
Abstract
The therapeutic use of messenger RNA (mRNA) has fueled great hope to combat a wide range of incurable diseases. Recent rapid advances in biotechnology and molecular medicine have enabled the production of almost any functional protein/peptide in the human body by introducing mRNA as a vaccine or therapeutic agent. This represents a rising precision medicine field with great promise for preventing and treating many intractable or genetic diseases. In addition, in vitro transcribed mRNA has achieved programmed production, which is more effective, faster in design and production, as well as more flexible and cost-effective than conventional approaches that may offer. Based on these extraordinary advantages, mRNA vaccines have the characteristics of the swiftest response to large-scale outbreaks of infectious diseases, such as the currently devastating pandemic COVID-19. It has always been the scientists’ desire to improve the stability, immunogenicity, translation efficiency, and delivery system to achieve efficient and safe delivery of mRNA. Excitingly, these scientific dreams have gradually been realized with the rapid, amazing achievements of molecular biology, RNA technology, vaccinology, and nanotechnology. In this review, we comprehensively describe mRNA-based therapeutics, including their principles, manufacture, application, effects, and shortcomings. We also highlight the importance of mRNA optimization and delivery systems in successful mRNA therapeutics and discuss the key challenges and opportunities in developing these tools into powerful and versatile tools to combat many genetic, infectious, cancer, and other refractory diseases.
Similar content being viewed by others
Introduction
Messenger RNA (mRNA) is a type of single-stranded ribonucleic acid that is transcribed from a strand of DNA, which carries the coding information for protein synthesis to be further transcribed and processed into functional proteins.1 In vitro transcription (IVT) mRNA was successfully transcribed and expressed in mouse skeletal muscle cells, which establishes the feasibility of mRNA therapy.2 mRNA-based therapeutics were proposed when mRNA could be successfully transfected and produce an immune response in a dose-dependent manner by direct injection into mice to express therapeutic proteins.3 An mRNA-based approach can theoretically produce any protein/peptide via the protein synthesis machine processed in the transfected cell in vitro or in vivo.4 Unlike DNA-based drugs, mRNA transcripts have a relatively high transfection efficiency and low toxicity because they do not need to enter the nucleus to be functional.5 Importantly, mRNA has no potential risk of accidental infection or opportunistic insertional mutagenesis.6 In addition, mRNA has broad potential for treating diseases requiring protein expression and higher therapeutic efficacy due to its continuous translation into encoded proteins/peptides to trigger long-lasting expression compared to transient traditional protein/peptide drugs.7 Apparently, these advantages of mRNA over DNA or protein/peptide enable the rapid entry of mRNA-based technology and products into various branches of the biomedical fields, which will benefit all aspects of human life, especially millions of patients suffering from incurable diseases.
Nevertheless, insufficient knowledge of mRNA structure instability and immunogenicity has dampened some of the promises and impeded the pace of mRNA-based therapeutics to combat diseases.8 mRNA is a negatively charged macromolecule that is susceptible to ubiquitous RNases. Hence, it is quite difficult for mRNA to pass through the anionic cell membrane and translate functional proteins in the cytoplasm (<1/10,000 mRNAs of the initial input).9 In addition, mRNA can also induce an immune response with associated toxicity, which greatly restricts the development of mRNA-based therapeutics.10 Engineering precision carriers for mRNA-based drug delivery reveal a critical role in improving immunogenicity and instability and overcoming cellular barriers.11 Recently, based on the important role of mRNA vaccines in controlling the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic, humans benefited from a large number of mRNA vaccines for infectious diseases on structural and chemical modifications, which have also greatly fueled enthusiastic efforts in the development of mRNA-based therapeutics to improve their stability, translation efficiency and immune response12 (Fig. 1). Simultaneously, mRNA can be successfully delivered into a variety of cells with continuous breakthroughs of delivery carriers.13 Numerous technologies have also been developed to improve mRNA therapeutic efficacy and the instability of mRNAs. Hence, it is necessary to draw a comprehensive landscape of the current status and analyze the general design approaches of mRNA-based drugs.

Key discoveries and advances in mRNA-based therapeutics. The development of mRNA-based therapeutics can be divided into three stages. Phase 1, mRNA discovery, in vitro synthesis and nucleic acid delivery system construction (1961–1990), including discovery mRNA523 and using protamine for RNA delivery,524 in vitro translation of isolated mRNA,525 mRNA cap was discovered,526 Liposome-entrapped mRNA delivery,527 Cap analog commercialized, T7 RNA polymerases commercialized, Cationic lipid-mediated mRNA delivery,528 Naked mRNA is translated in vivo by direct injection.529 Phase2 (1990-2019), accumulated knowledge with the continuous attempts and diverse applications, especially protein replacement therapies and vaccination approaches for cancer and infectious diseases, including using mRNAs for cancer immunotherapy,5 mRNA-based company founded and 3′-UTR regulates mRNA localization,530 antitumor T cell response induced by mRNA,531 first clinical trial with mRNA using ex vivo transfected DCs,532 mRNA-based immunotherapy for human cancer,533 preclinical study with intranodally injected DC-targeted mRNA,534 protective mRNAs vaccination in influenza240 and respiratory syncytial virus,98 CRISPR–Cas9 mRNA for gene editing,535 personalized mRNA cancer vaccine for clinical trials.330 Phase 3, mRNA-based therapeutics, as a disruptive therapeutic technology, is becoming powerful and versatile tools for therapy diseases (2019 to present), including clinical trials of mRNA vaccines for cancer and infectious disease, mRNA-1273,536 and BNT162b emergency use for SARS-CoV-2 pandemic537
Our lab has been committed to promoting mRNA-based therapeutics to become powerful and versatile tools to combat diseases, especially in gene therapy and immunotherapy.14 We have developed diverse novel targeted delivery nanoparticles15 and constructed receptor-binding domain (RBD)-encoding mRNA formulated in liposomes to prevent and treat the SARS-CoV-2 pandemic.16 In this review, we comprehensively summarize the recent progress towards mRNA design and synthesis, as well as the enabling of mRNA delivery technologies. Likewise, we point out the key issues and challenges facing the future of the platform, including mRNA optimization and application in specific diseases and populations, offering novel insight into the design, test, and development of mRNA therapeutics.
mRNA design and manufacture
The development of mRNA-based therapeutics mainly includes mRNA design, synthesis, mRNA entrapment, pharmacodynamics, pharmacokinetics, safety evaluation in vivo and in vitro, manufacturing, and clinical trials (Fig. 2). mRNA design and synthesis are crucial steps in mRNA-based medicines. mRNA features five functional regions, including the 5′ cap, the 3′ poly(A) tail, the open reading frame (ORF) flanking, and 3′ untranslated regions (UTRs), whose elements mediate the translation efficacy and decay rate of mRNA.6 Notably, obtaining highly biologically active RNA usually depends on reliable design and preparation.17 In this section, we focus on recent advances and discuss the challenges of mRNA design and preparation. In addition, nucleoside modification and purification are also reviewed (Table 1), which are widely applied to adjust the different demands for mRNA immune-stimulation in various therapies.

mRNA drugs production pipeline. The encoding of peptide/protein is designed and inserted into a plasmid DNA construct. Plasmid DNA is transcribed into mRNA by bacteriophage polymerases in vitro, and mRNA transcripts are purified by high-performance liquid chromatography (HPLC) or nanoprecipitation to remove contaminants and reactants. Subsequently, purified mRNA is entrapped in various vehicles. The interactions between vehicles and mRNA can be divided into three types: (a) electrostatic adsorption with phosphate ions of the ribonucleotides; (b) complementary paired hydrogen bonding with bases of the ribonucleotides; and (c) coordination with the phosphate ions. Thus, vehicles for mRNA delivery consist of the following categories: cationic compounds, such as cationic lipids, ionizable lipids, and cationic polymers. Nucleoside-based lipids, e.g., DNCA, or nucleoside-based amphiphilic polymers, e.g., Chol(+)-oligoRNA. Metal-based compounds provide vacant orbitals to coordinate with phosphate ions. Furthermore, the efficacy, pharmacology, and safety of mRNA drugs were evaluated in vaccinated mice and primates. Finally, the scale-up manufacturing of mRNA therapeutics is conducted and followed by clinical trials262
The structural elements of mRNA
mRNA is produced by the transcription process. The precursor mRNA is synthesized in eukaryotes when RNA polymerase converts genes into primary mRNA transcripts in vivo, which usually still contains noncoding sequence introns, and are further removed to become mature mRNA by mRNA processing, including 5′ mRNA capping, modifications, splicing, and A-to-I editing.18 IVT mature mRNA preparation includes several steps, linear DNA template obtainment, IVT, 5′ capping, and poly(A) tail adding. After the mRNA is transferred into the cell, poly(A)-binding protein (PABP) binds to the poly(A) tail and interacts with eukaryotic translation initiation factors (eIFs). The interaction of eIFs with the 5′ cap, UTRs, PABP, initiator methionyl transfer RNA (tRNA), and 40S ribosomal subunit, render mRNA circularization and the formation of an initiation complex. After 40S ribosomal subunit scans the transcription initiation codon, 60S ribosomal subunits are recruited and eIFs are released to start amino acid chain extension.19 Mature mRNA includes the coding region, UTR, the poly(A) tails, and the 5′ cap that can be recognized by ribosomes and carried by tRNA to create proteins. As in DNA, genetic information in mRNA is contained in the sequence of nucleotides that are arranged into codons consisting of three ribonucleotides each. Accordingly, IVT mRNA is performed to complete the transcription of RNA in vitro by stimulating the mechanism of eukaryotic mRNA synthesis to ensure the expression of mRNA in vivo (Fig. 3). Therefore, the optimization of mRNA is essential for successful mRNA-based therapeutics.

In vitro transcribed (IVT) mRNA and translation initiation.IVT mRNA preparation includes several steps, plasmid cloning, plasmid linearization, in vitro transcription, 5′ capping, and the poly(A) tail adding. Transcription, capping and the tail adding can combine into one, two or three steps that depend on the design of synthesis routes.2 After entering into the cell, mRNA translation can be initiated in an eIF4F-dependent manner to recruit a preinitiation complex (PIC). The 43S PIC is formed by 40S ribosomal subunit, the eukaryotic translation initiation factors (eIF, including eIF1, eIF1A, eIF3, eIF5) and the ternary complex, including a trimeric complex comprising eIF2 that contains α-, β-, and γ-subunits, initiating methionyl tRNA (tRNAiMet), and GTP. eIF4F is a complex composed of eIF4A, eIF4E and eIF4G. eIF4E binds to mRNA cap. eIF4G interacts with eIF3 and poly(A)-binding protein (PABP) that binds to the 3′ poly(A) tail. These interactions result in mRNA circularization and 48S PIC assembly. The 48S PIC ribosomal subunit scans and finds the start codon with the help of eIF4A helicase to resolve secondary mRNA structure in the 5′ UTR. Then, eIFs are released and 60S ribosomal subunit joins to initiate translation elongation by forming 80S ribosome21
mRNA translation and decay
Eukaryotic mRNA translation initiation is an exquisitely regulated process involving the assembly of a multiprotein–RNA complex that directs ribosomes to the initiation codon.20 Generally, Cap-dependent translation begins with the cap recognition by eukaryotic initiation factor 4F (eIF4F) and the assembly of the preinitiation complex (PIC), which consists of the ternary complex, the 40S ribosomal subunit, eIF1, eIF1A, eIF3 and eIF5.21 eIF4F consists of eIF4A, eIF4E and eIF4G, which facilitates PIC recruitment by eIF4E–cap and eIF4G–eIF3 interactions. eIF4F renders mRNA circularization by interacting with the 5′ cap through eIF4E and the PABP that binds with the poly(A) tail.22 40S ribosomal scans the 5′-UTR and recognizes initiation codon with the help of eIF4A to unwind the secondary structure of the 5′-UTR, subsequent, 40S ribosomal subunit scans the transcription initiation codon, 60S ribosomal subunits are recruited and eIFs are released to start amino acid chain extension.23 Then, mRNA is decoded in a ribosome to produce a specific amino acid chain or polypeptide. There is a balance between the processes of translation and mRNA decay24,25 (Fig. 4). It has previously been implicated that these structural elements that are being actively translated also intimately connect to mRNA decay, especially the 5′ cap and the poly(A) tail.26 The 5′ cap protects mRNA from 5′ to 3′ exoribonucleases,27 and the length of the poly(A) tail determines the 3′ to 5′ exonucleolytic decay.28 Based on the vital importance of these functional elements, numerous studies have focused on the optimization of mRNA structure, such as developing a series of 5′ cap analogs, changing the poly(A) tail length, screening feature UTRs and encoding various functional peptides or viral replication machinery in ORFs.29

Mechanisms of mRNA decay. Degradation of messenger mRNA plays an essential role in regulating sustained mRNA expression. mRNA is generally degraded in the following three pathways: ① Deadenylation-dependent mRNA decay: The poly(A) tail is removed by deadenylase activity (such as CCR4, CAF1 or PARN). The LSM1-7 complex associates with the 3′-end of the mRNA transcript to induce decapping by the Dcp1–Dcp2 complex and is then degraded by exoribonuclease XRN1. Alternatively, deadenylated mRNA can be degraded by exosomes. ② Endonuclease-mediated mRNA decay: The mRNA is cleaved into two fragments, and then the fragments are degraded by XRN1 and exosomes.538 ③ Deadenylation-independent pathways require recruitment of the decapping machinery. RPS28B interacts with the enhancer of decapping-3 (Edc3) to engage the decapping enzyme. Subsequently, the mRNA is degraded by XRN1538
mRNA design
The 5′ cap
The 5′ caps are located at the 5′ terminus of mRNA with different degrees of methylation.30 5′ caps (m7G ppp) contain a 7-methylguanosine (m7G) attaching the following nucleotide through a 5′-5′ triphosphate bridge (ppp) in eukaryotes31,32 (Fig. 5). The cap combines eIF4E via the hydrophobic cation–π interactions of m7G and the negative electrostatic charge of the triphosphate bridge during translation initiation.33 For cap removal, the triphosphate bridge is the major target mRNA decapping enzyme in eukaryotic cells. Dcp1/2 and DcpS: Dcp1/2 cleave α- and β-phosphate, and DcpS cleaves β- and γ-phosphates.34,35 Therefore, numerous strategies for mRNA structure optimization have been applied to optimize m7G or the triphosphate bridge to achieve cap analogs with high affinity for eIF4E and low susceptibility for decapping enzymes.36 Rydzik et al. increased the cap resistance to decapping by substituting the oxygen atom of triphosphates with dihalogenmethylenebisphosphonate.37 In addition, the modification of m7G is an essential approach to improve mRNA translation. It has previously been reported that the translation efficiency is significantly enhanced by replacing the 7-methylated guanosine (m7G) with 7-benzylated guanosine38 and further increased by 2-fold by attaching the m7G with another m7G via tetraphosphate (m7Gppppm7G), whose analogs have a higher affinity for eIF4E compared to natural eukaryotic 5′ caps.39 The bridged oxygen atoms between α-β or β-γ phosphate were, respectively, replaced with methylene to give rise to m7GpCH2ppG or m7GppCH2pG to prevent mRNA from Dcp1/2 or DcpS degradation.40 Dithiodiphosphate modification are also introduced to the tri- or tetraphosphate bridge, which decreased cap sensitivity to Dcp1/2, and improved mRNA translation.36 In addition, phosphorothioate cap analogs increase the stability and translational efficiency of RNA vaccines in immature dendritic cells (DCs).41 Notably, phosphorothioate substitution is position sensitive, which is possibly associated with stereochemistry in catalysis.36

Commercialization and commonly used Cap. The 5′ cap of mRNA is critical to improve mRNA stability and promote translation efficiency. Modification of the 5′-5′ phosphate bridge can increase the resistance to DcpS and Dcp1/Dcp2, but the translation efficiency may not necessarily increase (such as the introduction of methylene groups on the phosphate bridge). The modification of ribose nucleosides also plays essential functions in mRNA translation by recruiting translation initiation factors, such as the methylation modification on the N7 position of the guanosine cap and the ribose-2′O position of the first nucleotide (Cap 1), increasing the affinity for eIF4E and thereby improving translation efficiency116,539
The poly(A) tail
Poly(A) tails generally consist of 10–250 adenine ribonucleotides. Poly(A) tails are dynamic additions to mRNA that their length plays a crucial role in regulating mRNA translation efficacy and protein expression.42,43 Mechanically, the 3′ -end poly(A) tail combines with PABPs and subsequently interacts with the 5′ cap through the translation initiation factors eIF4G and eIF4E, which promotes a “closed-loop structure” and regulates the translation efficiency of mRNA.44 Mockey et al. are the first to observe a positive correlation between the length of poly(A) tails and translation efficacy by adding a poly(A) tail of 100 instead of 64 adenosines in cis, increasing the protein level by approximately 35-fold.45 Similarly, the poly(A) of 120 units is more conducive to the formation of stable and efficient translation mRNA compared to the 51 nt and 42 nt tails,46 and the 325-nucleotide poly(A) tail shows higher efficacy than the 172-nucleotide tail.47 Interestingly, the length of poly(A) is not always positively correlated with mRNA instability and attenuation. Traditionally, it was considered necessary for poly(A) tails to contain at least 20 nt to achieve sufficient mRNA translation, but the poly(A) tails of stabilizing β-actin are less than 20 nucleotides, and the poly(A) tails of 425 nt and 525 nt merely contribute to transfection efficiency than 120 nt poly(A) tails in human primary T cells.47,48,49
5′-UTRs and 3′-UTRs
The UTRs at the 3′ and 5′ terminals of mRNAs do not directly encode proteins but play important roles in regulating mRNA translation and protein expression.50 UTRs participate in the subcellular localization of mRNA, and regulating translation efficiency and mRNA stability.51 Both the 5′-UTR and 3′-UTR regulate protein expression levels, and the 5′-UTR is primarily involved in initiating the translation process,52 while the 3′-UTR mostly affects the stability and half-life of the mRNA.53 The 5′ cap triggers ribosome binding and subsequently recognizes the initiation sequence for protein synthesis during translation. Furthermore, the internal ribosome entry site in the 5′-UTR can also recruit ribosomes and initiate translation in a cap- and eIF4E-independent manner.54 The strongest Kozak sequence is widely used to improve mRNA translation. Foroughmand et al. improved protein expression by replacing the Kozak sequence of the human beta-globin 5′-UTR with the strongest sequence.55 A library of 10 UTR variants was constructed to validate the effect of UTR on the expression of therapeutic mRNA, and found that 5'UTRs containing the complement factor 3 (C3) and cytochrome p4502E1 significantly increased protein translation regardless of 3'UTR modifications.56 Similarly, optimization of the 3′-UTR can also enhance mRNA stability and translation duration. The stability of the mRNA is enhanced due to the discontinuous pyrimidine-rich sequence in the 3′-UTR of α-globin, and the β-globin in mRNA contributes to the increased duration of protein expression.57,58 More efficacious strategies are developed for increasing protein production and mRNA stability by adding two consecutive β-globin 3′-UTRs arranged head-to-tail to mRNA compared to one β-globin 3′-UTR. Notably, the improvement is cell-type dependent, which significantly increases protein expression in mature DCs but slightly immature DCs.46 Conversely, eGFP mRNA with two repeated β-globin 3′-UTRs produces less protein than mRNA with β-globin 5′-UTRs in human pluripotent stem cells (PSCs).59 However, two repeated cytochrome b-245 alpha polypeptide (CYBA) 3′-UTRs had lower protein production in A549 cells, compared to the single 3′-UTR.60 Moreover, the 5′-UTR and 3′-UTR influence each other on protein expression.56 Taken together, the 5′-UTR contributes to the regulation of protein expression depending on the systems and cell types.
Trepotec et al. designed a series of short 5′-UTRs by inserting or altering less than two ribonucleosides based on the Kozak sequence. Two short 5′-UTRs were either better or equally effective than the human alpha globin 5′-UTR.61 Ferizi et al. evaluated UTRs from five natural long-lived mRNAs and found that the UTRs from human CYBA have the highest and most stable protein expression in NIH3T3 cells and A549 cells.60 Schrom et al. compared the effectiveness of a minimal 5′-UTR, a human alpha globin 5′-UTR and CYBA 5′-UTR, which resulted in higher protein expression by optimizing coding.62 Segovia et al. tried to reduce the immune stimulation of mRNA using the 5′-UTR from the Venezuelan equine encephalitis (VEE) virus.63 Asrani et al. used plasmids and IVT mRNA to screen effective UTRs, while they found different protein expressions driven by plasmids and IVT mRNA in HepG2 cells.56 Notably, researchers tried to design effective UTRs with the help of bioinformation and machine learning.64,65
The open reading frame
The design of the ORF has largely focused on codon optimization and the introduction of functional peptides as well as replication processes.66 Codon optimization is an extensively used but controversial approach for translation improvement.67 mRNA translation efficiency was improved by replacing rare codons with synonymous codons decoded by tRNA with higher abundance in ORF,68,69 but it may change protein conformation and give rise to novel peptides with unknown biological activity in vivo.68,70 Increasing the GC content by replacing rare codons in ORFs protects mRNA from endoribonuclease degradation and enhances mRNA protein expression in vivo.71,72 In addition, functional peptides are crucial for mRNA drugs and the signal peptides encoded by mRNAs are necessary for proteins that exert functions outside of the cells.73 Accordingly, optimization of mRNA for improving the function of therapeutic mRNA by introducing signal peptides to ORF regions is required. Trafficking signal peptides and protein segments are also widely applied for the improvement of antigen presentation in mRNA vaccines.74 Kreiter et al. improved the trafficking property of protein antigens by encoding a secretion signal and the transmembrane cytoplasmic domain of the MHC I molecule in the ORF, which increased antigen presentation by ~10-fold in DCs and improved the antitumor efficacy of mRNA vaccines in mice.75 Other functional peptides are also used to enhance cytoplasmic expression: the β2-microglobulin of MHC I molecules and the signal peptide of DC lysosomal-associated membrane protein.76 Together, the quality control of mRNA at each step is directly related to its efficacy; therefore, mRNA production and preparation is the key to mRNA-based therapeutics.
RNA chemical formula design
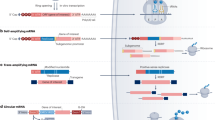
Self-amplifying RNA
Compared to conventional mRNA, self-amplifying RNA (saRNA) is another kind of mRNA molecule with a different structure.77,78 saRNA primarily originates from alphavirus structures and is constructed by replacing the gene sequence coding for virus structural proteins with the gene sequence of interest.79 Alphaviruses are positive-sense, single-stranded RNA viruses with self-amplifying ability, which is performed by a sequence of nucleotides coding for nonstructural proteins (nsP1-4).80 These nonstructural polyproteins function as replicases and replicate virus structural proteins through RNA-dependent RNA synthesis.81 Therefore, saRNA can produce a large amount of protein of interest in an effective way by using the innate nature of alphaviruses.
The basic elements of saRNA are the 5′ cap, 5′-UTR, sequence coding for nsP1-4, subgenomic promoter sequence, ORF with GOI, 3′-UTR, and 3′ poly(A) tail.82 The major difference between saRNA and conventional mRNA is the replicase sequence. The functions of individual nsP1-4 have been partially revealed: nsP1 plays a role in capping, nsP2 gains helicase activity, nsP3 is essential in the assembly of the replication complex and may interact with other proteins to prevent host cell-inhibiting pressure, and nsP4 obtains RNA-dependent RNA polymerase activity.83,84,85,86,87 All of the nonstructural proteins play an essential role in the function of saRNA. After saRNA is transfected into the cell, the sequence of nsP1-4 is translated into the nsP1-4 polyprotein, which functions as the precursor of the replicase complex, and subsequently, the nsP1-4 polyprotein is cleaved by nsP2, producing the nsP1-3 polyprotein and nsP4.85 This generated early phase replicase complex transcribes the original positive-sensed RNA strand into a negative-sensed RNA strand, and the latter strand is then used as the template for subsequent replication.88,89 After the nsP1-3 polyprotein is further cleaved into individual nsP1, nsP2, nsP3, together with nsP4, they form the cleaved replicase predominantly involved in the production of positive-strand synthesis.90,91
The greatest advantage of saRNA is the “dose-sparing” effect. Researchers in Imperial College London formulated the saRNA coding for S protein in the lipid nanoparticle (LNP) as vaccines against SARS-CoV-2, showing high efficiency in inducing neutralizing antibody titers.92 The same effect has also been shown in mRNA vaccines against ZIKV93 and influenza.94 However, the main challenge for saRNA is its longer sequence (usually 9–12 kb) compared to conventional mRNA. Some researchers have made some efforts to address this issue. Beissert et al. developed a novel bipartite vector system using trans-amplifying RNA.95 The vector system splits into two strands; one codes for the replicase with its enzyme activity provided by the second strand, and the other codes for the GOI that will be transamplified by the first strand.96 This work on saRNA structure showed the same efficacy as the single vector system while providing an easy, time- and cost-efficient manufacturing process. Li et al. optimized the replicon by identifying six mutations in nonstructural proteins of the VEE replicon that promoted subgenome expression in cells.97 Overall, saRNA is an attractive tool for transient expression of the target protein, generating stable cell lines expressing heterologous proteins from continuously replicating RNA, and developing recombinant vaccines.79,98 For example, Li et al. used saRNA to code the light and heavy chains of neutralizing anti-SARS-CoV-2 CB6 antibody simultaneously under the control of two identical subgenomic promoters.99 Together, saRNA has great absolute advantages in the continuous expression of proteins and long-lasting efficacy compared with other RNA chemical formula design, but the large nucleic acid sequence limits its application. Therefore, it still remains challenging for this promising technology.
Circular RNA, noncoding RNAs, and competitive endogenous RNA
Circular RNAs (circRNAs) are single-stranded, covalently closed RNA molecules that are ubiquitous in species ranging from viruses to mammals. CircRNAs, act as protein decoys, scaffolds and recruiters, exert biological functions by acting as transcriptional regulators, microRNA sponges, and protein templates. CircRNA is generated by back-splicing, in which the 3′-end of an exon ligates to the 5′-end of its own or an upstream exon through a 3′,5′-phosphodiester bond, forming a closed structure.100 The unique structure of circRNAs gives them greater stability, longer half-life, and greater RNase R resistance, which are linear mRNAs deficient and desired.101 Noncoding RNA (ncRNA) is an RNA molecule that is not translated into a protein, but affects normal gene expression and disease progression, including microRNA, intronic RNA, repetitive RNA, and long ncRNA.102 LncRNAs function as competing endogenous RNAs (ceRNAs) by competitively occupying shared binding sequences for miRNAs.103 CircRNA Cdr1as functions as a competitive endogenous RNA to promote hepatocellular carcinoma (HCC) progression.104 Research has shown that the complicated circRNA-miRNA-mRNA network revealed an important role in regulating Hantaan virus infection.105 circRNA-lncRNA-miRNA-mRNA ceRNA regulatory network was identified as novel prognostic markers for acute myeloid leukemia (AML).106 Currently, ncRNA-based therapeutics mainly regulates the expression of key proteins to treat diseases. The therapeutic potential of ncRNA has been recognized for more than forty years, few drugs have received approval due to high off-target effects.107 Although there is no report on the combination therapy strategy of mRNA and circRNA or ncRNAs. It may be an important means to achieve precise and individualized treatment by co-delivering them to form a regulatory network or complex, which is worthy of further exploration.
mRNA manufacture
mRNA synthesis and optimization
IVT mRNA is performed with linearizing plasmid DNA templates or PCR templates requiring at least a promoter and the corresponding mRNA construct sequence.2,108 IVT mRNA is carried out by adding polymerases (T7, T3, or SP6) but requires additional capping.108 Uncapped mRNA is rapidly degraded by RNase and contains a 5′-ppp group, which causes greater immune stimulation and can be treated with phosphatase to reduce undesirable efficacy.109,110 Two methods are implemented for the capping of IVT mRNA: co-transcriptional capping and posttranscriptional capping.111,112 Cap dinucleotide mixtures containing four other nucleoside triphosphates (NTPs) are incorporated at the 5′ end of the RNA with RNA polymerase during co-transcriptional capping.113 A label-free method was described to identify the 5′-end cap and the orientation of mRNA.114 Co-transcriptional capping processing has permitted coordinated transcription with mRNA capping, but its disadvantages are the competitive incorporation of GTP nucleosides, which impairs capping efficiency.111
Intriguingly, GTP first binds to RNA chains via a 5′-5′ triphosphate bond and then 7-methylation of the 5′ terminal guanosine in posttranscriptional capping.115 Capping enzymes from vaccinia virus are widely used to cap mRNA, have high end-capping efficiency and are able to completely cap mRNA with cap-0.116 Furthermore, it is necessary to consider mRNA immune stimulation, and cap-specific 2′-O methyltransferase is used to produce cap 1 or cap 2 based on cap 0, which reduces mRNA immunogenicity.117,118 The polymerase initiates transcription through the nucleophilic attack of the 3′-OH of the guanosine in m7G in the α-phosphate of the next nucleoside triphosphate specified by the DNA template when the mRNA is capped and generates m7GpppGpG.119 Notably, m7GpppGpG is formed when this attack occurs on the 3′-OH of m7G, resulting in a reversed linkage, which causes approximately 50 percent of mRNAs to be capped in the reverse direction and cannot be recognized by the ribosome and hinders overall mRNA translation activity.120,121,122,123,124 Generally, anti-reverse cap analogs are synthesized to modify the m7G part of caps at the 2′ or 3′ position (2′-O-Methyl, 3′-O-methyl, 3′-H), which initiates exclusive cap incorporation in the correct direction and enhances translation efficiency.125
Poly(A) tails of IVT mRNAs are normally encoded in the DNA template or attached to IVT mRNA by enzymatic polyadenylation, and the former has more precise control of the length of poly(A) tails.2,46 Notably, a type II restriction enzyme for linearization of the plasmid template was used to contribute to an overhang at the 3′ end of the poly(A) tail when the poly(A) tail stretch was encoded in the template vector, which hampered the translational efficacy of IVT mRNA. This needs to be avoided by replacing the type II restriction enzyme with type IIS restriction enzymes.46,126
mRNA purification
IVT mRNAs are mixed with RNA polymerase and DNA templates after synthesis; thus, it is essential to purify IVT mRNA, including removing immunostimulatory contaminants, free ribonucleotides, short mRNA and DNA templates.127 Generally, DNase is used to degrade excess DNA templates. Commercial purification kits are often used to purify and separate the synthesized mRNA, followed by precipitation using ethanol or isopropanol, which can remove most contaminants and obtain high purity mRNA, and then the mRNA is precipitated with high concentrations of LiCl or alcohol-based precipitation, chromatographic methods (molecular exclusion chromatography, ion-exchange chromatography, or affinity chromatography with immobilized oligo-dT), or elution from a silica membrane column, which removes proteins, free nucleotides or other components but not dsRNA impurities.128 To remove dsRNA contaminants from the transcription reaction solution, Kariko et al. used reversed-phase HPLC to purify mRNA, which contributed to a dramatic increase in protein expression by 1,000-fold and completely eliminated the immune response of modified mRNA. However, it is unsuitable for scalable or larger mRNA production.108,129
RNase III, a novel purification method, has been proposed to eliminate dsRNA contaminants and has been shown to significantly reduce the immunogenicity of mRNAs and increase the cytotoxic killing efficacy of CAR T cells by electroporation of RNase III into CAR T cells. The potential drawback is that RNase III may cleave the double-stranded secondary structure formed by single-stranded RNA.130 Recently, cellulose chromatography was proposed to purify IVT mRNAs from micrograms to milligrams and produce large mRNAs up to 4 kb without any special equipment or toxicity, and its materials are all disposable, which poses no risk of cross-contamination compared to HPLC. Furthermore, cellulose chromatography showed higher efficiency in recovering and purifying IVT mRNA. Finally, short RNAs can be removed by denaturing polyacrylamide gel electrophoresis, and long RNAs can be separated by denaturing agarose gel electrophoresis.108,131 In summary, a variety of methods may be chosen to purify mRNA with different purity requirements and scales, which should be decided by the purpose of the research or application. Apparently, regardless of the method used for purification, strict mRNA quality control standards are the core to ensure the maximum benefits of mRNA therapeutics.
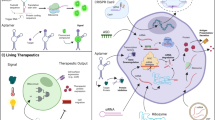
mRNA delivery systems
Researchers initially demonstrated a negative attitude to the therapeutic potential of mRNA due to its instability in early explorations.132 mRNA delivery remains a great challenge for current mRNA-based therapeutics. Primarily, mRNA, as a negatively charged macromolecule (approximately 1–15 kb), has difficulty crossing the anionic cell membrane.13 Second, the median intracellular half-life of mRNA is only approximately 7 h.133 Furthermore, large amounts of mRNA are trapped in endosomes after entry and are unable to leak into the cytoplasm to exert translation functions, although naked mRNA is difficult to internalize via scavenger-receptor mediated endocytosis.134 Suitable delivery systems are required to achieve ideal mRNA potency, provide mRNA with protection and facilitate its cellular uptake as well as endosome escape, such as liposomes and polymers. Likewise, it should have low toxicity and immunogenicity.135 Inspiringly, mRNA can be accurately delivered to hepatocytes, Kupffer cells, and endothelial cells in the liver.8
mRNA-loading mechanisms likely involve electrostatic interactions, hydrogen bonds, or coordination interactions by thin-film hydration, nanoprecipitation, or microfluidic mixing. To enhance mRNA delivery, various vectors have been designed and synthesized, including LNPs, polymetric nanoparticles, cationic nanoemulsions (CNEs), and other delivery systems136 (Fig. 6). Optimization of mRNA delivery systems would significantly improve mRNA transfection efficiency and activity, which are integral steps for the development of mRNA drugs. Yang et al. constructed LNPs using cholesterol with modification of cationic peptide DP7 (VQWRIRVAVIRK), which improved intracellular mRNA delivery and the immune stimulation of DCs.137 Wang et al. used graphene oxide and polyethyleneimine (PEI) to form an injectable hydrogel, which carried mRNA-encoding ovalbumin and the adjuvant R848. The mRNA vaccine inhibited tumor growth in the B16-OVA melanoma model.138 Phua et al. used a mesoporous-silica nanoparticle to encapsulate mRNA and the inhibitor of RNA-activated protein kinase, C16. C16 enhanced the translation of mRNA, and the vaccine significantly inhibited tumor growth.139 Huang et al. utilized mRNA encoding a constitutively active mutation of the stimulator of stimulator of interferon genes (STING), which amplified the immune response induced by mRNA vaccines.140

Positively charged lipids in mRNA-loaded lipid nanoparticles. The most widely used carrier of mRNA preparations is LNPs. Positively charged lipids play a vital role in LNPs because LNPs encapsulate mRNA through electrostatic adsorption between lipids and mRNA. These lipids can be classified into cationic lipids and ionizable lipids according to the generation of a positive charge. Furthermore, ionizable lipids can be divided into single-charged lipids and multicharged lipids. Here, we listed the representative lipids used in LNPs, including DOTMA, DOTAP, DSTAP, DMTAP, DDA, DOBAQ, DC-Chol,8,171 DLin-MC3-DMA,540 SM-102,67 A6,163 ALC-0315,541 and Lipid 5.151 Multicharged lipids in LNPs include C12-200,512 5A2-SC8,166 cKK-E12,542 G0-C14,151 OF-2,157 306Oi10,154 OF-Deg-Lin,158 92-O17S,160 OF-C4-Deg-Lin,543 A18-Iso5-2DC18,165 TT3,544 BAMEA-O16B,545 FTT5,546 Vc-Lipid,546 C14-4,161 Lipid 14,287 4A3-Cit,547 and ssPalmO-Phe548
Due to the extensive literature, we only briefly introduce the current developments in mRNA delivery vectors. We listed some typical vectors that bind mRNA with different interactions and form formulations by different preparation methods and summarized delivery vectors and adjuvants, payload mRNA, transfection efficiency, disease model or indication(s), routes of administration, and barriers to mRNA delivery.
Lipid nanoparticles
Cationic lipid nanoparticles
Cationic lipids have been broadly used in mRNA delivery, including N-[1-(2,3-dioleoyloxy)propyl]-N,N,N-trimethylammonium chloride (DOTMA), 1,2-dioleoyloxy-3-trimethylammonium propane chloride (DOTAP), 1,2-stearoyl-3-trimethylammonium-propane (DSTAP), and 1,2-dimyristoyl-3-trimethylammonium-propane (DMTAP).141 Co-delivered mRNA and gardiquimod by a poly (lactic-co-glycolic acid, PLGA) -core/DOTAP-shell hybrid nanoparticle vector not only improved mRNA transfection efficiency but also aroused a strong immune response in the spleen and thereby inhibited tumor growth in mice with B16-OVA melanoma tumors.142 The research showed that using cationic lipids dimethyldioctadecylammonium (DDA), DOTAP, DMTAP, DSTAP, N-(4-carboxybenzyl)-N,Ndimethyl-2,3-bis (oleoyloxy) propan-1-aminium (DOBAQ) and 3ß-[N-(N’,N’-dimethylaminoethane)-carbamoyl] cholesterol (DC-Chol) in combination with 1,2-dioleoyl-sn-3-phosphoethanolamine (DOPE) to form LNPs delivered RVG mRNA, including inducing strong humoral and cellular-mediated immune responses in mice.143 DOTAP/Chol/DSPE-polyethylene glycol (PEG) cationic liposomes were employed to encapsulate cytokeratin 19 mRNA that provoked a strong cellular immune response and inhibited tumor growth in an aggressive Lewis lung cancer model by intranasal immunization.144 DOTAP liposomes modified with mannose targets were used to evoke humoral and cellular immune responses to treat the H1N1 influenza virus.145 The tremendous advantages associated with lipid-nanoparticle-based mRNA delivery systems, including their high stability, transfection efficiency, efficacy, safety, and low-cost manufacturing processes, have allowed the development of mRNA vaccines and drugs at unprecedented speed, and provide a powerful disease-fighting tool.146
Ionizable lipid nanoparticles
The ionizable amino lipid Dlin-MC3-DMA (MC3) has been used to deliver siRNA clinically for the treatment of transthyretin-mediated amyloidosis. Further research showed that the compound prescription of MC3 and lipidosis (DSPC, cholesterol, DMG-PEG2000, and DSPE-PEG2000) was applied for the delivery of IL-10 mRNA as an inflammatory bowel disease therapeutic, which expressed the anti-inflammatory cytokine IL-10 in Ly6c+ inflammatory leukocytes and alleviated symptoms in a dextran sodium sulfate colitis model.147 Correcting the genetic variance of cystic fibrosis transmembrane conductance regulator (CFTR) is an efficacy target to cure cystic fibrosis. Robinson et al. loaded CFTR mRNA in an MC3 delivery system into patient-derived bronchial epithelial cells and rescued the primary function of CFTR as a chloride channel.148 Clinically relevant LNPs composed of MC3, DSPC, cholesterol, DMG-PEG2000, and mRNA were transfected into 30 cell lines, and these data demonstrated that different transfection efficacies of different cell lines depended on an early and narrow endosomal escape window.149 Li et al. also employed MC3 LNPs covalently conjugated with αPV1 antibody-encapsulated mRNA to specifically target the lung by binding plasma vesicle-associated protein.150
Sabnis et al. developed and synthesized a new series of amino lipids similar to MC3 for delivering mRNA efficiently after single and repeat dosing by introducing ester linkages in the lipid tails and changing the position of ester linkages to achieve optimal chemical stability, tissue clearance, and mRNA delivery efficiency.151 Kimberly et al. synthesized ionizable lipids with high tolerability and reduced innate immune stimulation for mRNA by i.m. administration, these data indicated that different administration routes would result in different protein expression.152 In addition, degradable or nondegradable lipoids have been designed and investigated for intravenous or local delivery of mRNA to targeted tissues and cells. A small library lipoid using 3,3′-diamino-N-methyldipropylamine was designed to react with 11 saturated alkyl acrylate tails varying in length from 6 to 18, showing that the lipoid 306Oi10 with a one-carbon branch in the tail conferred a tenfold improvement over the lipoid 306O10 with the straight tail, whose nanoparticle-containing 306Oi10 efficacy ionizes at endosomal pH 5.0, thereby benefiting mRNA delivery.153 Both mRNA and siRNA were encapsulated in a lipoid nanoparticle composed of 306Oi10, cholesterol, DSPC, DOPE, and PEG-lipid, whose codelivery of mRNA and siRNA not only can improve improved gene silencing of siRNA but can also facilitate protein expression of mRNA.154
Nanoparticles containing cKK-E12 and nine different cholesterol variants were prepared for delivering mRNA, and the results revealed that the oxidative position of cholesterol influences nanoparticle targeting by adsorbing different protein coronas onto LNPs and that nanoparticles including 20α-OH cholesterol can target the liver.155 In addition, the cKK-E12 delivery system protected trastuzumab mRNA from degradation and enabled efficient in vivo delivery, which significantly delayed the growth of HER2-positive breast cancer.156 OF-02, which was obtained by altering the lipid tails of cKK-E12, produced twofold higher erythropoietin than cKK-E12.157 OF-Deg-Lin, an ionizable lipid that changes the local structure of OF-02 from 1,2-amino-alcohol to degradable ester linkage, delivers mRNA into the spleen, inducing protein expression in the B cell population.158 OF-C4-Deg-Lin was synthesized by altering the carbon linker lengths of OF-Deg-Lin specifically targeting the spleen.159 It is well known that most mRNA delivery systems have low transfection efficacy in primary T lymphocytes. The imidazole-based lipoids that were screened from a library of lipidosis combinations of amine heads and degradable tails containing S/S-S/Se/Se-Se could deliver mRNA into primary T lymphocytes.160
Similarly, a series of piperazine-centered compounds were synthesized and selected as CAR mRNA vectors for primary human T cells.161 For novelty, a battery of cationic lipid-modified aminoglycosides centering on commercially available aminoglycosides were synthesized to specifically deliver Luc mRNA to the liver.162 Many degradable and biocompatible cholesterol derivatives (OCholB lipids) containing disulfide bonds in the tail were constructed to target the lung and spleen.163 Likewise, lipidomic materials (A1-A6) containing alkyne and ester groups in the tails were obtained by changing the structure of Dlin-MC3-DMA to increase the tumorigenicity and facilitate endosomal escape, which co-formulated lipidomic materials to efficiently treat renal anemia.163 An ionizable LNP that was based on iBL0713 lipid for delivering EPO mRNA demonstrated comparable efficacy to Dlin-MC3-DMA-based formulations in the liver.164
Lipid nanoparticles with immunostimulatory potency
Miao et al. developed lipidoses with cyclic amino head groups that activate the intracellular STING pathway, and LNPs composed of STING-activatable cyclic lipoids and OVA mRNA significantly prolonged survival and enhanced antitumor efficacy.165
Using 5A2-SC8-based dendrimer LNPs to encapsulate therapeutic FAH mRNA to produce FAH protein significantly increases the survival rate of FAH knockout mice suffering from HT-1.166 Choosing C12-(2-3-2)-based LNPs to encapsulate mRNA encoding angiotensin-converting enzyme 2 (ACE2) significantly improved liver and lung fibrosis.62 A redox-responsive NP platform consisting of G0-C14, a hydrophobic redox-responsive cysteine-based poly(disulfide amide) (PDSA), and lipid-PEG was used to deliver mRNA encoding p53, a critical tumor suppressor gene, to treat HCCs and non–small cell lung cancers (NSCLCs).167 A series of SS-cleavable proton-activated lipid-like materials based on vitamin E have also been applied to deliver mRNA to brain neuronal cells and astrocytes.168 Furthermore, TT3 lipid-like nanoparticles (TT3 LLNs) were used to codeliver mRNA and MRI contrast agents.169 Similarly, a theragnostic dendrimer-based LNP system formulated 4A3-SC8, pH-responsive PEGylated BODIPY dyes (PBD)-lipid and PBD were constructed for delivering mRNA and expressing protein in the liver, which was a promising delivery system for diagnosing and treating liver diseases and cancer.170
Polymetric nanoparticles
Polymeric compounds and their derivatives can be synthesized from natural or synthetic materials, allowing for a wide variety of possible structures and characteristics.171 PEI is one of the most potent nonviral vectors for gene delivery. However, PEI is highly toxic and nonbiodegradable, limiting its application, so PEI-g-PEG with different PEG terminal groups and PEG grafting degrees were synthesized and achieved satisfactory potency for the delivery of mRNA to the lung.172 Dunn et al. also showed the polymers PEI1800-LinA5-PEG0.3 by modifying PEI-encapsulated mRNA and targeting the pulmonary microvascular endothelium.173 Poly (β-amino esters) (PBAE), a biocompatible and biodegradable polymer, were synthesized and used to deliver mRNA to target lung endothelium and pulmonary immune cells.174 A series of oligopeptide end-modified PBAEs (OM-PBAEs) with endosomal escape and cytoplasm penetration functions for transfecting mRNA were applied for specific liver tissue targeting.175 Polymers of hyperbranched poly (beta-amino esters) (hPBAEs) were applied to deliver mRNA to the lung epithelium via inhalation and produced sufficient protein in the lung with safety and compatibility.176 Similarly, a novel PCL-based PBAE was constructed to deliver mRNA into the spleen via intravenous injection.177 APE LNPs can deliver mRNA into the lung endothelium, liver hepatocytes, and splenic antigen-presenting cells (APCs) with high transfection efficiency.177 Charge-altering releasable transporters (CARTs), a kind of cost-efficiency and biodegradable polymer, were initially positively charged polymers that can load mRNA efficiently and improve physical properties through a degradative, charge-neutralizing intramolecular rearrangement, thus releasing functional mRNA and translating protein in cells.178 CARTs applied for mRNA delivery not only target professional APCs but also target local APCs.179 CARTs were employed to deliver mRNA that (coding costimulatory and immune-modulating factors, including OX40 L-, CD80-, and CD86-encoding) significantly inhibited tumor growth in both A20 and CT26 tumor models.180 Moreover, Schumann adopted PEG[Glu(DET)]2 polymer protected and delivered FS-344 mRNA that could express FS-315 follistatin protein to cure muscle atrophy via subcutaneous administration.181 A series of amphiphilic polyaspartamide derivatives PAsp (DET/R) were synthesized to deliver mRNA to Ai9 mouse brains via intracerebroventricular and intrathecal injection.182 PEG polyamino acid block copolymer PEG-PAsp (DET) was designed to deliver brain-derived neurotrophic factor mRNA to treat spinal cord injury with satisfactory recovery.183 In addition, some peptide-derived materials were used to deliver mRNA. For instance, PEG12KL4/mRNA complexes were formulated into dry powder by spray-drying and spray freeze-drying techniques for intratracheal administration;184 RALA, a cell-penetrating peptide, was applied to deliver antigen-encoding mRNA to the immune system.185 An advanced lip polyplex containing TriMan-lip (a trimannosyl diether lipid), Lip1, Lip2, and PEG HpK was developed to deliver mRNA to inhibit tumor growth and prolong the survival of mice.186
Cationic nanoemulsion
CNEs were proposed as a potential nucleic acid delivery system in 1990187 and thus far have been proven to effectively deliver nucleic acids for the treatment of various diseases. The addition of cationic lipids to the formulation is essential for nucleic acid complexation through electrostatic interactions, which is also essential to improve the stability and transfection efficiency of nucleic acids and protect them from degradation by nucleases.188 Research shows that the self-amplifying mRNA (saRNA) CNE delivery system enhanced the local immune environment by recruiting immune cells and induced cellular responses to antibodies and T-primates at relatively low doses (75 µg).189
Other mRNA delivery systems
Other types of vectors were developed to deliver mRNA, including protamine-condensed mRNA, exosomes, extracellular vesicles (EVs), mesoporous silica, CaP and so on.190 Reactive astrocyte-derived exosomes were used to deliver MGMT mRNA to MGMT-negative glioma cells and inhibited temozolomide resistance.181 EVs with a high-affinity anti-HER2 scFv antibody (ML39) were also applied to deliver HchrR6 mRNA to recipient cells and tumors.191 Tetrasulfide-incorporated large-pore dendritic mesoporous organosilicon nanoparticles were constructed to consume intracellular GSH, thereby enhancing mRNA translation.192 Lipid-coated calcium phosphate NPs containing CaP core, DOPA, DOTAP, and DSPE-PEG for delivering MUC1 mRNA with anti-CTLA-4 monoclonal antibody were designed to treat triple-negative breast cancer.191,193 Nucleoside lipids for delivering mRNA have attracted public attention because mRNA can be loaded inside lipids through the hydrogen bonding interaction of base complementary pairings with good compatibility and safety. Uchida et al. hybridized a PEG-conjugated oligonucleotide (PEG-oligoRNA) with mRNA through hydrogen bond complementarity (20:1) to obtain PEGylated mRNA, which was then loaded with Lipofectamine LTX, and the delivery system maintained a high degree of structural stability in vivo.194 Polyplex micelles were developed by combining ω-cholesteryl (ω-Chol)-poly (ethyleneglycol) (PEG)-polycation block copolymers with mRNA prehybridized with cholesterol (Chol)-tethered RNA oligonucleotides (Chol (+)-OligoRNA) to improve the tolerance of mRNA nucleases and the stability of mRNA.195 Furthermore, an RNA linker that connected 10 nt oligo-adenine nucleotides (OligoA) with two 17 nt oligonucleotides was designed to improve the stability of mRNA to ribonuclease.196 Generally, most of the reported delivery vectors deliver mRNA through electrostatic interactions or hydrogen bond interactions. Novel delivery vectors have also emerged for further application, such as self-assembled core–shell nanoscale coordination polymer nanoparticles that were used to deliver siRNA, microRNA or DNA through coordination interactions.197,198,199 Overall, among mRNA delivery platforms, LNPs have been approved for clinical use and have been shown unique advantages, and potential nanomaterial candidates are still emerging. The choice of mRNA delivery system depends on the size of the delivered mRNA molecule, the charge, and the organ to be targeted. There are advantages and disadvantages to different delivery materials.
In vitro and in vivo barriers to mRNA delivery
It has always been the focus of our thinking by increasing cell uptake, facilitating lysosomal escape, and speeding up translation to maximize the availability of mRNA.200 Nanoparticle-based delivery systems provide a promising approach to improve cell uptake and lysosomal escape, which are also widely researched in the field of mRNA delivery.201 Multiple steps are involved in mRNAs entering the cytoplasm with the help of nanoparticles: endocytosis, lysosomal escape, and mRNA release. The cell membrane is a dynamic and formidable barrier to intracellular transport.201 Nanoparticles interact with cell membranes through various mechanisms, including clathrin-dependent endocytosis, caveolae-dependent endocytosis, and micropinocytosis,202 so particle properties, including particle shape, size, material composition, and surface charge, are involved in cellular uptake.
It is a prerequisite for efficient mRNA delivery to comprehend the mechanism of mRNA cellular uptake. It has been reported that naked mRNA is internalized by scavenger receptors without delivery materials and subsequently accumulates in lysosomes; minimally, mRNA escapes into the cytosol and expresses proteins, so it is necessary to use vectors for the intracellular delivery of mRNA and overcome the initial energy barrier to mRNA uptake.203 Stimulating scavenger receptor activity to increase the uptake of mRNA and promoting endosomal escape could boost the availability of mRNA in the cytoplasm.204 mRNA needs to be released from lysosomes and egressed to cytosol to translate encoding protein and was inevitably inhaled to lysosomes following micropinocytosis and clathrinid-mediated endocytosis, where acidic and enzyme-rich environment is prone to degradation of nanocarrier and mRNA, so lysosome degradation is another delivery barrier for mRNA.205 At present, electroporation is used for clinically delivering mRNA ex vivo, but its disadvantage is that membrane destruction by electroporation may lead to the loss of cytoplasmic content with significant cytotoxicity.206
Notably, endosome/lysosome formation is essential for exogenous mRNA function because the mammalian target of rapamycin on the lysosomal surface involves several cellular processes, including protein expression and mRNA transfection efficiency. The rapid rate at which nanoparticles are engulfed by lysosomes is directly affected by the properties of nanoparticles, so as quickly as possible to escape lysosomes is necessary for mRNA translation.149,207,208,209 Nanoparticle materials achieve lysosome escape through conductivity, such as DOPE, MM27, and DLinDMA, which are widely applied to the cell membrane in an acid-mediated manner.200,201,210 In addition, pH-responsive cell-penetrating peptides promoted endosome membrane disruption and enhanced protein expression.211 Recently, research showed preassembling an mRNA translation initiation structure called ribonucleoproteins through an intrinsic molecular recognition between m7G-capped mRNA and eIF4E protein, thereby mimicking the first step of intracellular protein synthesis, and subsequent ribonucleoproteins electrostatically stabilized with structurally adjustable cationic carriers to form nanoplexes. This approach significantly improved mRNA transfection efficiency by enhancing intracellular mRNA stability and protein synthesis.200 Collectively, engineering precision nanoparticle delivery systems for mRNA-based therapeutics is the key to determining mRNA translation efficiency and enhancing the expression of mRNA.
There is also a substantial challenge for mRNA delivery in vivo.212 Nude mRNA is directly used for mRNA-based therapeutics; however, it is vulnerable to the widely distributed RNase in vivo. Therefore, a delivery system is essential for mRNA-based therapeutics.213 Research on siRNA vectors is relatively mature. Regrettably, these vectors for siRNA and pDNA delivery may be unsuitable for mRNA delivery owing to their different characteristics.214 Therefore, it is urgent to develop new delivery vectors to achieve favorable loaded mRNA circulation, specific target organs or cells, cytomembrane penetration, lysosome escape, and mRNA and protein expression.215
There have been many reports on the enhancement of mRNA encoding antigen uptake by DCs through cell receptor modification of nanoparticles.208 There are still numerous barriers to uptake and intracellular trafficking that determine mRNA-based therapeutic efficiency.216 DCs play key roles in immunotherapy, which can efficiently take up, process, and present antigens and subsequently induce humoral and cellular immunity against various infectious diseases and cancers.217 DC-based vaccines are a potent immunotherapeutic strategy. Autologous DCs are used to load antigens by pulsing in vitro and are then administered back to the patient to initiate the immune response.218 There are several strategies to deliver mRNA into the cytoplasm of DCs, including electroporation, lipofection, and sonoporation.219 Electroporation is possibly the most diffusely used method for mRNA introduction, which rapidly introduces tumor-associated antigen (TAA)-encoding mRNA by using a relatively weak electric pulse, greatly avoids the degradation of mRNA by ubiquitous extracellular ribonuclease, and mediates mRNA cellular processing and presentation on the DC surface.220 Lipofection encapsulates and delivers mRNA into DCs by forming mRNA lipoplexes, which are subsequently taken up via cell endocytosis, and then the lipid fuses with the endosomal membrane to release mRNA into the cytoplasm.221 For the sonoporation strategy, mRNA is loaded in microbubbles and directly crosses the cytoplasm membrane via temporary pores, which are created by oscillating microbubbles and imploding them using ultrasound.222 The transfection and expression efficiency of mRNA drugs in DCs is the key to therapeutic efficacy. Different delivery strategies contribute to distinct mRNA transfection efficiency, namely, electroporation (90%), lipofection (5–50%) and sonporation (5–50%).223,224,225 Importantly, electroporation has high transfection efficiency and is used to treat various tumors in clinical studies, including melanoma,226,227,228 AML,76 ovarian cancer, and infectious diseases (human immunodeficiency virus [HIV]).229 In addition, previous research showed that lipofection provides the high expression of antigen and is more effective in expanding CD8+ T cells in DCs, indicating that lipofection has potent immune stimulation activity. However, the reproducibility of transfection efficiency makes GMP-standard manufacture implementation difficult and restricts lipofection clinical application.230 Collectively, focusing on optimized delivery strategies that overcome DC barriers is the key to mRNA-based immunotherapy.
The in vitro and in vivo efficiency of mRNA drugs is not always consistent. The transfection efficiency of alkyne lipids outperformed MC3, cKK-E12, and C12-200 in vitro but not in vivo.163 In addition, encapsulation of different mRNAs delivered extracellular displayed different distributions; OF-Deg-Lin LNPs loaded with Cy5 mRNA were transported predominantly to the liver, whereas OF-Deg-Lin LNPs encapsulated FLuc mRNA expressed protein in the spleen.158 We speculated that the abovementioned inconsistencies may be caused by the complicated internal environment, including the immune system, variable blood flow, heterogeneous vasculature, and off-target cells, and the specific mechanisms still need to be further explored.
Tissue-targeted delivery of mRNA-based therapeutics is essential for efficient in vivo delivery of mRNA.67 Delivery systems can provide much more effective and targeted delivery of mRNA drugs, including drug release that is triggered by the specific microenvironment and the physicochemical properties of mRNA vectors that play important roles in their systemic delivery and biodistribution.231 Engineering precision nanoparticles for mRNA-based drug delivery has expanded into a broad range of clinical applications and has been developed to navigate biological barriers.171 Nanoparticles are rapidly recognized by mononuclear phagocytic systems in the liver and spleen by binding to serum proteins, and encapsulated mRNA is released to target cells.221 The majority of the current most widely used mRNA-based delivery of LNPs specifically targets the liver, and LNPs continue to focus on optimizing delivery platforms in other tissue-targeted delivery.232 Recently, selective organ targeting has emerged as a therapeutic strategy to precisely and predictably optimize LNPs and allow them to deliver mRNA and Cas9 mRNA/single guide RNA and Cas9 ribonucleoprotein complexes to target tissues via intravenous injection into the liver and lung.233 In addition, cell-targeted delivery of mRNA-based therapeutics, especially DCs and APCs, plays crucial roles in shaping immune responses by delivering requisite signals to T cells and activating expansion and differentiation T cells.210 The field of mRNA-based therapeutics is currently focused on the development of novel materials and formulations that can potentially enhance transfection efficiency and therapeutic efficacy.2
The adjuvant activity of mRNA delivery systems
Cationic liposomes themselves act as adjuvants, and their main function is to protect the antigen from being eliminated and deliver the antigen to professional APCs.234 The RNActive (CureVac AG) vaccine platform relies on its carrier to provide adjuvant activity, and the adjuvant activity is provided by the codelivery of RNA complexed with protamine (a polycationic peptide) by inducing an adaptive response,235,236,237 which has elicited a favorable immune response in multiple preclinical animal studies against cancer and infectious diseases.238,239,240,241 Mechanistically, the adjuvant properties of the RNActive vaccine showed a potent TLR7/8-dependent immune response, including activation of TLR7 (in mouse and human cells) and TLR8 (in human cells), type I interference, cytokines, and chemokines.235 However, mRNA-mediated activation of type I interferon may cause protein translation and CD8+ T cell activation to be inhibited, which may be related to the kinetics of type I interferon signaling relative to TCR activation.242,243 The codelivery of mRNA and hydrophobic TLR7 adjuvant (gardiquimod) is achieved by a PLGA core/lipid-shell hybrid nanoparticle system, in which PLGA allows incorporation of the adjuvant into the nucleus and the lipid shell loads the mRNA through electrostatic interactions. The nanoparticle realizes a strong antigen-specific immune response and highly effective antitumor activity.142
The effect of administration routes on delivery efficiency
The administration routes play a vital role in the mRNA delivery system because some specific diseases require specific routes of administration, although intravenous administration can meet the needs of most diseases. For instance, inhaled administration or intratracheal administration is suitable for pulmonary diseases;184 cerebral diseases may be cured by intracerebroventricular injection or intrathecal injection;182 and liver diseases may be treated via intravenous, intraperitoneal, subcutaneous, or intramuscular administration.244 In addition, different delivery vectors will have different distributions or expressions under different administration routes. For example, LNPs containing lipidoid 306Oi10 targeted and expressed protein predominantly in the liver via i.v. injection, while the LNPs accumulated in the pancreas (11%), kidneys (12%), and lungs (15%) and expressed protein in the liver (67%), pancreas (17%), and spleen (13%); similarly, the LNPs drained through capillaries and the lymphatic system when administered via s.c. and i.m.244 It has been reported that cholesteryl-based disulfide bond-containing biodegradable cationic liposome nanoparticles OCholB LNPs have demonstrated the successful delivery of mRNA molecules in the skeletal muscle (via intramuscular injection), lung and spleen (via intravenous injection), and brain (via lateral ventricle infusion).162 CARTs preferentially targeted professional APCs in secondary lymphoid organs upon i.v. injections and targeted local APCs upon s.c. injection.179 Therefore, the optimal therapeutic efficacy can only be achieved by selecting the appropriate mRNA delivery vectors and routes of administration. Collectively, LNP–mRNA therapeutics (good manufacturing practices, stability, storage, and safety) have great potential in the treatment of infectious diseases, cancer, and genetic diseases. The development of mRNA delivery systems with high efficiency and safety is of great significance for the wide application of mRNA-based therapeutics.
Application
mRNA-based therapeutics are expected to become a powerful therapy for a variety of refractory diseases, including infectious diseases, metabolic genetic diseases, cancer, cardiovascular and cerebrovascular diseases, and other diseases (Fig. 7). A large number of studies have shown that mRNA cannot only mediate better transfection efficiency and longer protein expression but also has greater advantages than DNA and traditional protein drugs; mRNA initiates protein transient translation when reaching the cytoplasm without inserting into the genome, which has a lower insertion risk compared with traditional protein and DNA drugs. Importantly, mRNA is easily synthesized through the IVT process, is relatively easy to manufacture and can be quickly applied to various therapies. In addition, the two most concerning issues in mRNA, immunogenicity and stability, are controlled by the chemical modification of selected nucleotides. mRNA therapy has attracted billions of dollars, and an increasing number of well-funded biotechnology companies have been established, such as Moderna, CureVac, BioNTech, Argos Therapeutics, RaNA, Translate Bio, Ethris, Arcturus, and Acuitas (Table 2). Apparently, mRNA has become one of the most attractive areas for drug development, which is definitely worth exploring in the long term. In this section, we comprehensively summarize the latest developments in the current state of mRNA-based drug technologies and their applications.

Strategies and potential application of mRNA-based therapeutics. mRNA drugs have yielded numerous inspiring treatments for refractory or previously incurable diseases, including infectious diseases, genetic diseases, cancers, and cardiovascular diseases. In particular, the mRNA vaccine has shown a strong advantage in the prevention of SARS-CoV-2 infection and may also be a potential approach against the infection of other viruses and pathogenic microorganisms, including malaria, respiratory syncytial virus, and HIV13
mRNA therapeutics that are directly based on the encoding molecules
The aforementioned mRNA-based immunotherapy achieves promising outcomes by expressing antigens and then initiating immune responses,245 which is defined as an indirect therapy that does not target the virus or tumor cells with mRNA encoding therapeutic proteins.246 mRNAs encoding proteins/peptides directly target viruses, bacteria, or cancer cells. In contrast, mRNA therapeutics directly treating diseases by delivering mRNA-based functional proteins are considered a direct strategy, including missing or downregulated endogenous proteins, functional foreign proteins or antibodies, and proteins for gene editing tools.247 In addition, the strategy of directly expressing proteins in “cell factories” can also be used to engineer cells, such as engineered T cells.161 mRNA-based protein replacement therapeutics have already entered the clinical stage despite the limited number of clinical trials vs. mRNA vaccines.248,249
mRNA-based monoclonal antibodies
Antibody-based drugs have achieved rapid progress in biopharmaceutics, but the worldwide application of monoclonal antibodies (mAbs) is limited by their vulnerable properties and the high cost of production, storage, transportation, and distribution.250 Nucleic acid-encoded mAbs, especially mRNA-based monoclonal antibodies, have rendered great hope for improving antibody therapy efficacy, and targeted cells are expropriated as factories to translate nucleic acids into functional mAbs.251 Plasmid DNA-encoded mAbs are usually concentrated in the area of infectious diseases, and some have already entered the clinical stage, while studies on mRNA-based mAbs (mRNA-mAbs) have relatively lagged. Here, we focus on the application of mRNA-mAbs, which are mostly concentrated on the treatment of infection and tumors.252 The broadly neutralizing anti-HIV-1 antibody VRC01 was decoded into nucleoside-modified mRNA, and systemic administration of the LNP-encapsulated mRNA successfully produced VRC01 at the efficacy level and protected humanized mice from intravenous HIV-1 challenge.253 For human RSV, Tiwari et al. developed the existing drug palivizumab into engineered mRNA encoding membrane-anchored neutralizing antibodies, which displayed higher efficiency than palivizumabs and significantly inhibited RSV 7 days post-transfection.254 Isolated neutralizing mAbs (CHKV-24) from the B cells of a survivor of natural chikungunya virus infection were successfully encoded by mRNA, expressed at biologically significant levels in vivo, and protected mice from arthritis and musculoskeletal tissue infection with reduced viremia at undetectable levels after 2 days of inoculation.255 A nanostructured lipid carrier was exploited to transfer replicon RNA encoding ZIKV-117 mAb in situ by intramuscular delivery, which contributed to high levels of mAb expression and protected mice from lethal ZIKV infection.256 In addition, the strategy of mRNA-based mAbs is adopted in the treatment of tumors. Various mRNA-based antibodies against cancer were designed and induced rapid and sustained serum antibody titers in vivo, which allowed mice to survive the challenge of non-Hodgkin’s lymphoma tumor incubation.257 Anti-HER2 antibody (trastuzumab) was systemically delivered using IVT mRNA LNPs and synthesized in vivo, which improved the pharmacokinetic profile in comparison with directly injecting trastuzumab protein.156 In addition, Zhou et al. reported a novel method for rapidly delivering the nanobody/variable domain of the heavy chain from an antibody by introducing its coding mRNA.258 Bispecific T cell-engaging antibody (bsAb) has emerged as a promising approach to treat malignancy, although this is somewhat impeded by manufacturing difficulty and short serum half-life. Endogenously synthesized and durable bsAbs through systemic administration (mRNA-based bsAbs) efficiently inhibited tumor growth.259 Ye et al. developed a saRNA encoding an anti-SARS-CoV-2 antibody with an alphavirus vector.99 However, the virus vector showed poor safety in the development of the SARS-CoV-2 mRNA vaccine.260,261
mRNA-based immunotherapy
Immunotherapies have yielded numerous inspiring treatments for refractory or previously untreatable diseases, including infectious diseases, cancers, autoimmune diseases, and allergies.262,263,264,265,266 Vaccine research progress has fueled a great deal of enthusiasm and promise for immunotherapy approaches against pandemic infectious diseases, including attenuated vaccines, inactivated vaccines, and protein subunit vaccines.267 Recently, nucleic acid vaccines have emerged as innovative vaccines, including DNA vaccines and RNA vaccines. Notably, mRNA-based therapeutics have emerged as a safe and efficacious strategy to protect patients from infectious diseases and cancers due to their extraordinary advantages, including high efficiency, a relatively low severity of side efficacy, and ease of manufacture.1,262 Here, we reveiwed the applications of mRNA-based drugs, focusing on clinical trials of prophylactic and therapeutic vaccines for infectious diseases and cancers (Fig. 8).

mRNA drugs elicit immunity using disease-specific targeting antigen strategies. mRNA drugs mainly go through the following three aspects from synthesis to initiate immune protection, including mRNA synthesis, intracellular processing, and initiating immune protection. Briefly, IVT mRNA drugs are encapsulated into carriers (such as nanoparticles) and are endocytosed by antigen-presenting cells (①-②); mRNA is released into the cytoplasm after escaping from endosomes and then translated into antigenic proteins by ribosomes (③). Subsequently, endogenous antigens are degraded into polypeptides by the proteasome and are presented by MHC I and activate cytotoxic T cells (CD8+ T cells) (④-⑥). In addition, secreted antigens can be taken up by cells, degraded inside endosomes, and presented on the cell surface to helper T cells by MHC class II proteins (⑦-⑨). Finally, helper T cells (CD4+ T cells) stimulate B cells to produce neutralizing antibodies against pathogens382
mRNA vaccines against infectious diseases
SARS-CoV-2 mRNA vaccines
SARS-CoV-2 emerged in 2019268 and then caused pandemics worldwide.269 To date, there have been more than 228 million confirmed cases of COVID-19, including ~6.14 million deaths according to the WHO report (covid19.who.int). The first COVID-19 vaccine (Pfizer-BioNTech COVID-19 Vaccine; BNT162b2) was approved by the FDA for emergency use authorization and subsequently for the Moderna COVID-19 vaccine (mRNA-1273). These vaccines provide ~90% effectiveness prevention of infection for full vaccination and 80% for partial vaccination,270,271,272,273 However, neutralization antibodies against the SARS-CoV-2 Omicron variant are undetectable in the sera of most mRNA-1273 or BNT162b recipients, while additional mRNA vaccine dose seems to improve the neutralization.274
SARS-CoV-2 consists of structural proteins, spike (S), nucleocapsid (N), envelope (E), and membrane (M).275 The coronavirus S protein or the RBD of the S protein mediates receptor binding and fusion of the viral and cellular membranes, and entry of virions into target cells has emerged as an antigen therapeutic strategy to design vaccines.276 N proteins of SARS-CoV-2 can induce immune responses to inhibit viral infection, while E proteins and M proteins are generally not taken into account for the lack of immunogenicity.277,278 Several strategies have been used to improve the COVID-19 vaccine effect; prefusion S protein was formed by mutation of two proline residues of the spike protein to stabilize it in the prefusion conformation, and BNT162b2 and mRNA-1273 both used 1-methyl-3′-pseudouridylyl modified mRNA (m1Ψ mRNA) encoding prefusion S protein.271,279 SARS-CoV-2 spike RBD, as the binding site for hACE2, facilitates virus entry into target cells and is a promising target to design candidate vaccines.280 However, monomeric RBD antigens have limited ability on engaging interactions with B cell receptors thereby facilitating the generation of high-affinity antibodies.281,282,283 Various strategies have been developed to increase RBD protein immunogenicity, thus enhancing antibody titers, including conformation dimers, trimers or polymers, by adding humanized IgG Fc,284 T4 trimerization (Foldon)285 or ferritin286 to antigen (Fig. 9). mRNA that encodes the C-terminal fold or Helicobacter pylori ferritin rendered a multimeric conformation of RBDs and induced robust and durable humoral immunity.286 mRNA encoding RBD-conjugated Fc induces a stronger immune response.287 Furthermore, mRNA drugs can also effectively block the binding of the RBD to the human ACE2 receptor by encoding high-affinity truncated ACE2 variants.288

SARS-CoV-2 mRNA antigen immunogenicity and vaccine design. Full-length S-protein or RBD as a vaccine immunogen has been widely confirmed to induce high-affinity neutralizing antibodies. SARS-CoV-2 S protein is intrinsically metastable and can be stabilized in a prefusion conformation by structure-based design.549,550 Prefusion-stabilized SARS-CoV-2 Spike immunogen induces potent humoral and cellular immune responses.551,552 The RBD peptide is one of the most promising targets to design candidate vaccines. However, RBD has a low molecular weight, which leads to its weak immunogenicity, and can be further improved by forming multimers. Multimerization of RBD protein using humanized IgG Fc,284 T4 trimerization (FD)285 or Ferritin286 have been shown to induce higher neutralizing antibody compared to monomeric antigens, which will provide us with new ideas for designing powerful mRNA vaccines
Several SARS-CoV-2 variants have emerged with the global COVID-19 pandemic.289 Fortunately, chimeras of the viral S protein were developed to prevent SARS-CoV-2 variants,270 and BNT162b2 and mRNA-1273 can still effectively prevent SARS-CoV-2 variants infections, including Delta (B.1.617.2), Alpha (B.1.1.7) and Gamma (P.1) variants in adults.272,290,291 Interestingly, there is a large difference in the mRNA dosages of COVID-19 mRNA vaccines. The approved dosage of one dose of BNT162b2 is 30 μg mRNA, and mRNA-1273 is 100 μg (www.fda.gov/). The first 100 μg BNT162b1 vaccination lacked meaningfully increased immunogenicity compared with the first 30 μg vaccination.292 Nevertheless, dose-dependent responses were observed in the vaccinations of mRNA-1273 (25, 100, and 250 μg) and ARCoV (100 and 1000 μg).276 Notably, a saRNA vaccine encoding the S protein and the VEE virus replicase for self-amplification, called LUNAR-COV19, were designed and showed that a single 2 μg vaccination protected mice from lethal SARS-CoV-2 infection.293
The duration of the COVID-19 mRNA vaccine and its effectiveness in special populations necessitate further investigation into long-term protection, especially for patients with existing conditions and a pandemic pathogen with mutations. The anti-SARS-CoV-2 humoral immunity continuously declined for several months following full BNT162b2 or mRNA-1273 vaccination.294,295,296,297 BNT162b1 induced weaker humoral immunity in older adults than in younger adults.285,298 Fortunately, BNT162b2 vaccination appears to be safe for pregnant women and can reduce the risk of SARS-CoV-2 infection.299,300,301,302 Likewise, anti-SARS-CoV-2 antibodies can be transferred to neonates in pregnancy.303 BNT162b2 and mRNA-1273 appear to be well tolerated and induce a weaker but significant immune response in patients with immunocompromising conditions, including hemodialysis,304 hematological disorders,305,306 malignancy,307,308 chronic inflammatory disease309 and HIV infection (only BNT162b2 evaluated).310 BNT162b2 showed weaker but significant immunogenicity in patients with autoimmune diseases, including rheumatic diseases,311,312,313 multiple sclerosis,311,314,315,316 myasthenia gravis,317 and musculoskeletal diseases.318 Notably, mRNA-1273 and BNT162b2 showed impaired immunogenicity in solid organ transplant recipients.319,320,321,322,323
Various pathogens cause serious human infections, including viruses, bacteria, fungi, and parasites.324 Viruses have caused a series of public health emergencies: the H1N1 influenza pandemic in 2009–2010,325 Zika virus infection in 2015–2016,326 and the current COVID-19 pandemic.327 Vaccines are a vital tool in the battle against infectious diseases.328 mRNA vaccine candidates have shown similar safety and reactogenicity profiles to inactivated vaccines approved by the European Union and Americans, but acute and chronic infections account for 15% of all deaths worldwide due to unreasonable vaccine distribution in resource-limited areas and insufficient response to infectious outbreaks.329 mRNA vaccines are an ideal approach to overcome these challenges and fulfill the urgent need for vaccines during epidemics in a timely manner.330 Currently, mRNA vaccines have been intensively researched and developed to combat highly contagious SARS-CoV-2, influenza virus, Zika virus, rabies virus, and HIV, and corresponding clinical results are summarized (Table 3).331 mRNA vaccine candidates were rapidly generated 8 days after the publication of hemagglutinin and neuraminidase genes of H7N9 influenza virus. An mRNA vaccine (NCT03014089) showed 47% placentas from Zika virus infection in comparison with 91% infected placentas of placebo-vaccinated mice, and protective humoral immunity was also confirmed in rhesus macaques.332 Likewise, mRNA-1273 successfully decreased the viral load in the lungs of mice and rhesus macaques challenged with SARS-CoV-2 and evoked a Th1-biased immune response in healthy adults (NCT04283461, NCT04470427).333 An mRNA vaccine (CV7201) was developed by using mRNA encoding the glycoprotein of rabies virus to treat rabies, which showed temperature stability and successfully elicited a WHO-specified antibody response in >70% of participants via three rounds of intradermal (i.d.) vaccination (NCT02241135).334 Despite extensive efforts in design and testing, scientists failed to generate an effective preventive HIV vaccine. Unlike the prophylactic vaccines above, the mRNA vaccines for HIV not only aim to prevent but also aspire to cure infection. Anti-HIV mRNA vaccine (NCT02888756) and DC-based mRNA vaccine (AGS-004, NCT00672191) have entered clinical trials,335 but no antiviral efficacy has been observed in clinical trials.336 Vibcinated patients had similar plasma virus levels to placebo-treated controls (NCT00672191), and all participants restarted antiretroviral therapy for unsuccessful control of acute HIV infection (NCT00672191).336 There are several mRNA vaccines against bacteria and parasites,337 but they are still under preclinical evaluation.338 Collectively, these studies indicated that mRNA vaccination is a promising strategy against infectious diseases, although further research and development are urgently required for some of these diseases, such as AIDS.
Influenza virus mRNA vaccine
Nachbagauer et al. selected the conserved HA stalk domain, matrix-2 ion channel, nucleoprotein, and broadly reactive neuraminidase as antigens to provide universal protection against the influenza virus. The vaccines used LNP to deliver m1Ψ mRNA and protected mice from challenge with H1N1 virus at 500-fold the median lethal dose (intradermally, an ionizable cationic lipid/phosphatidylcholine/cholesterol/PEG-lipid (50:10:38.5:1.5 mol/mol)).339
HIV mRNA vaccine
Mariano Esteban used vaccinia virus Ankara vectors to load unmodified and 1-methyl-3′-pseudouridylyl modified mRNA (m1Ψ mRNA) encoding HIV-1 Gag, Pol and Nef proteins (an ionizable cationic lipid/phosphatidylcholine/cholesterol/PEG/lipid (50:10:38.5:1.5 mol/mol)).340,341
RSV mRNA vaccine
Respiratory syncytial virus mRNA vaccine mRNA-1777 showed safety and tolerability in a phase I clinical trial.342 Bett et al. used LNP to deliver mRNA encoding full-length wild-type F protein, a full-length mutated F protein, a truncated secreted trimeric form of F protein, a secreted prefusion-stabilized F protein, and the full-length wild-type and prefusion-stabilized forms evoked a higher immune response (LNP formulation: asymmetric ionizable amino lipid, DSPC, cholesterol, and poly(ethyleneglycol) 2000-dimyristoylglycerol (PEG2000-DMG) in a molar ratio of 58:30:10:2, respectively).343
HSV mRNA vaccine
Friedman et al. developed a trivalent mRNA vaccine targeting herpes simplex virus type 2 glycoproteins C, D, and E. Compared to a trivalent protein vaccine, a m1Ψ-modified mRNA vaccine provided better protection.344,345 Friedman et al. compared the HSV mRNA vaccine and protein vaccine that used the same antigens (glycoproteins C2, D2, E2), and the former induced a stronger immune response and memory.346
VZV mRNA vaccine
Vora et al. used LNP to deliver m1Ψ mRNA encoding varicella-zoster virus (VZV) gE antigen, which showed an effect comparable to that of a protein vaccine adjuvanted with AS01B (LNP formulation, ionizable lipid: DSPC:cholesterol:PEG-lipid, 50:10:38.5:1.5).347
Human cytomegalovirus (HCMV) vaccines mRNA vaccine
Permar et al. used LNP to deliver m1Ψ mRNA encoding full-length glycoprotein B protein that evoked a more durable immune response than the protein vaccine adjuvanted with MF59 (LNP formulation, an ionizable cationic lipid (proprietary to Acuitas), phosphatidylcholine, cholesterol, and PEG-lipid (50:10:38.5:1.5, mol/mol).348 Similarly, they developed HCMV vaccines using mRNA encoding glycoprotein B and the pentameric complex that induced significant immune responses in nonhuman primates with preexisting immunity against HCMV.349
Rabies virus mRNA vaccine
Rabies virus causes a zoonotic infection, imposing an estimated 59,000 deaths each year. Despite effective vaccines, rabies remains one of the most distressing diseases worldwide, owing to unobtainable treatment and complicated vaccine regimens (requiring 4 doses). CureVac AG developed a rabies virus (RABV) mRNA vaccine, CV7201, that uses the cationic protein protamine to encapsulate mRNA encoding the glycoprotein of rabies virus to treat rabies. CV7201 was temperature stable and successfully elicited a WHO-specified antibody response in 70.3% of participants via three i.d. vaccination (NCT02241135).330 Based on CV7201, CureVac AG optimized the LNP formulation and developed CV7202, which uses the same mRNA antigen as CV7201. The optimized LNP includes an ionizable amino lipid, a PEG-modified lipid, phospholipid, and cholesterol.350 CV7202 showed good tolerance in a clinical trial (NCT03713086).351 Luis-Alexander Rodriguez used a CNE to encapsulate saRNA encoding alphavirus RNA-dependent RNA polymerase and the rabies glycoprotein G.352
Dengue virus mRNA vaccine
Richner et al. used LNP to encapsulate mRNA encoding the envelope and membrane structural proteins of Dengue virus serotype 1.353
Other mRNA vaccines
Spiropoulou et al. used LNP to encapsulate mRNA encoding the soluble Hendra virus glycoprotein, which protected 70% of Syrian hamsters from lethal NiV challenge.354 Sigal et al. developed an mRNA vaccine against ectromelia virus using mRNA encoding EVM158.355
mRNA cancer vaccines
Immunotherapy has been an evolving and promising cancer treatment by stimulating the immune system, including immune checkpoint blockade (ICB), chimeric antigen receptor T cells (CAR-T cells), and vaccines.356 Unlike ICB releasing immunosuppression and CAR-T cells directly killing tumor cells, a cancer vaccine initiates and amplifies the antitumor immune response by APCs, especially DCs.357 mRNA cancer vaccine platforms have been developed and have achieved encouraging outcomes based on their unique efficacy in pushing the cancer immunity cycle and safety. mRNA vaccines for castration-resistant prostate cancer and non-small-cell lung cancer were clinically evaluated.358 Meanwhile, mRNA vaccines for melanoma, glioblastoma, AML, and renal cell carcinoma (RCC) demonstrated an active response to immunotherapy, which deserves intensive further exploration in the mRNA vaccine field.359
Melanoma
Three non-DC-based and seven DC-based mRNA vaccines have been tested clinically. Among them, one non-DC-based360 and one DC-based mRNA vaccine361 used complete mRNAs from tumor cells, and other vaccines selected TAAs and encoded them into mRNAs. Notably, all DC-based mRNA vaccines failed to significantly improve clinical outcome in metastatic melanoma patients, and more than half of the participants developed disease progression during clinical trials, and intranodal (i.n.) vaccination failed to improve the efficacy of DC-based mRNA vaccines and had a lower response rate than i.d. vaccination (NCT01278940).226,361,362,363,364,365 TriMix-mRNA (containing mRNAs coding immunostimulatory molecules: CD40 L, CD70, and caTLR4) was implemented to improve DC-based vaccine efficacy.362,364 In addition, the BioNTech company developed a personal mRNA vaccine for metastatic melanoma, had no detectable lesions on radiology, and remained recurrence-free after 23 months of i.n. vaccination (NCT02035956)366 and exploited LNP to generate an anti-melanoma mRNA vaccine, which attributed to regression of a suspected metastasis in an intravenously vaccinated patient (NCT02410733).367 Due to the inconsistent data, further research may help confirm that mRNA vaccines can serve as an immunotherapy for melanoma.
Glioblastoma
mRNA vaccination has been considered a promising strategy to treat glioblastoma.368 DC-based mRNA vaccines were generated by using mRNA copies of glioblastoma in patients and prolonged progression-free survival 2.9 times compared with matched controls (NCT00961844).369 Likewise, pre-conditioning the vaccine site with a potent recall antigen such as tetanus/diphtheria (Td) toxoid can significantly improve the efficacy of tumor-antigen-specific DCs, thus increasing DC migration bilaterally and significantly improving glioblastoma patients survival.370 A DC-based mRNA vaccine was developed to improve mRNA-pulsed DC homing to lymph organs (NCT00639639, relevant results have not yet been announced).
Acute myeloid leukemia
Two DC-based mRNA vaccines have been developed to reduce the relapse risk of AML patients with complete remission (NCT00510133 and NCT00965224). Electroporation DCs with WT1 mRNAs improved relapse-free survival in vaccination responders compared with nonresponders. Another study exploited mRNA encoding human telomerase reverse transcriptase, and i.d. vaccinations resulted in 11 of 19 patients in complete remission with a 52-month median follow-up.74,371 Notably, mRNA vaccines may be unsuitable for patients with processive AML because they depend on the immune system to exert function, while AML can impair patients’ immune system.76
Renal cell carcinoma (RCC)
RCC continues to have high mortality rates, and two mRNA vaccines have been developed to treat RCC. DC-based mRNA vaccines showed moderate efficacy (NCT00678119) for advanced RCC treatment.372 Another anti-RCC mRNA vaccine is directly administered to patients via the i.d. route, and the vaccine-specific immune response seems to be related to the long-term survival of RCC patients.373
Tolerance to mRNA cancer vaccines
Tumors boast many mechanisms to evade efficacy immunosurveillance by upregulating immunosuppressive molecules and corresponding cells under the antitumor pressure of immunotherapy, resulting in the induction of peripheral tolerance and central tolerance and significantly impairing immunotherapy efficacy.374 The treatment strategies of ICBs are widely exploited to break immune tolerance, including anti-PD-1 antibodies,366 anti-CTLA-4 antibodies,375 and PD-L1 siRNA.376 Unlike ICBs, natural killer (NK) cells may be favorable for overcoming the tolerance mechanism, which is related to NK cells eliminating tumor cells without the presentation of MHC I molecules.366,377 TAAs, as self-antigens, have central tolerance due to the clonal deletion of autoreactive lymph cells during ontogenesis.378 Neoepitopes can bypass central tolerance with high immunogenicity because they are never present in normal tissues and generate the accumulation of gene mutations in cancer cells (including driver mutations and passenger mutations).379 Therefore, neoepitopes were applied to overcome the central tolerance of cancer vaccines and address the issue of tumor heterogeneity. The personal mRNA vaccine has shown relatively favorable clinical efficacy, but some patients were unavailable for vaccination due to disease progression, and merely a portion of neoepitopes successfully induced a specific immune response in patients.366 Recently, several clinical trials have been launched to further evaluate the antitumor efficacy of personal mRNA vaccines (NCT03313778, NCT02316457, and NCT03468244, relevant results have not yet been announced).380,381 Collectively, based on the complexity of tumor pathogenesis, codelivery of multiple therapeutic mRNAs has great potential to defeat cancer.
The safety of mRNA vaccines
mRNA vaccines have sufficient safety with good tolerance, and their adverse events (AEs) are generally mild to moderate, including injection site reactions such as pain, swelling, erythema, and influenza-like illnesses such as fatigue, myalgia, pyrexia, and chills.382,383 In particular, the antirabies mRNA vaccine CV7201 caused unexpected grade 2 Bell’s palsy in a healthy adult with intramuscular (i.m.) vaccination,330 and CV9130 caused urinary retention in three patients with prostate cancer, while urinary retention is also a common symptom in prostate cancer.384 The CV9201 vaccination also caused a grade 3 asthma attack in 1 patient, abnormal thyroid-stimulating hormone in nine patients, and increased antinuclear antibody in five patients.358 DC vaccines seldom caused grade 3 AEs.361 The severity of AEs relates to the administration route and dosage.352,385 Notably, it seems that i.d. vaccination has a higher AE frequency than i.m. administration: CV7201 caused 7 of 10 grade 3 AEs in the i.d. groups (64 participants), only 3 AEs in the i.m. group (37 participants).330 mRNA vaccines are a practical platform to improve the safety of vaccines by changing antigen sequences and modifying protein structures. Antibody-dependent enhancement (ADE) is a phenomenon in which preexisting antibodies promote viral infection of host cells and lead to increased virulence.386 mRNA encoding an E protein mutation without a conserved fusion-loop epitope was employed to enhance the safety of the anti-Zika mRNA vaccine and avoid potential ADE risk.387 Furthermore, mRNA encoding the RBD instead of its parental protein reduced the harmful immune response induced by vaccines.388
Adjuvants for mRNA vaccines
Adjuvants are essential for mRNA-based therapeutics, especially mRNA vaccines, which can amplify and direct immune responses and modulate the magnitude and type of certain subsets of T helper, IgG subclasses, or mucosal antibody responses. There are a few adjuvants approved by the FDA for use in humans, including aluminum salts, MF59, AS01, AS03, AS04, and CpG 10181.389 For mRNA vaccines, the sources of adjuvants mainly include the following five categories: (1) the self-adjuvant efficacy of IVT mRNA; (2) the immune-activating protein encoded by the mRNA (e.g., CD70, CD40 L and TriMix-DC); (3) direct-acting adjuvants: pathogen-associated molecular patterns and danger-associated molecular patterns (e.g., TLRs, helicases, NODs, and inflammasome agonists); (4) mRNAs complexed with specific reagents (protamine, lipid reagent); and (5) adjuvants that can promote DC recruitment, proliferation, and cross-presentation, such as GM-CSF and Fms-like tyrosine kinase 3 ligand (FLT3 L).390,391,392 Exogenous mRNAs have an inherent immunostimulatory effect due to their recognition by a variety of innate immune receptors, which allow them to stimulate the innate immune response in favor of vaccination, but they induce mRNA degradation and inhibit antigen expression, which are detrimental to maintaining the activity of mRNA therapeutics.78,393 Previous research has indicated that nucleoside modifications improved mRNA translation efficiency (Ψ, 5mC, Ψ/5mC or N1-methyl-pseudouridine/5-methylcytidine), and the pseudouridine/5-methylcytidine (Ψ/5mC)-modified mRNA partly suppressed the innate immune activation by mRNA vaccines and increased the encoding protein levels (firefly luciferase) up to 100-fold in vitro and 20-fold in the spleen of mice.394,395 Paradoxically, studies also showed that Ψ modification increased the immune stimulation function of mRNAs and failed to enhance mRNA translation efficiency.396,397 This opposite conclusion may be related to variations in RNA sequence optimization, stringency of removal of dsRNA contaminants by mRNA purification, and the level of innate immune sensing in targeted cell types.6 Another efficacious adjuvant strategy is to encode immunomodulatory proteins used as adjuvants with mRNAs, such as TriMix, which encodes a combination of three immune-activating proteins: CD70, CD40 ligand (CD40 L), and constitutively active TLR4 (caTLR4).398 Numerous cancer vaccine studies have shown that TriMix mRNA is associated with the stimulation of DC maturation and the generation of potent cytotoxic T lymphocyte (CTL) responses.399 DCs electroporated with mRNA encoding the costimulatory molecule 4-1BB ligand (4-1BBL) and CD40 L enhanced the proliferation and function of HIV-specific CD8+ T cells and increased the secretion of cytokines.400 Other costimulatory molecules, including CD83 and tumor necrosis factor receptor superfamily member 4 (TNFRSF4; also known as OX40), can also be encoded by mRNA and electroporated DCs, resulting in a significant increase in the immunostimulatory activity of DCs.401,402 Recently, a novel mRNA vaccine against SARS-CoV-2 also incorporated the costimulatory molecule CD40 L as an adjuvant to activate professional APC.403 Pattern recognition receptor ligands act as adjuvants to induce innate immunity and target APCs, thereby influencing the adaptive immune response. Pam3, a lipopeptide adjuvant recognized by TLR1 and TLR2, was incorporated into LNP, which enhanced mRNA-mediated cancer immunotherapy by stimulating different TLR subclasses.404 Double-stranded RNA (dsRNA) that is produced during the replication of viruses can powerfully induce natural immunity. Poly (I: C), a synthetic analog of dsRNA, is considered to be a TLR3 agonist that induces the production of IL-12 and type I IFN, promotes antigen cross-presentation to MHC class II molecules, and improves the generation of cytotoxic T cells.405 However, nucleic acid adjuvants have certain restrictions related to instability and easy degradation after drug administration, so delivery systems are generally considered to optimize them. Recently, an anionic poly I:C-derived double-stranded RNA adjuvant was complexed with chitosan to synthesize polyplexes to stimulate DC maturation, promote antigen presentation, and initiate cytotoxic T cells, which showed certain therapeutic efficacy in cancer treatment.406 Monophosphorylate lipid A activates the immune system via TLR4 without affecting mRNA translation.407 Synthetic CpG oligodeoxynucleotides (ODNs) are TLR-9 agonists that can induce the production of type I IFN and proinflammatory cytokines and generate Th1-type cellular and humoral immune responses.408,409 The hepatitis B vaccine HBsAg-1018 (HEPLISAV-B™) containing CpG-ODN as an adjuvant has been approved by the US Food and Drug Administration for use in adults.410 RNAdjuvant® (CureVac AG), an RNA-based TLR-7/8/RIG-I agonist consisting of a single-stranded, noncoding, cap-free RNA sequence containing multiple poly(U) repeat sequences, is a potent Th1-driven adjuvant that induces high levels of IFN-γ and has played a role in multiple tumor treatment studies.411,412
Other adjuvants that promote DC recruitment, proliferation, and cross-presentation, such as GM-CSF, were combined with naked mRNA to induce mainly a Th1 immune response, while naked mRNA alone induced a Th2 response.413 FLT3 L plays an important role in in situ vaccination, and the confounding protein FLT3 L also improves therapeutic immunity induced by naked mRNA.414,415 Overall, adjuvants reveal a critical role in mRNA-encoding antigens expression and initiating durable protective immunity, and have huge application prospects in mRNA-based therapeutics.
mRNA-based protein replacement therapies
Protein replacement treatment has an extensive application in replacing missing or defective proteins with favorable proteins.50 mRNA-based therapeutics have become a new pillar for protein replacement therapy, which has been extensively explored in various fields, including cardiac diseases,416 lung diseases,417 hematologic diseases,418 metabolic diseases,419 cancer,420 orthopedic diseases,421 neurogenic disorders,422,423 muscle atrophy, and so on.50,424 However, the majority of mRNA-based therapies for protein replacement are in the preclinical status, and only mRNA drugs encoding vascular endothelial growth factor (VEGF, NCT03370887) and CFTR (NCT03375047) have entered clinical development. To date, the most extensive efforts have been made in protein replacement therapeutics for cardiac diseases, focusing on heart failure and myocardial infarction.416 VEGFA mRNA treatment (AZD8601) protected mice from heart failure and significantly reduced apoptosis of myocardial cells with increased capillary density,425 and corresponding efficacy evaluation is ongoing in clinical trials (NCT03370887).426 However, testing an mRNA-based therapeutic also encouraged its application in protein replacement therapies for various lung diseases, especially genetic lung diseases.417
Cystic fibrosis
Cystic fibrosis is a life-limiting autosomal-recessive disease caused by mutations in the CFTR gene, while CFTR-mRNA transfection markedly restores impaired CFTR function in vitro.427 Nasally administered LNPs-CFTR mRNA was reported to result in recovery of up to 55% of the net chloride efflux characteristic in healthy mice.428 Furthermore, MRT5005, as an mRNA-based CFTR protein, has entered phase I/II clinical research.148
Hematologic diseases
Preclinical studies on mRNA-based protein replacement therapy have tested hematologic diseases.429 Hemophilia is a group of bleeding disorders for blood coagulation factor deficiency, including hemophilia A (factor VIII deficiency) and hemophilia B (factor IX deficiency).430 mRNA-based protein replacement can correct hematologic disorders by delivering corresponding factors in the template for mRNA. LNPs encapsulated mRNAs encoding different FVIII variants (F8 LNP) had rapid induction and durable FVIII expression in hemophilia A mice.431 FIX mRNA was delivered to FIX-knockout mice by using a series of lipidoids named TTs (corresponding lipid-like nanoparticles named TT-LLNs), which restored FIX function in FIX-knockout mice.432 Termed lipid-enabled LUNAR LNPs encapsulating hFIX mRNA were developed to treat hemophilia B mice, contributing to a rapid pulse of FIX within 4–6 h and a stable duration for up to 4–6 days.433
Metabolic diseases
The application of mRNAs also represents a promising solution for metabolic diseases that currently lack efficacious treatments, such as hepatorenal tyrosinemia, acute intermittent porphyria, Fabry disease, glycogen storage disease type 1 A, Crigler-Najjar syndrome type 1, and ornithine transcarboxylase deficiency.418,419 Hepatorenal tyrosinemia is a rare genetic metabolic disease caused by tyrosine degradation disorder due to a fumarylacetoacetate-hydrolase mutation, which can result in multiple organ damage.434 Cheng et al. designed and optimized 5A2-SC8 mRNA-loaded dendrimer LNPs to carry fumarylacetoacetate-hydrolase mRNA, which rendered FAH knockout mice statistically significant for liver function, similar to wild-type C57BL/6 mice.166 Acute intermittent porphyria is caused by the haploinsufficiency of porphobilinogen deaminase, which induces neurovisceral attacks associated with increased hepatic heme demand.435 LNP-encapsulated mRNA was used to induce dose-dependent expression of human porphobilinogen deaminase in mouse hepatocytes.435 This replacement therapy rapidly normalized urine porphyrin precursor excretion and counteracted porphyria attack in deficient mice, rabbits, and nonhuman primates. Methylmalonic acidemia, a genetic metabolic disease primarily caused by the loss of methylmalonyl-CoA mutase activity, results in approximately 20% mortality.436 LNP-encapsulated mRNA was delivered to systemically express functional mitochondrial methylmalonyl-CoA mutase in methylmalonic acidemia mice with a reduction of 75%-85% in plasma methylmalonic acid.437 A hybrid mRNA technology delivery system was exploited to load ornithine transcarboxylase mRNA, which restored the levels of plasma ammonia and urinary orotic acid and prolonged the survival of relatively deficient mice.438 Fabry disease is a lysosomal storage disorder caused by the deficiency of α-galactosidase A, resulting in cardiomyopathy and end-stage renal disease. Fabry disease can be improved by using nanoparticles sustainably to deliver α-galactosidase A mRNA into a mouse and nonhuman primate.439 Mutation of the SERPINA1 gene leads to alpha 1-antitrypsin (AAT) deficiency and damages the liver where the AAT protein is produced. Karadagi et al. identified mRNA encoding human AAT in primary human hepatocytes and developed it into LNP formulations. An in vivo study showed that secreted AAT protein increased from 1.14 to 3.43 µg/mL in media from primary human hepatocytes.440 mRNA-based protein replacement also provides an alternative to tumor treatment. Phosphatase and tensin homologue deleted on chromosome 10 (PTEN) is a potent tumor suppressor gene that is missing or mutated in many human cancers. PTEN inhibited the PI3K-AKT pathway and enhanced apoptosis of prostate cancer cells.441 Polymer–lipid hybrid nanoparticles were employed to systemically deliver PTEN mRNA and significantly inhibited the growth of disseminated metastatic and intratibial orthotopic prostate cancer in PTEN-null mice.442 Similarly, polymer–lipid hybrid nanoparticles were modified with the redox-responsive polymer PDSA and applied to transmit p53 mRNA (another gene encoding a tumor suppressor), and the results showed that the p53 mRNA NPs arrested the cell cycle and induced apoptosis, contributing to significant growth inhibition of p53-null HCCs and NSCLCs and improving the sensitivity of tumor cells to rapamycin inhibitors.167 In addition, mRNA encoding an anti-angiogenic protein, soluble fms-like tyrosine kinase 1,443 also efficiently inhibited pancreatic tumors; the liposome-protamine-IL-22BP mRNA complex strongly inhibited C26 tumor growth in both a peritoneal metastasis model and subcutaneous xenograft model.444
mRNA encoding a peptide/protein
The function of a peptide/protein encoded by mRNA is the key factor in the selection of therapeutics targeting cells, which directly influences mRNA therapeutic design.445 Precise delivery is required to target cells with appropriate protein convertase or endoprotease for the peptide that needs posttranslational modification to assemble them into functional types.446 Proteins need to be secreted outside of the cells to exert their function. Thus, mRNAs need to be conveyed to cells with natural secretion functions; otherwise, it is necessary to insert the mRNA sequence of the corresponding signal peptide near the ORF of the secretory protein.447 Encoded peptide/protein antigens can also give rise to a heterogeneous immune response even if they are involved in the same vaccine.448 A trivalent vaccine using three mRNAs was generated to encode different proteins, while these three antigens contributed to different IgG levels.94 Similarly, Sahin et al. designed neopeptide-encoded mRNAs, while the magnitude of the immune response varied from peptide to peptide, which indicates that the mRNA vaccine can be improved by selecting strongly responsive antigens; however, the underlying mechanism is far from fully clear, and it is difficult to ensure that encoded peptides/proteins all possess high immunogenicity.366 Moreover, the encoded peptide/protein greatly impacts its sustained expression. Holtkamp et al. observed that a fluorescent protein sustained high-level expression up to 120 h in mRNA format, while expression duration was dramatically reduced using immunodominant peptide from OVA in a similar mRNA format.46,379 Notably, the duration of protein expression plays a role in mRNA therapeutic efficacy. For example, migratory DCs in the skin need to spend 48 h trafficking to the T cell zoo and another couple of hours evoking a de novo CD8+ T cell response after delivery of mRNA into DCs.449,450,451 Therefore, mRNA encoding a peptide/protein theoretically needs to be present on the surface of migratory DCs for at least 48 h for mRNA vaccines with subcutaneous or i.d. administration. Note that in most cancer mRNA vaccines, there is a sharp drop in peptide/protein expression at approximately 24 h after DC transfection or vaccine immunization.46,452,453 However, it remains unclear whether a longer duration is needed, which requires further research on the relationship between the kinetics of peptide/protein expression and mRNA vaccine (or therapeutic) efficacy.454
mRNA-based gene editing therapeutics
Gene editing has a torn pace of application in various fields driven by the rapid development of programmable nucleases,423,424 especially for cancer, infectious diseases, primary defects of the immune system, muscular dystrophy, and hematological disease.455 mRNA is widely used to deliver programmable nucleases.456 The three most important programmable nucleases, zinc finger nucleases (ZFNs),457 transcription activator-like effector nucleases (TALENs),458,459 and the clustered regularly interspaced short palindromic repeat (CRISPR)-associated protein (CRISPR/Cas) nuclease system,460 have all achieved efficient transfection and manipulated insertions/deletions mutations in the form of mRNA. mRNA is an attractive approach in gene editing therapy due to its transient expression without mutant risk, and currently, several clinical trials based on mRNA genetic editing are in progress.461 Here, we discuss the application of mRNA-based gene editing, as well as its future prospects and challenges.
CRISPR/Cas nuclease system
The advance of artificial endonucleases renders high-speed development of mRNA-based gene editing. mRNA drugs modulate cellular genomic information by encoding artificial endonucleases, such as ZFNs, TALENs, and more recently CRISPR/Cas nuclease systems.462 Generally, the three mRNA-encoded endonucleases were designed to achieve insertions/deletions (indels) and mutations by introducing a targeting DNA double-stranded break, followed by DNA repair through nonhomologous end joining or homology-directed repair pathways.463 CRISPR/Cas9 systems are currently the most frequently used gene-editing technology because of their convenience for design and implementation among three gene-editing tools.
mRNA-based T lymphocyte therapeutics
T lymphocytes are an intriguing target for their tremendous potential against cancer and infectious diseases, and electroporation is the main way to transform endonuclease-encoding mRNA into T cells in vitro.464,465,466 The main consideration is about the efficiency, specificity, and safety of engineering T lymphocytes via mRNA transfection, chemically modified sgRNAs and Cas9 mRNAs increased genome editing efficiency via electroporation into human primary T cells in vitro.467 Moreover, the delivery of Cas9 mRNA improved genome editing and reduced toxicity compared with DNA-based editing.468 In addition, TALEN endonuclease achieved high specificity and efficient genome editing in primary T cells. TALEN mRNA was electroporated into primary T cells and contributed to more than 50% CCR5 (HIV coreceptor) knockout with low off-target activity.459 Furthermore, the TCR knockout rate reached up to 81% in primary T cells after electroporation with TALEN mRNA and five guide RNAs from the CRISPR/cas9 system.469
mRNA-based autologous T cell therapeutics
Engineering T lymphocytes by mRNA electroporation ex vivo provides an efficient platform for the treatment of both viral infections and cancers without safety concerns associated with viral carriers.470 Generally, T cells acquire the ability to recognize tumor antigens via transgenic expression of a CAR or a high-affinity T cell receptor and subsequently exert therapeutic efficacy post infusion.471 Adoptive transfer of autologous T cells is a promising cancer immunotherapy but requires a high quantity and quality of autologous T cells, such as CAR-T cells.472 Nevertheless, genetic modification is a powerful approach to address these issues. Third-party donor T cells were electroporated with TCRa constant (TRAC) TALEN mRNA to develop large-scale manufacturing of T cells. Moreover, researchers disrupted the TRAC gene to avoid graft-versus-host reactions.458 To further improve the efficacy of CAR T cells, alemtuzumab, a chemotherapeutic agent, was administered to downregulate CD52 genes and synergistically promote engraftment by mediating lymphodepletion and immunosuppression, and it endowed TCR/CD52-deficient CD19 CAR T cells (dKO-CART19) with potent antitumor activity in an orthotopic CD19+ lymphoma murine model.458 Recently, the CRISPR/Cas system has emerged as a potential genome engineering tool for CAR T cell therapy. CAR and CRISPR were delivered by using lentiviral-loaded and electroporated mRNA, respectively, to engineer CAR T cells with HLA class I molecule, PD1 and TCR deficiency, and the CRISPR/Cas9 mRNA-disrupted allogeneic CAR T cells showed both efficient antitumor activity in vitro and in vivo.473 A hybrid ΔU3-sgRNA was designed and incorporated into the ΔU3-3′-long terminal repeat of a self-inactivating lentiviral vector, resulting in targeted TRAC locus cleavage and enrichment of highly homogeneous (>96%) CAR+ (>99%) TCR- populations and potent antileukemic activity of TCR-depleted CAR19 T cells in a human: murine chimeric tumor model.474 Together, CRISPR/Cas9 systems overcome allo-recognition and provide an alternative strategy to autologous T cells. Successful genome engineering was achieved by electroporation of mRNA coding for a CD19-CAR, with 94% CAR expression in > 80% viable T cells.475,476 The CTLs electroporated with mRNA encoding a CAR against CD19 exhibited significant CD19-specific antitumor activity after tail vein injection.477 Multiple infusions of CD19-directed RNA CAR T cells resulted in improved survival and sustained antitumor responses in a robust leukemia xenograft model preceded by lymphodepleting chemotherapy.478 In contrast to gene editing, Zhao and colleagues electroporated autologous T cells with mRNA encoding a CAR against mesothelin overexpression in pancreatic cancer, ovarian cancer, and mesothelioma.
Robust antitumor efficacy was demonstrated in a human disseminated mesothelioma xenograft model with multiple injections.479 However, inefficient trafficking to tumors has hindered ex vivo mRNA-based T cell treatment in clinical trials.480 Research has shown that T cell migration is improved by transfecting tumor-infiltrating T cells with mRNA encoding the chemokine receptor CXCR2.481 Recently, further clinical application of mRNA electroporated CAR-T cells was promoted by establishing clinical-scale production, and the mRNA encoding chondroitin sulfate proteoglycan to treat melanoma patients is under full GMP compliance, suggesting a potential value of the further clinical application.482 Currently, several studies of mRNA-based engineered CAR T cells have entered clinical safety and efficacy evaluations (NCT01837602, NCT02624258 and NCT03060356).473 Nonviral vectors have recently been designed for ex vivo mRNA delivery to human T cells considering the electroporation cytotoxicity. Olden et al. explored a series of cationic pHEMA-g-pDMAEMA polymers to deliver mRNA to CD4+ and CD8+ primary human T cells in vitro, which resulted in 25% transfection efficiency with high cell viability.483 Library screening approaches have been utilized to develop lipid/polymer-based mRNA delivery systems and provide a quick and easy method to recognize potential mRNA delivery systems for both preclinical and clinical engineering T lymphocytes. Billingsley et al. synthesized a library of 24 ionizable lipids and formulated them into LNPs, whose top-performing LNP renders CAR mRNA expression comparable to electroporation.161 McKinlay et al. generated a library of oligonucleotide transporters containing various lipid domains, which facilitated efficient mRNA release using amphiphilic CARTs and achieved a ninefold mRNA translation enhancement (80%) in lymphocytes in vitro compared to Lipofectamine 2000.484
mRNA-based CD4+ T cell therapeutics
To date, there is only one completed phase I study of CD4+ T cells modified at the CCR5 gene by ZFN mRNA in HIV-infected patients (NCT02388594).485 Challenges remain in cytotoxic gene delivery of the viral or electroporation methods, complex and expensive manipulations, and off-target efficacy of the gene-editing system. Encouragingly, very strong efforts have been made to explore nonviral and in vivo mRNA delivery for efficient and safe gene editing, which is worth looking forwards to in the future.485
mRNA-based stem cell therapeutics
mRNA-based genome editing has also been successfully applied to stem cells for many disease treatments.486 Previously, ZFN protein, mRNA, and DNA were delivered to a human cell line and mouse embryonic stem cells via a retrovirus vector and disrupted the targeted gene at frequencies of 15%, 15%, and >50%, respectively, indicating the universality of retroviral vectors.487 Kohn et al. further examined the efficiency, specificity, and mutational signatures of ZFN mRNA, TALEN mRNA, and CRISPR/Cas9 mRNA, which were electroporated into primary human hematopoietic stem and progenitor cells, and analyses revealed that ZFN mRNA has higher specificity than the other two endonucleases mRNA.488 ZFN mRNA enabled CD34+ to engraft in NOD-PrkdcSCID-IL2Rγ null mice with reserved multilineage potential compared with TALEN mRNA editing.488 For plasmid gRNA and Cas9 mRNA, their codelivery showed similar acute cytotoxicity with separate plasmid delivery, highlighting the need for further optimization of CRISPR/Cas9 delivery in primary human hematopoietic stem cells.489 Genome-editing approaches that innovatively transfect hematopoietic stem and progenitor cells with macaque-specific CCR5 ZFN mRNA ex vivo first modified multilineage and long-term repopulating cells in a large animal model and resulted in persistent in vivo tracking of genome-edited hematopoietic stem cells in a mutation-specific manner.490 Strategies for the transfection of stem cells are worth investigating for the ex vivo and in vivo delivery of endonuclease mRNA to facilitate clinical applications.
Ex vivo delivery of mRNA to stem cells has been explored for various purposes. Electroporation was used to transfer mRNA encoding EGFP into mesenchymal stem cells and H9 human embryonic stem (H9 hES) cells, both of which achieved 90% transgene efficiency.491,492 To provide a great alternative to pDNA, cationic carriers were explored to deliver mRNA encoding CXCR4 into mesenchymal stem cells and resulted in 80% positive expression rates of the target protein.493 In addition, numerous researchers have focused on improving the efficiency of mRNA transfection of stem cells. In vitro mRNA transcription was performed to characterize histone variant distribution in human embryonic stem cells.494 Researchers have successfully transdifferentiated insulin-producing cells to treat diabetes by using in vitro duodenal transcription factor 1 mRNA to transform the mouse pancreas into mesenchymal stem cells.432 Recently, HIV-1 Tat mRNA was delivered into bone marrow mesenchymal stem cells (BMSCs), confirming the inhibitory effect of HIV-1 Tat protein on the hematopoietic support function of hBMSCs.495
mRNA-based pluripotent stem (iPS) therapeutics
Genome editing of induced pluripotent stem (iPS) cells holds great promise in cell therapy and disease modeling.496,497 Many efforts have been made for genome editing of iPSCs using the CRISPR/Cas9 system.498,499,500 Transient delivery of Cas9 mRNA or protein is preferable for iPS clinical applications without mutation risk. Delivery of Cas9 in the form of mRNA has several advantages over direct protein delivery, including considerable protein molecule production from a single mRNA molecule and versatile mRNA engineering. A workflow capitalizing on the transient delivery of CRISPR/Cas9 mRNAs was presented to support the high-throughput development of gene-edited iPSCs. Subsequently, iPSCs can be differentiated into representative specific cell types of embryonic lineages for further research or potential clinical application. In addition, it was also applied to other gene-editing tools, such as ZFN mRNA and TALEN mRNA.501 However, RNA instability and off-target efficacy are challenging for clinical application.502 Hence, future efforts will pay attention to safe and efficacious delivery strategies of mRNA for further therapeutic purposes.
Combination therapeutics based on mRNA drugs
Recently, combined therapeutics have emerged as a powerful modality to treat malignancy, contributing to synergetic efficacy.503 ICB,504 CAR T cells 265 and cancer vaccines are three important immunotherapies for cancer treatment. ICBs can release the brake of T cell activation and function,504 but durable clinical benefit is only achieved in a minority of patients.505 The combination of ICBs and cancer vaccines has attracted considerable attention.506 The cancer vaccine can expand ICB efficacy by evoking a tumor-specific CD8+ T cell response to treat patients who lack preexisting CTLs and respond to ICBs507,508 and improve mRNA cancer vaccine efficacy.366,509,510 Recently, mRNA vaccines were amplified by CAR-T cells over 2 orders of magnitude by mimicking the dynamics of the secondary response following the initial reaction of T cells, which significantly increased median survival and contributed to the complete rejection of solid tumors in 6 of 10 mice compared to a single administration of CAR T cells.511 Apparently, mRNA-based therapeutics mainly focused on tumor immunotherapy and infectious disease, exploration of its potential and mechanism in other diseases is the next priority. Undoubtedly, mRNA-based therapeutics have become powerful and versatile tools to combat diseases.
Conclusion and prospects
mRNA-based therapeutics have made great strides, achieving remarkable improvement in mRNA stability, function, and production during the past 30 years.2 mRNA drugs exploit cells as factories for antigen or functional protein production with promising efficacy and sufficient safety.512 Currently, a great deal of research focuses on varied applications of mRNA therapeutics, and a series of clinical trials are ongoing. mRNA vaccines have drawn considerable attention due to the important role of mRNA vaccines in controlling the SARS-CoV-2 pandemic.513 For vaccines against infection, the humoral immune response plays an important role in mRNA vaccine efficacy, especially IgG magnitude.514 The mRNA vaccine completely protected mice from influenza virus challenges with undetectable hemagglutination inhibition titers.94 Notably, mucosal immunity has also contributed significantly to defending against infectious diseases because many infections start from mucous membranes.513,515,516 Patel et al. systematically reviewed clinical trials of rotavirus vaccines following PRISMA guidelines, which displayed a consistent relationship between serum IgA and vaccine protection.517 Meanwhile, mucosal immunity may provide a wider protection than humoral immunity. The influenza virus vaccine with a higher nasal IgA level provided stronger protection than a lower IgA response, although the two vaccines had a similar serum IgG magnitude,518 and Tamura et al. also observed superior cross-reactivity of nasal IgA against heterologous influenza viruses compared to IgG.519 Moreover, mucosa immunity may play an important role in preventing the transmission of infection, and serum IgG possibly tends to prevent severe infectious diseases but no disease transmission.515,520,521 It is vital for vaccines to prevent COVID-19 transmission caused by asymptomatic carriers to counteract the current pandemic, which has demonstrated huge success. Currently, an intranasal vaccine was developed by regulating mucosal immunity against SARS-CoV-2, while the role of mucosal immunity is unclear in the prevention of SARS-CoV-2 transmission, which warrants further research to reveal the relationship between mucosal immunity and mRNA vaccines.276 Intriguingly, many mRNA vaccines tend to induce a Th-1-biased immune response through interferon signals, which may be related to mRNA delivery into the cytoplasm and translate antigen proteins that are largely processed on MHC I molecules and specifically activate the CD8+ T cell response.522 Together, mRNA vaccines have shown potent efficacy in defending against infectious diseases by humoral immune mucosal immunity, but cellular immunity needs to be assessed in detail in the future.
In recent decades, especially the last few years, we have witnessed great scientific advances in mRNA-based therapeutics. Current clinical efforts encompassing mRNA-based drugs are directed toward infectious disease vaccines, cancer immunotherapies, therapeutic protein replacement therapies, and genetic disease treatment. Opportunities and challenges in mRNA-based therapeutics coexist, and there are a large number of questions requiring clarification. (1) How can mRNA macromolecules be better delivered? (2) How can its inherent instability and degradation be improved by structure-based antigen design and delivery system-optimization? (3) How can its activation of the immune system be regulated? In essence, the clinical translation of mRNA-based therapeutics requires delivery technologies that can ensure stabilization of mRNA under physiological conditions. Improving the optimization technology of mRNA structure and engineering precision nanoparticles for mRNA-based therapeutics are also crucial points for the development of mRNA drugs as powerful and versatile tools to combat diseases. Built on the highly fueled interest and potential, we have full confidence to predict an accelerated pace in mRNA therapy studies and development in the next decade, possibly providing many solutions for the prevention and treatment of currently incurable diseases.
References
Mascola, J. R. & Fauci, A. S. Novel vaccine technologies for the 21st century. Nat. Rev. Immunol. 20, 87–88 (2020).
Sahin, U., Kariko, K. & Tureci, O. mRNA-based therapeutics–developing a new class of drugs. Nat. Rev. Drug Discov. 13, 759–780 (2014).
Boczkowski, D., Nair, S. K., Snyder, D. & Gilboa, E. Dendritic cells pulsed with RNA are potent antigen-presenting cells in vitro and in vivo. J. Exp. Med. 184, 465–472 (1996).
Kallen, K. J. & Thess, A. A development that may evolve into a revolution in medicine: mRNA as the basis for novel, nucleotide-based vaccines and drugs. Ther. Adv. Vaccines. 2, 10–31 (2014).
Conry, R. M. et al. Characterization of a messenger RNA polynucleotide vaccine vector. Cancer Res. 55, 1397–1400 (1995).
Pardi, N., Hogan, M. J., Porter, F. W. & Weissman, D. mRNA vaccines – a new era in vaccinology. Nat. Rev. Drug Discov. 17, 261–279 (2018).
Deal, C. E., Carfi, A. & Plante, O. J. Advancements in mRNA encoded antibodies for passive immunotherapy. Vaccines (Basel) 9, 108 (2021).
Weng, Y. et al. The challenge and prospect of mRNA therapeutics landscape. Biotechnol. Adv. 40, 107534 (2020).
Dowdy, S. F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 35, 222–229 (2017).
Sergeeva, O. V., Koteliansky, V. E. & Zatsepin, T. S. mRNA-based therapeutics – advances and perspectives. Biochemistry (Mosc.). 81, 709–722 (2016).
Lin, Y. X. et al. RNA nanotechnology-mediated cancer immunotherapy. Theranostics 10, 281–299 (2020).
Chung, J. Y., Thone, M. N. & Kwon, Y. J. COVID-19 vaccines: the status and perspectives in delivery points of view. Adv. Drug Deliv. Rev. 170, 1–25 (2021).
Reichmuth, A. M. et al. mRNA vaccine delivery using lipid nanoparticles. Ther. Deliv. 7, 319–334 (2016).
Song, X. et al. Delivery of CRISPR/Cas systems for cancer gene therapy and immunotherapy. Adv. Drug Deliv. Rev. 168, 158–180 (2021).
Zheng, Q. et al. mRNA-loaded lipid-like nanoparticles for liver base editing via the optimization of central composite design. Adv. Funct. Mater. 31, 2011068 (2021).
Huang, H. et al. The investigation of mRNA vaccines formulated in liposomes administrated in multiple routes against SARS-CoV-2. J. Control Release 335, 449–456 (2021).
Guevara, M. L., Persano, F. & Persano, S. Advances in lipid nanoparticles for mRNA-based cancer immunotherapy. Front Chem. 8, 589959 (2020).
Desterro, J., Bak-Gordon, P. & Carmo-Fonseca, M. Targeting mRNA processing as an anticancer strategy. Nat. Rev. Drug Discov. 19, 112–129 (2020).
Fabbri, L., Chakraborty, A., Robert, C. & Vagner, S. The plasticity of mRNA translation during cancer progression and therapy resistance. Nat. Rev. Cancer 21, 558–577 (2021).
Jackson, R. J., Hellen, C. U. & Pestova, T. V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 11, 113–127 (2010).
Bhat, M. et al. Targeting the translation machinery in cancer. Nat. Rev. Drug Discov. 14, 261–278 (2015).
Hinnebusch, A. G. Structural insights into the mechanism of scanning and start codon recognition in eukaryotic translation initiation. Trends Biochem Sci. 42, 589–611 (2017).
Yourik, P. et al. Yeast eIF4A enhances recruitment of mRNAs regardless of their structural complexity. Elife 6, e31476 (2017).
Pelechano, V., Wei, W. & Steinmetz, L. M. Widespread co-translational RNA decay reveals ribosome dynamics. Cell 161, 1400–1412 (2015).
Jacobson, A. & Peltz, S. W. Interrelationships of the pathways of mRNA decay and translation in eukaryotic cells. Annu Rev. Biochem. 65, 693–739 (1996).
Gallie, D. R. The role of the poly(A) binding protein in the assembly of the Cap-binding complex during translation initiation in plants. Translation (Austin). 2, e959378 (2014).
Coller, J. & Parker, R. Eukaryotic mRNA decapping. Annu Rev. Biochem. 73, 861–890 (2004).
Wilusz, C. J., Wormington, M. & Peltz, S. W. The cap-to-tail guide to mRNA turnover. Nat. Rev. Mol. Cell Biol. 2, 237–246 (2001).
Leppek, K. et al. Combinatorial optimization of mRNA structure, stability, and translation for RNA-based therapeutics. Preprint at bioRxiv (2021).
Furuichi, Y. Discovery of m(7)G-cap in eukaryotic mRNAs. Proc. Jpn Acad. Ser. B Phys. Biol. Sci. 91, 394–409 (2015).
Warminski, M., Sikorski, P. J., Kowalska, J. & Jemielity, J. Applications of phosphate modification and labeling to study (m)RNA caps. Top. Curr. Chem. (Cham). 375, 16 (2017).
Ramanathan, A., Robb, G. B. & Chan, S. H. mRNA capping: biological functions and applications. Nucleic Acids Res. 44, 7511–7526 (2016).
Niedzwiecka, A. et al. Biophysical studies of eIF4E cap-binding protein: recognition of mRNA 5’ cap structure and synthetic fragments of eIF4G and 4E-BP1 proteins. J. Mol. Biol. 319, 615–635 (2002).
Charenton, C. & Graille, M. mRNA decapping: finding the right structures. Philos. Trans. R. Soc. Lond. B Biol. Sci. 373, 20180164 (2018).
Taverniti, V. & Seraphin, B. Elimination of cap structures generated by mRNA decay involves the new scavenger mRNA decapping enzyme Aph1/FHIT together with DcpS. Nucleic Acids Res. 43, 482–492 (2015).
Strenkowska, M. et al. Cap analogs modified with 1,2-dithiodiphosphate moiety protect mRNA from decapping and enhance its translational potential. Nucleic Acids Res. 44, 9578–9590 (2016).
Rydzik, A. M. et al. mRNA cap analogues substituted in the tetraphosphate chain with CX2: identification of O-to-CCl2 as the first bridging modification that confers resistance to decapping without impairing translation. Nucleic Acids Res. 45, 8661–8675 (2017).
Kasprzyk, R. & Jemielity, J. Enzymatic assays to explore viral mRNA capping machinery. Chembiochem 22, 3236–3253 (2021).
Grudzien, E. et al. Novel cap analogs for in vitro synthesis of mRNAs with high translational efficiency. RNA 10, 1479–1487 (2004).
Kalek, M. et al. Synthesis and biochemical properties of novel mRNA 5’ cap analogs resistant to enzymatic hydrolysis. Nucleosides Nucleotides Nucleic Acids 24, 615–621 (2005).
Kuhn, A. N. et al. Phosphorothioate cap analogs increase stability and translational efficiency of RNA vaccines in immature dendritic cells and induce superior immune responses in vivo. Gene Ther. 17, 961–971 (2010).
Nicholson, A. L. & Pasquinelli, A. E. Tales of detailed poly(A) tails. Trends Cell Biol. 29, 191–200 (2019).
Jalkanen, A. L., Coleman, S. J. & Wilusz, J. Determinants and implications of mRNA poly(A) tail size–does this protein make my tail look big? Semin Cell Dev. Biol. 34, 24–32 (2014).
Pelletier, J. & Sonenberg, N. The organizing principles of eukaryotic ribosome recruitment. Annu Rev. Biochem. 88, 307–335 (2019).
Mockey, M. et al. mRNA transfection of dendritic cells: synergistic effect of ARCA mRNA capping with Poly(A) chains in cis and in trans for a high protein expression level. Biochem. Biophys. Res. Commun. 340, 1062–1068 (2006).
Holtkamp, S. et al. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood 108, 4009–4017 (2006).
Grier, A. E. et al. pEVL: a linear plasmid for generating mRNA IVT templates with extended encoded poly(A) sequences. Mol. Ther. Nucleic Acids 5, e306 (2016).
Meijer, H. A. et al. A novel method for poly(A) fractionation reveals a large population of mRNAs with a short poly(A) tail in mammalian cells. Nucleic Acids Res. 35, e132 (2007).
Choi, Y. H. & Hagedorn, C. H. Purifying mRNAs with a high-affinity eIF4E mutant identifies the short 3’ poly(A) end phenotype. Proc. Natl Acad. Sci. USA. 100, 7033–7038 (2003).
Kwon, H. et al. Emergence of synthetic mRNA: in vitro synthesis of mRNA and its applications in regenerative medicine. Biomaterials 156, 172–193 (2018).
Wadhwa, A. et al. Opportunities and challenges in the delivery of mRNA-based vaccines. Pharmaceutics 12, 102 (2020).
Hinnebusch, A. G., Ivanov, I. P. & Sonenberg, N. Translational control by 5’-untranslated regions of eukaryotic mRNAs. Science 352, 1413–1416 (2016).
Matoulkova, E., Michalova, E., Vojtesek, B. & Hrstka, R. The role of the 3’ untranslated region in post-transcriptional regulation of protein expression in mammalian cells. RNA Biol. 9, 563–576 (2012).
Gomez-Aguado, I. et al. Nanomedicines to deliver mRNA: state of the art and future perspectives. Nanomaterials (Basel) 10, 364 (2020).
Zarghampoor, F. et al. Improved translation efficiency of therapeutic mRNA. Gene 707, 231–238 (2019).
Asrani, K. H. et al. Optimization of mRNA untranslated regions for improved expression of therapeutic mRNA. RNA Biol. 15, 756–762 (2018).
Jiang, Y., Xu, X. S. & Russell, J. E. A nucleolin-binding 3’ untranslated region element stabilizes beta-globin mRNA in vivo. Mol. Cell Biol. 26, 2419–2429 (2006).
Emery, I. et al. Characterization of T-cell immune responses of Echinococcus multilocularis-infected C57BL/6J mice. Parasite Immunol. 18, 463–472 (1996).
Sultana, N. et al. Optimization of 5’ untranslated region of modified mRNA for use in cardiac or hepatic ischemic injury. Mol. Ther. Methods Clin. Dev. 17, 622–633 (2020).
Ferizi, M. et al. Human cellular CYBA UTR sequences increase mRNA translation without affecting the half-life of recombinant RNA transcripts. Sci. Rep. 6, 39149 (2016).
Trepotec, Z. et al. Maximizing the translational yield of mRNA therapeutics by minimizing 5’-UTRs. Tissue Eng. Part A. 25, 69–79 (2019).
Schrom, E. et al. Translation of angiotensin-converting enzyme 2 upon liver- and lung-targeted delivery of optimized chemically modified mRNA. Mol. Ther. Nucleic Acids 7, 350–365 (2017).
Quintana-Bustamante, O. et al. Gene editing of PKLR gene in human hematopoietic progenitors through 5’ and 3’ UTR modified TALEN mRNA. PLoS One 14, e0223775 (2019).
Sample, P. J. et al. Human 5’ UTR design and variant effect prediction from a massively parallel translation assay. Nat. Biotechnol. 37, 803–809 (2019).
Zeng, C. et al. Leveraging mRNA sequences and nanoparticles to deliver SARS-CoV-2 antigens in vivo. Adv. Mater. 32, e2004452 (2020).
Alexaki, A. et al. Effects of codon optimization on coagulation factor IX translation and structure: Implications for protein and gene therapies. Sci. Rep. 9, 15449 (2019).
Kowalski, P. S., Rudra, A., Miao, L. & Anderson, D. G. Delivering the messenger: advances in technologies for therapeutic mRNA delivery. Mol. Ther. 27, 710–728 (2019).
Mauro, V. P. & Chappell, S. A. A critical analysis of codon optimization in human therapeutics. Trends Mol. Med. 20, 604–613 (2014).
Presnyak, V. et al. Codon optimality is a major determinant of mRNA stability. Cell 160, 1111–1124 (2015).
Weissman, D. mRNA transcript therapy. Expert Rev. Vaccines. 14, 265–281 (2015).
Mauger, D. M. et al. mRNA structure regulates protein expression through changes in functional half-life. Proc. Natl Acad. Sci. USA. 116, 24075–24083 (2019).
Kudla, G. et al. High guanine and cytosine content increases mRNA levels in mammalian cells. PLoS Biol. 4, e180 (2006).
Schlake, T. et al. mRNA: a novel avenue to antibody therapy? Mol. Ther. 27, 773–784 (2019).
Khoury, H. J. et al. Immune responses and long-term disease recurrence status after telomerase-based dendritic cell immunotherapy in patients with acute myeloid leukemia. Cancer 123, 3061–3072 (2017).
Kreiter, S. et al. Increased antigen presentation efficiency by coupling antigens to MHC class I trafficking signals. J. Immunol. 180, 309–318 (2008).
Anguille, S. et al. Dendritic cell vaccination as postremission treatment to prevent or delay relapse in acute myeloid leukemia. Blood 130, 1713–1721 (2017).
Bloom, K., van den Berg, F. & Arbuthnot, P. Self-amplifying RNA vaccines for infectious diseases. Gene Ther. 28, 117–129 (2021).
Minnaert, A. K. et al. Strategies for controlling the innate immune activity of conventional and self-amplifying mRNA therapeutics: getting the message across. Adv. Drug Deliv. Rev. 176, 113900 (2021).
Lundstrom, K. Self-amplifying RNA viruses as RNA vaccines. Int. J. Mol. Sci. 21, 5130 (2020).
Ballesteros-Briones, M. C. et al. A new generation of vaccines based on alphavirus self-amplifying RNA. Curr. Opin. Virol. 44, 145–153 (2020).
Demoulins, T. et al. Self-replicating RNA vaccine delivery to dendritic cells. Methods Mol. Biol. 1499, 37–75 (2017).
McCullough, K. C. et al. Self-amplifying replicon RNA vaccine delivery to dendritic cells by synthetic nanoparticles. Vaccines (Basel). 2, 735–754 (2014).
Schuchman, R. et al. Comparative characterization of the Sindbis virus proteome from mammalian and invertebrate hosts identifies nsP2 as a component of the virion and sorting Nexin 5 as a significant host factor for alphavirus replication. J. Virol. 92, e00694–18 (2018).
Sawicki, D. L. & Sawicki, S. G. Alphavirus positive and negative strand RNA synthesis and the role of polyproteins in formation of viral replication complexes. Arch. Virol. Suppl. 9, 393–405 (1994).
Pietila, M. K., Hellstrom, K. & Ahola, T. Alphavirus polymerase and RNA replication. Virus Res. 234, 44–57 (2017).
Jones, R., Bragagnolo, G., Arranz, R. & Reguera, J. Capping pores of alphavirus nsP1 gate membranous viral replication factories. Nature 589, 615–619 (2021).
Gotte, B., Liu, L. & McInerney, G. M. The enigmatic alphavirus non-structural protein 3 (nsP3) revealing its secrets at last. Viruses. 10, 105 (2018).
Lello, L. S. et al. nsP4 is a major determinant of alphavirus replicase activity and template selectivity. J. Virol. 95, e0035521 (2021).
Rupp, J. C., Sokoloski, K. J., Gebhart, N. N. & Hardy, R. W. Alphavirus RNA synthesis and non-structural protein functions. J. Gen. Virol. 96, 2483–2500 (2015).
Hellstrom, K. et al. Partially uncleaved alphavirus replicase forms spherule structures in the presence and absence of RNA template. J. Virol. 91, e00787–17 (2017).
Jose, J., Snyder, J. E. & Kuhn, R. J. A structural and functional perspective of alphavirus replication and assembly. Future Microbiol. 4, 837–856 (2009).
McKay, P. F. et al. Self-amplifying RNA SARS-CoV-2 lipid nanoparticle vaccine candidate induces high neutralizing antibody titers in mice. Nat. Commun. 11, 3523 (2020).
Luisi, K. et al. Development of a potent Zika virus vaccine using self-amplifying messenger RNA. Sci. Adv. 6, eaba5068 (2020).
Vogel, A. B. et al. Self-amplifying RNA vaccines give equivalent protection against influenza to mRNA vaccines but at much lower doses. Mol. Ther. 26, 446–455 (2018).
Beissert, T. et al. A trans-amplifying RNA vaccine strategy for induction of potent protective immunity. Mol. Ther. 28, 119–128 (2020).
Beissert, T. et al. Improvement of in vivo expression of genes delivered by self-amplifying RNA using vaccinia virus immune evasion proteins. Hum. Gene Ther. 28, 1138–1146 (2017).
Li, Y. et al. In vitro evolution of enhanced RNA replicons for immunotherapy. Sci. Rep. 9, 6932 (2019).
Geall, A. J. et al. Nonviral delivery of self-amplifying RNA vaccines. Proc. Natl Acad. Sci. USA. 109, 14604–14609 (2012).
Li, J. Q. et al. Intranasal delivery of replicating mRNA encoding neutralizing antibody against SARS-CoV-2 infection in mice. Signal Transduct. Target Ther. 6, 369 (2021).
Chen, L. L. & Yang, L. Regulation of circRNA biogenesis. RNA Biol. 12, 381–388 (2015).
Zhou, W. Y. et al. Circular RNA: metabolism, functions and interactions with proteins. Mol. Cancer 19, 172 (2020).
Matsui, M. & Corey, D. R. Non-coding RNAs as drug targets. Nat. Rev. Drug Discov. 16, 167–179 (2017).
Wang, L. et al. Long noncoding RNA (lncRNA)-mediated competing endogenous RNA networks provide novel potential biomarkers and therapeutic targets for colorectal cancer. Int. J. Mol. Sci. 20, 5758 (2019).
Su, Y. et al. CircRNA Cdr1as functions as a competitive endogenous RNA to promote hepatocellular carcinoma progression. Aging (Albany NY). 11, 8183–8203 (2019).
Lu, S. et al. RNA-Seq revealed a circular RNA-microRNA-mRNA regulatory network in hantaan virus infection. Front Cell Infect. Microbiol. 10, 97 (2020).
Cheng, Y. et al. Identification of circRNA-lncRNA-miRNA-mRNA competitive endogenous RNA network as novel prognostic markers for acute myeloid leukemia. Genes (Basel). 11, 868 (2020).
Panni, S., Lovering, R. C., Porras, P. & Orchard, S. Non-coding RNA regulatory networks. Biochim. Biophys. Acta Gene Regul. Mech. 1863, 194417 (2020).
Baiersdorfer, M. et al. A facile method for the removal of dsRNA contaminant from in vitro-transcribed mRNA. Mol. Ther. Nucleic Acids 15, 26–35 (2019).
Pichlmair, A. & Reis e Sousa, C. Innate recognition of viruses. Immunity 27, 370–383 (2007).
Hornung, V. et al. 5’-Triphosphate RNA is the ligand for RIG-I. Science 314, 994–997 (2006).
Muttach, F., Muthmann, N. & Rentmeister, A. Synthetic mRNA capping. Beilstein J. Org. Chem. 13, 2819–2832 (2017).
Ouranidis, A. et al. mRNA therapeutic modalities design, formulation and manufacturing under pharma 4.0 principles. Biomedicines 10, 50 (2021).
Senthilvelan, A. et al. Trinucleotide cap analogue bearing a locked nucleic acid moiety: synthesis, mRNA modification, and translation for therapeutic applications. Org. Lett. 23, 4133–4136 (2021).
Beverly, M., Dell, A., Parmar, P. & Houghton, L. Label-free analysis of mRNA capping efficiency using RNase H probes and LC-MS. Anal. Bioanal. Chem. 408, 5021–5030 (2016).
Bentley, D. L. Coupling mRNA processing with transcription in time and space. Nat. Rev. Genet. 15, 163–175 (2014).
Decroly, E., Ferron, F., Lescar, J. & Canard, B. Conventional and unconventional mechanisms for capping viral mRNA. Nat. Rev. Microbiol. 10, 51–65 (2011).
Jackson, N. A. C. et al. The promise of mRNA vaccines: a biotech and industrial perspective. NPJ Vaccines. 5, 11 (2020).
Galloway, A. & Cowling, V. H. mRNA cap regulation in mammalian cell function and fate. Biochim. Biophys. Acta Gene Regul. Mech. 1862, 270–279 (2019).
Pasquinelli, A. E., Dahlberg, J. E. & Lund, E. Reverse 5’ caps in RNAs made in vitro by phage RNA polymerases. RNA 1, 957–967 (1995).
Stepinski, J. et al. Synthesis and properties of mRNAs containing the novel “anti-reverse” cap analogs 7-methyl(3’-O-methyl)GpppG and 7-methyl (3’-deoxy)GpppG. RNA 7, 1486–1495 (2001).
Peng, Z. H., Sharma, V., Singleton, S. F. & Gershon, P. D. Synthesis and application of a chain-terminating dinucleotide mRNA cap analog. Org. Lett. 4, 161–164 (2002).
Kore, A. R., Shanmugasundaram, M. & Vlassov, A. V. Synthesis and application of a new 2’,3’-isopropylidene guanosine substituted cap analog. Bioorg. Med Chem. Lett. 18, 4828–4832 (2008).
Kore, A. R. et al. Synthesis and application of 2’-fluoro-substituted cap analogs. Bioorg. Med. Chem. Lett. 17, 5295–5299 (2007).
Jemielity, J. et al. Novel “anti-reverse” cap analogs with superior translational properties. RNA 9, 1108–1122 (2003).
Kocmik, I. et al. Modified ARCA analogs providing enhanced translational properties of capped mRNAs. Cell Cycle 17, 1624–1636 (2018).
Kisiala, M. et al. Restriction endonucleases that cleave RNA/DNA heteroduplexes bind dsDNA in A-like conformation. Nucleic Acids Res. 48, 6954–6969 (2020).
Granados-Riveron, J. T. & Aquino-Jarquin, G. Engineering of the current nucleoside-modified mRNA-LNP vaccines against SARS-CoV-2. Biomed. Pharmacother. 142, 111953 (2021).
Martins, R., Queiroz, J. A. & Sousa, F. Ribonucleic acid purification. J. Chromatogr. A. 1355, 1–14 (2014).
Kariko, K., Muramatsu, H., Ludwig, J. & Weissman, D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 39, e142 (2011).
Foster, J. B. et al. Purification of mRNA encoding chimeric antigen receptor is critical for generation of a robust T-cell response. Hum. Gene Ther. 30, 168–178 (2019).
Aviv, H. & Leder, P. Purification of biologically active globin messenger RNA by chromatography on oligothymidylic acid-cellulose. Proc. Natl Acad. Sci. USA. 69, 1408–1412 (1972).
Zhang, C., Maruggi, G., Shan, H. & Li, J. Advances in mRNA vaccines for infectious diseases. Front Immunol. 10, 594 (2019).
Schlake, T., Thess, A., Fotin-Mleczek, M. & Kallen, K. J. Developing mRNA-vaccine technologies. RNA Biol. 9, 1319–1330 (2012).
Muntjes, K., Devan, S. K., Reichert, A. S. & Feldbrugge, M. Linking transport and translation of mRNAs with endosomes and mitochondria. EMBO Rep. 22, e52445 (2021).
Revia, R. A., Stephen, Z. R. & Zhang, M. Theranostic nanoparticles for RNA-based cancer treatment. Acc. Chem. Res. 52, 1496–1506 (2019).
Ho, W. et al. Next-generation vaccines: nanoparticle-mediated DNA and mRNA delivery. Adv. Health. Mater. 10, e2001812 (2021).
Zhang, R. et al. DP7-C-modified liposomes enhance immune responses and the antitumor effect of a neoantigen-based mRNA vaccine. J. Control Release 328, 210–221 (2020).
Yin, Y. et al. In situ transforming RNA nanovaccines from polyethylenimine functionalized graphene oxide hydrogel for durable cancer immunotherapy. Nano Lett. 21, 2224–2231 (2021).
Zhang, W., Liu, Y., Min Chin, J. & Phua, K. K. L. Sustained release of PKR inhibitor C16 from mesoporous silica nanoparticles significantly enhances mRNA translation and anti-tumor vaccination. Eur. J. Pharm. Biopharm. 163, 179–187 (2021).
Tse, S. W. et al. mRNA-encoded, constitutively active STING(V155M) is a potent genetic adjuvant of antigen-specific CD8(+) T cell response. Mol. Ther. 29, 2227–2238 (2021).
Schoenmaker, L. et al. mRNA-lipid nanoparticle COVID-19 vaccines: structure and stability. Int J. Pharm. 601, 120586 (2021).
Yang, J. et al. Hybrid nanovaccine for the co-delivery of the mRNA antigen and adjuvant. Nanoscale 11, 21782–21789 (2019).
Lou, G. et al. Delivery of self-amplifying mRNA vaccines by cationic lipid nanoparticles: the impact of cationic lipid selection. J. Control Release 325, 370–379 (2020).
Mai, Y. et al. Intranasal delivery of cationic liposome-protamine complex mRNA vaccine elicits effective anti-tumor immunity. Cell Immunol. 354, 104143 (2020).
Zhuang, X. et al. mRNA vaccines encoding the HA protein of influenza A H1N1 virus delivered by cationic lipid nanoparticles induce protective immune responses in mice. Vaccines (Basel). 8, 123 (2020).
Tang, Z. et al. Insights from nanotechnology in COVID-19 treatment. Nano Today 36, 101019 (2021).
Veiga, N. et al. Cell specific delivery of modified mRNA expressing therapeutic proteins to leukocytes. Nat. Commun. 9, 4493 (2018).
Robinson, E. et al. Lipid nanoparticle-delivered chemically modified mRNA restores chloride secretion in cystic fibrosis. Mol. Ther. 26, 2034–2046 (2018).
Sayers, E. J. et al. Endocytic profiling of cancer cell models reveals critical factors influencing LNP-mediated mRNA delivery and protein expression. Mol. Ther. 27, 1950–1962 (2019).
Li, Q. et al. Engineering caveolae-targeted lipid nanoparticles to deliver mRNA to the lungs. ACS Chem. Biol. 15, 830–836 (2020).
Sabnis, S. et al. A novel amino lipid series for mRNA delivery: improved endosomal escape and sustained pharmacology and safety in non-human primates. Mol. Ther. 26, 1509–1519 (2018).
Hassett, K. J. et al. Optimization of lipid nanoparticles for intramuscular administration of mRNA vaccines. Mol. Ther. Nucleic Acids 15, 1–11 (2019).
Hajj, K. A. et al. Branched-tail lipid nanoparticles potently deliver mRNA in vivo due to enhanced ionization at endosomal pH. Small 15, e1805097 (2019).
Ball, R. L. et al. Lipid nanoparticle formulations for enhanced co-delivery of siRNA and mRNA. Nano Lett. 18, 3814–3822 (2018).
Paunovska, K. et al. Nanoparticles containing oxidized cholesterol deliver mRNA to the liver microenvironment at clinically relevant doses. Adv. Mater. 31, e1807748 (2019).
Rybakova, Y. et al. mRNA delivery for therapeutic anti-HER2 antibody expression in vivo. Mol. Ther. 27, 1415–1423 (2019).
Fenton, O. S. et al. Bioinspired alkenyl amino alcohol ionizable lipid materials for highly potent in vivo mRNA delivery. Adv. Mater. 28, 2939–2943 (2016).
Fenton, O. S. et al. Synthesis and biological evaluation of ionizable lipid materials for the in vivo delivery of messenger RNA to B lymphocytes. Adv. Mater. 29 (2017).
Fenton, O. S. et al. Customizable lipid nanoparticle materials for the delivery of siRNAs and mRNAs. Angew. Chem. 130, 13770–13774 (2018).
Zhao, X. et al. Imidazole-based synthetic lipidoids for in vivo mRNA delivery into primary T lymphocytes. Angew. Chem. Int. Ed. Engl. 59, 20083–20089 (2020).
Billingsley, M. M. et al. Ionizable lipid nanoparticle-mediated mRNA delivery for human CAR T cell engineering. Nano Lett. 20, 1578–1589 (2020).
Li, Y. et al. Protein and mRNA delivery enabled by cholesteryl-based biodegradable lipidoid nanoparticles. Angew. Chem. Int. Ed. Engl. 59, 14957–14964 (2020).
Miao, L. et al. Synergistic lipid compositions for albumin receptor mediated delivery of mRNA to the liver. Nat. Commun. 11, 2424 (2020).
Yang, T. et al. Efficient hepatic delivery and protein expression enabled by optimized mRNA and ionizable lipid nanoparticle. Bioact. Mater. 5, 1053–1061 (2020).
Miao, L. et al. Delivery of mRNA vaccines with heterocyclic lipids increases anti-tumor efficacy by STING-mediated immune cell activation. Nat. Biotechnol. 37, 1174–1185 (2019).
Cheng, Q. et al. Dendrimer-based lipid nanoparticles deliver therapeutic FAH mRNA to normalize liver function and extend survival in a mouse model of hepatorenal tyrosinemia type I. Adv. Mater. 30, e1805308 (2018).
Kong, N. et al. Synthetic mRNA nanoparticle-mediated restoration of p53 tumor suppressor sensitizes p53-deficient cancers to mTOR inhibition. Sci. Transl. Med. 11, eaaw1565 (2019).
Tanaka, H. et al. In vivo introduction of mRNA encapsulated in lipid nanoparticles to brain neuronal cells and astrocytes via intracerebroventricular administration. Mol. Pharm. 15, 2060–2067 (2018).
Luo, X. et al. Dual-functional lipid-like nanoparticles for delivery of mRNA and MRI contrast agent. Nanoscale 9, 1575–1579 (2017).
Xiong, H. et al. Theranostic dendrimer-based lipid nanoparticles containing PEGylated BODIPY dyes for tumor imaging and systemic mRNA delivery in vivo. J. Control Release 325, 198–205 (2020).
Mitchell, M. J. et al. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 20, 101–124 (2021).
Ke, X. et al. Surface-functionalized PEGylated nanoparticles deliver messenger RNA to pulmonary immune cells. ACS Appl Mater. Interfaces 12, 35835–35844 (2020).
Dunn, A. W., Kalinichenko, V. V. & Shi, D. Highly efficient in vivo targeting of the pulmonary endothelium using novel modifications of polyethylenimine: an importance of charge. Adv. Health. Mater. 7, e1800876 (2018).
Kaczmarek, J. C. et al. Optimization of a degradable polymer-lipid nanoparticle for potent systemic delivery of mRNA to the lung endothelium and immune cells. Nano Lett. 18, 6449–6454 (2018).
Fornaguera, C. et al. mRNA delivery system for targeting antigen-presenting cells in vivo. Adv. Health. Mater. 7, e1800335 (2018).
Patel, A. K. et al. Inhaled nanoformulated mRNA polyplexes for protein production in lung epithelium. Adv. Mater. 31, e1805116 (2019).
Kowalski, P. S. et al. Ionizable amino-polyesters synthesized via ring opening polymerization of tertiary amino-alcohols for tissue selective mRNA delivery. Adv. Mater e1801151 (2018).
McKinlay, C. J. et al. Charge-altering releasable transporters (CARTs) for the delivery and release of mRNA in living animals. Proc. Natl Acad. Sci. USA. 114, E448–E456 (2017).
Haabeth, O. A. W. et al. mRNA vaccination with charge-altering releasable transporters elicits human T cell responses and cures established tumors in mice. Proc. Natl Acad. Sci. USA. 115, E9153–E9161 (2018).
Haabeth, O. A. W. et al. Local delivery of Ox40l, Cd80, and Cd86 mRNA kindles global anticancer immunity. Cancer Res. 79, 1624–1634 (2019).
Schumann, C. et al. Increasing lean muscle mass in mice via nanoparticle mediated hepatic delivery of follistatin mRNA. Theranostics 8, 5276–5288 (2018).
Kim, H. J. et al. Fine-tuning of hydrophobicity in amphiphilic polyaspartamide derivatives for rapid and transient expression of messenger RNA directed toward genome engineering in brain. ACS Cent. Sci. 5, 1866–1875 (2019).
Crowley, S. T. et al. Enhancement of motor function recovery after spinal cord injury in mice by delivery of brain-derived neurotrophic factor mRNA. Mol. Ther. Nucleic Acids 17, 465–476 (2019).
Qiu, Y. et al. Effective mRNA pulmonary delivery by dry powder formulation of PEGylated synthetic KL4 peptide. J. Control Release 314, 102–115 (2019).
Udhayakumar, V. K. et al. Arginine-rich peptide-based mRNA nanocomplexes efficiently instigate cytotoxic T cell immunity dependent on the amphipathic organization of the peptide. Adv. Healthc. Mater. 6 (2017).
Van der Jeught, K. et al. Dendritic cell targeting mRNA lipopolyplexes combine strong antitumor T-cell immunity with improved inflammatory safety. ACS Nano. 12, 9815–9829 (2018).
Teixeira, H. et al. Submicron cationic emulsions as a new delivery system for oligonucleotides. Pharm. Res. 16, 30–36 (1999).
Teixeira, H. F. et al. Cationic nanoemulsions as nucleic acids delivery systems. Int J. Pharm. 534, 356–367 (2017).
Brito, L. A. et al. A cationic nanoemulsion for the delivery of next-generation RNA vaccines. Mol. Ther. 22, 2118–2129 (2014).
O’Brien, K. et al. RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat. Rev. Mol. Cell Biol. 21, 585–606 (2020).
Wang, Y. et al. mRNA vaccine with antigen-specific checkpoint blockade induces an enhanced immune response against established melanoma. Mol. Ther. 26, 420–434 (2018).
Wang, Y. et al. Functional nanoparticles with a reducible tetrasulfide motif upregulate mRNA translation and enhance transfection in hard to-transfect cells. Chem. Int. Ed. Engl. 59, 2695–2699 (2020).
Liu, L. et al. Combination immunotherapy of MUC1 mRNA nano-vaccine and CTLA-4 blockade effectively inhibits growth of triple negative breast cancer. Mol. Ther. 26, 45–55 (2018).
Kurimoto, S. et al. PEG-OligoRNA hybridization of mRNA for developing sterically stable lipid nanoparticles toward in vivo administration. Molecules 24, 1303 (2019).
Yoshinaga, N. et al. Induced packaging of mRNA into polyplex micelles by regulated hybridization with a small number of cholesteryl RNA oligonucleotides directed enhanced in vivo transfection. Biomaterials 197, 255–267 (2019).
Yoshinaga, N. et al. Bundling mRNA strands to prepare nano-assemblies with enhanced stability towards RNase for in vivo delivery. Angew. Chem. Int Ed. Engl. 58, 11360–11363 (2019).
Poddar, A. et al. Encapsulation, visualization and expression of genes with biomimetically mineralized zeolitic imidazolate framework-8 (ZIF-8). Small 15, e1902268 (2019).
Oshima, G. et al. In vivo delivery and therapeutic effects of a microRNA on colorectal liver metastases. Mol. Ther. 25, 1588–1595 (2017).
He, C. et al. Nanoscale coordination polymers codeliver chemotherapeutics and siRNAs to eradicate tumors of cisplatin-resistant ovarian cancer. J. Am. Chem. Soc. 138, 6010–6019 (2016).
Cullis, P. R. & Hope, M. J. Lipid nanoparticle systems for enabling gene therapies. Mol. Ther. 25, 1467–1475 (2017).
Midoux, P. & Pichon, C. Lipid-based mRNA vaccine delivery systems. Expert Rev. Vaccines. 14, 221–234 (2015).
Hillaireau, H. & Couvreur, P. Nanocarriers’ entry into the cell: relevance to drug delivery. Cell Mol. Life Sci. 66, 2873–2896 (2009).
Stewart, M. P., Langer, R. & Jensen, K. F. Intracellular delivery by membrane disruption: mechanisms, strategies, and concepts. Chem. Rev. 118, 7409–7531 (2018).
Lorenz, C. et al. Protein expression from exogenous mRNA: uptake by receptor-mediated endocytosis and trafficking via the lysosomal pathway. RNA Biol. 8, 627–636 (2011).
Fujiwara, Y., Wada, K. & Kabuta, T. Lysosomal degradation of intracellular nucleic acids-multiple autophagic pathways. J. Biochem. 161, 145–154 (2017).
Cools, N., Van Camp, K., Van Tendeloo, V. & Berneman, Z. mRNA electroporation as a tool for immunomonitoring. Methods Mol. Biol. 969, 293–303 (2013).
Wang, F. et al. Optimization of the linker length of mannose-cholesterol conjugates for enhanced mRNA delivery to dendritic cells by liposomes. Front Pharmacol. 9, 980 (2018).
Yanez Arteta, M. et al. Successful reprogramming of cellular protein production through mRNA delivered by functionalized lipid nanoparticles. Proc. Natl Acad. Sci. USA. 115, E3351–E3360 (2018).
Zhdanov, V. P. Intracellular RNA delivery by lipid nanoparticles: diffusion, degradation, and release. Biosystems 185, 104032 (2019).
Guevara, M. L., Persano, S. & Persano, F. Lipid-based vectors for therapeutic mRNA-based anti-cancer vaccines. Curr. Pharm. Des. 25, 1443–1454 (2019).
Coolen, A. L. et al. Poly(lactic acid) nanoparticles and cell-penetrating peptide potentiate mRNA-based vaccine expression in dendritic cells triggering their activation. Biomaterials 195, 23–37 (2019).
Zeng, C., Zhang, C., Walker, P. G. & Dong, Y. Formulation and delivery technologies for mRNA vaccines. Curr. Top. Microbiol. Immunol. 10, 1–40 (2020).
Liu, T., Liang, Y. & Huang, L. Development and delivery systems of mRNA vaccines. Front Bioeng. Biotechnol. 9, 718753 (2021).
Bholakant, R. et al. Recent advances of polycationic siRNA vectors for cancer therapy. Biomacromolecules 21, 2966–2982 (2020).
Martini, P. G. V. & Guey, L. T. A new era for rare genetic diseases: messenger RNA therapy. Hum. Gene Ther. 30, 1180–1189 (2019).
Crooke, S. T. et al. Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol. 35, 230–237 (2017).
Gu, Y. Z., Zhao, X. & Song, X. R. Ex vivo pulsed dendritic cell vaccination against cancer. Acta Pharm. Sin. 41, 959–969 (2020).
Benteyn, D. et al. mRNA-based dendritic cell vaccines. Expert Rev. Vaccines. 14, 161–176 (2015).
Copur, M. Messenger RNA vaccines: beckoning of a new era in cancer immunotherapy. Oncology (Williston Park). 35, 190–198 (2021).
Van Nuffel, A. M. et al. Dendritic cells loaded with mRNA encoding full-length tumor antigens prime CD4+ and CD8+ T cells in melanoma patients. Mol. Ther. 20, 1063–1074 (2012).
Aldosari, B. N., Alfagih, I. M. & Almurshedi, A. S. Lipid nanoparticles as delivery systems for RNA-based vaccines. Pharmaceutics 13, 206 (2021).
Bouakaz, A., Zeghimi, A. & Doinikov, A. A. Sonoporation: concept and mechanisms. Adv. Exp. Med Biol. 880, 175–189 (2016).
De Haes, W. et al. Lipoplexes carrying mRNA encoding Gag protein modulate dendritic cells to stimulate HIV-specific immune responses. Nanomedicine (Lond.). 8, 77–87 (2013).
Markov, O. O. et al. Novel cationic liposomes provide highly efficient delivery of DNA and RNA into dendritic cell progenitors and their immature offsets. J. Control Release 160, 200–210 (2012).
Tateshita, N. et al. Development of a lipoplex-type mRNA carrier composed of an ionizable lipid with a vitamin E scaffold and the KALA peptide for use as an ex vivo dendritic cell-based cancer vaccine. J. Control Release 310, 36–46 (2019).
Borch, T. H. et al. mRNA-transfected dendritic cell vaccine in combination with metronomic cyclophosphamide as treatment for patients with advanced malignant melanoma. Oncoimmunology 5, e1207842 (2016).
Chung, D. J. et al. Langerhans-type dendritic cells electroporated with TRP-2 mRNA stimulate cellular immunity against melanoma: results of a phase I vaccine trial. Oncoimmunology 7, e1372081 (2017).
Derdelinckx, J. et al. Clinical and immunological control of experimental autoimmune encephalomyelitis by tolerogenic dendritic cells loaded with MOG-encoding mRNA. J. Neuroinflammation. 16, 167 (2019).
Gandhi, R. T. et al. Immunization of HIV-1-infected persons with autologous dendritic cells transfected with mRNA encoding HIV-1 Gag and Nef: results of a randomized, placebo-controlled clinical trial. J. Acquir Immune Defic. Syndr. 71, 246–253 (2016).
Schaft, N. et al. CD8(+) T-cell priming and boosting: more antigen-presenting DC, or more antigen per DC? Cancer Immunol. Immunother. 62, 1769–1780 (2013).
Tarach, P. & Janaszewska, A. Recent advances in preclinical research using PAMAM dendrimers for cancer gene therapy. Int. J. Mol. Sci. 22, 2912 (2021).
Shobaki, N., Sato, Y. & Harashima, H. Mixing lipids to manipulate the ionization status of lipid nanoparticles for specific tissue targeting. Int J. Nanomed. 13, 8395–8410 (2018).
Cheng, Q. et al. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-Cas gene editing. Nat. Nanotechnol. 15, 313–320 (2020).
Christensen, D. et al. Cationic liposomes as vaccine adjuvants. Expert Rev. Vaccines. 6, 785–796 (2007).
Edwards, D. K. et al. Adjuvant effects of a sequence-engineered mRNA vaccine: translational profiling demonstrates similar human and murine innate response. J. Transl. Med. 15, 1 (2017).
Kallen, K. J. et al. A novel, disruptive vaccination technology: self-adjuvanted RNActive((R)) vaccines. Hum. Vaccin Immunother. 9, 2263–2276 (2013).
Rauch, S. et al. RNActive(R) technology: generation and testing of stable and immunogenic mRNA vaccines. Methods Mol. Biol. 1499, 89–107 (2017).
Fotin-Mleczek, M. et al. Messenger RNA-based vaccines with dual activity induce balanced TLR-7 dependent adaptive immune responses and provide antitumor activity. J. Immunother. 34, 1–15 (2011).
Kowalczyk, A. et al. Self-adjuvanted mRNA vaccines induce local innate immune responses that lead to a potent and boostable adaptive immunity. Vaccine 34, 3882–3893 (2016).
Petsch, B. et al. Protective efficacy of in vitro synthesized, specific mRNA vaccines against influenza A virus infection. Nat. Biotechnol. 30, 1210–1216 (2012).
Schnee, M. et al. An mRNA vaccine encoding rabies virus glycoprotein induces protection against lethal infection in mice and correlates of protection in adult and newborn pigs. PLoS Negl. Trop. Dis. 10, e0004746 (2016).
Crouse, J., Kalinke, U. & Oxenius, A. Regulation of antiviral T cell responses by type I interferons. Nat. Rev. Immunol. 15, 231–242 (2015).
De Beuckelaer, A., Grooten, J. & De Koker, S. Type I interferons modulate CD8(+) T cell immunity to mRNA vaccines. Trends Mol. Med. 23, 216–226 (2017).
Hajj, K. A. et al. A potent branched-tail lipid nanoparticle enables multiplexed mRNA delivery and gene editing in vivo. Nano Lett. 08, 7 (2020).
Heine, A., Juranek, S. & Brossart, P. Clinical and immunological effects of mRNA vaccines in malignant diseases. Mol. Cancer 20, 52 (2021).
Schlake, T., Thess, A., Thran, M. & Jordan, I. mRNA as novel technology for passive immunotherapy. Cell Mol. Life Sci. 76, 301–328 (2019).
Datu, A. K. & Bag, J. Enhanced translation of mRNAs encoding proteins involved in mRNA translation during recovery from heat shock. PLoS One 8, e64171 (2013).
Sontag, E. M., Vonk, W. I. M. & Frydman, J. Sorting out the trash: the spatial nature of eukaryotic protein quality control. Curr. Opin. Cell Biol. 26, 139–146 (2014).
Lee, S. et al. Effect of recombinant alpha1-antitrypsin Fc-fused (AAT-Fc)protein on the inhibition of inflammatory cytokine production and streptozotocin-induced diabetes. Mol. Med. 19, 65–71 (2013).
Shepard, H. M., Phillips, G. L., D Thanos, C. & Feldmann, M. Developments in therapy with monoclonal antibodies and related proteins. Clin. Med. (Lond.). 17, 220–232 (2017).
Castelli, M. S., McGonigle, P. & Hornby, P. J. The pharmacology and therapeutic applications of monoclonal antibodies. Pharm. Res Perspect. 7, e00535 (2019).
Patel, A., Bah, M. A. & Weiner, D. B. In vivo delivery of nucleic acid-encoded monoclonal antibodies. BioDrugs 34, 273–293 (2020).
Pardi, N. et al. Administration of nucleoside-modified mRNA encoding broadly neutralizing antibody protects humanized mice from HIV-1 challenge. Nat. Commun. 8, 14630 (2017).
Tiwari, P. M. et al. Engineered mRNA-expressed antibodies prevent respiratory syncytial virus infection. Nat. Commun. 9, 3999 (2018).
Kose, N. et al. A lipid-encapsulated mRNA encoding a potently neutralizing human monoclonal antibody protects against chikungunya infection. Sci. Immunol. 4, eaaw6647 (2019).
Erasmus, J. H. et al. Intramuscular delivery of replicon RNA encoding ZIKV-117 human monoclonal antibody protects against zika virus infection. Mol. Ther. Methods Clin. Dev. 18, 402–414 (2020).
Thran, M. et al. mRNA mediates passive vaccination against infectious agents, toxins, and tumors. EMBO Mol. Med. 9, 1434–1447 (2017).
Zhou, X. et al. Rapid delivery of nanobodies/VHHs into living cells via expressing in vitro-transcribed mRNA. Mol. Ther. Methods Clin. Dev. 17, 401–408 (2020).
Stadler, C. R. et al. Elimination of large tumors in mice by mRNA-encoded bispecific antibodies. Nat. Med. 23, 815–817 (2017).
Rosenblum, H. G. et al. Use of COVID-19 vaccines after reports of adverse events among adult recipients of Janssen (Johnson & Johnson) and mRNA COVID-19 vaccines (Pfizer-BioNTech and Moderna): update from the Advisory Committee on Immunization Practices - United States, July 2021. MMWR Morb. Mortal. Wkly Rep. 70, 1094–1099 (2021).
Greinacher, A. et al. Thrombotic thrombocytopenia after ChAdOx1 nCov-19 vaccination. N. Engl. J. Med. 384, 2092–2101 (2021).
Chaudhary, N., Weissman, D. & Whitehead, K. A. mRNA vaccines for infectious diseases: principles, delivery and clinical translation. Nat. Rev. Drug Discov. 20, 817–838 (2021).
Ribas, A. & Wolchok, J. D. Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355 (2018).
Richardson, N., Ng, S. T. H. & Wraith, D. C. Antigen-specific immunotherapy for treatment of autoimmune liver diseases. Front Immunol. 11, 1586 (2020).
Schuster, S. J. et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N. Engl. J. Med. 377, 2545–2554 (2017).
Stower, H. Immunotherapy for heart injury. Nat. Med. 25, 1799 (2019).
Nabel, G. J. Designing tomorrow’s vaccines. N. Engl. J. Med. 368, 551–560 (2013).
Wu, F. et al. A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269 (2020).
Scudellari, M. How the pandemic might play out in 2021 and beyond. Nature 584, 22–25 (2020).
Guijarro, C. et al. SARS-CoV-2 new infections among health-care workers after the first dose of the BNT162b2 mRNA COVID-19 vaccine. A hospital-wide cohort study. Clin. Microbiol. Infect. 27, 1699.e1–1699.e4 (2021).
Polack, F. P. et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N. Engl. J. Med. 383, 2603–2615 (2020).
Thompson, M. G. et al. Interim estimates of vaccine effectiveness of BNT162b2 and mRNA-1273 COVID-19 vaccines in preventing SARS-CoV-2 infection among health care personnel, first responders, and other essential and frontline workers - eight U.S. locations, December 2020-March 2021. MMWR Morb. Mortal. Wkly Rep. 70, 495–500 (2021).
Zacay, G. et al. BNT162b2 vaccine effectiveness in preventing asymptomatic infection with SARS-CoV-2 virus: a nationwide historical cohort study. Open Forum Infect. Dis. 8, ofab262 (2021).
Garcia-Beltran, W. F. et al. mRNA-based COVID-19 vaccine boosters induce neutralizing immunity against SARS-CoV-2 Omicron variant. Cell 185, 457–466.e4 (2022).
V’Kovski, P. et al. Coronavirus biology and replication: implications for SARS-CoV-2. Nat. Rev. Microbiol. 19, 155–170 (2021).
Laczko, D. et al. A single immunization with nucleoside-modified mRNA vaccines elicits strong cellular and humoral immune responses against SARS-CoV-2 in mice. Immunity 53, 724–732.e727 (2020).
Dagotto, G., Yu, J. & Barouch, D. H. Approaches and challenges in SARS-CoV-2 vaccine development. Cell Host Microbe 28, 364–370 (2020).
Jeyanathan, M. et al. Immunological considerations for COVID-19 vaccine strategies. Nat. Rev. Immunol. 20, 615–632 (2020).
Corbett, K. S. et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature 586, 567–571 (2020).
Tai, W. et al. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell Mol. Immunol. 17, 613–620 (2020).
Routhu, N. K. et al. SARS-CoV-2 RBD trimer protein adjuvanted with Alum-3M-052 protects from SARS-CoV-2 infection and immune pathology in the lung. Nat. Commun. 12, 3587 (2021).
Walls, A. C. et al. Elicitation of potent neutralizing antibody responses by designed protein nanoparticle vaccines for SARS-CoV-2. Cell 183, 1367–1382.e1317 (2020).
Kato, Y. et al. Multifaceted effects of antigen valency on B cell response composition and differentiation in vivo. Immunity 53, 548–563.e548 (2020).
Liu, Z. et al. RBD-Fc-based COVID-19 vaccine candidate induces highly potent SARS-CoV-2 neutralizing antibody response. Signal Transduct. Target Ther. 5, 282 (2020).
Yang, L. et al. A recombinant receptor-binding domain in trimeric form generates protective immunity against SARS-CoV-2 infection in nonhuman primates. Innov. (N. Y). 2, 100140 (2021).
Sun, W. et al. The self-assembled nanoparticle-based trimeric RBD mRNA vaccine elicits robust and durable protective immunity against SARS-CoV-2 in mice. Signal Transduct. Target Ther. 6, 340 (2021).
Elia, U. et al. Design of SARS-CoV-2 hFc-conjugated receptor-binding domain mRNA vaccine delivered via lipid nanoparticles. ACS Nano. 15, 9627–9637 (2021).
Li, M. et al. Secreted expression of mRNA-encoded truncated ACE2 variants for SARS-CoV-2 via lipid-like nanoassemblies. Adv. Mater. 33, e2101707 (2021).
Gupta, R. K. Will SARS-CoV-2 variants of concern affect the promise of vaccines? Nat. Rev. Immunol. 21, 340–341 (2021).
Tan, C. W. et al. Pan-sarbecovirus neutralizing antibodies in BNT162b2-immunized SARS-CoV-1 survivors. N. Engl. J. Med. 385, 1401–1406 (2021).
Skowronski, D. M. et al. Single-dose mRNA vaccine effectiveness against SARS-CoV-2, including Alpha and Gamma variants: a test-negative design in adults 70 years and older in British Columbia, Canada. Clin. Infect. Dis. 74, 1158–1165 (2021).
Mulligan, M. J. et al. Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults. Nature 586, 589–593 (2020).
de Alwis, R. et al. A single dose of self-transcribing and replicating RNA-based SARS-CoV-2 vaccine produces protective adaptive immunity in mice. Mol. Ther. 29, 1970–1983 (2021).
McDade, T. W. et al. Durability of antibody response to vaccination and surrogate neutralization of emerging variants based on SARS-CoV-2 exposure history. Sci. Rep. 11, 17325 (2021).
Favresse, J. et al. Antibody titres decline 3-month post-vaccination with BNT162b2. Emerg. Microbes Infect. 10, 1495–1498 (2021).
Self, W. H. et al. Comparative effectiveness of Moderna, Pfizer-BioNTech, and Janssen (Johnson & Johnson) vaccines in preventing COVID-19 hospitalizations among adults without immunocompromising conditions - United States, March-August 2021. MMWR Morb. Mortal. Wkly Rep. 70, 1337–1343 (2021).
Ferre, V. M. et al. Decreasing humoral response among healthcare workers up to 4 months after two doses of BNT162b2 vaccine. J. Infect. 84, 248–288 (2021).
Padoan, A. et al. Antibody response to first and second dose of BNT162b2 in a cohort of characterized healthcare workers. Clin. Chim. Acta 519, 60–63 (2021).
Bertrand, K., Honerkamp-Smith, G. & Chambers, C. D. Maternal and child outcomes reported by breastfeeding women following messenger RNA COVID-19 vaccination. Breastfeed. Med. 16, 697–701 (2021).
Dagan, N. et al. Effectiveness of the BNT162b2 mRNA COVID-19 vaccine in pregnancy. Nat. Med. 27, 1693–1695 (2021).
Goldshtein, I. et al. Association between BNT162b2 vaccination and incidence of SARS-CoV-2 infection in pregnant women. JAMA 326, 728–735 (2021).
Theiler, R. N. et al. Pregnancy and birth outcomes after SARS-CoV-2 vaccination in pregnancy. Am. J. Obstet. Gynecol. Mfm. 3, 100467 (2021).
Beharier, O. et al. Efficient maternal to neonatal transfer of antibodies against SARS-CoV-2 and BNT162b2 mRNA COVID-19 vaccine. J. Clin. Invest. 131, e150319 (2021).
Zitt, E. et al. The safety and immunogenicity of the mRNA-BNT162b2 SARS-CoV-2 vaccine in hemodialysis patients. Front Immunol. 12, 704773 (2021).
Benda, M. et al. Serological SARS-CoV-2 antibody response, potential predictive markers and safety of BNT162b2 mRNA COVID-19 vaccine in haematological and oncological patients. Br. J. Haematol. 195, 523–531 (2021).
Pimpinelli, F. et al. Lower response to BNT162b2 vaccine in patients with myelofibrosis compared to polycythemia vera and essential thrombocythemia. J. Hematol. Oncol. 14, 119 (2021).
Addeo, A. et al. Immunogenicity of SARS-CoV-2 messenger RNA vaccines in patients with cancer. Cancer Cell. 39, 1091–1098.e1092 (2021).
Harrington, P. et al. Single dose of BNT162b2 mRNA vaccine against SARS-CoV-2 induces high frequency of neutralising antibody and polyfunctional T-cell responses in patients with myeloproliferative neoplasms. Leukemia 35, 3573–3577 (2021).
Deepak, P. et al. Effect of immunosuppression on the immunogenicity of mRNA vaccines to SARS-CoV-2: a prospective cohort study. Ann. Intern. Med. 174, 1572–1585 (2021).
Schmidt, K. G. et al. Characterization of serum and mucosal SARS-CoV-2-antibodies in HIV-1-infected subjects after BNT162b2 mRNA vaccination or SARS-CoV-2 infection. Viruses 14, 651 (2022).
Achiron, A. et al. Humoral immune response to COVID-19 mRNA vaccine in patients with multiple sclerosis treated with high-efficacy disease-modifying therapies. Ther. Adv. Neurol. Disord. 14, 17562864211012835 (2021).
Furer, V. et al. Immunogenicity and safety of the BNT162b2 mRNA COVID-19 vaccine in adult patients with autoimmune inflammatory rheumatic diseases and in the general population: a multicentre study. Ann. Rheum. Dis. 80, 1330–1338 (2021).
Picchianti-Diamanti, A. et al. Immunosuppressive therapies differently modulate humoral- and T-cell-specific responses to COVID-19 mRNA vaccine in rheumatoid arthritis patients. Front Immunol. 12, 740249 (2021).
Ali, A. et al. Characterization of humoral response to COVID mRNA vaccines in multiple sclerosis patients on disease modifying therapies. Vaccine 39, 6111–6116 (2021).
Drulovic, J. et al. Humoral response to SARS-CoV-2 COVID-19 vaccines in patients with multiple sclerosis treated with immune reconstitution therapies. Mult. Scler. Relat. Disord. 54, 103150 (2021).
Gallo, A. et al. Preliminary evidence of blunted humoral response to SARS-CoV-2 mRNA vaccine in multiple sclerosis patients treated with ocrelizumab. Neurol. Sci. 42, 3523–3526 (2021).
Plymate, L. C., Pepper, G., Krist, M. P. & Koelle, D. M. Immunogenicity of repeat COVID-19 mRNA vaccinations in a patient with myasthenia gravis receiving mycophenolate, prednisone, and eculizumab. J. Transl. Autoimmun. 4, 100114 (2021).
Connolly, C. M. et al. Disease flare and reactogenicity in patients with rheumatic and musculoskeletal diseases following two-dose SARS-CoV-2 messenger RNA vaccination. Arthritis Rheumatol. 74, 28–32 (2021).
Boyarsky, B. J. et al. Immunogenicity of a single dose of SARS-CoV-2 messenger RNA vaccine in solid organ transplant recipients. JAMA 325, 1784–1786 (2021).
Chenxi Song, C. et al. Early experience with SARs-CoV-2 mRNA vaccine breakthrough among kidney transplant recipients. Transpl. Infect. Dis. 23, e13654 (2021).
Havlin, J. et al. Immunogenicity of BNT162b2 mRNA COVID-19 vaccine and SARS-CoV-2 infection in lung transplant recipients. J. Heart Lung Transplant. 40, 754–758 (2021).
Holden, I. K. et al. Immunogenicity of SARS-CoV-2 mRNA vaccine in solid organ transplant recipients. J. Intern. Med. 290, 1264–1267 (2021).
Stumpf, J. et al. Humoral and cellular immunity to SARS-CoV-2 vaccination in renal transplant versus dialysis patients: a prospective, multicenter observational study using mRNA-1273 or BNT162b2 mRNA vaccine. Lancet Reg. Health Eur. 9, 100178 (2021).
McArthur, D. B. Emerging infectious diseases. Nurs. Clin. North Am. 54, 297–311 (2019).
Fineberg, H. V. Pandemic preparedness and response–lessons from the H1N1 influenza of 2009. N. Engl. J. Med. 370, 1335–1342 (2014).
Kazmi, S. S., Ali, W., Bibi, N. & Nouroz, F. A review on Zika virus outbreak, epidemiology, transmission and infection dynamics. J. Biol. Res (Thessalon.). 27, 5 (2020).
Wollina, U. Challenges of COVID-19 pandemic for dermatology. Dermatol Ther. 33, e13430 (2020).
Peck, M. et al. Global routine vaccination coverage, 2018. MMWR Morb. Mortal. Wkly Rep. 68, 937–942 (2019).
Disease, G. B. D., Injury, I. & Prevalence, C. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 392, 1789–1858 (2018).
Alberer, M. et al. Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: an open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. Lancet 390, 1511–1520 (2017).
Van Craenenbroeck, A. H. et al. Induction of cytomegalovirus-specific T cell responses in healthy volunteers and allogeneic stem cell recipients using vaccination with messenger RNA-transfected dendritic cells. Transplantation 99, 120–127 (2015).
Hekele, A. et al. Rapidly produced SAM((R)) vaccine against H7N9 influenza is immunogenic in mice. Emerg. Microbes Infect. 2, e52 (2013).
Corbett, K. S. et al. Evaluation of the mRNA-1273 Vaccine against SARS-CoV-2 in Nonhuman Primates. N. Engl. J. Med. 383, 1544–1555 (2020).
Armbruster, N., Jasny, E. & Petsch, B. Advances in RNA vaccines for preventive indications: a case study of a vaccine against rabies. Vaccines (Basel). 7, 132 (2019).
Leal, L. et al. Phase I clinical trial of an intranodally administered mRNA-based therapeutic vaccine against HIV-1 infection. AIDS 32, 2533–2545 (2018).
de Jong, W. et al. iHIVARNA phase IIa, a randomized, placebo-controlled, double-blinded trial to evaluate the safety and immunogenicity of iHIVARNA-01 in chronically HIV-infected patients under stable combined antiretroviral therapy. Trials 20, 361 (2019).
Versteeg, L., Almutairi, M. M., Hotez, P. J. & Pollet, J. Enlisting the mRNA vaccine platform to combat parasitic infections. Vaccines (Basel). 7, 122 (2019).
Maruggi, G. et al. Immunogenicity and protective efficacy induced by self-amplifying mRNA vaccines encoding bacterial antigens. Vaccine 35, 361–368 (2017).
Freyn, A. W. et al. A multi-targeting, nucleoside-modified mRNA influenza virus vaccine provides broad protection in mice. Mol. Ther. 28, 1569–1584 (2020).
Gomez, C. E. et al. Enhancement of the HIV-1-specific immune response induced by an mRNA vaccine through boosting with a poxvirus MVA vector expressing the same antigen. Vaccines (Basel). 9, 959 (2021).
Awasthi, S. et al. Nucleoside-modified mRNA encoding HSV-2 glycoproteins C, D, and E prevents clinical and subclinical genital herpes. Sci. Immunol. 4, eaaw7083 (2019).
Aliprantis, A. O. et al. A phase 1, randomized, placebo-controlled study to evaluate the safety and immunogenicity of an mRNA-based RSV prefusion F protein vaccine in healthy younger and older adults. Hum. Vaccin Immunother. 17, 1248–1261 (2021).
Espeseth, A. S. et al. Modified mRNA/lipid nanoparticle-based vaccines expressing respiratory syncytial virus F protein variants are immunogenic and protective in rodent models of RSV infection. NPJ Vaccines. 5, 16 (2020).
LaTourette, P. C. II et al. Protection against herpes simplex virus type 2 infection in a neonatal murine model using a trivalent nucleoside-modified mRNA in lipid nanoparticle vaccine. Vaccine 38, 7409–7413 (2020).
Egan, K. P. et al. An HSV-2 nucleoside-modified mRNA genital herpes vaccine containing glycoproteins gC, gD, and gE protects mice against HSV-1 genital lesions and latent infection. PLoS Pathog. 16, e1008795 (2020).
Awasthi, S. et al. Trivalent nucleoside-modified mRNA vaccine yields durable memory B cell protection against genital herpes in preclinical models. J. Clin. Invest. 131, e152310 (2021).
Monslow, M. A. et al. Immunogenicity generated by mRNA vaccine encoding VZV gE antigen is comparable to adjuvanted subunit vaccine and better than live attenuated vaccine in nonhuman primates. Vaccine 38, 5793–5802 (2020).
Nelson, C. S. et al. Human cytomegalovirus glycoprotein B nucleoside-modified mRNA vaccine elicits antibody responses with greater durability and breadth than MF59-adjuvanted gB protein immunization. J. Virol. 94, e00186–20 (2020).
Webster, H. et al. Pre-existing immunity to cytomegalovirus in macaques influences human CMV vaccine responses in preclinical models. Vaccine 39, 5358–5367 (2021).
Lutz, J. et al. Unmodified mRNA in LNPs constitutes a competitive technology for prophylactic vaccines. NPJ Vaccines. 2, 29 (2017).
Aldrich, C. et al. Proof-of-concept of a low-dose unmodified mRNA-based rabies vaccine formulated with lipid nanoparticles in human volunteers: a phase 1 trial. Vaccine 39, 1310–1318 (2021).
Stokes, A. et al. Nonclinical safety assessment of repeated administration and biodistribution of a novel rabies self-amplifying mRNA vaccine in rats. Regul. Toxicol. Pharmacol. 113, 104648 (2020).
Wollner, C. J. et al. A Dengue virus serotype 1 mRNA-LNP vaccine elicits protective immune responses. J. Virol. 95, e02482–20 (2021).
Lo, M. K. et al. Evaluation of a single-dose nucleoside-modified messenger RNA vaccine encoding hendra virus-soluble glycoprotein against lethal nipah virus challenge in syrian hamsters. J. Infect. Dis. 221, S493–S498 (2020).
Knudson, C. J. et al. Lipid-nanoparticle-encapsulated mRNA vaccines induce protective memory CD8 T cells against a lethal viral infection. Mol. Ther. 29, 2769–2781 (2021).
Riley, R. S., June, C. H., Langer, R. & Mitchell, M. J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 18, 175–196 (2019).
Cafri, G. et al. mRNA vaccine-induced neoantigen-specific T cell immunity in patients with gastrointestinal cancer. J. Clin. Invest. 130, 5976–5988 (2020).
Sebastian, M. et al. A phase I/IIa study of the mRNA-based cancer immunotherapy CV9201 in patients with stage IIIB/IV non-small cell lung cancer. Cancer Immunol. Immunother. 68, 799–812 (2019).
Liu, M. A. comparison of plasmid DNA and mRNA as vaccine technologies. Vaccines (Basel). 7, 37 (2019).
Weide, B. et al. Results of the first phase I/II clinical vaccination trial with direct injection of mRNA. J. Immunother. 31, 180–188 (2008).
Kyte, J. A. et al. Immune response and long-term clinical outcome in advanced melanoma patients vaccinated with tumor-mRNA-transfected dendritic cells. Oncoimmunology 5, e1232237 (2016).
Jansen, Y. et al. A randomized controlled phase II clinical trial on mRNA electroporated autologous monocyte-derived dendritic cells (TriMixDC-MEL) as adjuvant treatment for stage III/IV melanoma patients who are disease-free following the resection of macrometastases. Cancer Immunol. Immunother. 69, 2589–2598 (2020).
Boudewijns, S. et al. Autologous monocyte-derived DC vaccination combined with cisplatin in stage III and IV melanoma patients: a prospective, randomized phase 2 trial. Cancer Immunol. Immunother. 69, 477–488 (2020).
Bol, K. F. et al. Intranodal vaccination with mRNA-optimized dendritic cells in metastatic melanoma patients. Oncoimmunology 4, e1019197 (2015).
Aarntzen, E. H. et al. Vaccination with mRNA-electroporated dendritic cells induces robust tumor antigen-specific CD4+ and CD8+ T cells responses in stage III and IV melanoma patients. Clin. Cancer Res. 18, 5460–5470 (2012).
Sahin, U. et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547, 222–226 (2017).
Kranz, L. M. et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 534, 396–401 (2016).
Lim, M., Xia, Y., Bettegowda, C. & Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 15, 422–442 (2018).
Vik-Mo, E. O. et al. Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol. Immunother. 62, 1499–1509 (2013).
Mitchell, D. A. et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 519, 366–369 (2015).
de Lima, M. et al. Proceedings from the National Cancer Institute’s Second International Workshop on the Biology, Prevention, and Treatment of Relapse After Hematopoietic Stem Cell Transplantation: part III. Prevention and treatment of relapse after allogeneic transplantation. Biol. Blood Marrow Transplant. 20, 4–13 (2014).
Amin, A. et al. Survival with AGS-003, an autologous dendritic cell-based immunotherapy, in combination with sunitinib in unfavorable risk patients with advanced renal cell carcinoma (RCC): Phase 2 study results. J. Immunother. Cancer 3, 14 (2015).
Rittig, S. M. et al. Long-term survival correlates with immunological responses in renal cell carcinoma patients treated with mRNA-based immunotherapy. Oncoimmunology 5, e1108511 (2016).
Wculek, S. K. et al. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 20, 7–24 (2020).
Fotin-Mleczek, M. et al. Highly potent mRNA based cancer vaccines represent an attractive platform for combination therapies supporting an improved therapeutic effect. J. Gene Med. 14, 428–439 (2012).
Spitzer, M. H. et al. Systemic immunity is required for effective cancer immunotherapy. Cell 168, 487–502.e415 (2017).
Karre, K., Ljunggren, H. G., Piontek, G. & Kiessling, R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 319, 675–678 (1986).
Klein, L., Hinterberger, M., Wirnsberger, G. & Kyewski, B. Antigen presentation in the thymus for positive selection and central tolerance induction. Nat. Rev. Immunol. 9, 833–844 (2009).
Sahin, U. & Tureci, O. Personalized vaccines for cancer immunotherapy. Science 359, 1355–1360 (2018).
Ott, P. A. et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221 (2017).
Carreno, B. M. et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 348, 803–808 (2015).
Kim, J., Eygeris, Y., Gupta, M. & Sahay, G. Self-assembled mRNA vaccines. Adv. Drug Deliv. Rev. 170, 83–112 (2021).
Hussain, A. et al. mRNA vaccines for COVID-19 and diverse diseases. J. Control Release 345, 314–333 (2022).
Kubler, H. et al. Self-adjuvanted mRNA vaccination in advanced prostate cancer patients: a first-in-man phase I/IIa study. J. Immunother. Cancer 3, 26 (2015).
Feldman, R. A. et al. mRNA vaccines against H10N8 and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in phase 1 randomized clinical trials. Vaccine 37, 3326–3334 (2019).
Bournazos, S., Gupta, A. & Ravetch, J. V. The role of IgG Fc receptors in antibody-dependent enhancement. Nat. Rev. Immunol. 20, 633–643 (2020).
Richner, J. M. et al. Modified mRNA vaccines protect against zika virus infection. Cell 169, 176 (2017).
Tai, W. et al. A novel receptor-binding domain (RBD)-based mRNA vaccine against SARS-CoV-2. Cell Res. 30, 932–935 (2020).
Del Giudice, G., Rappuoli, R. & Didierlaurent, A. M. Correlates of adjuvanticity: a review on adjuvants in licensed vaccines. Semin Immunol. 39, 14–21 (2018).
Chen, N. et al. RNA sensors of the innate immune system and their detection of pathogens. IUBMB Life. 69, 297–304 (2017).
Tanji, H. et al. Toll-like receptor 8 senses degradation products of single-stranded RNA. Nat. Struct. Mol. Biol. 22, 109–115 (2015).
Zhang, Z. et al. Structural analysis reveals that Toll-like receptor 7 is a dual receptor for guanosine and single-stranded RNA. Immunity 45, 737–748 (2016).
Svitkin, Y. V. et al. N1-methyl-pseudouridine in mRNA enhances translation through eIF2alpha-dependent and independent mechanisms by increasing ribosome density. Nucleic Acids Res. 45, 6023–6036 (2017).
Kariko, K. et al. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 16, 1833–1840 (2008).
Kariko, K., Buckstein, M., Ni, H. & Weissman, D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23, 165–175 (2005).
Kauffman, K. J. et al. Efficacy and immunogenicity of unmodified and pseudouridine-modified mRNA delivered systemically with lipid nanoparticles in vivo. Biomaterials 109, 78–87 (2016).
Thess, A. et al. Sequence-engineered mRNA without chemical nucleoside modifications enables an effective protein therapy in large animals. Mol. Ther. 23, 1456–1464 (2015).
Bonehill, A. et al. Enhancing the T-cell stimulatory capacity of human dendritic cells by co-electroporation with CD40L, CD70 and constitutively active TLR4 encoding mRNA. Mol. Ther. 16, 1170–1180 (2008).
Bialkowski, L. et al. Adjuvant-enhanced mRNA vaccines. Methods Mol. Biol. 1499, 179–191 (2017).
De Keersmaecker, B. et al. The combination of 4-1BBL and CD40L strongly enhances the capacity of dendritic cells to stimulate HIV-specific T cell responses. J. Leukoc. Biol. 89, 989–999 (2011).
Aerts-Toegaert, C. et al. CD83 expression on dendritic cells and T cells: correlation with effective immune responses. Eur. J. Immunol. 37, 686–695 (2007).
Dannull, J. et al. Enhancing the immunostimulatory function of dendritic cells by transfection with mRNA encoding OX40 ligand. Blood 105, 3206–3213 (2005).
Ahammad, I. & Lira, S. S. Designing a novel mRNA vaccine against SARS-CoV-2: an immunoinformatics approach. Int J. Biol. Macromol. 162, 820–837 (2020).
Lee, K. et al. Adjuvant incorporated lipid nanoparticles for enhanced mRNA-mediated cancer immunotherapy. Biomater. Sci. 8, 1101–1105 (2020).
Coffman, R. L., Sher, A. & Seder, R. A. Vaccine adjuvants: putting innate immunity to work. Immunity 33, 492–503 (2010).
Choi, J. J. et al. High molecular weight chitosan-complexed RNA nanoadjuvant for effective cancer immunotherapy.Pharmaceutics 11, 680 (2019).
Verbeke, R. et al. Co-delivery of nucleoside-modified mRNA and TLR agonists for cancer immunotherapy: restoring the immunogenicity of immunosilent mRNA. J. Control Release 266, 287–300 (2017).
Vollmer, J. & Krieg, A. M. Immunotherapeutic applications of CpG oligodeoxynucleotide TLR9 agonists. Adv. Drug Deliv. Rev. 61, 195–204 (2009).
Bode, C. et al. CpG DNA as a vaccine adjuvant. Expert Rev. Vaccines. 10, 499–511 (2011).
Jackson, S. et al. Immunogenicity of a two-dose investigational hepatitis B vaccine, HBsAg-1018, using a toll-like receptor 9 agonist adjuvant compared with a licensed hepatitis B vaccine in adults. Vaccine 36, 668–674 (2018).
Mauriello, A. et al. Immunological effects of adjuvants in subsets of antigen presenting cells of cancer patients undergoing chemotherapy. J. Transl. Med. 18, 34 (2020).
Circelli, L. et al. Immunological effects of a novel RNA-based adjuvant in liver cancer patients. Cancer Immunol. Immunother. 66, 103–112 (2017).
Carralot, J. P. et al. Polarization of immunity induced by direct injection of naked sequence-stabilized mRNA vaccines. Cell Mol. Life Sci. 61, 2418–2424 (2004).
Hammerich, L. et al. Systemic clinical tumor regressions and potentiation of PD1 blockade with in situ vaccination. Nat. Med. 25, 814–824 (2019).
Kreiter, S. et al. FLT3 ligand enhances the cancer therapeutic potency of naked RNA vaccines. Cancer Res. 71, 6132–6142 (2011).
Magadum, A., Kaur, K. & Zangi, L. mRNA-based protein replacement therapy for the heart. Mol. Ther. 27, 785–793 (2019).
Sahu, I. et al. Recent developments in mRNA-based protein supplementation therapy to target lung diseases. Mol. Ther. 27, 803–823 (2019).
Trepotec, Z. et al. Delivery of mRNA therapeutics for the treatment of hepatic diseases. Mol. Ther. 27, 794–802 (2019).
Berraondo, P., Martini, P. G. V., Avila, M. A. & Fontanellas, A. Messenger RNA therapy for rare genetic metabolic diseases. Gut 68, 1323–1330 (2019).
Vallazza, B. et al. Recombinant messenger RNA technology and its application in cancer immunotherapy, transcript replacement therapies, pluripotent stem cell induction, and beyond. Wiley Interdiscip. Rev. RNA. 6, 471–499 (2015).
Badieyan, Z. S. et al. Transcript-activated collagen matrix as sustained mRNA delivery system for bone regeneration. J. Control Release 239, 137–148 (2016).
Uchida, S. et al. In vivo messenger RNA introduction into the central nervous system using polyplex nanomicelle. PLoS One 8, e56220 (2013).
Nabhan, J. F. et al. Intrathecal delivery of frataxin mRNA encapsulated in lipid nanoparticles to dorsal root ganglia as a potential therapeutic for Friedreich’s ataxia. Sci. Rep. 6, 20019 (2016).
Schumann, C. et al. Increasing lean muscle mass in mice via nanoparticle-mediated hepatic delivery of follistatin mRNA. Theranostics 8, 5276–5288 (2018).
Lui, K. O. et al. Driving vascular endothelial cell fate of human multipotent Isl1+ heart progenitors with VEGF modified mRNA. Cell Res. 23, 1172–1186 (2013).
Anttila, V. et al. Synthetic mRNA encoding VEGF-A in patients undergoing coronary artery bypass grafting: design of a Phase 2a clinical trial. Mol. Ther. Methods Clin. Dev. 18, 464–472 (2020).
Kreda, S. M., Davis, C. W. & Rose, M. C. CFTR, mucins, and mucus obstruction in cystic fibrosis. Cold Spring Harb. Perspect. Med. 2, a009589 (2012).
Bangel-Ruland, N. et al. Cystic fibrosis transmembrane conductance regulator-mRNA delivery: a novel alternative for cystic fibrosis gene therapy. J. Gene Med. 15, 414–426 (2013).
Van Hoecke, L. & Roose, K. How mRNA therapeutics are entering the monoclonal antibody field. J. Transl. Med. 17, 54 (2019).
Chen, C. Y. et al. Treatment of hemophilia A using factor VIII messenger RNA lipid nanoparticles. Mol. Ther. Nucleic Acids 20, 534–544 (2020).
Russick, J. et al. Correction of bleeding in experimental severe hemophilia A by systemic delivery of factor VIII-encoding mRNA. Haematologica 105, 1129–1137 (2020).
Guo, X. R. et al. PDX-1 mRNA-induced reprogramming of mouse pancreas-derived mesenchymal stem cells into insulin-producing cells in vitro. Clin. Exp. Med. 15, 501–509 (2015).
Ramaswamy, S. et al. Systemic delivery of factor IX messenger RNA for protein replacement therapy. Proc. Natl Acad. Sci. USA. 114, E1941–E1950 (2017).
Kitagawa, T. Hepatorenal tyrosinemia. Proc. Jpn Acad. Ser. B Phys. Biol. Sci. 88, 192–200 (2012).
Jiang, L. et al. Systemic messenger RNA as an etiological treatment for acute intermittent porphyria. Nat. Med. 24, 1899–1909 (2018).
Cosson, M. A. et al. Long-term outcome in methylmalonic aciduria: a series of 30 French patients. Mol. Genet Metab. 97, 172–178 (2009).
An, D. et al. Systemic messenger RNA therapy as a treatment for methylmalonic acidemia. Cell Rep. 21, 3548–3558 (2017).
Prieve, M. G. et al. Targeted mRNA therapy for ornithine transcarbamylase deficiency. Mol. Ther. 26, 801–813 (2018).
DeRosa, F. et al. Improved efficacy in a Fabry disease model using a systemic mRNA liver depot system as compared to enzyme replacement therapy. Mol. Ther. 27, 878–889 (2019).
Karadagi, A. et al. Systemic modified messenger RNA for replacement therapy in alpha 1-antitrypsin deficiency. Sci. Rep. 10, 7052 (2020).
Islam, M. A. et al. Restoration of tumour-growth suppression in vivo via systemic nanoparticle-mediated delivery of PTEN mRNA. Nat. Biomed. Eng. 2, 850–864 (2018).
Islam, M. A. et al. Author Correction: restoration of tumour-growth suppression in vivo via systemic nanoparticle-mediated delivery of PTEN mRNA. Nat. Biomed. Eng. 2, 968 (2018).
Uchida, S. et al. Systemic delivery of messenger RNA for the treatment of pancreatic cancer using polyplex nanomicelles with a cholesterol moiety. Biomaterials 82, 221–228 (2016).
Zhang, R. et al. Delivery of a modified mRNA encoding IL-22 binding protein (IL-22BP) for colon cancer gene therapy. J. Biomed. Nanotechnol. 14, 1239–1251 (2018).
Kamalinia, G. et al. Directing evolution of novel ligands by mRNA display. Chem. Soc. Rev. 50, 9055–9103 (2021).
Granot-Matok, Y. et al. Therapeutic mRNA delivery to leukocytes. J. Control Release 305, 165–175 (2019).
Bicknell, A. A. & Ricci, E. P. When mRNA translation meets decay. Biochem Soc. Trans. 45, 339–351 (2017).
Purcell, A. W., McCluskey, J. & Rossjohn, J. More than one reason to rethink the use of peptides in vaccine design. Nat. Rev. Drug Discov. 6, 404–414 (2007).
Tomura, M. et al. Tracking and quantification of dendritic cell migration and antigen trafficking between the skin and lymph nodes. Sci. Rep. 4, 6030 (2014).
Kitano, M. et al. Imaging of the cross-presenting dendritic cell subsets in the skin-draining lymph node. Proc. Natl Acad. Sci. USA. 113, 1044–1049 (2016).
Eisenbarth, S. C. Dendritic cell subsets in T cell programming: location dictates function. Nat. Rev. Immunol. 19, 89–103 (2019).
Orlandini von Niessen, A. G. et al. Improving mRNA-based therapeutic gene delivery by expression-augmenting 3’ UTRs identified by cellular library screening. Mol. Ther. 27, 824–836 (2019).
Benteyn, D. et al. Design of an Optimized Wilms’ Tumor 1 (WT1) mRNA construct for enhanced WT1 expression and improved immunogenicity in vitro and in vivo. Mol. Ther. Nucleic Acids 2, e134 (2013).
Turner, J. S. et al. SARS-CoV-2 mRNA vaccines induce persistent human germinal centre responses. Nature 596, 109–113 (2021).
Cornu, T. I., Mussolino, C. & Cathomen, T. Refining strategies to translate genome editing to the clinic. Nat. Med. 23, 415–423 (2017).
Porteus, M. H. A new class of medicines through DNA editing. N. Engl. J. Med. 380, 947–959 (2019).
Wang, J. et al. Homology-driven genome editing in hematopoietic stem and progenitor cells using ZFN mRNA and AAV6 donors. Nat. Biotechnol. 33, 1256–1263 (2015).
Poirot, L. et al. Multiplex genome-edited T-cell manufacturing platform for “Off-the-Shelf” adoptive T-cell immunotherapies. Cancer Res. 75, 3853–3864 (2015).
Mock, U. et al. mRNA transfection of a novel TAL effector nuclease (TALEN) facilitates efficient knockout of HIV co-receptor CCR5. Nucleic Acids Res. 43, 5560–5571 (2015).
Miller, J. B. et al. Non-viral CRISPR/Cas gene editing in vitro and in vivo enabled by synthetic nanoparticle co-delivery of Cas9 mRNA and sgRNA. Angew. Chem. Int Ed. Engl. 56, 1059–1063 (2017).
Zhang, H. X., Zhang, Y. & Yin, H. Genome editing with mRNA encoding ZFN, TALEN, and Cas9. Mol. Ther. 27, 735–746 (2019).
Gaj, T., Gersbach, C. A. & Barbas, C. F. III ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 31, 397–405 (2013).
Li, H. et al. Applications of genome editing technology in the targeted therapy of human diseases: mechanisms, advances and prospects. Signal Transduct. Target Ther. 5, 1 (2020).
Restifo, N. P., Dudley, M. E. & Rosenberg, S. A. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat. Rev. Immunol. 12, 269–281 (2012).
Prasad, V. Immunotherapy: tisagenlecleucel – the first approved CAR-T-cell therapy: implications for payers and policy makers. Nat. Rev. Clin. Oncol. 15, 11–12 (2018).
Maude, S. L. et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 371, 1507–1517 (2014).
Hendel, A. et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 33, 985–989 (2015).
Lino, C. A., Harper, J. C., Carney, J. P. & Timlin, J. A. Delivering CRISPR: a review of the challenges and approaches. Drug Deliv. 25, 1234–1257 (2018).
Knipping, F. et al. Genome-wide specificity of highly efficient TALENs and CRISPR/Cas9 for T cell receptor modification. Mol. Ther. Methods Clin. Dev. 4, 213–224 (2017).
Parayath, N. N. et al. In vitro-transcribed antigen receptor mRNA nanocarriers for transient expression in circulating T cells in vivo. Nat. Commun. 11, 6080 (2020).
Lim, W. A. & June, C. H. The principles of engineering immune cells to treat cancer. Cell 168, 724–740 (2017).
Singh, N., Perazzelli, J., Grupp, S. A. & Barrett, D. M. Early memory phenotypes drive T cell proliferation in patients with pediatric malignancies. Sci. Transl. Med. 8, 320ra323 (2016).
Tchou, J. et al. Safety and efficacy of intratumoral injections of chimeric antigen receptor (CAR) T cells in metastatic breast cancer. Cancer Immunol. Res. 5, 1152–1161 (2017).
Georgiadis, C. et al. Long terminal repeat CRISPR-CAR-coupled “Universal” T cells mediate potent anti-leukemic effects. Mol. Ther. 26, 1215–1227 (2018).
Moore, J. K. & Haber, J. E. Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol. Cell Biol. 16, 2164–2173 (1996).
Almasbak, H. et al. Transiently redirected T cells for adoptive transfer. Cytotherapy 13, 629–640 (2011).
Barrett, D. M. et al. Treatment of advanced leukemia in mice with mRNA engineered T cells. Hum. Gene Ther. 22, 1575–1586 (2011).
Barrett, D. M. et al. Regimen-specific effects of RNA-modified chimeric antigen receptor T cells in mice with advanced leukemia. Hum. Gene Ther. 24, 717–727 (2013).
Zhao, Y. et al. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 70, 9053–9061 (2010).
Pockaj, B. A. et al. Localization of 111indium-labeled tumor infiltrating lymphocytes to tumor in patients receiving adoptive immunotherapy. Augmentation with cyclophosphamide and correlation with response. Cancer 73, 1731–1737 (1994).
Idorn, M., Thor Straten, P., Svane, I. M. & Met, O. Transfection of tumor-infiltrating T cells with mRNA encoding CXCR2. Methods Mol. Biol. 1428, 261–276 (2016).
Wiesinger, M. et al. Clinical-scale production of CAR-T cells for the treatment of melanoma patients by mRNA transfection of a CSPG4-specific CAR under full GMP compliance.Cancers (Basel) 11, 1198 (2019).
Olden, B. R., Cheng, Y., Yu, J. L. & Pun, S. H. Cationic polymers for non-viral gene delivery to human T cells. J. Control Release 282, 140–147 (2018).
McKinlay, C. J. et al. Enhanced mRNA delivery into lymphocytes enabled by lipid-varied libraries of charge-altering releasable transporters. Proc. Natl Acad. Sci. USA. 115, E5859–E5866 (2018).
Tebas, P. et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 370, 901–910 (2014).
Urnov, F. D. et al. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 11, 636–646 (2010).
Bobis-Wozowicz, S. et al. Non-integrating gamma-retroviral vectors as a versatile tool for transient zinc-finger nuclease delivery. Sci. Rep. 4, 4656 (2014).
Hoban, M. D. et al. CRISPR/Cas9-mediated correction of the sickle mutation in human CD34+ cells. Mol. Ther. 24, 1561–1569 (2016).
Bjurstrom, C. F. et al. Reactivating fetal hemoglobin expression in human adult erythroblasts through BCL11A knockdown using targeted endonucleases. Mol. Ther. Nucleic Acids 5, e351 (2016).
Peterson, C. W. et al. Long-term multilineage engraftment of autologous genome-edited hematopoietic stem cells in nonhuman primates. Blood 127, 2416–2426 (2016).
Smits, E. et al. RNA-based gene transfer for adult stem cells and T cells. Leukemia 18, 1898–1902 (2004).
Ponsaerts, P. et al. Highly efficient mRNA-based gene transfer in feeder-free cultured H9 human embryonic stem cells. Cloning Stem Cells 6, 211–216 (2004).
Rejman, J. et al. mRNA transfection of cervical carcinoma and mesenchymal stem cells mediated by cationic carriers. J. Control Release 147, 385–391 (2010).
Shaw, M. L., Williams, E. J., Hawes, S. & Saffery, R. Characterisation of histone variant distribution in human embryonic stem cells by transfection of in vitro transcribed mRNA. Mol. Reprod. Dev. 76, 1128–1142 (2009).
Yuan, Y. et al. HIV-1 Tat protein inhibits the hematopoietic support function of human bone marrow mesenchymal stem cells. Virus Res. 273, 197756 (2019).
Kondo, T. et al. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Abeta and differential drug responsiveness. Cell. Stem Cell. 12, 487–496 (2013).
Goyenvalle, A. et al. Functional correction in mouse models of muscular dystrophy using exon-skipping tricyclo-DNA oligomers. Nat. Med. 21, 270–275 (2015).
Takahashi, K. et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 (2007).
Miyoshi, N. et al. Reprogramming of mouse and human cells to pluripotency using mature microRNAs. Cell. Stem Cell. 8, 633–638 (2011).
Polo, J. M. et al. A molecular roadmap of reprogramming somatic cells into iPS cells. Cell 151, 1617–1632 (2012).
Kehler, J. et al. RNA-generated and gene-edited induced pluripotent stem cells for disease modeling and therapy. J. Cell Physiol. 232, 1262–1269 (2017).
Winkle, M., El-Daly, S. M., Fabbri, M. & Calin, G. A. Noncoding RNA therapeutics – challenges and potential solutions. Nat. Rev. Drug Discov. 20, 629–651 (2021).
Phung, C. D. et al. Rational combination immunotherapeutic approaches for effective cancer treatment. J. Control Release 294, 114–130 (2019).
Pardoll, D. M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264 (2012).
Sharma, P., Hu-Lieskovan, S., Wargo, J. A. & Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707–723 (2017).
Ramachandran, M., Dimberg, A. & Essand, M. The cancer-immunity cycle as rational design for synthetic cancer drugs: novel DC vaccines and CAR T-cells. Semin Cancer Biol. 45, 23–35 (2017).
Tumeh, P. C. et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571 (2014).
Eroglu, Z. et al. High response rate to PD-1 blockade in desmoplastic melanomas. Nature 553, 347–350 (2018).
Wilgenhof, S. et al. Phase II study of autologous monocyte-derived mRNA electroporated dendritic cells (TriMixDC-MEL) plus ipilimumab in patients with pretreated advanced melanoma. J. Clin. Oncol. 34, 1330–1338 (2016).
De Keersmaecker, B. et al. TriMix and tumor antigen mRNA electroporated dendritic cell vaccination plus ipilimumab: link between T-cell activation and clinical responses in advanced melanoma. J. Immunother. Cancer 8, e000329 (2020).
Reinhard, K. et al. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science 367, 446–453 (2020).
Kauffman, K. J. et al. Optimization of lipid nanoparticle formulations for mRNA delivery in vivo with fractional factorial and definitive screening designs. Nano Lett. 15, 7300–7306 (2015).
Li, M. et al. Mucosal vaccines: strategies and challenges. Immunol. Lett. 217, 116–125 (2020).
Bahl, K. et al. Preclinical and clinical demonstration of immunogenicity by mRNA vaccines against H10N8 and H7N9 influenza viruses. Mol. Ther. 25, 1316–1327 (2017).
Lycke, N. Recent progress in mucosal vaccine development: potential and limitations. Nat. Rev. Immunol. 12, 592–605 (2012).
Breedveld, A. & van Egmond, M. IgA and FcalphaRI: pathological roles and therapeutic opportunities. Front Immunol. 10, 553 (2019).
Patel, M. et al. A systematic review of anti-rotavirus serum IgA antibody titer as a potential correlate of rotavirus vaccine efficacy. J. Infect. Dis. 208, 284–294 (2013).
Gustin, K. M. et al. Comparative immunogenicity and cross-clade protective efficacy of mammalian cell-grown inactivated and live attenuated H5N1 reassortant vaccines in ferrets. J. Infect. Dis. 204, 1491–1499 (2011).
Tamura, S. et al. Functional role of respiratory tract haemagglutinin-specific IgA antibodies in protection against influenza. Vaccine 8, 479–485 (1990).
Solans, L. & Locht, C. The role of mucosal immunity in pertussis. Front Immunol. 9, 3068 (2018).
Sano, K., Ainai, A., Suzuki, T. & Hasegawa, H. Intranasal inactivated influenza vaccines for the prevention of seasonal influenza epidemics. Expert Rev. Vaccines. 17, 687–696 (2018).
Guermonprez, P. et al. ER-phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature 425, 397–402 (2003).
Brenner, S., Jacob, F. & Meselson, M. An unstable intermediate carrying information from genes to ribosomes for protein synthesis. Nature 190, 576–581 (1961).
Smull, C. E., Mallette, M. F. & Ludwig, E. The use of basic proteins to increase the infectivity of enterovirus ribonucleic acid. Biochem. Biophys. Res. Commun. 5, 247–249 (1961).
Gurdon, J. B., Lane, C. D., Woodland, H. R. & Marbaix, G. Use of frog eggs and oocytes for the study of messenger RNA and its translation in living cells. Nature 233, 177–182 (1971).
Muthukrishnan, S., Both, G. W., Furuichi, Y. & Shatkin, A. J. 5’-Terminal 7-methylguanosine in eukaryotic mRNA is required for translation. Nature 255, 33–37 (1975).
Dimitriadis, G. J. Translation of rabbit globin mRNA introduced by liposomes into mouse lymphocytes. Nature 274, 923–924 (1978).
Malone, R. W., Felgner, P. L. & Verma, I. M. Cationic liposome-mediated RNA transfection. Proc. Natl Acad. Sci. USA. 86, 6077–6081 (1989).
Wolff, J. A. et al. Direct gene transfer into mouse muscle in vivo. Science 247, 1465–1468 (1990).
Rajagopalan, L. E. & Malter, J. S. Regulation of eukaryotic messenger RNA turnover. Prog. Nucleic Acid Res Mol. Biol. 56, 257–286 (1997).
Zhou, W. Z. et al. RNA melanoma vaccine: induction of antitumor immunity by human glycoprotein 100 mRNA immunization. Hum. Gene Ther. 10, 2719–2724 (1999).
Heiser, A. et al. Autologous dendritic cells transfected with prostate-specific antigen RNA stimulate CTL responses against metastatic prostate tumors. J. Clin. Invest. 109, 409–417 (2002).
Weide, B. et al. Direct injection of protamine-protected mRNA: results of a phase 1/2 vaccination trial in metastatic melanoma patients. J. Immunother. 32, 498–507 (2009).
Kreiter, S. et al. Intranodal vaccination with naked antigen-encoding RNA elicits potent prophylactic and therapeutic antitumoral immunity. Cancer Res. 70, 9031–9040 (2010).
Hwang, W. Y. et al. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 31, 227–229 (2013).
Baden, L. R. et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med. 384, 403–416 (2021).
Vogel, A. B. et al. BNT162b vaccines protect rhesus macaques from SARS-CoV-2. Nature 592, 283–289 (2021).
Garneau, N. L., Wilusz, J. & Wilusz, C. J. The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol. 8, 113–126 (2007).
Covelo-Molares, H., Bartosovic, M. & Vanacova, S. RNA methylation in nuclear pre-mRNA processing. Wiley Interdiscip. Rev. RNA. 9, e1489 (2018).
Jayaraman, M. et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem. Int Ed. Engl. 51, 8529–8533 (2012).
Morais, P., Adachi, H. & Yu, Y. T. The critical contribution of pseudouridine to mRNA COVID-19 vaccines. Front Cell Dev. Biol. 9, 789427 (2021).
Dong, Y. et al. Lipopeptide nanoparticles for potent and selective siRNA delivery in rodents and nonhuman primates. Proc. Natl Acad. Sci. USA. 111, 3955–3960 (2014).
Fenton, O. S. et al. Customizable lipid nanoparticle materials for the delivery of siRNAs and mRNAs. Angew. Chem. Int Ed. Engl. 57, 13582–13586 (2018).
Li, B. et al. An orthogonal array optimization of lipid-like nanoparticles for mRNA delivery in vivo. Nano Lett. 15, 8099–8107 (2015).
Liu, J. et al. Fast and efficient CRISPR/Cas9 genome editing in vivo enabled by bioreducible lipid and messenger RNA nanoparticles. Adv. Mater. 31, e1902575 (2019).
Hou, X. et al. Vitamin lipid nanoparticles enable adoptive macrophage transfer for the treatment of multidrug-resistant bacterial sepsis. Nat. Nanotechnol. 15, 41–46 (2020).
Lee, S. M. et al. A systematic study of unsaturation in lipid nanoparticles leads to improved mRNA transfection in vivo. Angew. Chem. Int Ed. Engl. 60, 5848–5853 (2021).
Akita, H. Development of an SS-cleavable pH-activated lipid-like material (ssPalm) as a nucleic acid delivery device. Biol. Pharm. Bull. 43, 1617–1625 (2020).
McCallum, M. et al. Structure-guided covalent stabilization of coronavirus spike glycoprotein trimers in the closed conformation. Nat. Struct. Mol. Biol. 27, 942–949 (2020).
Juraszek, J. et al. Stabilizing the closed SARS-CoV-2 spike trimer. Nat. Commun. 12, 244 (2021).
Xu, C. et al. Conformational dynamics of SARS-CoV-2 trimeric spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM. Sci. Adv. 7, eabe5575 (2021).
Bangaru, S. et al. Structural analysis of full-length SARS-CoV-2 spike protein from an advanced vaccine candidate. Science 370, 1089–1094 (2020).
Baden, L. R. et al. Phase 3 trial of mRNA-1273 during the Delta-variant surge. N. Engl. J. Med. 385, 2485–2487 (2021).
Goldberg, Y. et al. Waning immunity after the BNT162b2 vaccine in Israel. N. Engl. J. Med. 385, e85 (2021).
Vasileiou, E. et al. Interim findings from first-dose mass COVID-19 vaccination roll-out and COVID-19 hospital admissions in Scotland: a national prospective cohort study. Lancet 397, 1646–1657 (2021).
Walsh, E. E. et al. Safety and immunogenicity of two RNA-based Covid-19 vaccine candidates. N. Engl. J. Med. 383, 2439–2450 (2020).
Choi, A. et al. Safety and immunogenicity of SARS-CoV-2 variant mRNA vaccine boosters in healthy adults: an interim analysis. Nat. Med. 27, 2025–2031 (2021).
Wu, K. et al. Variant SARS-CoV-2 mRNA vaccines confer broad neutralization as primary or booster series in mice. Vaccine 39, 7394–7400 (2021).
Kalnin, K. V. et al. Immunogenicity and efficacy of mRNA COVID-19 vaccine MRT5500 in preclinical animal models. NPJ Vaccines. 6, 61 (2021).
Mallapaty, S. The COVID vaccine pioneer behind southeast Asia’s first mRNA shot. Nature 594, 163 (2021).
Gay, C. L. et al. Immunogenicity of AGS-004 dendritic cell therapy in patients treated during acute HIV infection. AIDS Res Hum. Retroviruses. 34, 111–122 (2018).
DiGiusto, D. L. et al. Preclinical development and qualification of ZFN-mediated CCR5 disruption in human hematopoietic stem/progenitor cells. Mol. Ther. Methods Clin. Dev. 3, 16067 (2016).
Papachristofilou, A. et al. Phase Ib evaluation of a self-adjuvanted protamine formulated mRNA-based active cancer immunotherapy, BI1361849 (CV9202), combined with local radiation treatment in patients with stage IV non-small cell lung cancer. J. Immunother. Cancer 7, 38 (2019).
Svoboda, J. et al. Nonviral RNA chimeric antigen receptor-modified T cells in patients with Hodgkin lymphoma. Blood 132, 1022–1026 (2018).
Gillmore, J. D. et al. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N. Engl. J. Med. 385, 493–502 (2021).
Gan, L. M. et al. Intradermal delivery of modified mRNA encoding VEGF-A in patients with type 2 diabetes. Nat. Commun. 10, 871 (2019).
Acknowledgements
This work was financially supported by the Sichuan Province Science and Technology Support Program (2021YFH0003, 2021YFSY008, 2020YFH0065, and 2020YJ0238) and the Chengdu Key S&T Innovation Projects (2019-YF08-00139-GX). Some icons or graphic elements in Figs. 2, 6, 7, and 9 are adapted from BioRender.com (2022), retrieved from https://app.biorender.com/. The structure and multimerization in Fig. 8 are from https://pdbj.org/. Final schematic illustrations were created and integrated by our original design.
Author information
Authors and Affiliations
Contributions
X.S. conceived, supervised, and revised the paper. S.Q. and X.T. organized figures and formatted the paper. S.Q., X.T., Y.C., K.C., N.F., W.X., Q.Z., G.L., Y.T., M.W., and X.S. participated in different parts of writing.
Corresponding author
Ethics declarations
Competing interests
The authors have no financial conflict of interest. X.S. and M.W. are members of the editorial board; they have not been involved In the process of the manuscript handling.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Qin, S., Tang, X., Chen, Y. et al. mRNA-based therapeutics: powerful and versatile tools to combat diseases. Sig Transduct Target Ther 7, 166 (2022). https://doi.org/10.1038/s41392-022-01007-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-022-01007-w
This article is cited by
-
Breaking the mold with RNA—a “RNAissance” of life science
npj Genomic Medicine (2024)
-
Strategies to reduce the risks of mRNA drug and vaccine toxicity
Nature Reviews Drug Discovery (2024)
-
Claudin 18.2 as a novel therapeutic target
Nature Reviews Clinical Oncology (2024)
-
Durable cross-protective neutralizing antibody responses elicited by lipid nanoparticle-formulated SARS-CoV-2 mRNA vaccines
npj Vaccines (2024)
-
Characterization of mRNA Lipid Nanoparticles by Electron Density Mapping Reconstruction: X-ray Scattering with Density from Solution Scattering (DENSS) Algorithm
Pharmaceutical Research (2024)
