Genetic reduction of mitochondrial complex I function does not lead to loss of dopamine neurons in vivo. Neurobiol. Aging 36, 2617–2627 (2015).
González-Rodríguez, P. et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature 599, 650–656 (2021).
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The Hallmarks of Aging. Cell 153, 1194–1217 (2013).
Trifunovic, A. et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417–423 (2004).
Shigenaga, M. K., Hagen, T. M. & Ames, B. N. Oxidative damage and mitochondrial decay in aging. Proc. Natl. Acad. Sci. 91, 10771–10778 (1994).
Schriner, S. E. et al. Extension of Murine Life Span by Overexpression of Catalase Targeted to Mitochondria. Science 308, 1909–1911 (2005).
Harman, D. Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 11, 298–300 (1956).
Velarde, M. C., Flynn, J. M., Day, N. U., Melov, S. & Campisi, J. Mitochondrial oxidative stress caused by Sod2 deficiency promotes cellular senescence and aging phenotypes in the skin. Aging (Albany NY) 4, 3–12 (2012).
St Sauver, J. L. et al. Biomarkers of cellular senescence and risk of death in humans. Aging Cell 22, e14006 (2023).
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. Hallmarks of aging: An expanding universe. Cell 186, 243–278 (2023).
Gorgoulis, V. et al. Cellular Senescence: Defining a Path Forward. Cell 179, 813–827 (2019).
Holloway, G. P. et al. Age-Associated Impairments in Mitochondrial ADP Sensitivity Contribute to Redox Stress in Senescent Human Skeletal Muscle. Cell Rep. 22, 2837–2848 (2018).
Wang, Y. & Hekimi, S. Mitochondrial dysfunction and longevity in animals: Untangling the knot. Science 350, 1204–1207 (2015).
Remmen, H. V. et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol. Genom. 16, 29–37 (2003).
Raamsdonk, J. M. V. & Hekimi, S. Deletion of the Mitochondrial Superoxide Dismutase sod-2 Extends Lifespan in Caenorhabditis elegans. PLoS Genet. 5, e1000361 (2009).
Wang, Y., Oxer, D. & Hekimi, S. Mitochondrial function and lifespan of mice with controlled ubiquinone biosynthesis. Nat. Commun. 6, 6393 (2015).
Spinelli, J. B. & Haigis, M. C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 20, 745–754 (2018).
Alberti, K. G. M. M., Zimmet, P. & Shaw, J. Metabolic syndrome—a new world‐wide definition. A Consensus Statement from the International Diabetes Federation. Diabet. Med. 23, 469–480 (2006).
Cornier, M.-A. et al. The Metabolic Syndrome. Endocr. Rev. 29, 777–822 (2008).
James, A. M., Collins, Y., Logan, A. & Murphy, M. P. Mitochondrial oxidative stress and the metabolic syndrome. Trends Endocrinol. Metab. 23, 429–434 (2012).
Schrauwen, P. & Hesselink, M. K. C. Oxidative Capacity, Lipotoxicity, and Mitochondrial Damage in Type 2 Diabetes. Diabetes 53, 1412–1417 (2004).
Ren, J., Pulakat, L., Whaley-Connell, A. & Sowers, J. R. Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. J. Mol. Med 88, 993–1001 (2010).
Mootha, V. K. et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet 34, 267–273 (2003).
Short, K. R., Nair, K. S. & Stump, C. S. Impaired Mitochondrial Activity and Insulin-Resistant Offspring of Patients with Type 2 Diabetes. N. Engl. J. Med. 350, 2419–2421 (2004).
Bhatti, J. S., Bhatti, G. K. & Reddy, P. H. Mitochondrial dysfunction and oxidative stress in metabolic disorders — A step towards mitochondria based therapeutic strategies. Biochimica Et. Biophysica Acta Bba - Mol. Basis Dis. 1863, 1066–1077 (2017).
Rovira-Llopis, S. et al. Mitochondrial dynamics in type 2 diabetes: Pathophysiological implications. Redox Biol. 11, 637–645 (2017).
Kelley, D. E., He, J., Menshikova, E. V. & Ritov, V. B. Dysfunction of Mitochondria in Human Skeletal Muscle in Type 2 Diabetes. Diabetes 51, 2944–2950 (2002).
Hesselink, M. K. C., Schrauwen-Hinderling, V. & Schrauwen, P. Skeletal muscle mitochondria as a target to prevent or treat type 2 diabetes mellitus. Nat. Rev. Endocrinol. 12, 633–645 (2016).
Yu, J., Marsh, S., Hu, J., Feng, W. & Wu, C. The Pathogenesis of Nonalcoholic Fatty Liver Disease: Interplay between Diet, Gut Microbiota, and Genetic Background. Gastroent. Res Pr. 2016, 2862173 (2016).
Szendroedi, J. et al. Role of diacylglycerol activation of PKCθ in lipid-induced muscle insulin resistance in humans. Proc. Natl Acad. Sci. 111, 9597–9602 (2014).
Dresner, A. et al. Effects of free fatty acids on glucose transport and IRS-1–associated phosphatidylinositol 3-kinase activity. J. Clin. Invest 103, 253–259 (1999).
Samuel, V. T., Petersen, K. F. & Shulman, G. I. Lipid-induced insulin resistance: unravelling the mechanism. Lancet 375, 2267–2277 (2010).
Erion, D. M. & Shulman, G. I. Diacylglycerol-mediated insulin resistance. Nat. Med 16, 400–402 (2010).
Koliaki, C. et al. Adaptation of Hepatic Mitochondrial Function in Humans with Non-Alcoholic Fatty Liver Is Lost in Steatohepatitis. Cell Metab. 21, 739–746 (2015).
Koves, T. R. et al. Mitochondrial Overload and Incomplete Fatty Acid Oxidation Contribute to Skeletal Muscle Insulin Resistance. Cell Metab. 7, 45–56 (2008).
Perreault, L. et al. Intracellular localization of diacylglycerols and sphingolipids influences insulin sensitivity and mitochondrial function in human skeletal muscle. JCI Insight 3, e96805 (2018).
Anderson, E. J. et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Investig. 119, 573–581 (2009).
Lee, H.-Y. et al. Mitochondrial-Targeted Catalase Protects Against High-Fat Diet–Induced Muscle Insulin Resistance by Decreasing Intramuscular Lipid Accumulation. Diabetes 66, 2072–2081 (2017).
Liesa, M. & Shirihai, O. S. Mitochondrial Dynamics in the Regulation of Nutrient Utilization and Energy Expenditure. Cell Metab. 17, 491–506 (2013).
Jheng, H.-F. et al. Mitochondrial Fission Contributes to Mitochondrial Dysfunction and Insulin Resistance in Skeletal Muscle. Mol. Cell Biol. 32, 309–319 (2012).
Younossi, Z. et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 15, 11–20 (2018).
Herman, M. A. & Samuel, V. T. The Sweet Path to Metabolic Demise: Fructose and Lipid Synthesis. Trends Endocrinol. Metab. 27, 719–730 (2016).
Petersen, K. F. et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc. Natl Acad. Sci. 104, 12587–12594 (2007).
Petersen, M. C. & Shulman, G. I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 98, 2133–2223 (2018).
Fromenty, B. & Roden, M. Mitochondrial alterations in fatty liver diseases. J. Hepatol. 78, 415–429 (2023).
Rector, R. S. et al. Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model. J. Hepatol. 52, 727–736 (2010).
Patterson, R. E. et al. Lipotoxicity in steatohepatitis occurs despite an increase in tricarboxylic acid cycle activity. Am. J. Physiol.-endoc M 310, E484–E494 (2016).
Lima, N. et al. Inhibition of ATG3 ameliorates liver steatosis by increasing mitochondrial function. J. Hepatol. 76, 11–24 (2022).
Zweers, H., Janssen, M. C. H., Leij, S. & Wanten, G. Patients With Mitochondrial Disease Have an Inadequate Nutritional Intake. J. Parenter. Enter. Nutr. 42, 581–586 (2018).
Zweers, H., Smit, D., Leij, S., Wanten, G. & Janssen, M. C. H. Individual dietary intervention in adult patients with mitochondrial disease due to the m.3243 A>G mutation. Nutrition 69, 110544 (2020).
Wesselink, E., Koekkoek, W. A. C., Grefte, S., Witkamp, R. F. & van Zanten, A. R. H. Feeding mitochondria: Potential role of nutritional components to improve critical illness convalescence. Clin. Nutr. 38, 982–995 (2019).
Mantle, D. & Hargreaves, I. P. Mitochondrial Dysfunction and Neurodegenerative Disorders: Role of Nutritional Supplementation. Int. J. Mol. Sci. 23, 12603 (2022).
Bomer, N. et al. Micronutrient deficiencies in heart failure: Mitochondrial dysfunction as a common pathophysiological mechanism? J. Intern. Med. 291, 713–731 (2022).
Laat et al. Dysphagia, malnutrition and gastrointestinal problems in patients with mitochondrial disease caused by the m3243A>G mutation. Neth. J. Med. 73, 30–36 (2015).
Brass, E. P. Supplemental carnitine and exercise. Am. J. Clin. Nutr. 72, 618S–623S (2000).
El-Hattab, A. W. & Scaglia, F. Disorders of carnitine biosynthesis and transport. Mol. Genet. Metab. 116, 107–112 (2015).
Hernández-Camacho, J. D., Bernier, M., López-Lluch, G. & Navas, P. Coenzyme Q10 Supplementation in Aging and Disease. Front. Physiol. 9, 44 (2018).
Kazak, L. & Cohen, P. Creatine metabolism: energy homeostasis, immunity and cancer biology. Nat. Rev. Endocrinol. 16, 421–436 (2020).
Depeint, F., Bruce, W. R., Shangari, N., Mehta, R. & O’Brien, P. J. Mitochondrial function and toxicity: Role of the B vitamin family on mitochondrial energy metabolism. Chem. -Biol. Interact. 163, 94–112 (2006).
Kolb, H. et al. Ketone bodies: from enemy to friend and guardian angel. BMC Med. 19, 313 (2021).
Bedi, K. C. Jr et al. Evidence for Intramyocardial Disruption of Lipid Metabolism and Increased Myocardial Ketone Utilization in Advanced Human Heart Failure. Circulation 133, 706–716 (2016).
Kang, H., Lee, Y., Kim, H. D., Lee, J. S. & Slama, A. Safe and Effective Use of the Ketogenic Diet in Children with Epilepsy and Mitochondrial Respiratory Chain Complex Defects. Epilepsia 48, 82–88 (2007).
Yin, J., Han, P., Tang, Z., Liu, Q. & Shi, J. Sirtuin 3 Mediates Neuroprotection of Ketones against Ischemic Stroke. J. Cereb. Blood Flow. Metab. 35, 1783–1789 (2015).
Yin, J. X. et al. Ketones block amyloid entry and improve cognition in an Alzheimer’s model. Neurobiol. Aging 39, 25–37 (2016).
Tieu, K. et al. D-β-Hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J. Clin. Investig. 112, 892–901 (2003).
Frey, S. et al. The addition of ketone bodies alleviates mitochondrial dysfunction by restoring complex I assembly in a MELAS cellular model. Biochim. Biophys. Acta (BBA) - Mol. Basis Dis. 1863, 284–291 (2017).
Horton, J. L. et al. The failing heart utilizes 3-hydroxybutyrate as a metabolic stress defense. JCI Insight 4, e124079 (2019).
Seira, O. et al. Ketogenesis controls mitochondrial gene expression and rescues mitochondrial bioenergetics after cervical spinal cord injury in rats. Sci. Rep. 11, 16359 (2021).
Hughes, S. D. et al. The ketogenic diet component decanoic acid increases mitochondrial citrate synthase and complex I activity in neuronal cells. J. Neurochem. 129, 426–433 (2014).
Xu, S. et al. Ketogenic diets inhibit mitochondrial biogenesis and induce cardiac fibrosis. Signal Transduct. Target. Ther. 6, 54 (2021).
Parikh, S. et al. Patient care standards for primary mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet. Med. 19, 1380–1397 (2017).
Cadenas, S. ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free Radic. Bio Med 117, 76–89 (2018).
Bedard, K. & Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 87, 245–313 (2007).
Tjahjono, E., Kirienko, D. R. & Kirienko, N. V. The emergent role of mitochondrial surveillance in cellular health. Aging Cell 21, e13710 (2022).
Wagner, G. R. & Hirschey, M. D. Nonenzymatic Protein Acylation as a Carbon Stress Regulated by Sirtuin Deacylases. Mol. Cell 54, 5–16 (2014).
Stefanatos, R. & Sanz, A. The role of mitochondrial ROS in the aging brain. Febs Lett. 592, 743–758 (2018).
Zhao, M. et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics 11, 1845–1863 (2021).
Zorov, D. B., Juhaszova, M. & Sollott, S. J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 94, 909–950 (2014).
Castro, L., Tórtora, V., Mansilla, S. & Radi, R. Aconitases: Non-redox Iron–Sulfur Proteins Sensitive to Reactive Species. Acc. Chem. Res. 52, 2609–2619 (2019).
Detaille, D., Pasdois, P., Sémont, A., Santos, P. D. & Diolez, P. An old medicine as a new drug to prevent mitochondrial complex I from producing oxygen radicals. PLoS ONE 14, e0216385 (2019).
Banba, A., Tsuji, A., Kimura, H., Murai, M. & Miyoshi, H. Defining the mechanism of action of S1QELs, specific suppressors of superoxide production in the quinone-reaction site in mitochondrial complex I. J. Biol. Chem. 294, 6550–6561 (2019).
Roubenne, L. et al. OP2113, a new drug for chronic hypoxia‐induced pulmonary hypertension treatment in rat. Br. J. Pharmacol. 180, 2802–2821 (2023).
Beyrath, J. et al. KH176 Safeguards Mitochondrial Diseased Cells from Redox Stress-Induced Cell Death by Interacting with the Thioredoxin System/Peroxiredoxin Enzyme Machinery. Sci. Rep. 8, 6577 (2018).
Pastore, A. et al. Glutathione: A redox signature in monitoring EPI-743 therapy in children with mitochondrial encephalomyopathies. Mol. Genet. Metab. 109, 208–214 (2013).
Huang, Y. et al. Effectiveness of idebenone nanorod formulations in the treatment of Alzheimer’s disease. J. Control. Release 336, 169–180 (2021).
Briston, T., Selwood, D. L., Szabadkai, G. & Duchen, M. R. Mitochondrial Permeability Transition: A Molecular Lesion with Multiple Drug Targets. Trends Pharm. Sci. 40, 50–70 (2019).
Sorrentino, V., Menzies, K. J. & Auwerx, J. Repairing Mitochondrial Dysfunction in Disease. Annu Rev. Pharm. 58, 1–37 (2016).
Smith, R. A. J., Porteous, C. M., Gane, A. M. & Murphy, M. P. Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl Acad. Sci. 100, 5407–5412 (2003).
Murphy, M. P. Targeting lipophilic cations to mitochondria. Biochimica Et. Biophysica Acta Bba - Bioenerg. 1777, 1028–1031 (2008).
Graham, D. et al. Mitochondria-Targeted Antioxidant MitoQ10 Improves Endothelial Function and Attenuates Cardiac Hypertrophy. Hypertension 54, 322–328 (2009).
Petrov, A., Perekhvatova, N., Skulachev, M., Stein, L. & Ousler, G. SkQ1 Ophthalmic Solution for Dry Eye Treatment: Results of a Phase 2 Safety and Efficacy Clinical Study in the Environment and During Challenge in the Controlled Adverse Environment Model. Adv. Ther. 33, 96–115 (2016).
Birk, A. V. et al. The Mitochondrial-Targeted Compound SS-31 Re-Energizes Ischemic Mitochondria by Interacting with Cardiolipin. J. Am. Soc. Nephrol. 24, 1250–1261 (2013).
Fernandez-Marcos, P. J. & Auwerx, J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 93, S884–S890 (2011).
Russell, O. M., Gorman, G. S., Lightowlers, R. N. & Turnbull, D. M. Mitochondrial Diseases: Hope for the Future. Cell 181, 168–188 (2020).
Spaulding, H. R. & Yan, Z. AMPK and the Adaptation to Exercise. Annu. Rev. Physiol. 84, 209–227 (2022).
Cantó, C. & Auwerx, J. PGC-1α, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 20, 98–105 (2009).
Golubitzky, A. et al. Screening for Active Small Molecules in Mitochondrial Complex I Deficient Patient’s Fibroblasts, Reveals AICAR as the Most Beneficial Compound. PLoS ONE 6, e26883 (2011).
Dagorn, P. G. et al. A novel direct adenosine monophosphate kinase activator ameliorates disease progression in preclinical models of Autosomal Dominant Polycystic Kidney Disease. Kidney Int 103, 917–929 (2023).
Ota, S. et al. Metformin suppresses glucose-6-phosphatase expression by a complex I inhibition and AMPK activation-independent mechanism. Biochem. Biophys. Res. Commun. 388, 311–316 (2009).
Ljubicic, V., Burt, M., Lunde, J. A. & Jasmin, B. J. Resveratrol induces expression of the slow, oxidative phenotype in mdx mouse muscle together with enhanced activity of the SIRT1-PGC-1α axis. Am. J. Physiol. -Cell Physiol. 307, C66–C82 (2014).
Bogacka, I., Xie, H., Bray, G. A. & Smith, S. R. Pioglitazone Induces Mitochondrial Biogenesis in Human Subcutaneous Adipose Tissue In Vivo. Diabetes 54, 1392–1399 (2005).
Yatsuga, S. & Suomalainen, A. Effect of bezafibrate treatment on late-onset mitochondrial myopathy in mice. Hum. Mol. Genet. 21, 526–535 (2012).
Chan, D. C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol.: Mech. Dis. 15, 1–25 (2019).
Mishra, P. & Chan, D. C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 212, 379–387 (2016).
Rappold, P. M. et al. Drp1 inhibition attenuates neurotoxicity and dopamine release deficits in vivo. Nat. Commun. 5, 5244 (2014).
Ong, S.-B. et al. Inhibiting Mitochondrial Fission Protects the Heart Against Ischemia/Reperfusion Injury. Circulation 121, 2012–2022 (2010).
Abudupataer, M. et al. Aorta smooth muscle-on-a-chip reveals impaired mitochondrial dynamics as a therapeutic target for aortic aneurysm in bicuspid aortic valve disease. eLife 10, e69310 (2021).
Chen, Z. et al. Cardiomyocyte-Restricted Low Density Lipoprotein Receptor-Related Protein 6 (LRP6) Deletion Leads to Lethal Dilated Cardiomyopathy Partly Through Drp1 Signaling. Theranostics 8, 627–643 (2018).
Coronado, M. et al. Physiological Mitochondrial Fragmentation Is a Normal Cardiac Adaptation to Increased Energy Demand. Circ. Res. 122, 282–295 (2018).
Bordt, E. A. et al. The Putative Drp1 Inhibitor mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev. Cell 40, 583–594.e6 (2017).
Demine, S., Renard, P. & Arnould, T. Mitochondrial Uncoupling: A Key Controller of Biological Processes in Physiology and Diseases. Cells 8, 795 (2019).
Perry, R. J. et al. Reversal of Hypertriglyceridemia, Fatty Liver Disease, and Insulin Resistance by a Liver-Targeted Mitochondrial Uncoupler. Cell Metab. 18, 740–748 (2013).
Perry, R. J., Zhang, D., Zhang, X.-M., Boyer, J. L. & Shulman, G. I. Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science 347, 1253–1256 (2015).
Noureddin, M. et al. Safety and efficacy of once-daily HU6 versus placebo in people with non-alcoholic fatty liver disease and high BMI: a randomised, double-blind, placebo-controlled, phase 2a trial. Lancet Gastroenterol. Hepatol. 8, 1094–1105 (2023).
Masuzawa, A. et al. Transplantation of autologously derived mitochondria protects the heart from ischemia-reperfusion injury. Am. J. Physiol.-heart C. 304, H966–H982 (2013).
Sun, X. et al. Alda-1 treatment promotes the therapeutic effect of mitochondrial transplantation for myocardial ischemia-reperfusion injury. Bioact. Mater. 6, 2058–2069 (2021).
Moskowitzova, K. et al. Mitochondrial transplantation prolongs cold ischemia time in murine heart transplantation. J. Hear Lung Transpl. 38, 92–99 (2019).
Lin, H.-C., Liu, S.-Y., Lai, H.-S. & Lai, I.-R. Isolated Mitochondria Infusion Mitigates Ischemia-Reperfusion Injury of the Liver in Rats. Shock 39, 304–310 (2013).
Huang, P.-J. et al. Transferring Xenogenic Mitochondria Provides Neural Protection against Ischemic Stress in Ischemic Rat Brains. Cell Transpl. 25, 913–927 (2015).
Orfany, A. et al. Mitochondrial transplantation ameliorates acute limb ischemia. J. Vasc. Surg. 71, 1014–1026 (2020).
Ma, H. et al. Transplantation of platelet-derived mitochondria alleviates cognitive impairment and mitochondrial dysfunction in db/db mice. Clin. Sci. 134, 2161–2175 (2020).
Chang, J.-C. et al. Allogeneic/xenogeneic transplantation of peptide-labeled mitochondria in Parkinson’s disease: restoration of mitochondria functions and attenuation of 6-hydroxydopamine-induced neurotoxicity. Transl. Res 170, 40–56.e3 (2016).
Shi, X., Zhao, M., Fu, C. & Fu, A. Intravenous administration of mitochondria for treating experimental Parkinson’s disease. Mitochondrion 34, 91–100 (2017).
Wang, Y. et al. Mitochondrial transplantation attenuates lipopolysaccharide- induced depression-like behaviors. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 93, 240–249 (2019).
Shi, C., Guo, H. & Liu, X. Platelet Mitochondria Transplantation Rescues Hypoxia/Reoxygenation-Induced Mitochondrial Dysfunction and Neuronal Cell Death Involving the FUNDC2/PIP3/Akt/FOXO3a Axis. Cell Transpl. 30, 096368972110242 (2021).
Zhao, J. et al. Mitochondria transplantation protects traumatic brain injury via promoting neuronal survival and astrocytic BDNF. Transl. Res. 235, 102–114 (2021).
Xia, L. et al. AdMSC-derived exosomes alleviate acute lung injury via transferring mitochondrial component to improve homeostasis of alveolar macrophages. Theranostics 12, 2928–2947 (2022).
Sun, C. K. et al. Systemic combined melatonin-mitochondria treatment improves acute respiratory distress syndrome in the rat. J. Pineal Res 58, 137–150 (2015).
Yu, Z. et al. The effect of mitochondrial transplantation therapy from different gender on inhibiting cell proliferation of malignant melanoma. Int J. Biol. Sci. 17, 2021–2033 (2021).
Sun, C. et al. Endocytosis-mediated mitochondrial transplantation: Transferring normal human astrocytic mitochondria into glioma cells rescues aerobic respiration and enhances radiosensitivity. Theranostics 9, 3595–3607 (2019).
Murphy, E. & Steenbergen, C. Mechanisms Underlying Acute Protection From Cardiac Ischemia-Reperfusion Injury. Physiol. Rev. 88, 581–609 (2008).
Murphy, M. P. How mitochondria produce reactive oxygen species. Biochem J. 417, 1–13 (2009).
Ikeda, G. et al. Mitochondria-Rich Extracellular Vesicles From Autologous Stem Cell-Derived Cardiomyocytes Restore Energetics of Ischemic Myocardium. J. Am. Coll. Cardiol. 77, 1073–1088 (2021).
Chang, C.-Y., Liang, M.-Z. & Chen, L. Current progress of mitochondrial transplantation that promotes neuronal regeneration. Transl. Neurodegener. 8, 17 (2019).
Poewe, W. et al. Parkinson disease. Nat. Rev. Dis. Prim. 3, 17013 (2017).
Chang, J.-C. et al. Intranasal delivery of mitochondria for treatment of Parkinson’s Disease model rats lesioned with 6-hydroxydopamine. Sci. Rep.-uk 11, 10597 (2021).
Nitzan, K. et al. Mitochondrial Transfer Ameliorates Cognitive Deficits, Neuronal Loss, and Gliosis in Alzheimer’s Disease Mice. J. Alzheimer’s Dis. 72, 587–604 (2019).
Bobkova, N. V., Zhdanova, D. Y., Belosludtseva, N. V., Penkov, N. V. & Mironova, G. D. Intranasal administration of mitochondria improves spatial memory in olfactory bulbectomized mice. Exp. Biol. Med 247, 416–425 (2021).
Zhao, Z., Yu, Z., Hou, Y., Zhang, L. & Fu, A. Improvement of cognitive and motor performance with mitotherapy in aged mice. Int J. Biol. Sci. 16, 849–858 (2020).
Javani, G., Babri, S., Farajdokht, F., Ghaffari-Nasab, A. & Mohaddes, G. Mitochondrial transplantation improves anxiety- and depression-like behaviors in aged stress-exposed rats. Mech. Ageing Dev. 202, 111632 (2022).
Kim, Y. et al. Mitochondria, Metabolism, and Redox Mechanisms in Psychiatric Disorders. Antioxid. Redox Sign 31, 275–317 (2019).
Burtscher, J. et al. The interplay of hypoxic and mental stress: implications for anxiety and depressive disorders. Neurosci. Biobehav Rev. 138, 104718 (2022).
Alexander, J. F. et al. Nasal administration of mitochondria reverses chemotherapy-induced cognitive deficits. Theranostics 11, 3109–3130 (2021).
Liu, D. et al. Brain-derived neurotrophic factor–mediated projection-specific regulation of depressive-like and nociceptive behaviors in the mesolimbic reward circuitry. Pain 159, 175 (2018).
O’Mahony, S. M., Clarke, G., Borre, Y. E., Dinan, T. G. & Cryan, J. F. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav. Brain Res 277, 32–48 (2015).
Nassir, F. & Ibdah, J. A. Role of Mitochondria in Nonalcoholic Fatty Liver Disease. Int J. Mol. Sci. 15, 8713–8742 (2014).
Ciaula, A. D. et al. Nonalcoholic Fatty Liver Disease (NAFLD). Mitochondria as Players and Targets of Therapies? Int J. Mol. Sci. 22, 5375 (2021).
Fu, A., Shi, X., Zhang, H. & Fu, B. Mitotherapy for Fatty Liver by Intravenous Administration of Exogenous Mitochondria in Male Mice. Front Pharm. 8, 241 (2017).
Manibusan, M. K., Odin, M. & Eastmond, D. A. Postulated Carbon Tetrachloride Mode of Action: A Review. J. Environ. Sci. Heal Part C. 25, 185–209 (2007).
Zhao, Z., Hou, Y., Zhou, W., Keerthiga, R. & Fu, A. Mitochondrial transplantation therapy inhibit carbon tetrachloride‐induced liver injury through scavenging free radicals and protecting hepatocytes. Bioeng. Transl. Med. 6, e10209 (2020).
Shi, X. et al. Treatment of acetaminophen-induced liver injury with exogenous mitochondria in mice. Transl. Res 196, 31–41 (2018).
Laffey, J. G. & Matthay, M. A. Fifty Years of Research in ARDS.Cell-based Therapy for Acute Respiratory Distress Syndrome. Biology and Potential Therapeutic Value. Am. J. Resp. Crit. Care 196, 266–273 (2017).
Muraca, M. et al. Mesenchymal stromal cells and their secreted extracellular vesicles as therapeutic tools for COVID-19 pneumonia? J. Control Release 325, 135–140 (2020).
Curley, G. F. et al. Mesenchymal stem cells enhance recovery and repair following ventilator-induced lung injury in the rat. Thorax 67, 496 (2012).
Millar, J. E. et al. Combined Mesenchymal Stromal Cell Therapy and ECMO in ARDS: A Controlled Experimental Study in Sheep. Am. J. Resp. Crit. Care 0, 383–392 (2019).
Islam, M. N. et al. Mitochondrial transfer from bone-marrow–derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med 18, 759–765 (2012).
Silva, J. D. et al. Mesenchymal stromal cell extracellular vesicles rescue mitochondrial dysfunction and improve barrier integrity in clinically relevant models of ARDS. Eur. Respir. J. 58, 2002978 (2021).
Yao, Y. et al. Connexin 43-Mediated Mitochondrial Transfer of iPSC-MSCs Alleviates Asthma Inflammation. Stem Cell Rep. 11, 1120–1135 (2018).
Su, Y. et al. Mitochondrial Transplantation Attenuates Airway Hyperresponsiveness by Inhibition of Cholinergic Hyperactivity. Theranostics 6, 1244–1260 (2016).
Jackson, M. V. et al. Mitochondrial Transfer via Tunneling Nanotubes is an Important Mechanism by Which Mesenchymal Stem Cells Enhance Macrophage Phagocytosis in the In Vitro and In Vivo Models of ARDS. Stem Cells Dayt. Ohio 34, 2210–2223 (2016).
Morrison, T. J. et al. Mesenchymal Stromal Cells Modulate Macrophages in Clinically Relevant Lung Injury Models by Extracellular Vesicle Mitochondrial Transfer. Am. J. Resp. Crit. Care 196, 1275–1286 (2017).
Rabchevsky, A. G., Michael, F. M. & Patel, S. P. Mitochondria focused neurotherapeutics for spinal cord injury. Exp. Neurol. 330, 113332 (2020).
Kuo, C.-C. et al. Prevention of Axonal Degeneration by Perineurium Injection of Mitochondria in a Sciatic Nerve Crush Injury Model. Neurosurgery 80, 475–488 (2017).
Lin, M.-W. et al. Mitochondrial Transplantation Attenuates Neural Damage and Improves Locomotor Function After Traumatic Spinal Cord Injury in Rats. Front Neurosci.-switz. 16, 800883 (2022).
Li, H. et al. Mitochondrial Transfer from Bone Marrow Mesenchymal Stem Cells to Motor Neurons in Spinal Cord Injury Rats via Gap Junction. Theranostics 9, 2017–2035 (2019).
Xu, J. et al. Targeted transplantation of engineered mitochondrial compound promotes functional recovery after spinal cord injury by enhancing macrophage phagocytosis. Bioact. Mater. 32, 427–444 (2024).
Levoux, J. et al. Platelets Facilitate the Wound-Healing Capability of Mesenchymal Stem Cells by Mitochondrial Transfer and Metabolic Reprogramming. Cell Metab. 33, 283–299.e9 (2021).
Peruzzotti-Jametti, L. et al. Neural stem cells traffic functional mitochondria via extracellular vesicles. Plos Biol. 19, e3001166 (2021).
Lee, J. M. et al. Mitochondrial Transplantation Modulates Inflammation and Apoptosis, Alleviating Tendinopathy Both In Vivo and In Vitro. Antioxidants 10, 696 (2021).
Lee, A. R. et al. Mitochondrial Transplantation Ameliorates the Development and Progression of Osteoarthritis. Immune Netw. 22, e14 (2022).
Bruning, U. et al. Impairment of Angiogenesis by Fatty Acid Synthase Inhibition Involves mTOR Malonylation. Cell Metab. 28, 866–880.e15 (2018).
Zhao, Q. et al. Targeting Mitochondria-Located circRNA SCAR Alleviates NASH via Reducing mROS Output. Cell 183, 76–93.e22 (2020).
Yamada, Y. et al. Power of mitochondrial drug delivery systems to produce innovative nanomedicines. Adv. Drug Deliv. Rev. 154–155, 187–209 (2020).
Hayashida, K. et al. Mitochondrial transplantation therapy for ischemia reperfusion injury: a systematic review of animal and human studies. J. Transl. Med 19, 214 (2021).
Dong, L.-F. et al. Mitochondria on the move: Horizontal mitochondrial transfer in disease and health. J. Cell Biol. 222, e202211044 (2023).
Picard, M. et al. Mitochondrial Structure and Function Are Disrupted by Standard Isolation Methods. Plos One 6, e18317 (2011).
Yamada, Y. et al. Challenges in Promoting Mitochondrial Transplantation Therapy. Int J. Mol. Sci. 21, 6365 (2020).
Chen, P. et al. A plant-derived natural photosynthetic system for improving cell anabolism. Nature 612, 546–554 (2022).
Fecher, C. et al. Cell-type-specific profiling of brain mitochondria reveals functional and molecular diversity. Nat. Neurosci. 22, 1731–1742 (2019).
Benador, I. Y. et al. Mitochondria Bound to Lipid Droplets Have Unique Bioenergetics, Composition, and Dynamics that Support Lipid Droplet Expansion. Cell Metab. 27, 869–885.e6 (2018).
Rodriguez, A.-M., Nakhle, J., Griessinger, E. & Vignais, M.-L. Intercellular mitochondria trafficking highlighting the dual role of mesenchymal stem cells as both sensors and rescuers of tissue injury. Cell Cycle 17, 1–25 (2018).
McCully, J. D., del Nido, P. J. & Emani, S. M. Mitochondrial transplantation for organ rescue. Mitochondrion 64, 27–33 (2022).
Pacak, C. A. et al. Actin-dependent mitochondrial internalization in cardiomyocytes: evidence for rescue of mitochondrial function. Biol. Open 4, 622–626 (2015).
Kitani, T., Kami, D., Matoba, S. & Gojo, S. Internalization of isolated functional mitochondria: involvement of macropinocytosis. J. Cell Mol. Med 18, 1694–1703 (2014).
Garrett, W. S. & Mellman, I. Dendritic Cells (Second Edition). In 213–VII (2001).
Qin, Y. et al. The Functions, Methods, and Mobility of Mitochondrial Transfer Between Cells. Front. Oncol. 11, 672781 (2021).
Rustom, A., Saffrich, R., Markovic, I., Walther, P. & Gerdes, H.-H. Nanotubular Highways for Intercellular Organelle Transport. Science 303, 1007–1010 (2004).
Matula, Z. et al. Stromal Cells Serve Drug Resistance for Multiple Myeloma via Mitochondrial Transfer: A Study on Primary Myeloma and Stromal Cells. Cancers 13, 3461 (2021).
Díaz-Carballo, D. et al. Cytotoxic stress induces transfer of mitochondria-associated human endogenous retroviral RNA and proteins between cancer cells. Oncotarget 8, 95945–95964 (2017).
Tiash, S., Brestoff, J. R. & Crewe, C. A guide to studying mitochondria transfer. Nat. Cell Biol. 25, 1551–1553 (2023).
Zhang, Z. et al. Muscle-derived autologous mitochondrial transplantation: A novel strategy for treating cerebral ischemic injury. Behav. Brain Res 356, 322–331 (2019).
Doulamis, I. P. et al. Mitochondrial transplantation for myocardial protection in diabetic hearts. Eur. J. Cardio-thorac. 57, 836–845 (2019).
Shin, B. et al. A Novel Biological Strategy for Myocardial Protection by Intracoronary Delivery of Mitochondria: Safety and Efficacy. Jacc Basic Transl. Sci. 4, 871–888 (2019).
Zhao, M. et al. Mesenchymal Stem Cell-Derived Extracellular Vesicles Attenuate Mitochondrial Damage and Inflammation by Stabilizing Mitochondrial DNA. Acs Nano 15, 1519–1538 (2020).
Lu, T. et al. Extracellular vesicles derived from mesenchymal stromal cells as nanotherapeutics for liver ischaemia–reperfusion injury by transferring mitochondria to modulate the formation of neutrophil extracellular traps. Biomaterials 284, 121486 (2022).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (82002339 to Junjie Gao), and Shanghai Frontiers Science Center of Degeneration and Regeneration in Skeletal System (BJ1-9000-22-4002). BioRender (https://www.biorender.com/) was used to create the Figs. 2–4.
Author information
Authors and Affiliations
Contributions
Yao Zong, Hao Li and Junjie Gao drafted and conceptualized the initial manuscript. Yao Zong, Long Chen, Peng Liao, Yongqiang Zheng, Yao Pan designed and organized the figure and tables. Supervision and strategic direction were provided by Junjie Gao, Changqing Zhang, Delin Liu, and Minghao Zheng, who also contributed key ideas that shaped the manuscript. Hao Li, Delin Liu, Junjie Gao, and Minghao Zheng refined the manuscript. All authors have read and approved the review article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zong, Y., Li, H., Liao, P. et al. Mitochondrial dysfunction: mechanisms and advances in therapy. Sig Transduct Target Ther 9, 124 (2024). https://doi.org/10.1038/s41392-024-01839-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-024-01839-8
