
Abstract
Mitochondria have been primarily considered intracellular organelles that are responsible for generating energy for cell survival. However, accumulating evidence suggests that mitochondria are secreted into the extracellular space under physiological and pathological conditions, and these secreted mitochondria play diverse roles by regulating metabolism, the immune response, or the differentiation/maturation in target cells. Furthermore, increasing amount of research shows the therapeutic effects of local or systemic administration of mitochondria in various disease models. These findings have led to growing interest in exploring mitochondria as potential therapeutic agents. Here, we discuss the emerging roles of mitochondria as extracellularly secreted organelles to shed light on their functions beyond energy production. Additionally, we provide information on therapeutic outcomes of mitochondrial transplantation in animal models of diseases and an update on ongoing clinical trials, underscoring the potential of using mitochondria as a novel therapeutic intervention.
Similar content being viewed by others
Introduction
Mitochondria are multifaceted organelles that perform various functions to regulate cellular homeostasis1. Despite being the most well-known site of energy production or the “powerhouse” of the cell, mitochondria play many other pivotal roles, such as controlling the biosynthesis of molecules needed for cell growth and regulation of apoptosis2, intracellular calcium level3, redox balance4, the immune response5, cell stemness6, and interorganelle communication7. Mitochondria are also highly dynamic organelles, continuously changing their shapes through fusion and fission events8,9. The coordinated remodeling of mitochondrial morphology is tightly coupled with the major mitochondrial functions listed above, and imbalances in mitochondrial dynamics lead to mitochondrial dysfunction and pathological conditions9. A notable feature of mitochondria is their ability to generate mitochondrial-derived vesicles (MDVs) that transport mitochondrial components to lysosomes10, endosomes11, or peroxisomes12 for communication13. Emerging evidence suggests that MDVs and mitochondria may also be involved in intercellular communication or systemic regulation of cellular function14,15,16, the mechanisms of which are currently under active investigation.
Cells release diverse membrane-bound vesicles into the extracellular space to eliminate or transfer specific compounds or communicate with other cells17. Depending on their size and biogenesis pathway, these extracellular vesicles (EVs) are subcategorized as exosomes (50–150 nm in diameter, multivesicular body-derived), ectosomes (less than 0.1 μm to several μm in diameter, plasma membrane-derived), microvesicles (0.1-1 μm in diameter), large oncosomes (>1 μm in diameter), apoptotic bodies (>1 μm in diameter, apoptotic cell-derived), migrasomes (0.5–3 μm in diameter, migrating cell-derived), and the newly identified exomers (<50 nm in diameter, biogenesis unclear)17,18,19,20. However, simple categorization based on size and biogenesis pathway does not fully reflect the heterogeneity of the cellular origins, cargos, and functions of EVs. In this regard, the relatively recent discovery of extracellular mitochondria and EVs containing mitochondrial components that are secreted by many cell types adds a new level of complexity to EV biology21. The characterization, sorting and secretory mechanisms, and biological effects of extracellular mitochondria and EVs containing mitochondrial components under physiological or pathological conditions are currently under intense research.
Accumulating evidence suggests that extracellularly secreted mitochondria are transferred to recipient cells to induce therapeutic responses, suggesting that exogenous supplementation with mitochondria isolated from proper donor cells or tissues could be a therapeutic strategy. Local or systemic delivery of isolated mitochondria or mitochondrial transplantation has shown promising outcomes in animal models under various conditions, and several clinical trials involving mitochondrial transplantation to treat myocardial ischemia, cerebral ischemia, or inflammatory muscle diseases have been initiated. Despite significant attention and efforts aimed at developing mitochondrial transfer/transplantation strategies, research on the mechanisms of mitochondrial transfer is still in its early stages, and many critical questions remain unanswered. Importantly, understanding the mechanisms and biological effects of the extracellular secretion and transfer of mitochondria in vivo, as well as the mechanisms of recipient cell contact and uptake of extracellular mitochondria, will aid in the selection of appropriate sources for mitochondrial isolation and improve target specificity for successful mitochondrial transplantation therapy with minimal adverse effects.
In the first part of the review, we will discuss the evidence, mechanism and outcomes of extracellular mitochondrial secretion, focusing on the release of whole mitochondria due to its relevance with mitochondrial transplantation therapy involving the isolation and administration of intact mitochondria. We will not discuss the release of selective mitochondrial components (mitochondrial proteins, lipids, RNAs and/or DNA) or other modes of intercellular mitochondrial transfer such as tunnelling nanotubes (TNTs) as EVs containing mitochondrial components and TNT-mediated mitochondrial transfer have been previously reviewed21,22,23. In the second part, we will review the therapeutic effects of mitochondrial transfer in animal models under pathological conditions and current updates on human trials involving mitochondrial transplantation.
Extracellular mitochondrial secretion and its biological effects
Evidence and mechanisms of extracellular mitochondrial secretion
Mitochondria have been reported to be secreted extracellularly by many cell types, including mesenchymal stem cells (MSCs), astrocytes, neural stem cells, platelets, adipocytes, hepatocytes, cardiomyocytes, endothelial progenitor cells, osteoblasts, and various cell lines (Table 1). In this section, we briefly discuss key evidence and major mechanisms of mitochondrial extrusion.
ARRDC1-mediated microvesicles
Extracellular release of mitochondria was reported as early as 200624 in human MSCs under normal conditions in vitro. Vesicles containing fluorescently labelled mitochondria were released by MSCs onto tissue culture plates and contacted the plasma membranes of nearby cells24. Since then, a number of reports have demonstrated that human, mouse, and rat MSCs actively release microvesicles containing mitochondria into the extracellular space (Table 1). Regarding the mechanism of mitochondrial secretion, Phinney et al. showed that under standard culture conditions, MSCs manage oxidative stress by extracellularly releasing depolarized mitochondria through arrestin domain-containing protein 1 (ARRDC1)-mediated microvesicles (ARMMs)25. Live cell imaging showed that mitochondria travel toward the cell periphery and are included in the outward budding blebs of the plasma membrane25. Electron microscopy confirmed the presence of microvesicles containing mitochondria in MSC-conditioned media25.
CD38/cADPR/calcium signaling
In addition to MSCs, astrocytes have been shown to secrete mitochondria into the extracellular space through a calcium-dependent mechanism (Table 1). In 2016, Hayakawa et al. reported the presence of extracellular particles (0.3-1.1 μm in diameter) containing functional mitochondria released from rat cortical astrocytes26. A high percentage of extracellular mitochondria-containing particles were β1-integrin- and CD63-positive and were released via CD38/cyclic ADP-ribose (cADPR)/calcium signaling in astrocytes26. Similarly, our group recently demonstrated that mature osteoblasts secrete CD63-positive mitochondria-containing vesicles (>0.2 μm in diameter) into the extracellular space partly through CD38/cADPR signaling27. CD38 is highly expressed in osteoblasts28 and has been suggested to play a critical role during bone formation, and Cd38-knockout mice exhibit an osteoporotic phenotype29,30. Our group showed that the Cd38 expression pattern in differentiating osteoblasts coincided with the pattern of mitochondrial secretion, and knockdown of Cd38 significantly impaired mitochondrial release27. However, whether CD38/cADPR signaling is specific to mitochondrial release or regulates mitochondrial secretion by other cell types requires further investigation.
Actin polymerization
Activated platelets can release functional mitochondria into the extracellular space through actin dynamics (Table 1). Boudreau et al. demonstrated that intact free mitochondria or microparticles containing mitochondria were present in the supernatant of thrombin-activated human platelets16. The group used actin polymerization inhibitors (cytochalasin B, D, E, or latrunculin A) or tubulin polymerization inhibitor (nocodazole) and found that the extrusion of free mitochondria and microparticles containing mitochondria was significantly decreased by the addition of actin inhibitors but not the tubulin inhibitor, suggesting that mitochondrial secretion requires intact actin but not microtubule dynamics16. However, the release of microparticles without mitochondria also significantly decreased in response to the actin inhibitors and not the tubulin inhibitor, suggesting that the mechanism may not be mitochondria-specific but may apply to microparticle secretion in general. The involvement of actin polymerization in mitochondrial extrusion was also demonstrated in cellular FLICE-like inhibitory protein (cFLIP)-deficient mouse embryonic fibroblasts (MEFs) stimulated with tumor necrosis factor alpha (TNF-α)31. Unlike in platelets, exposure to an actin polymerization inhibitor (cytochalasin D) or tubulin destabilizer (paclitaxel) impaired cytoplasmic vacuole formation and the subsequent secretion of free mitochondria by MEFs, indicating that both intact actin and tubulin dynamics are essential for mitochondrial release by MEFs31.
Changes in mitochondrial morphology
Specific alterations in mitochondrial morphology have been suggested as a mechanism leading to extracellular mitochondrial secretion27,31. Nakajima et al. showed that cytoplasmic vacuoles within cFLIP-deficient MEFs engulfed fragmented mitochondria but not elongated mitochondria and released them into the extracellular space in response to TNF-α stimulation, indicating that mitochondrial fragmentation is a prerequisite for their extracellular release31. Likewise, our group recently reported that mitochondrial fragmentation and donut formation, which actively produce MDVs, stimulated mitochondrial extrusion from osteoblasts27. We demonstrated that inducing mitochondrial fission and donut formation by knocking down Opa1, which mediates mitochondrial fusion, or overexpressing Fis1, which promotes mitochondrial fission and MDV formation8, significantly increased the extracellular release of mitochondria, while treatment with the mitochondrial fusion promoter M1 prevented mitochondrial secretion by osteoblasts27. These results indicate that mitochondrial dynamics may play direct and critical roles in mediating mitochondrial extrusion. Close examination of mitochondrial morphology in different cell types that secrete mitochondria will help determine whether this mechanism applies universally.
Secretory autophagy
Extracellular release of mitochondria may be a mitochondrial quality control (MQC) process alternative to mitophagy32,33,34. Nicolas-Avila et al. reported that healthy or stressed mouse cardiomyocytes ejected defective mitochondria into the extracellular space through LC3-positive membrane vesicles called exophers, which are distinct from classical EVs in that they are larger in size (3.5 \(\pm\) 0.1 μm in mean diameter), contain large organelles such as mitochondria and are driven by the autophagy machinery34. The group showed that extracellular secretion of damaged mitochondria through exophers was a mechanism of mitochondrial quality control34. In the same year, Choong et al. suggested that extracellular release of mitochondria was an alternative MQC system to maintain mitochondrial homeostasis in rat PC12 cells and several human cell lines33. In support of this conclusion, genetic deletion or knockdown of autophagy/mitophagy genes significantly increased mitochondrial secretion into the extracellular environment through direct budding from the plasma membrane to compensate for defective mitophagy33. Likewise, Tan et al. reported that during PINK1-Parkin-mediated mitophagy, damaged mitochondria could still be cleared (through extracellular secretion) without the mammalian ATG8 (mATG8)-conjugation system, which is a crucial step in autophagy that leads to lysosomal degradation32. The group suggested that mitochondria were extruded through the secretory autophagy pathway that releases secretory cargos within autophagosomes, which was supported by data showing that genetic inhibition of autophagosome formation or autophagosome-plasma membrane fusion decreased mitochondrial secretion32. Through biochemical analysis and proteinase K protection analysis of the EV fraction of ATG7-knockout HeLa cells, the group demonstrated that secreted mitochondria were present as free organelles and were not enclosed by EVs32. Overall, these findings indicate that different mechanisms of mitochondrial secretion exist in a variety of cell types. However, further investigation is necessary to determine whether the various mechanisms discussed above act in conjunction with each other or if there is a distinct molecular mechanism that is specific to the secretion of mitochondria other than general EV secretion.
Biological effects of extracellular mitochondria on target cells
Extracellularly secreted mitochondria have been shown to target recipient cells to modulate various metabolic, immune, or differentiation/maturation responses (Table 1). In the following section, we review the major biological effects of secreted mitochondria on recipient cells.
Metabolic effects
The most widely reported biological effects of extracellular mitochondria are their incorporation into recipient cells and subsequent regulation of metabolism, such as mitochondrial bioenergetics and the oxidative stress response. For instance, extracellular mitochondria secreted by MSCs are taken up by macrophages through phagocytosis and fuse with host mitochondria to enhance the macrophage oxygen consumption rate (mitochondrial respiration)25. MSC-derived extracellular mitochondria are also internalized by IEC-6 intestinal epithelial cells and fused with host mitochondria to improve mitochondrial function35. Hayakawa and colleagues demonstrated that extracellular mitochondria secreted by astrocytes entered neurons and enhanced intracellular ATP levels and neuronal viability26. Similarly, mitochondria extruded by neural stem cells were taken up by mononuclear phagocytes by endocytosis and integrated into the endogenous mitochondrial network to restore oxidative phosphorylation36. Furthermore, activated platelets secrete functional mitochondria that are subsequently incorporated into MSCs to stimulate the tricarboxylic acid (TCA) cycle and de novo fatty acid synthesis, thereby triggering the secretion of proangiogenic factors37. In addition to regulating mitochondrial bioenergetics in recipient cells, extracellular mitochondria have been shown to regulate the oxidative stress response. Small EVs containing oxidatively damaged mitochondria released by adipocytes were internalized into cardiomyocytes to induce a burst of reactive oxygen species (ROS), triggering a compensatory antioxidant response and protecting cardiomyocytes through hormesis38. Likewise, exosomes containing polarized mitochondria (MitoTracker Green-positive) secreted by proinflammatory human leukocyte antigen-antigen D related (HLA-DR)-positive airway myeloid-derived regulatory cells (MDRCs) were taken up by peripheral T cells, and MitoTracker Green-positive mitochondria were integrated into the host mitochondrial network, possibly affecting T-cell differentiation and function through ROS generation39.
Anti- or proinflammatory effects
In addition to modulating metabolism, extracellular mitochondria have been shown to trigger anti- or proinflammatory responses in recipient cells. The transfer of EVs containing mitochondria derived from MSCs to human macrophages enhanced phagocytosis and downregulated proinflammatory cytokine secretion by macrophages40. Similarly, mitochondria and mitochondrial DNA (mtDNA) transfer through exosomes released by adipose-derived MSCs restored mitochondrial integrity in macrophages and induced their shift to an anti-inflammatory phenotype by suppressing proinflammatory cytokine (IL-1β and TNF-α) secretion and upregulating anti-inflammatory cytokine (IL-10 and Arg-1) production41. The expression of proinflammatory genes (Il1β, Nos2, and Il6) was also significantly downregulated in mononuclear phagocytes in response to extracellular mitochondria released by neural stem cells36. Although further research is necessary, the reported anti-inflammatory effects of extracellular mitochondria appear to be secondary responses to improvements in mitochondrial integrity and function.
Secreted mitochondria have also been reported to stimulate proinflammatory responses in recipient cells. For example, extracellular mitochondria released by activated platelets were shown to act as endogenous substrates of secreted phospholipase A2 IIA (sPLA2-IIA), which hydrolyzes the mitochondrial membrane and leads to the generation of inflammatory mediators such as lysophospholipids, fatty acids, and mtDNAs that induce a proinflammatory response in neutrophils16. Furthermore, extracellular mitochondria or mtDNA enclosed by microparticles released from hepatocytes activate Toll-like receptor 9 (TLR9) and the proinflammatory response in lysozyme-expressing cells such as neutrophils, monocytes, and macrophages42. Damaged mitochondria released extracellularly by HeLa cells via the secretory autophagy pathway induced a proinflammatory response in recipient HeLa cells by activating the cGAS-STING pathway, possibly through mtDNA32. Based on these reports, mtDNA, which is a well-known and potent damage-associated molecular pattern (DAMP), appears to be largely responsible for the proinflammatory response triggered by extracellular mitochondria. Importantly, specific mechanisms that induce or prevent the proinflammatory effects of mtDNAs on recipient cells warrant further investigation.
Cell differentiation/maturation effects
Extracellular mitochondria have been shown to regulate target cell differentiation or maturation. Recently, Rosina and colleagues reported that brown adipocytes released EVs containing MDVs with damaged mitochondrial parts, which were incorporated into recipient brown adipocytes and activated AMP-activated protein kinase (AMPK) to suppress adipocyte differentiation and thermogenic potential by downregulating the expression of Cd36, Fabp4, and Ucp114. The group suggested that oxidized materials and high levels of AMP contained within EVs could trigger AMPK in target cells to prevent adipogenesis14. Regarding the effects of extracellularly secreted mitochondria on stimulating target cell maturation, mitochondria released by necroptotic, Fas-associated protein with death domain (FADD)-deficient Jurkat cells were engulfed by human monocyte-derived dendritic cells (MDDCs) and promoted their maturation, inducing the cell surface markers CD80, CD83, and CD8643. Furthermore, our group recently reported that mitochondria secreted from mature osteoblasts enhanced the osteogenic maturation of osteoprogenitor cells without affecting mitochondrial respiration through the delivery of cargo proteins27. We showed that the incorporation of intact extracellular mitochondria into recipient cells was not required for this effect as treatment with secreted mitochondria after a repeated freeze/thaw cycle did not abrogate the increase in the maturation of osteoprogenitors. Instead, exposure of extracellular mitochondria to proteinase K abolished their ability to stimulate osteoprogenitor maturation, indicating that specific proteins within mitochondria were responsible for this effect. Whether the proteins activate surface receptors or are taken up by target cells needs further investigation, but the results suggest a mechanism that does not involve direct metabolic effects mediated by the integration of intact mitochondria into recipient cells27.
Mitochondrial transplantation therapy
Therapeutic effects of mitochondrial transplantation on animal models
The beneficial effects of extracellularly secreted mitochondria on the metabolism and function of recipient cells indicate that exogenous supplementation with mitochondria could induce curative responses through similar mechanisms. As with those secreted extracellularly by donor cells, injected mitochondria have been shown to enter recipient cells to regulate target cell metabolism, inflammatory response, or their differentiation/maturation in vivo (Table 2). Through these effects, mitochondrial transplantation has been shown to repair damaged tissues, including but not limited to the heart, brain, spinal cord, liver, lungs, kidney, musculoskeletal tissues, and intestine, in animal models of critical illnesses (Table 2). Currently, several clinical trials are recruiting or selecting patients to investigate the effects of mitochondrial treatment on myocardial ischemia, cerebral ischemia, or inflammatory muscle diseases such as polymyositis and dermatomyositis (Table 3). Thus, we will briefly review the major therapeutic effects of mitochondrial transplantation on animal models of cardiac ischemia, cerebral ischemia, and decline in skeletal muscle function or mass. Descriptions of mitochondrial transplantation in animal models of other diseases are provided in Table 2.
Cardiac ischemia
The first therapeutic evaluation of mitochondrial transplantation in an animal model dates back to 2009 when McCully et al. demonstrated that directly injecting viable mitochondria isolated from a nonischemic heart into the ischemic zone of cardiac tissue promoted myocardial functional recovery and cell viability in rabbits subjected to ischemia/reperfusion (I/R) injury44. The authors emphasized that the isolated mitochondria must be fresh, viable, and respiration-competent to induce cardioprotective effects, since frozen mitochondria or separated mitochondrial components failed to do so44. Since then, McCully’s team has actively investigated mitochondrial transplantation therapy for cardiac ischemia in rabbits, pigs, and rats (Table 2). Masuzawa et al. reported that injecting mitochondria isolated from autologous skeletal muscle into the ischemic heart attenuated myocardial injury and improved cardiac function in rabbits subjected to I/R injury45. The group showed that the injected mitochondria were taken up by cardiomyocytes 2 hours after administration, and this uptake resulted in an increase in the oxygen consumption rate and ATP production, while also leading to a significant decrease in serum inflammatory markers45. Notably, the authors suggested that the transplanted mitochondria also exerted extracellular effects (without internalization by cardiomyocytes) as cardioprotection was apparent within 10 minutes of reperfusion when mitochondria were present in the interstitial space outside of cardiomyocytes45. The authors also highlighted the importance of transplanting viable mitochondria45. A few years later, McCully’s team reported similar findings in a porcine model of I/R, suggesting that injecting mitochondria derived from autologous skeletal muscle into the ischemic area of cardiac tissue improved myocardial cell viability46, and recently reported the cardioprotective effects of mitochondrial transplantation on diabetic rat hearts subject to warm global ischemia47.
Cerebral ischemia
Mitochondrial transplantation has also exerted beneficial effects on animal models of cerebral ischemia. In 2016, Huang et al. first demonstrated that local intracerebral or systemic intra-arterial injection of xenogenic hamster mitochondria (isolated from the BHK-21 hamster cell line) significantly rescued motor performance and attenuated infarct size and neuronal cell death in a rat ischemic stroke model induced by middle central artery occlusion (MCAO)48. The group detected the internalization of transplanted mitochondria into neurons and astrocytes but with low efficacy, suggesting that cellular uptake of exogenous mitochondria may not be necessary for their neuroprotective effects48. A few years later, Zhang et al. used the same rat stroke model of MCAO and showed that intracerebroventricular (ICV) injection of mitochondria isolated from autologous skeletal muscle led to their internalization by neurons, especially in the ischemic penumbra, thereby increasing ATP levels and neurogenesis while decreasing oxidative stress, apoptosis, reactive astrogliosis, and brain infarct volume49. Nakamura et al. also showed that in a mouse MCAO model, intravenous infusion of mitochondria isolated from cryopreserved placentas significantly reduced infarction volume50. The infused mitochondria crossed the blood‒brain barrier and were distributed in the ischemic brain and peripheral organs such as the lung, liver, kidney, and heart, the effects of which are yet to be determined50. Xie et al. and Lee et al. further confirmed the therapeutic potential of mitochondrial transplantation, which decreased infarct volume and improved cell survival in a rat MCAO model of stroke51,52.
Loss of skeletal muscle function or mass
The therapeutic effects of mitochondrial transplantation on skeletal muscle tissues have only recently been investigated. In 2020, Zhao et al. demonstrated that tail vein injection of mitochondria isolated from young mouse livers significantly improved skeletal muscle function in aged mice (18-month-old mice with compromised muscle function), which was assessed by the forced swimming test for muscular endurance and rotarod test for muscular coordination53. Mitochondrial treatment significantly increased pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, NADH dehydrogenase, and ATP levels in aged skeletal muscles while decreasing ROS levels53. In the same year, McCully’s group evaluated the therapeutic potential of mitochondrial transplantation in a mouse model of acute limb ischemia (ALI), which leads to loss of muscle viability and function54. The injection of mitochondria isolated from donor mouse skeletal muscle into the hindlimb muscles of ALI-induced mice resulted in their distribution within myofibers, a decrease in apoptosis, and an increase in hindlimb function and power, as measured by DigiGait54. Recently, Alway et al. showed that in a mouse model of muscle injury induced by BaCl2, injecting liver-derived mitochondria through the tail vein resulted in preferential uptake by injured myofibers, which was possibly facilitated by the damaged sarcolemma. This uptake significantly improved muscle regeneration, increased muscle weight and fiber size and restored maximal muscle force55. Kim et al. also reported that intramuscular injection of mitochondria obtained from human umbilical cord-derived mesenchymal stem cells (UC-MSCs) significantly increased muscle mass by 1.5-fold and reduced the expression of muscle-specific ubiquitin E3-ligases in a dexamethasone-induced rat model of muscle atrophy56. Although research on skeletal muscle-targeted mitochondrial transplantation is still in its early stage, the results thus far suggest that mitochondrial treatment is a promising therapeutic strategy to accelerate the recovery of muscle function and mass.
Clinical trials
The encouraging results of mitochondrial transplantation in animal models of diseases have led to several human trials. Here, we identified registered clinical trials based on the information provided by ClinicalTrials.gov with search terms for intervention/treatment, including “mitochondria transplantation” or “mitochondria injection”. At the time that this manuscript was revised, another review discussing clinical trials using mitochondrial transplantation has been published57. To date, two human trials involving mitochondrial transplantation have been completed, which include the microinjection of mitochondria into oocytes to treat infertility and the infusion of mitochondria-transplanted/augmented hematopoietic stem cells to treat Pearson syndrome (Table 3). More importantly, four trials involving systemic or tissue-specific injection of isolated mitochondria, which align more closely with the focus of this review, are currently recruiting or enrolling by invitation (Table 3). We will introduce these 4 clinical trials in the following paragraphs.
A clinical trial sponsored by Boston Children’s Hospital is recruiting participants to assess the effects of autologous mitochondrial transplantation on myocardial I/R injury and extracorporeal membrane oxygenation (ECMO) complications (NCT02851758). The mitochondria will be isolated from the skeletal muscle of the chest wall and administered to the ischemic myocardium by direct injection (for surgical reoperation subjects) or intracoronary infusion (for catheterization subjects). The outcomes will be determined by safety, improvements in ventricular function, and the ability to be removed from ECMO support.
A clinical trial held at the University of Washington is recruiting subjects to investigate the effects of autologous mitochondrial transplantation on brain ischemia for the first time (NCT04998357). The mitochondria will be isolated from muscle tissues adjacent to the surgical access site and will be infused into an artery in the brain by a microcatheter during standard-of-care endovascular reperfusion therapy. For the outcome measures, investigators will monitor adverse effects associated with mitochondrial infusion and check for a reduction in infarct volume.
A human trial in South Korea sponsored by Paean Biotechnology Inc. is enrolling by invitation to evaluate the safety, tolerability, and efficacy of the transplantation of PN-101 (mitochondria isolated from allogeneic UC-MSCs) in subjects with refractory polymyositis or dermatomyositis (NCT04976140). In mice, PN-101 was shown to reduce pathological inflammatory responses by blocking the nuclear factor kappa B (NFκB) signaling pathway58. The mitochondria will be administered intravenously, and the primary outcome will be measured by dose-limiting toxicity (DLT) and International Myositis Assessment and Clinical Studies Group-total improvement score (IMACS-TIS).
Finally, investigators at Tehran University of Medical Sciences are recruiting patients to determine the therapeutic effects of transplanting autologous mitochondria and/or MSC-derived exosomes on myocardial infarction, ischemia, and stunning (NCT05669144). Exosomes-only, mitochondria-only, exosomes plus mitochondria, or placebo will be administered through intracoronary and intramyocardial injection. The primary outcome measures include left ventricle ejection fraction and allergic reactions.
Conclusion and future perspectives
Over the past several years, it has become evident that mitochondria are spontaneously secreted by various cells into the extracellular space and are transferred to recipient cells (Fig. 1). The field of extracellular mitochondrial secretion and transfer has increasingly gained attention partly because (1) a significant advancement in imaging technology has led to a more qualitative way to detect extracellular mitochondrial release by different cell types; (2) the development of various mitochondrial reporter mice, such as PHaM mitoDendra259, C57BL/6Jsu9-dsRed237, adipo-mitoFlag38, MitoFat60, and Col1a1-Cre; Igs1CKI-mitoGFP/+27 (Table 1), which has allowed greater experimental and technical convenience in the analysis of mitochondrial secretion and transfer in vivo; and 3) the potential therapeutic applications because mitochondria released by donor cells have been shown to enter recipient cells to enhance mitochondrial bioenergetics and cellular functions. Similar to those secreted extracellularly, mitochondria that are transferred exogenously have been shown to regulate mitochondrial metabolism, the inflammatory response, or the differentiation and maturation of recipient cells. This regulation can occur through their integration into the host mitochondrial network, signaling by mitochondrial cargos, or other unspecified extracellular effects (Fig. 1). The promising outcomes of exogenous mitochondrial transfer in animal models of diseases have led to the development of clinical trials with mitochondrial transplantation-based therapeutic interventions. However, research on the mechanisms of the secretion and transfer of mitochondria is still in its early stages, and several critical questions remain to be answered in future investigations.

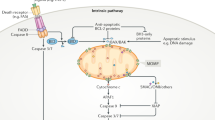
Donor cells extracellularly secrete microvesicles containing mitochondria through outward budding, exosomes containing mitochondrial-derived vesicles (MDVs) through the fusion of multivesicular bodies (MVBs) with the plasma membrane, free/naked mitochondria through an unclarified mechanism, or depolarized mitochondria through the secretory autophagy pathway. Mitochondrial fission and CD38/cADPR signaling have been suggested to mediate extracellular mitochondrial secretion. Mitochondrial transplantation involves the isolation of mitochondria from autologous tissues such as skeletal muscle or healthy cells such as mesenchymal stem cells (MSCs) via differential centrifugation or filtration methods and subsequent local or systemic administration. Although the administration of freeze-stored mitochondria has been described, the injection of freshly isolated mitochondria appears to be ideal. Mitochondria that are secreted extracellularly or introduced exogenously can be taken up by recipient cells through membrane fusion or endocytosis. Extracellular mitochondria may also interact with recipient cell surface receptors such as heparan sulfate proteoglycans (HSPGs) for uptake. Once inside the cells, the mitochondria integrate with the host mitochondrial network or activate signaling pathways mediated by their cargo. Although further investigation is needed, secreted or transplanted mitochondria have also been suggested to exert extracellular effects. Overall, these reactions elicit major biological effects on recipient cells, including increases in mitochondrial respiration and cell survival and the regulation of the oxidative stress response, the inflammatory response, and cell differentiation or maturation.
First, the molecular mechanisms specific to extracellular mitochondrial secretion are largely unknown. While CD38/cADPR signaling has been shown to promote extracellular mitochondrial release in astrocytes and osteoblasts26,27, whether it plays the same role in other cells that highly express CD38, such as immune cells, needs further examination. Future research could also focus on identifying additional pathways that commonly regulate mitochondrial secretion by different cell types. Furthermore, mechanisms that differentiate the secretion of free/naked mitochondria from vesicle-enclosed mitochondria also require additional investigation.
Second, it is still unclear whether there are mechanisms that allow extracellularly released mitochondria to target specific cell types. Recently, mitochondria released by adipocytes were shown to preferentially target macrophages among various clusters of cells in adipose tissues60, and cellular heparan sulfates were thought to act as receptors to mediate the specific uptake of extracellular mitochondria by macrophages61 and human HepG2 cells62 (Fig. 1). However, it is also possible that specific surface molecules or proteins are expressed on extracellularly secreted mitochondria to direct their interaction with the recipient cell membrane. Identifying these molecules may allow for the modification or genetic engineering of isolated mitochondria to improve the target specificity of mitochondrial transplantation therapies and minimize off-target effects.
Third, future research should focus on developing techniques to enhance the purity of isolated mitochondria for biological/biochemical analysis and exogenous delivery. While differential centrifugation and differential filtration are the most popular methods used to isolate mitochondria for therapeutic transfer because they are simple and quick processes, they likely also yield nonmitochondrial particles that may contribute to undesired effects. Flow cytometry-based sorting of mitochondria significantly increases purity and allows precise quantification63 but takes relatively longer to obtain a sufficient number of mitochondria and requires a specialized device, making it applicable for biochemical analysis of pure mitochondria but suboptimal for clinical situations where fresh mitochondria are expected to be rapidly isolated. The development of strategies to enhance the long-term storage of viable mitochondria for off-the-shelf use would partly overcome this limitation.
Finally, mitochondrial transplantation protocols, including methods, time point, frequency, and dose of administration, needs further establishment and optimization for long-term efficacy. Whether treatment with mitochondria plus other drugs can enhance mitochondrial transfer efficiency or function may also be investigated. The transplantation of cells that are naturally capable of mitochondrial transfer may prolong the effects of mitochondrial delivery and minimize the rejection of exogenous mitochondria. In this regard, in vivo transplantation of CD34+ hematopoietic stem and progenitor cells (HSPCs) augmented with normal exogenous mitochondria ex vivo induced long-term persistence (up to 4.5 months post transplantation) of exogenous mitochondrial transfer from HSPCs to myeloid and B cells in a mouse model of mitochondrial dysfunction64. Transplantation of CD34+ stem cells enriched with mitochondria is currently being examined in a clinical trial in pediatric patients with Pearson syndrome (Table 3). Additionally, whether artificially packaging mitochondria in vesicles for transplantation enhances mitochondrial stability or uptake may be tested in the future as mitochondria inside EVs have been suggested to be more resistant to the extracellular environment containing high levels of Ca2+ and oxidative stress than free isolated mitochondria65.
In conclusion, extracellular mitochondrial secretion, their transfer to recipient cells, and mitochondrial transplantation are increasingly gaining attention for their potential in a variety of therapeutic settings. Understanding the mechanisms and biological effects of the extracellular secretion and transfer of mitochondria, which still require extensive research, serves as a theoretical basis for the development of successful mitochondrial transplantation strategies. Therefore, future efforts should focus on unraveling the molecular and cellular mechanisms of extracellular mitochondrial secretion and transfer, as well as methods to improve the efficiency and efficacy of mitochondrial transplantation therapy.
References
Monzel, AS, Enriquez, JA & Picard, M Multifaceted mitochondria: moving mitochondrial science beyond function and dysfunction. Nat Metab 5, 546–562 (2023).
McArthur, K. et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 359, eaao6047 (2018).
Baughman, JM et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345 (2011).
Handy, DE & Loscalzo, J Redox regulation of mitochondrial function. Antioxid Redox Signal 16, 1323–1367 (2012).
Yasukawa, K et al. Mitofusin 2 inhibits mitochondrial antiviral signaling. Sci Signal 2, ra47 (2009).
Dohla, J et al. Metabolic determination of cell fate through selective inheritance of mitochondria. Nat Cell Biol 24, 148–154 (2022).
Csordas, G, Weaver, D & Hajnoczky, G Endoplasmic Reticulum-Mitochondrial Contactology: Structure and signaling functions. Trends Cell Biol. 28, 523–540 (2018).
Kleele, T et al. Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature 593, 435–439 (2021).
Giacomello, M, Pyakurel, A, Glytsou, C & Scorrano, L The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Biol 21, 204–224 (2020).
Soubannier, V et al. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol 22, 135–141 (2012).
McLelland, GL, Lee, SA, McBride, HM & Fon, EA Syntaxin-17 delivers PINK1/parkin-dependent mitochondrial vesicles to the endolysosomal system. J Cell Biol 214, 275–291 (2016).
Mohanty, A, Zunino, R, Soubannier, V & Dilipkumar, S A new functional role of mitochondria-anchored protein ligase in peroxisome morphology in mammalian cells. J Cell Biochem 122, 1686–1700 (2021).
Chaiyarit, S & Thongboonkerd, V Mitochondria-derived vesicles and their potential roles in kidney stone disease. J Transl Med. 21, 294 (2023).
Rosina, M et al. Ejection of damaged mitochondria and their removal by macrophages ensure efficient thermogenesis in brown adipose tissue. Cell Metab. 34, 533–548 e512 (2022).
Hayakawa, K et al. Protective effects of endothelial progenitor cell-derived extracellular mitochondria in brain endothelium. Stem Cells 36, 1404–1410 (2018).
Boudreau, LH et al. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood 124, 2173–2183 (2014).
van Niel, G, D’Angelo, G & Raposo, G Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 19, 213–228 (2018).
Dixson, AC, Dawson, TR, Di Vizio, D & Weaver, AM Context-specific regulation of extracellular vesicle biogenesis and cargo selection. Nat Rev Mol Cell Biol 24, 454–476 (2023).
Zhang, H et al. Identification of distinct nanoparticles and subsets of extracellular vesicles by asymmetric flow field-flow fractionation. Nat Cell Biol 20, 332–343 (2018).
Ma, L et al. Discovery of the migrasome, an organelle mediating release of cytoplasmic contents during cell migration. Cell Res 25, 24–38 (2015).
Zhou, X et al. MitoEVs: A new player in multiple disease pathology and treatment. J Extracell Vesic 12, e12320 (2023).
Liu, D et al. The existence and function of mitochondrial component in extracellular vesicles. Mitochondrion 54, 122–127 (2020).
Liu, Z, Sun, Y, Qi, Z, Cao, L & Ding, S Mitochondrial transfer/transplantation: an emerging therapeutic approach for multiple diseases. Cell Biosci. 12, 66 (2022).
Spees, JL, Olson, SD, Whitney, MJ & Prockop, DJ Mitochondrial transfer between cells can rescue aerobic respiration. Proc Natl Acad Sci USA 103, 1283–1288 (2006).
Phinney, DG et al. Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs. Nat Commun 6, 8472 (2015).
Hayakawa, K et al. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 535, 551–555 (2016).
Suh, J et al. Mitochondrial fragmentation and donut formation enhance mitochondrial secretion to promote osteogenesis. Cell Metab. 35, 345–360 e347 (2023).
Sun, L et al. A novel mechanism for coupling cellular intermediary metabolism to cytosolic Ca2+ signaling via CD38/ADP-ribosyl cyclase, a putative intracellular NAD+ sensor. FASEB J. 16, 302–314 (2002).
Moridera, K et al. Skeletal unloading reduces cluster of differentiation (CD) 38 expression in the bone marrow and osteoblasts of mice. J Orthop Sci. 25, 331–337 (2020).
Sun, L et al. Disordered osteoclast formation and function in a CD38 (ADP-ribosyl cyclase)-deficient mouse establishes an essential role for CD38 in bone resorption. FASEB J. 17, 369–375 (2003).
Nakajima, A, Kurihara, H, Yagita, H, Okumura, K & Nakano, H Mitochondrial Extrusion through the cytoplasmic vacuoles during cell death. J Biol Chem. 283, 24128–24135 (2008).
Tan, HWS et al. A degradative to secretory autophagy switch mediates mitochondria clearance in the absence of the mATG8-conjugation machinery. Nat Commun. 13, 3720 (2022).
Choong, CJ et al. Alternative mitochondrial quality control mediated by extracellular release. Autophagy 17, 2962–2974 (2021).
Nicolas-Avila, JA et al. A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell 183, 94–109 e123 (2020).
Zheng, D et al. Mesenchymal stem cell-derived microvesicles improve intestinal barrier function by restoring mitochondrial dynamic balance in sepsis rats. Stem Cell Res Ther. 12, 299 (2021).
Peruzzotti-Jametti, L et al. Neural stem cells traffic functional mitochondria via extracellular vesicles. PLoS Biol. 19, e3001166 (2021).
Levoux, J et al. Platelets Facilitate the Wound-Healing Capability of Mesenchymal Stem Cells by Mitochondrial Transfer and Metabolic Reprogramming. Cell Metab. 33, 283–299 e289 (2021).
Crewe, C et al. Extracellular vesicle-based interorgan transport of mitochondria from energetically stressed adipocytes. Cell Metab. 33, 1853–1868 e1811 (2021).
Hough, KP et al. Exosomal transfer of mitochondria from airway myeloid-derived regulatory cells to T cells. Redox Biol. 18, 54–64 (2018).
Morrison, TJ et al. Mesenchymal Stromal Cells Modulate Macrophages in Clinically Relevant Lung Injury Models by Extracellular Vesicle Mitochondrial Transfer. Am J Respir Crit Care Med. 196, 1275–1286 (2017).
Xia, L et al. AdMSC-derived exosomes alleviate acute lung injury via transferring mitochondrial component to improve homeostasis of alveolar macrophages. Theranostics 12, 2928–2947 (2022).
Garcia-Martinez, I et al. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J Clin Invest. 126, 859–864 (2016).
Maeda, A & Fadeel, B Mitochondria released by cells undergoing TNF-alpha-induced necroptosis act as danger signals. Cell Death Dis. 5, e1312 (2014).
McCully, JD et al. Injection of isolated mitochondria during early reperfusion for cardioprotection. Am J Physiol Heart Circ Physiol. 296, H94–H105 (2009).
Masuzawa, A et al. Transplantation of autologously derived mitochondria protects the heart from ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 304, H966–H982 (2013).
Kaza, AK et al. Myocardial rescue with autologous mitochondrial transplantation in a porcine model of ischemia/reperfusion. J Thorac Cardiovasc Surg 153, 934–943 (2017).
Doulamis, IP et al. Mitochondrial transplantation for myocardial protection in diabetic hearts. Eur J Cardiothorac Surg 57, 836–845 (2020).
Huang, PJ et al. Transferring Xenogenic Mitochondria Provides Neural Protection Against Ischemic Stress in Ischemic Rat Brains. Cell Transpl. 25, 913–927 (2016).
Zhang, Z et al. Muscle-derived autologous mitochondrial transplantation: A novel strategy for treating cerebral ischemic injury. Behav Brain Res 356, 322–331 (2019).
Nakamura, Y, Lo, EH & Hayakawa, K Placental Mitochondria Therapy for cerebral ischemia-reperfusion injury in mice. Stroke 51, 3142–3146 (2020).
Lee, EH et al. Primary astrocytic mitochondrial transplantation ameliorates ischemic stroke. BMB Rep. 56, 90–95 (2023).
Xie, Q et al. Mitochondrial transplantation attenuates Cerebral Ischemia-Reperfusion Injury: Possible involvement of mitochondrial component separation. Oxid Med. Cell Longev. 2021, 1006636 (2021).
Zhao, Z, Yu, Z, Hou, Y, Zhang, L & Fu, A Improvement of cognitive and motor performance with mitotherapy in aged mice. Int J Biol Sci. 16, 849–858 (2020).
Orfany, A et al. Mitochondrial transplantation ameliorates acute limb ischemia. J Vasc Surg. 71, 1014–1026 (2020).
Alway, SE et al. Mitochondria transplant therapy improves regeneration and restoration of injured skeletal muscle. J Cachexia Sarcopenia Muscle 14, 493–507 (2023).
Kim, MJ., Lee, JM, Min, K & Choi, YS. Xenogeneic transplantation of mitochondria induces muscle regeneration in an in vivo rat model of dexamethasone-induced atrophy. J Muscle Res Cell Motil https://doi.org/10.1007/s10974-023-09643-7. (2023)
Kim, JS, Lee, S, Kim, WK & Han, BS Mitochondrial transplantation: an overview of a promising therapeutic approach. BMB Rep. 56, 488–495 (2023).
Yu, SH et al. Human umbilical cord mesenchymal stem cell-derived mitochondria (PN-101) attenuate LPS-induced inflammatory responses by inhibiting NFkappaB signaling pathway. BMB Rep. 55, 136–141 (2022).
Thomas, MA et al. Human mesenchymal stromal cells release functional mitochondria in extracellular vesicles. Front Bioeng Biotechnol. 10, 870193 (2022).
Borcherding, N et al. Dietary lipids inhibit mitochondria transfer to macrophages to divert adipocyte-derived mitochondria into the blood. Cell Metab. 34, 1499–1513 e1498 (2022).
Brestoff, JR et al. Intercellular Mitochondria transfer to macrophages regulates white adipose tissue homeostasis and is impaired in obesity. Cell Metab. 33, 270–282 e278 (2021).
Kesner, EE, Saada-Reich, A & Lorberboum-Galski, H Characteristics of mitochondrial transformation into human cells. Sci Rep. 6, 26057 (2016).
MacDonald, JA et al. A nanoscale, multi-parametric flow cytometry-based platform to study mitochondrial heterogeneity and mitochondrial DNA dynamics. Commun Biol. 2, 258 (2019).
Jacoby, E et al. Mitochondrial augmentation of CD34(+) cells from healthy donors and patients with mitochondrial DNA disorders confers functional benefit. NPJ Regen Med 6, 58 (2021).
Ikeda, G et al. Mitochondria-rich extracellular vesicles from autologous stem cell-derived cardiomyocytes restore energetics of ischemic Myocardium. J. Am Coll Cardiol. 77, 1073–1088 (2021).
Jackson, MV et al. Mitochondrial transfer via tunneling nanotubes is an important mechanism by which mesenchymal stem cells enhance macrophage phagocytosis in the in vitro and in vivo models of ARDS. Stem Cells 34, 2210–2223 (2016).
Ko, JH, Kim, HJ, Jeong, HJ, Lee, HJ & Oh, JY Mesenchymal stem and stromal cells harness macrophage-derived amphiregulin to maintain tissue homeostasis. Cell Rep. 30, 3806–3820 e3806 (2020).
Wang, Y et al. Activation of astrocytic sigma-1 receptor exerts antidepressant-like effect via facilitating CD38-driven mitochondria transfer. Glia 68, 2415–2426 (2020).
D’Acunzo, P et al. Mitovesicles are a novel population of extracellular vesicles of mitochondrial origin altered in Down syndrome. Sci Adv 7, eabe5085 (2021).
Unuma, K, Aki, T, Funakoshi, T, Hashimoto, K & Uemura, K Extrusion of mitochondrial contents from lipopolysaccharide-stimulated cells: Involvement of autophagy. Autophagy 11, 1520–1536 (2015).
Cai, Y et al. Mitochondrial DNA-enriched microparticles promote acute-on-chronic alcoholic neutrophilia and hepatotoxicity. JCI Insight 2, e92634 (2017).
Leermakers, PA et al. Iron deficiency-induced loss of skeletal muscle mitochondrial proteins and respiratory capacity; the role of mitophagy and secretion of mitochondria-containing vesicles. FASEB J. 34, 6703–6717 (2020).
Puhm, F et al. Mitochondria are a subset of extracellular vesicles released by activated monocytes and induce Type I IFN and TNF responses in endothelial cells. Circ Res. 125, 43–52 (2019).
Abad, E & Lyakhovich, A Movement of Mitochondria with mutant DNA through extracellular vesicles helps cancer cells acquire Chemoresistance. ChemMedChem 17, e202100642 (2022).
Takenaga, K, Koshikawa, N & Nagase, H Intercellular transfer of mitochondrial DNA carrying metastasis-enhancing pathogenic mutations from high- to low-metastatic tumor cells and stromal cells via extracellular vesicles. BMC Mol Cell Biol. 22, 52 (2021).
Cowan, DB et al. Intracoronary delivery of mitochondria to the ischemic heart for cardioprotection. PLoS One 11, e0160889 (2016).
Moskowitzova, K et al. Mitochondrial transplantation prolongs cold ischemia time in murine heart transplantation. J Heart Lung Transpl. 38, 92–99 (2019).
Guariento, A et al. Preischemic autologous mitochondrial transplantation by intracoronary injection for myocardial protection. J Thorac Cardiovasc Surg. 160, e15–e29 (2020).
Weixler, V et al. Autogenous mitochondria transplantation for treatment of right heart failure. J Thorac Cardiovasc Surg. 162, e111–e121 (2021).
Alemany, VS et al. Mitochondrial transplantation preserves myocardial function and viability in pediatric and neonatal pig hearts donated after circulatory death. J Thorac Cardiovasc Surg. 167, e6–e21 (2023).
Mokhtari, B, Hamidi, M, Badalzadeh, R & Mahmoodpoor, A Mitochondrial transplantation protects against sepsis-induced myocardial dysfunction by modulating mitochondrial biogenesis and fission/fusion and inflammatory response. Mol Biol Rep 50, 2147–2158 (2023).
Babenko, VA et al. Miro1 enhances mitochondria transfer from multipotent Mesenchymal Stem Cells (MMSC) to neural cells and improves the efficacy of cell recovery. Molecules 23, 687 (2018).
Pourmohammadi-Bejarpasi, Z et al. Mesenchymal stem cells-derived mitochondria transplantation mitigates I/R-induced injury, abolishes I/R-induced apoptosis, and restores motor function in acute ischemia stroke rat model. Brain Res Bull. 165, 70–80 (2020).
Bamshad, C et al. Human umbilical cord-derived mesenchymal stem cells-harvested mitochondrial transplantation improved motor function in TBI models through rescuing neuronal cells from apoptosis and alleviating astrogliosis and microglia activation. Int Immunopharmacol. 118, 110106 (2023).
Chang, JC et al. Allogeneic/xenogeneic transplantation of peptide-labeled mitochondria in Parkinson’s disease: restoration of mitochondria functions and attenuation of 6-hydroxydopamine-induced neurotoxicity. Transl Res. 170, 40–56 e43 (2016).
Chang, JC et al. Intranasal delivery of mitochondria for treatment of Parkinson’s Disease model rats lesioned with 6-hydroxydopamine. Sci Rep 11, 10597 (2021).
Nitzan, K et al. Mitochondrial transfer ameliorates cognitive deficits, neuronal loss, and gliosis in Alzheimer’s disease mice. J Alzheimers Dis. 72, 587–604 (2019).
Wang, Y et al. Mitochondrial transplantation attenuates lipopolysaccharide- induced depression-like behaviors. Prog Neuropsychopharmacol Biol Psychiatry 93, 240–249 (2019).
Zhang, Z et al. Hippocampal mitochondrial transplantation alleviates age-associated cognitive decline via enhancing Wnt signaling and neurogenesis. Comput Intell Neurosci. 2022, 9325302 (2022).
Gollihue, JL et al. Effects of mitochondrial transplantation on bioenergetics, cellular incorporation, and functional recovery after spinal cord injury. J Neurotrauma 35, 1800–1818 (2018).
Li, H et al. Mitochondrial transfer from bone marrow mesenchymal stem cells to motor neurons in spinal cord injury rats via gap junction. Theranostics 9, 2017–2035 (2019).
Lin, MW et al. Mitochondrial transplantation attenuates neural damage and improves locomotor function after traumatic spinal cord injury in rats. Front Neurosci 16, 800883 (2022).
Lin, HC, Liu, SY, Lai, HS & Lai, IR Isolated mitochondria infusion mitigates ischemia-reperfusion injury of the liver in rats. Shock 39, 304–310 (2013).
Fu, A, Shi, X, Zhang, H & Fu, B Mitotherapy for fatty liver by intravenous administration of exogenous mitochondria in male mice. Front Pharmacol. 8, 241 (2017).
Shi, X et al. Treatment of acetaminophen-induced liver injury with exogenous mitochondria in mice. Transl Res. 196, 31–41 (2018).
Ulger, O et al. The effects of mitochondrial transplantation in acetaminophen-induced liver toxicity in rats. Life Sci. 279, 119669 (2021).
Lu, T et al. Extracellular vesicles derived from mesenchymal stromal cells as nanotherapeutics for liver ischaemia-reperfusion injury by transferring mitochondria to modulate the formation of neutrophil extracellular traps. Biomaterials 284, 121486 (2022).
Islam, MN et al. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med. 18, 759–765 (2012).
Zhang, F et al. TFAM-Mediated mitochondrial transfer of MSCs improved the permeability barrier in sepsis-associated acute lung injury. Apoptosis 28, 1048–1059 (2023).
Moskowitzova, K et al. Mitochondrial transplantation enhances murine lung viability and recovery after ischemia-reperfusion injury. Am J Physiol Lung Cell Mol Physiol. 318, L78–L88 (2020).
Huang, T et al. Iron oxide nanoparticles augment the intercellular mitochondrial transfer-mediated therapy. Sci Adv. 7, eabj0534 (2021).
Zou, X et al. Renal scattered tubular-like cells confer protective effects in the stenotic murine kidney mediated by release of extracellular vesicles. Sci Rep. 8, 1263 (2018).
Konari, N, Nagaishi, K, Kikuchi, S & Fujimiya, M Mitochondria transfer from mesenchymal stem cells structurally and functionally repairs renal proximal tubular epithelial cells in diabetic nephropathy in vivo. Sci Rep. 9, 5184 (2019).
Jabbari, H et al. Mitochondrial transplantation ameliorates ischemia/reperfusion-induced kidney injury in rat. Biochim Biophys. Acta Mol. Basis Dis. 1866, 165809 (2020).
Doulamis, IP et al. Mitochondrial transplantation by intra-arterial injection for acute kidney injury. Am J Physiol Ren Physiol. 319, F403–F413 (2020).
Zhao, M et al. Mesenchymal stem cell-derived extracellular vesicles attenuate mitochondrial damage and inflammation by stabilizing mitochondrial DNA. ACS Nano 15, 1519–1538 (2021).
Yuan, Y et al. Mitochondrial transfer from mesenchymal stem cells to macrophages restricts inflammation and alleviates kidney injury in diabetic nephropathy mice via PGC-1alpha activation. Stem Cells 39, 913–928 (2021).
Sun, J et al. High-efficiency quantitative control of mitochondrial transfer based on droplet microfluidics and its application on muscle regeneration. Sci Adv 8, eabp9245 (2022).
Guo, Y et al. Mitochondria transfer enhances proliferation, migration, and osteogenic differentiation of bone marrow mesenchymal stem cell and promotes bone defect healing. Stem Cell Res Ther. 11, 245 (2020).
Lee, AR et al. Mitochondrial transplantation ameliorates the development and progression of Osteoarthritis. Immune Netw. 22, e14 (2022).
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grants (NRF-2020R1A2C1010359, NRF-2018R1A5A2024418, and RS-2023-00246115).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Suh, J., Lee, YS. Mitochondria as secretory organelles and therapeutic cargos. Exp Mol Med 56, 66–85 (2024). https://doi.org/10.1038/s12276-023-01141-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-023-01141-7
