
Abstract
Like other classes of treatments described in this issue’s section, neuroactive steroids have been studied for decades but have risen as a new class of rapid-acting, durable antidepressants with a distinct mechanism of action from previous antidepressant treatments and from other compounds covered in this issue. Neuroactive steroids are natural derivatives of progesterone but are proving effective as exogenous treatments. The best understood mechanism is that of positive allosteric modulation of GABAA receptors, where subunit selectivity may promote their profile of action. Mechanistically, there is some reason to think that neuroactive steroids may separate themselves from liabilities of other GABA modulators, although research is ongoing. It is also possible that intracellular targets, including inflammatory pathways, may be relevant to beneficial actions. Strengths and opportunities for further development include exploiting non-GABAergic targets, structural analogs, enzymatic production of natural steroids, precursor loading, and novel formulations. The molecular mechanisms of behavioral effects are not fully understood, but study of brain network states involved in emotional processing demonstrate a robust influence on affective states not evident with at least some other GABAergic drugs including benzodiazepines. Ongoing studies with neuroactive steroids will further elucidate the brain and behavioral effects of these compounds as well as likely underpinnings of disease.
Similar content being viewed by others

Introduction
The recent development of rapid-acting antidepressants has changed the landscape for the treatment of depression. Rapid antidepressant actions offer the opportunity to re-envision treatment strategies for depression, enabling an episodic treatment approach rather than a prophylactic approach which would revolutionize treatment options for patients.
These newly developed, rapidly acting antidepressants have novel mechanisms of action which differ from the traditional antidepressant classes, such as SPRAVATO® which acts on NMDA receptors and ZULRESSO® (brexanolone) which is a neuroactive steroid (NAS) and synthetic formulation of allopregnanolone which acts as a positive allosteric modulator (PAM) at GABAA receptors (GABAARs). The antidepressant effects of these compounds offer new avenues for further treatment development targeting these novel targets as well as offer new insights into the pathophysiological mechanisms of disease.
To set the stage for discussion of risks and opportunities and mechanism of action, we briefly review the clinical data that resulted in recent approval of brexanolone for treatment of women with postpartum depression, and we give a status update of trials testing efficacy in men and women with depression. Brexanolone, an intravenous, synthetic formulation of allopregnanolone demonstrated antidepressant effects in women with severe postpartum depression (Hamilton Rating Scale for Depression [HAM-D] total score ≥26) in a double-blind, randomized, placebo-controlled trial across four different sites, demonstrating a significant reduction in HAM-D total score from baseline, albeit in a small number of patients (21: 10 brexanolone, 11 placebo) [1]. The antidepressant effects of brexanolone in women with postpartum depression was repeated in two multicenter, double-blind, randomized, placebo-controlled, phase 3 trials, both studies demonstrating a significant reduction in total HAM-D scores [2]. Brexanolone treatment was also demonstrated to achieve a more rapid and effective antidepressant effect as well as reduce anxiety and insomnia in patients [3]. It is interesting to note not only the rapid antidepressant effects of these compounds, but also the durability of the antidepressant effects, lasting at least 30 days [1,2,3]. On March 19, 2019, ZULRESSO® (brexanolone) was approved by the U.S. Food and Drug Administration (FDA) for the treatment of postpartum depression.
Based on the robust antidepressant effects of brexanolone, a similar, but orally available compound, zuranolone was developed and the antidepressant potential was evaluated in patients with postpartum depression or major depressive disorder. Similar to brexanolone, a 2-week treatment with zuranolone demonstrated a significant reduction in HAM-D score and Hamilton Rating Scale for Anxiety score in women with postpartum depression in a phase 3, double-blind, randomized, placebo-controlled clinical trial [4]. A follow-up phase 3, double-blind, randomized, placebo-controlled trial with a once-daily oral zuranolone treatment for 14 days increased concurrent remission of depressive and anxiety symptoms at days 3, 15, and 45 [5]. Zuranolone was shown to exert antidepressant effects in a double-blind, phase 2 trial in patients with major depressive disorder [6]. Improved quality of life was reported in patients with major depressive disorder in a randomized, placebo-controlled phase 2 trial [7]. Similar to brexanolone, zuranolone also exerted rapid and sustained antidepressant effects, lasting up to 45 days [4, 5]. On December 6, 2022, Biogen Inc. and Sage Therapeutics announced the submission of a New Drug Application (NDA) to the FDA for the use of zuranolone for the treatment of major depressive disorder and postpartum depression. This application was based on data from the LANDSCAPE program for major depressive disorder treatment, which includes five studies (MDD-201B, MOUNTAIN, SHORELINE, WATERFALL, and CORAL), and the NEST program for postpartum depression which included two studies (ROBIN and SKYLARK). At the time of this publication, the status of this application is unknown.
These are breakthrough studies which transform our current thinking regarding antidepressant treatment. It is critically important to understand how these compounds exert their rapid and persistent antidepressant action not only for the development of future treatments but also to obtain a better understanding of the underlying neurobiology of disease. In this section we will fully consider the advantages, disadvantages, and potential concerns regarding these rapid-acting antidepressants, discuss the unique features of GABAAR PAMs, possible non-GABAergic impacts of NAS, and potential role of network dynamics in the therapeutic properties of these compounds. Collectively, this contribution highlights potential novel mechanisms of action of these compounds which will inform further therapeutic development.
Neuroactive steroids: strengths, weaknesses, opportunities and threats to clinical use
Strengths
NAS are naturally-occurring compounds representing an attractive treatment strategy. NAS were approved in 2019 for treatment of postpartum depression based on a steroid replacement hypothesis [8]. Indeed, there is interest in this approach for treating other neuroendocrine disorders in women [9], and neurosteroid deficiency may characterize a subset of individuals with major depressive disorder in both sexes as described further below. At the same time, there has been an interest in exploiting glutamate and GABA transmitter systems in depressive disorders to circumvent limitations of SSRIs and other first-line treatments for depressive and mood disorders [10]. Given the especially highly potent and efficacious effects of NAS at GABAARs, the field is interested in expanding usage of NAS for disorders beyond neuroendocrine disorders. By targeting a mechanism distinct from conventional treatments, NAS, like ketamine and other novel approaches, might benefit patients who are not helped by traditional treatments (up to a third of patients). We are fortunate to have witnessed this turnaround given that only a decade ago, many warnings were given about the retreat of the pharmaceutical industry from neuropsychiatric indications [11,12,13].
Additional strengths of the NAS approach include rapidity of action. If indeed GABAAR PAM effects are important for behavioral effects, NAS readily pass the blood-brain barrier to have direct and immediate impact on signaling. By directly binding sites on heteropentameric GABAARs, NAS will immediately influence signaling, with no delay for second messenger signaling, transcription, and other slow processes. Despite the steroid structure, allopregnanolone and related NAS appear to have little to no effect on classical intracellular steroid receptors [14, 15]. Compared with benzodiazepines, NAS may target receptors of different subunit combinations (see below), thereby impacting different cell classes and different networks than benzodiazepines.
As noted above, a single administration of NAS has persisting, durable effects on nervous system function, well beyond the presence of drug in the brain. Benefit in women treated with 60 h infusion lasted up to 30 days (see above). Therefore, another strength of NAS is this longevity of action. The combination of rapidity and persistence has obvious advantageous clinical ramifications. However, it raises mechanistic questions. Do GABAAR effects trigger longer lasting changes to nervous system function that are responsible for maintaining the therapeutic benefit, or is the trigger related to a different target? After all, GABAARs are not especially well known for triggering plasticity. Similarly, do the effects responsible for maintaining the therapeutic benefit after drug has dissipated involve GABAARs, or are these effects independent of GABAAR signaling? These are important questions to address for optimizing effects of NAS: limiting side effects and maximizing benefit.
Weaknesses
A major weakness is alluded to immediately above. Without a detailed understanding of mechanism of action, it is difficult to optimize analogs to limit off-target effects and screen for effectiveness using mechanism-based assays. This gap also reveals limits in our understanding of disease mechanisms. This weakness is of course not unique to neurosteroids but plagues virtually all psychotropic drugs.
To date, brexanolone is approved for postpartum depression, but its utility for other psychiatric indications, including major depressive disorder, is unclear, as outlined above. Trials for seizures, insomnia and essential tremor are also underway [16].
Another weakness of NAS is that the approved compound brexanolone/ZULRESSO® utilizes a Captisol formulation and 60 h intravenous infusion. Thus, it is limited to inpatient treatment. Development of the oral compound Zuranolone, described above would mitigate this weakness [4, 17, 18]. However, the maintenance of therapeutic effect of Zuranolone has emerged as a contentious issue [19].
Opportunities
NAS in clinical development are similar to (Zuranolone) or identical to (brexanolone/allopregnanolone) endogenous neurosteroids. One opportunity for NAS that could circumvent formulation challenges is the targeting of enzymatic steps from cholesterol to allopregnanolone production. In principle, drugs or genetic therapies could be developed to alter neurosteroidogenesis, allowing the periphery and/or brain to produce its own drug. Another opportunity that exploits endogenous synthetic machinery is precursor loading. Systemic administration of the progesterone results in rapid anesthesia, attributable to progesterone’s conversion to allopregnanolone [20]. Precursors themselves could have beneficial effects [21] and thus could synergize with metabolites to maximize therapeutic impact.
Another opportunity involves the large catalog of synthetic NAS with GABAAR actions. The Covey group has designed and synthesized hundreds of analogs over the last few decades, and other groups are interested as well [22, 23]. An effective screening regimen could unearth compounds within this group that may have superior properties to the compounds tested to date, especially given the potential tunability of effects at GABAARs [24], a leading candidate to mediate benefit.
The present review focuses on mood disorders, including postpartum depression and major depressive disorder, since clinical trials are most advanced for these disorders. However, we note that GABAAR inhibition has been implicated in several other neuropsychiatric disorders (e.g., schizophrenia, epilepsy, intellectual and developmental disorders, substance use disorders). The neuroprotective properties of neurosteroids described below also suggest potential use in neurodegenerative disorders, such as Alzheimer’s disease, stroke, and traumatic brain injury. Compounds with similar GABAA receptor subunit selectivity to NAS have also been of interest for sleep [16]. Fittingly, there is interest in NAS and clinical trials are underway for many of these indications [16, 25,26,27] (see also ClinicalTrials.gov).
Threats
Targeting a transmitter system as widespread as the GABA system has inherent risk. By titrating dosage and fostering subunit selectivity or other unique mechanisms (described further below), detrimental effects such as sedation and abuse liability, which plague other GABAAR PAMs such as barbiturates and benzodiazepines, may be avoided. However, this magic shotgun approach [28] is not fully proven for NAS. In fact, allopregnanolone and pregnanolone in preclinical studies have shown reinforcing properties and indications of abuse liability, with tolerance related to the dosage and likely dosing frequency [29].
Use of brexanolone in women with postpartum depression increased dizziness and somnolence over the placebo arm and 4% of patients developed loss of consciousness. These side effects are worrisome in that they echo effects of benzodiazepines, with attendant abuse liability and other drawbacks. On the other hand, many patients in the clinical trial were co-medicated with other psychotropics, which could have contributed to the side effect. Caution has been encouraged in interpreting the “antidepressant” effects of brexanolone. Without additional study, antidepressant benefit is readily confused with reinforcing drug properties or effects on sleep (which can have direct impact on mood), a distinction that the clinical trials were not designed to make [30]. Dependence and withdrawal also remain understudied. Thus, while there is reason to think that GABAAR effects for benzodiazepines and NAS will differ based on the sub-classes of receptors targeted, liabilities of NAS remain unclear.
Comparison of NAS with other GABAAR PAMs
Allopregnanolone and other NAS may achieve mood altering effects through their action as GABAAR PAMs [22, 31,32,33]. To contextualize allopregnanolone’s difference from other GABAAR PAMs, we first briefly review GABAAR composition, anatomical localization, physiological/biophysical properties, and pharmacological selectivity. We focus on two populations of receptors, defined by receptor subunit differences that may be relevant to allopregnanolone selectivity.
GABAARs are heteropentameric chloride channels that are activated by the neurotransmitter GABA and are resident on the plasma membrane of virtually all neurons in the CNS. They play an essential role in maintaining circuit function [34,35,36,37]. In general, the opening of the chloride channel by GABA binding promotes inhibition through chloride influx through the channel. Dysfunction of GABAARs has been implicated in the pathogenesis of neurological and neuropsychiatric diseases including epilepsy, autism, and depression [38,39,40]. Nineteen GABAAR subunit genes exist (α1-6, β1-3, γ1-3, δ, ε, ρ1-3, π, and θ); however, most native receptors are composed of two α, two β, and a variable fifth subunit that is not required for channel activation [41, 42]. This fifth subunit is most typically a γ (usually γ2) or δ subunit. Subunit combination, including α and β subvariants but especially the fifth γ2 or δ subunit, strongly influences anatomical, physiological and pharmacologic properties of the receptor.
γ2-containing receptors are expressed in many cell types, but δ expression is restricted to relatively few cell classes, including pivotal circuit relays such as dentate granule neurons of the hippocampal formation, cerebellar granule neurons, and thalamocortical neurons. The subunit is also enriched in parvalbumin-expressing interneurons and some pyramidal cell classes of the neocortex [43]. δ subunits preferentially assemble with α4 or α6 subunits in cell types that express these α subunits [44, 45]. By contrast, in δ-expressing interneurons, the subunit appears to pair with the more ubiquitous α1 subunit since neither α4 nor α6 is expressed in this cell class [46,47,48,49].
Regarding sub-cellular localization, GABAARs containing a γ2 subunit dominate occupation of synaptic territory opposite GABAergic presynaptic terminals. The trafficking of γ2-containing receptors includes interaction of γ2 with anchoring proteins at the synapse [50,51,52]. By contrast, δ-containing receptors are excluded from synapses and occupy perisynaptic and extrasynaptic locations [53].
These differences in sub-cellular location (synaptic vs. perisynaptic) also appear to partly drive differences in function. Receptors containing γ2 tend to activate and decay more quickly than receptors containing δ subunits, reflecting in part different distances from release sites but also biophysical differences [54,55,56]. δ-Containing receptors typically exhibit responsivity to lower GABA concentrations than γ2-containing receptors (i.e., they have a lower EC50 for GABA) but blunted maximum responses to GABA (lower efficacy) [54, 57]. This lower efficacy may be important to the pharmacological effects of NAS and other PAMs, described further below.
The difference in location and higher sensitivity to GABA give δ-containing receptors a preferential role in generating a low-amplitude, tonic inhibition driven by low concentrations of ambient GABA. Despite the small amplitude of tonic current, tonic inhibition can robustly influence neuronal excitability [58,59,60]. By contrast, phasic (synaptic) inhibition is mainly generated by γ2-containing receptors at synapses.
Allopregnanolone can modulate both tonic and phasic inhibition through its ability to potentiate many different populations of GABAARs, but it may have preferential effects based on the properties of extrasynaptic δ-containing receptors [61, 62]. Here we compare allopregnanolone actions, including subunit selectivity, with actions of barbiturates and classical benzodiazepines, which perhaps represent more familiar classes of GABAAR PAMs.
Much evidence suggests that allopregnanolone may selectively modulate δ-containing receptors, thereby primarily enhancing tonic current at low concentrations presumably relevant for effects on mood [59, 61, 63]. This would make NAS effects distinct from benzodiazepines, which require a γ subunit [64, 65]. First, mice deficient in δ subunits show loss of hypnotic sensitivity (sleep time) to NAS but not to other GABAAR PAMs [66]. Second, deletion of the δ subunit reduces tonic current in many relevant neuronal classes [59].
On the other hand, some evidence from concatemeric receptors and pharmacoresistant receptors in mice suggests that δ-containing receptors do not preferentially underlie the actions of allopregnanolone [62, 67]. Furthermore, the preferential effect of NAS results from the low efficacy of GABA acting at δ containing receptors [68], and other PAMs including propofol, etomidate, and pentobarbital have this same characteristic [54, 69,70,71]. In summary, effects of allopregnanolone on GABAARs are distinct from benzodiazepines, but the distinction from other GABA-active anesthetics remains unclear.
If GABAAR PAM activity is important for clinical benefit, another approach to developing evermore improved NAS compounds to understand the binding sites on GABAAR for this class of compound. NAS appear to access transmembrane sites on the GABAAR multimer [72,73,74], with several functional intra-subunit and inter-subunit sites identified [75] (Fig. 1). Because the functional impact of binding each site differs [24], there is promise for a rich pharmacology that exploits differential access to the sites to customize pharmacodynamic effects. However, from the perspective of the receptor, relative affinities of the multiple sites for neurosteroids are not yet clear. However, mutagenesis studies have shown that a single neurosteroid site may suffice for full potentiation [76]. As broad spectrum potentiators, neurosteroids show relatively little difference in affinity for receptors of different subunit composition. As described elsewhere, differences attributed to the incorporation of the δ subunit appear mainly related to the low efficacy of GABA; NAS mainly increase agonist efficacy [54, 68].

Allopregnanolone (mesh structure) in the three neurosteroid binding sites (b–d) identified by photolabeling with photolabile NAS analogs. a (center): the six sites identified by photolabeling are grouped into three clusters: inter-subunit sites β3(+)/α1(−) (brown circle, detailed in d), β3 intra-subunit sites (blue circle, detailed in b), and α1 intra-subunit sites (red circle, detailed in c). Residues photolabeled by different analogs are colored red, green, and blue respectively. This figure is reproduced from [75].
From the ligand perspective, physiological pharmacology experiments have revealed important components of the pharmacophore that are important for full activity [31, 77,78,79]. These include a 3α hydrogen bond donor and a hydrogen bond acceptor at carbon 11 of the steroid. Interestingly, activity at GABAA receptors is not markedly altered by stereochemistry of the reduction at carbon 5. Potency and to some degree efficacy of steroid effects can be manipulated by alterations to many other regions of the steroid molecule.
Possible non-GABAAR targets of relevance
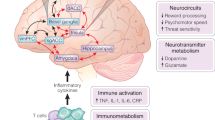
There is reason to think that NAS benefit may arise at least in part through non-GABAergic mechanisms. NAS have ameliorative behavioral effects in female mice lacking δ subunits [80]. Further, other GABAAR PAMs described above, including those with very similar GABAAR sub-selectivity, are not known to have the same clinical benefit as NAS. In direct studies, some NAS appear to engage other targets, as described below. Therefore, non-GABAAR targets should be considered when evaluating mechanistic underpinnings of benefit. We briefly review other ion channel targets, intracellular kinases, and protein expression mechanisms, with particular focus on anti-inflammatory mechanisms (Fig. 2).

Depicted is a neuron (left) and microglial cell (right). Some targets may be found in multiple cell classes, so the relevant cell type is not known in all cases. See text for details.
Some NAS classes negatively modulate NMDARs [81,82,83,84,85], which could impart clinical benefit akin to the NMDAR antagonist ketamine. A few compounds possess GABAAR PAM effects in combination with NMDAR negative modulation [84, 85]. It is tempting to consider that this constellation of effects might yield an advantageous brexanolone-like plus ketamine-like double hit. However, to date pharmacokinetic properties of such compounds have proven limiting.
Other work demonstrated that some NAS influence low-voltage activated (T-type) calcium channels. Allopregnanolone is an LVA inhibitor at sufficiently high concentrations [86, 87]. Medicinal chemistry efforts have resulted in NAS selectivity for LVA calcium channels over GABAARs [88]. It remains unclear how combined effects at LVA channels and GABAARs affect behaviors relevant to mood.
NAS can also exert indirect effects on numerous signaling pathways, including corticotropin releasing hormone (CRH) and monoaminergic signaling which may contribute to the antidepressant actions of these compounds. Previous reviews have addressed this topic in depth [89], therefore, we will provide an overview of potential indirect mechanisms contributing to the antidepressant effects of NAS. Dysfunction of the hypothalamic-pituitary-adrenal (HPA) axis and excessive glucocorticoid and CRH signaling has been implicated in affective disorders, including depression. Allopregnanolone has been shown to normalize HPA axis function and restore homeostasis in CRH signaling (for review see [89]). The HPA axis is under tight GABAergic regulation which may underlie the ability of NAS to normalize neuroendocrine stress signaling. Similarly, the GABAergic system also regulates the monoaminergic system. The monoamine hypothesis of depression remains the prevailing theory of the underlying pathophysiology of depression. However, emerging evidence implicates the GABAergic system in contributing to monoamine deficits associated with depression and the antidepressant effects of SSRIs. SSRIs have been shown to increase the levels of NAS which has been proposed to contribute to the antidepressant effects of these compounds [90]. Recently, allopregnanolone has been demonstrated to influence dopamine release [91]. Given the crosstalk between GABAergic, CRF, and monoaminergic signaling, it is difficult to tease apart the exact mediators of the antidepressant actions of NAS.
Fluorescent NAS and photoaffinity labels have shown that NAS accumulate readily within cells, interacting with Golgi and perhaps other organelles in a structurally selective manner [92, 93]. One might hypothesize that these intracellular targets could contribute to beneficial effects. NAS targets may include proteins important for cellular stress responses, including autophagy and neuroinflammation [94,95,96,97,98,99]. Unnatural enantiomers of allopregnanolone and related NAS can engage intracellular pathways without substantially affecting ion channel targets [96, 100,101,102,103,104]. These tool compounds offer the possibility of parsing behavioral effects related to different molecular mechanisms.
Anti-inflammatory effects of NAS are of interest given hypothesized roles for inflammation in dysfunction in neuropsychiatric illness [105,106,107,108]. Allopregnanolone and progesterone exhibit anti-inflammatory effects in preclinical models, [109] and clinical studies have recently demonstrated a correlation between anti-inflammatory effect of brexanolone and clinical benefit [99]. These effects may occur via toll-like receptors-4 and -7 (TLR4/7), including binding the TLR adapter protein MyD88, but not TLR2 and TLR3 [97, 98, 110]. Pregnenolone promotes loss of TIRAP, an adapter protein for TLR2 and TLR4, which reduces secretion of inflammatory cytokines [110]. Microtubules are another possible locus for NAS anti-inflammatory effects. NAS photolabeling compounds bind the microtubule protein, β-tubulin, at cysteine-354, a site for colchicine, a compound with anti-inflammatory effects [93]. Further, NAS can alter the function of microtubules, and these effects contribute to antidepressant-like effects in rodents [111].
Some NAS can promote expression and trafficking of GABAAR subunits, including extrasynaptic receptor subunits affecting tonic inhibition. Effects may involve phosphorylation by protein kinase C (PKC) and other kinases [112]. Interestingly, increased receptor expression is triggered by allopregnanolone but not by the closely related synthetic NAS, ganaxolone [113]. Membrane progesterone receptors (mPRs) represent a class of G-protein-coupled receptors in the CNS that are activated by allopregnanolone and linked to phosphorylation of GABAAR subunits [114]. The NAS ORG OD 02-2 may selectively activate mPRs (without acute effects on GABAARs). ORG OD 02-2 increases GABAAR phosphorylation via cyclic AMP-dependent protein kinase (PKA) and PKC, resulting in increased GABAAR expression and enhanced tonic inhibition [115].
Allopregnanolone and related NAS also promote expression of nuclear receptors, including pregnane xenobiotic receptors (PXR) and liver xenobiotic receptors (LXR) [105, 116,117,118,119]. PXR/LXR may modulate steroid synthesis, among other pathways, to regulate cellular stress mechanisms [120], thereby contributing to anti-inflammatory and neuroprotective actions [119]. NAS interaction with PXR/LXR could also promote BDNF synthesis and signaling [121,122,123]. Downstream, the BDNF receptor TrkB can interact with antidepressant drugs through cholesterol [124]. These observations are of potential import because of the longstanding observations that BDNF and the plasticity induced by this neurotrophic factor have a role in the antidepressant effects of other treatments [125, 126].
Taken together, post-translational modification and protein expression changes represent attractive mechanisms to explain the durable, persisting effects on brain function observed in patients.
NAS modulate emotional processing networks and behavioral states
Exogenous NAS have been shown to exert robust antidepressant and anxiolytic effects in both clinical and preclinical studies [22, 127, 128]. The role of neurosteroids in modulating affective states has been inferred from studies investigating the impact of neurosteroid deficits on mood. Levels of 5α-reductase, key rate limiting enzymes involved in neurosteroidogenesis, are reduced in the prefrontal cortex of individuals with major depressive disorder [129].
Cholesterol represents the upstream precursor of all neurosteroids, and the first step in neurosteroidogenesis is translocation of cholesterol from the plasma membrane to mitochondria by the actions of protein complexes that remain somewhat mysterious [130]. Both steroidogenic acute regulatory (StAR) proteins and translocator protein (TSPO), along with voltage-dependent anion channel (VDAC) interacting proteins, have been implicated. In the mitochondrion, P450 side-chain cleavage enzyme converts cholesterol to pregnenolone. The 3β-hydroxysteroid dehydrogenase enzyme converts pregnenolone to progesterone, which migrates across mitochondrial membranes to the cytoplasm, where 5α-reductase then 3α-hydroxysteroid dehydrogenase catalyze synthesis of allopregnanolone. As noted, 5α-reductase enzymes (of which there are three isoforms) are rate limiting so are a favored target for therapeutic development. Of experimental interest finasteride, dutasteride, and related compounds are 5α-reductase inhibitors. Ligands of TSPO are also enjoying interest as therapeutic agents in depression due to their potential impact on neurosteroidogenesis [131, 132].
One of the limitations impeding our knowledge regarding the role of endogenous neurosteroids is the difficulty in measuring endogenous neurosteroid levels and the inability to manipulate the enzymes involves in neurosteroid synthesis. Inhibition of 5α-reductase pharmacologically with finasteride induces behavioral deficits in animal models (for review see [128]). Human men treated with finasteride for male pattern baldness or benign prostatic hyperplasia can develop a condition termed post-finasteride syndrome. The syndrome is characterized by sexual dysfunction and negatively impacted mood [133], which is thought to involve impaired endogenous neurosteroidogenesis [134]. A recent study has demonstrated that knockdown of 5α-reductase type 1 and 2 in the basolateral amygdala (BLA) induces behavioral deficits in preclinical models with potential relevance to anxiety and depression [135]. These data suggest that endogenous 5α-reduced neurosteroids may play a role in setting a baseline affective tone.
As such, impaired endogenous neurosteroid synthesis has been implicated in the pathophysiology of both depression and anxiety (for review see [127]). A rapid decline in the endogenous levels of allopregnanolone during the peripartum period is thought to increase vulnerability to mood disorders during this period (for review see [136]). Thus, the mechanistic link to the established antidepressant effects of exogenous allopregnanolone analogs for the treatment of postpartum depression is clear. Treatment with exogenous allopregnanolone analogs is also showing robust antidepressant effects in major depressive disorder in which the mechanistic link is less clear. Recent preclinical studies have demonstrated that risk factors for psychiatric illnesses, such as chronic stress, alter the capacity for endogenous neurosteroid synthesis in preclinical models [128]. For example, social isolation (for review see [128]) and chronic unpredictable stress [135] decreased endogenous allopregnanolone levels and the levels of key neurosteroidogenic enzymes [137,138,139,140]. These data suggest that exogenous allopregnanolone treatment may exert its antidepressant effects potentially by supplementing deficits in allopregnanolone synthesis associated with the underlying neurobiology of disease.
As summarized above, the therapeutic efficacy of exogenous NAS treatment may be due to the evidence that deficits in endogenous neurosteroid signaling contributes to the underlying pathophysiology of mood disorders [141]. However, the mechanisms through which deficits in endogenous neurosteroidogenesis may contribute to mood disorders and the mechanisms mediating the antidepressant effects of exogenous NAS treatment remains unclear. Despite the fact that we have a good understanding of primary mechanisms of action of neurosteroids, how these mechanisms translate to antidepressant and anxiolytic effects is not clear.
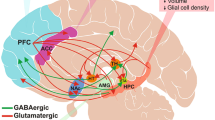
The persistent antidepressant effects observed in clinical trials with brexanolone and zuranolone treatment (see section “Clinical effectiveness of NAS for the treatment of depression”) suggest a mechanism of action other than actions as a GABAAR PAM. These sustained antidepressant effects may be mediated by the other known actions of NAS, such as the impact on autophagy, inflammation, other ion channels, or actions mediated by membrane progesterone receptors (see section “Possible non-GABAAR targets of relevance”). In addition, emerging evidence demonstrates that circuit network states drive behavioral states [142,143,144,145] and suggest that network-level changes play a role in the pathophysiology of psychiatric illnesses [146]. Psychiatric illnesses are increasingly acknowledged to involve network dysfunction [147]. In fact, many different approaches have demonstrated success in using EEG signatures as a diagnostic criterion for depression [148,149,150,151] as well as predict antidepressant response [152,153,154,155]. Commonalities observed in network-level changes associated with depression implicate specific brain regions contributing to the pathophysiology, including altered activity and connectivity within and between regions in the anterior cingulate cortex (ACC), dorsolateral prefrontal cortex, hippocampus, and amygdala (for review see [156]; Fig. 3). Similarly, studies in animal models demonstrate a unique role for the basolateral amygdala in mediating the impact of stress [157,158,159] and the anxiolytic and antidepressant effects of allopregnanolone [160,161,162,163]. These emerging studies point toward a role for NAS in modulating BLA network states to influence affective tone, such that deficits in NAS signaling in the BLA induces behavioral abnormalities and enhanced NAS signaling in the BLA improves behavioral outcomes. Consistent with this conceptual framework, preclinical studies have demonstrated altered network states (oscillations) associated with chronic stress-induced behavioral deficits [160] and the ability of NAS-based antidepressant treatments to alter network states across species [160, 164, 165] and improve behavioral outcomes following chronic stress [160]. It is important to note that the effects of NAS on network and behavioral states are unique from other GABAAR PAMs [160, 165, 166]. Recent evidence demonstrates that chronic stress reduces the capacity for endogenous neurosteroidogenesis [135] and that impaired endogenous neurosteroid signaling is sufficient to mimic the behavioral effects of chronic stress in mice [135]. This foundational preclinical work establishes a mechanism of action of the antidepressant effects of NAS-based treatments on network states and implicate impaired neurosteroidogenesis in the pathophysiological mechanisms underlying mood disorders.

The diagram depicts oscillatory rhythms affected by NAS compounds in brain areas believed to be key to mood.
Future research directions
NAS represent a novel class of rapid-acting antidepressants with a different mechanism of action from previous antidepressant treatments. The robust preclinical and clinical evidence supporting the antidepressant effects of NAS suggest new mechanisms contributing to the underlying neurobiology of depression as well as offers the potential to identify new targets for the next generation of antidepressant treatments, including enantiomers, novel formulations, and targeting endogenous neurosteroid synthesis. This article tempers the enthusiasm for the potential of these rapid-acting antidepressant actions to revolutionize treatment options for patients with a discussion of potential limitations for consideration. The mechanisms whereby NAS exert antidepressant effects are not fully understood but emerging evidence suggests the ability of these compounds to alter network states involved in emotional processing to influence affective states. Finally, the ability of NAS to modulate network and behavioral states relevant to depression is unique to this class of compounds and is unique from other GABA agonists, such as benzodiazepines. These emerging findings elucidate the antidepressant mechanisms of these compounds as well as provide information regarding the underlying neurobiology of disease.
References
Kanes S, Colquhoun H, Gunduz-Bruce H, Raines S, Arnold R, Schacterle A, et al. Brexanolone (SAGE-547 injection) in post-partum depression: a randomised controlled trial. Lancet. 2017;390:480–9.
Meltzer-Brody S, Colquhoun H, Riesenberg R, Epperson CN, Deligiannidis KM, Rubinow DR, et al. Brexanolone injection in post-partum depression: two multicentre, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet. 2018;392:1058–70.
Epperson CN, Rubinow DR, Meltzer-Brody S, Deligiannidis KM, Riesenberg R, Krystal AD, et al. Effect of brexanolone on depressive symptoms, anxiety, and insomnia in women with postpartum depression: pooled analyses from 3 double-blind, randomized, placebo-controlled clinical trials in the HUMMINGBIRD clinical program. J Affect Disord. 2023;320:353–9.
Deligiannidis KM, Meltzer-Brody S, Gunduz-Bruce H, Doherty J, Jonas J, Li S, et al. Effect of zuranolone vs placebo in postpartum depression: a randomized clinical trial. JAMA Psychiatry. 2021;78:951–9.
Deligiannidis KM, Citrome L, Huang MY, Acaster S, Fridman M, Bonthapally V, et al. Effect of zuranolone on concurrent anxiety and insomnia symptoms in women with postpartum depression. J Clin Psychiatry. 2023;84:22m14475.
Gunduz-Bruce H, Silber C, Kaul I, Rothschild AJ, Riesenberg R, Sankoh AJ, et al. Trial of SAGE-217 in patients with major depressive disorder. N Engl J Med. 2019;381:903–11.
Suthoff E, Kosinski M, Arnaud A, Hodgkins P, Gunduz-Bruce H, Lasser R, et al. Patient-reported health-related quality of life from a randomized, placebo-controlled phase 2 trial of zuranolone in adults with major depressive disorder. J Affect Disord. 2022;308:19–26.
Walton N, Maguire J. Allopregnanolone-based treatments for postpartum depression: why/how do they work? Neurobiol Stress. 2019;11:100198.
Reddy DS. Neurosteroid replacement therapy for catamenial epilepsy, postpartum depression and neuroendocrine disorders in women. J Neuroendocrinol. 2022;34:e13028. https://doi.org/10.1111/jne.13028.
Luscher B, Shen Q, Sahir N. The GABAergic deficit hypothesis of major depressive disorder. Mol Psychiatry. 2011;16:383–406. https://doi.org/10.1038/mp.2010.120.
Insel TR. Next-generation treatments for mental disorders. Sci Transl Med. 2012;4:155ps19.
Cressey D. Psychopharmacology in crisis. Nature. 2011. https://doi.org/10.1038/news.2011.367.
Insel TR, Sahakian BJ. A plan for mental illness. Nature. 2012;483:269. https://doi.org/10.1038/483269a.
Rupprecht R, Reul JM, Trapp T, van Steensel B, Wetzel C, Damm K, et al. Progesterone receptor-mediated effects of neuroactive steroids. Neuron. 1993;11:523–30.
Rupprecht R, Holsboer F. Neuroactive steroids: mechanisms of action and neuropsychopharmacological perspectives. Trends Neurosci. 1999;22:410–6. https://doi.org/10.1016/S0166-2236(99)01399-5.
Cerne R, Lippa A, Poe MM, Smith JL, Jin X, Ping X, et al. GABAkines—advances in the discovery, development, and commercialization of positive allosteric modulators of GABAA receptors. Pharm Ther. 2022;234:108035.
Deligiannidis K, Huang MY, Suthoff E, Acaster S, Fridman M, Gunduz-Bruce H, et al. Evaluation of insomnia symptoms in a double-blind, randomized, placebo-controlled phase 3 trial of zuranolone in postpartum depression. Biol Psychiatry. 2021;89:S91.
Arnaud A, Suthoff E, Stenson K, Werneburg B, Hodgkins P, Bonthapally V, et al. Number needed to treat and number needed to harm analysis of the zuranolone phase 2 clinical trial results in major depressive disorder. J Affect Disord. 2021;285:112–9.
ten Doesschate F, van Waarde JA, van Wingen GA. Non-superiority of zuranolone (SAGE-217) at the longer-term. J Affect Disord. 2021;291:329–30.
Reddy DS, Apanites LA. Anesthetic effects of progesterone are undiminished in progesterone receptor knockout mice. Brain Res. 2005;1033:96–101.
Bassani TB, Bartolomeo CS, Oliveira RB, Ureshino RP. Progestogen-mediated neuroprotection in central nervous system disorders. Neuroendocrinology. 2023;113:14–35.
Zorumski CF, Paul SM, Covey DF, Mennerick S. Neurosteroids as novel antidepressants and anxiolytics: GABA-A receptors and beyond. Neurobiol Stress. 2019;11:100196.
Adla SK, Slavikova B, Smidkova M, Tloustova E, Svoboda M, Vyklicky V, et al. Physicochemical and biological properties of novel amide-based steroidal inhibitors of NMDA receptors. Steroids. 2017;117:52–61.
Wang L, Covey DF, Akk G, Evers AS. Neurosteroid modulation of GABA A receptor function by independent action at multiple specific binding sites. Curr Neuropharmacol. 2021;20:886–90.
Hernandez GD, Solinsky CM, Mack WJ, Kono N, Rodgers KE, Wu CY, et al. Safety, tolerability, and pharmacokinetics of allopregnanolone as a regenerative therapeutic for Alzheimer’s disease: a single and multiple ascending dose phase 1b/2a clinical trial. Alzheimers Dement. 2020;6:e12107.
Tomaselli G, Vallée M. Stress and drug abuse-related disorders: the promising therapeutic value of neurosteroids focus on pregnenolone-progesterone-allopregnanolone pathway. Front Neuroendocrinol. 2019;55:100789.
Stein DG, Sayeed I. Repurposing and repositioning neurosteroids in the treatment of traumatic brain injury: a report from the trenches. Neuropharmacology. 2019;147:66–73.
Roth BL, Sheffer DJ, Kroeze WK. Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and schizophrenia. Nat Rev Drug Discov. 2004;3:353–9. https://doi.org/10.1038/nrd1346.
Gauvin DV, Zimmermann ZJ, Baird TJ. Preclinical assessment of abuse liability of biologics: in defense of current regulatory control policies. Regul Toxicol Pharmacol. 2015;73:43–54.
Kleinman RA, Schatzberg AF. Understanding the clinical effects and mechanisms of action of neurosteroids. Am J Psychiatry. 2021;178:221–3. https://doi.org/10.1176/appi.ajp.2020.20020134.
Majewska MD, Harrison NL, Schwartz RD, Barker JL, Paul SM. Steroid hormone metabolites are barbiturate-like modulators of the GABA receptor. Science. 1986;232:1004–7.
Khisti RT, Chopde CT, Jain SP. Antidepressant-like effect of the neurosteroid 3α-hydroxy-5α-pregnan-20-one in mice forced swim test. Pharm Biochem Behav. 2000;67:137–43.
Paul SM, Purdy RH. Neuroactive steroids. FASEB J. 1992;6:2311–22.
Haider B, Duque A, Hasenstaub AR, McCormick DA. Neocortical network activity in vivo is generated through a dynamic balance of excitation and inhibition. J Neurosci. 2006;26:4535–45.
Mann EO, Mody I. Control of hippocampal gamma oscillation frequency by tonic inhibition and excitation of interneurons. Nat Neurosci. 2010;13:205–12.
Mann EO, Paulsen O. Role of GABAergic inhibition in hippocampal network oscillations. Trends Neurosci. 2007;30:343–9.
Sieghart W. Structure, pharmacology, and function of GABAA receptor subtypes. Adv Pharm. 2006;54:231–63.
Eichler SA. E-I balance and human diseases—from molecules to networking. Front Mol Neurosci. 2008;1:2. https://doi.org/10.3389/neuro.02.002.2008.
van Bokhoven H, Selten M, Nadif Kasri N. Inhibitory control of the excitatory/inhibitory balance in psychiatric disorders. F1000Research. 2018;7:23. https://doi.org/10.12688/f1000research.12155.1.
Fee C, Banasr M, Sibille E. Somatostatin-positive Gamma-aminobutyric acid interneuron deficits in depression: cortical microcircuit and therapeutic perspectives. Biol Psychiatry. 2017;82:549–59.
Sieghart W, Fuchs K, Tretter V, Ebert V, Jechlinger M, Höger H, et al. Structure and subunit composition of GABAA receptors. Neurochem Int. 1999;34:379–85.
McKernan RM, Whiting PJ. Which GABAA-receptor subtypes really occur in the brain? Trends Neurosci. 1996;19:139–43.
Pirker S, Schwarzer C, Wieselthaler A, Sieghart W, Sperk G. GABAA receptors: immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience. 2000;101:815–50.
Sur C, Farrar SJ, Kerby J, Whiting PJ, Atack JR, McKernan RM. Preferential coassembly of α4 and δ subunits of the γ-aminobutyric acid A receptor in rat thalamus. Mol Pharm. 1999;56:110–5.
Jones A, Korpi ER, McKernan RM, Pelz R, Nusser Z, Mäkelä R, et al. Ligand-gated ion channel subunit partnerships: GABAA receptor α6 subunit gene inactivation inhibits δ subunit expression. J Neurosci. 1997;17:1350–62.
Glykys J, Peng Z, Chandra D, Homanics GE, Houser CR, Mody I. A new naturally occurring GABAA receptor subunit partnership with high sensitivity to ethanol. Nat Neurosci. 2007;10:40–8.
Hu H, Gan J, Jonas P. Fast-spiking, parvalbumin+ GABAergic interneurons: from cellular design to microcircuit function. Science. 2014;345:1255263.
Milenkovic I, Vasiljevic M, Maurer D, Höger H, Klausberger T, Sieghart W. The parvalbumin-positive interneurons in the mouse dentate gyrus express GABAA receptor subunits alpha1, beta2, and delta along their extrasynaptic cell membrane. Neuroscience. 2013;254:80–96.
McDonald AJ, Mascagni F. Parvalbumin-containing interneurons in the basolateral amygdala express high levels of the α1 subunit of the GABAA receptor. J Comp Neurol. 2004;473:137–46.
Martenson JS, Yamasaki T, Chaudhury NH, Albrecht D, Tomita S. Assembly rules for GABAA receptor complexes in the brain. Elife. 2017;6:e27443.
Essrich C, Lorez M, Benson JA, Fritschy JM, Luscher B. Postsynaptic clustering of major GABAA receptor subtypes requires the g2 subunit and gephyrin. Nat Neurosci. 1998;1:563–71.
Schweizer C, Balsiger S, Bluethmann H, Mansuy IM, Fritschy JM, Mohler H, et al. The gamma 2 subunit of GABA(A) receptors is required for maintenance of receptors at mature synapses. Mol Cell Neurosci. 2003;24:442–50.
Wei W, Zhang N, Peng Z, Houser CR, Mody I. Perisynaptic localization of δ subunit-containing GABAA receptors and their activation by GABA spillover in the mouse dentate gyrus. J Neurosci. 2003;23:10650–61.
Brown N, Kerby J, Bonnert TP, Whiting PJ, Wafford KA. Pharmacological characterization of a novel cell line expressing human α4β3δ GABAA receptors. Br J Pharm. 2002;136:965–74.
Sun M, Ziolkowski L, Mennerick S. δ subunit‐containing GABA A IPSCs are driven by both synaptic and diffusional GABA in mouse dentate granule neurons. J Physiol. 2020;598:1205–21.
Haas KF, Macdonald RL. GABAA receptor subunit gamma2 and delta subtypes confer unique kinetic properties on recombinant GABAA receptor currents in mouse fibroblasts. J Physiol. 1999;514:27–45.
Feng HJ, Macdonald RL. Multiple actions of propofol on αβγ and αβδ GABAA receptors. Mol Pharm. 2004;66:1517–24.
Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABA A receptors. Nat Rev Neurosci. 2005;6:215–29. https://doi.org/10.1038/nrn1625.
Lee V, Maguire J. The impact of tonic GABAA receptor-mediated inhibition on neuronal excitability varies across brain region and cell type. Front Neural Circuits. 2014;8:3.
Brickley SG, Mody I. Extrasynaptic GABA A receptors: their function in the CNS and implications for disease. Neuron. 2012;73:23–34. https://doi.org/10.1016/j.neuron.2011.12.012.
Herd MB, Belelli D, Lambert JJ. Neurosteroid modulation of synaptic and extrasynaptic GABAA receptors. Pharm Ther. 2007;116:20–34.
Lu X, Zorumski CF, Mennerick S. Lack of neurosteroid selectivity at δ vs. γ2-containing GABAA receptors in dentate granule neurons. Front Mol Neurosci. 2020;13:6.
Belelli D, Lambert JJ. Neurosteroids: endogenous regulators of the GABAA receptor. Nat Rev Neurosci. 2005;6:565–75.
Rudolph U, Mohler H. GABA-based therapeutic approaches: GABAA receptor subtype functions. Curr Opin Pharm. 2006;6:18–23.
Sigel E, Steinmann ME. Structure, function, and modulation of GABAA receptors. J Biol Chem. 2012;287:40224–31. https://doi.org/10.1074/jbc.R112.386664.
Mihalek RM, Banerjee PK, Korpi ER, Quinlan JJ, Firestone LL, Mi ZP, et al. Attenuated sensitivity to neuroactive steroids in g-aminobutyrate type A receptor d subunit knockout mice. Proc Natl Acad Sci USA. 1999;96:12905–10.
Shu HJ, Bracamontes J, Taylor A, Wu K, Eaton MM, Akk G, et al. Characteristics of concatemeric GABAA receptors containing α4/δ subunits expressed in Xenopus oocytes. Br J Pharm. 2012;165:2228–43.
Bianchi MT, Macdonald RL. Neurosteroids shift partial agonist activation of GABAA receptor channels from low- to high-efficacy gating patterns. J Neurosci. 2003;23:10934–43.
Meera P, Olsen RW, Otis TS, Wallner M. Etomidate, propofol and the neurosteroid THDOC increase the GABAA efficacy of recombinant a4b3d and a4b3 GABAA receptors expressed in HEK cells. Neuropharmacology. 2009;56:155–60.
Ahring PK, Bang LH, Jensen ML, Strøbæk D, Hartiadi LY, Chebib M, et al. A pharmacological assessment of agonists and modulators at α4β2γ2 and α4β2δ GABAA receptors: the challenge in comparing apples with oranges. Pharm Res. 2016;111:563–76.
Feng HJ, Bianchi MT, Macdonald RL. Pentobarbital differentially modulates α1β3δ and α1β3γ2L GABAA receptor currents. Mol Pharm. 2004;66:988–1003.
Akk G, Shu HJ, Wang C, Steinbach JH, Zorumski CF, Covey DF, et al. Neurosteroid access to the GABAA receptor. J Neurosci. 2005;25:11605–13.
Hosie AM, Clarke L, da Silva H, Smart TG. Conserved site for neurosteroid modulation of GABAA receptors. Neuropharmacology. 2009;56:149–54.
Hosie AM, Wilkins ME, da Silva HMA, Smart TG. Endogenous neurosteroids regulate GABAA receptors via two discrete transmembrane sites. Nature. 2006;444:486–9.
Chen ZW, Bracamontes JR, Budelier MM, Germann AL, Shin DJ, Kathiresan K, et al. Multiple functional neurosteroid binding sites on GABAAreceptors. PLoS Biol. 2019;17:e3000157.
Bracamontes J, McCollum M, Esch C, Li P, Ann J, Steinbach JH, et al. Occupation of either site for the neurosteroid allopregnanolone potentiates the opening of the GABAA receptor induced from either transmitter binding site. Mol Pharm. 2011;80:79–86.
Phillipps GH. Structure-activity relationships in steroidal anaesthetics. J Steroid Biochem. 1975;6:607–13.
Callachan H, Cottrell GA, Hather NY, Lambert JJ, Nooney JM, Peters JA. Modulation of the GABAA receptor by progesterone metabolites. Proc R Soc Lond B Biol Sci. 1987;231:359–69.
Covey DF, Evers AS, Mennerick S, Zorumski CF, Purdy RH. Recent developments in structure–activity relationships for steroid modulators of GABAA receptors. Brain Res Rev. 2001;37:91–7.
Melón L, Hammond R, Lewis M, Maguire J. A novel, synthetic, neuroactive steroid Is effective at decreasing depression-like behaviors and improving maternal care in preclinical models of postpartum depression. Front Endocrinol. 2018;9:703.
Korinek M, Kapras V, Vyklicky V, Adamusova E, Borovska J, Vales K, et al. Neurosteroid modulation of N-methyl-D-aspartate receptors: molecular mechanism and behavioral effects. Steroids. 2011;76:1409–18.
Vyklicky V, Smejkalova T, Krausova B, Balik A, Korinek M, Borovska J, et al. Preferential inhibition of tonically over phasically activated NMDA receptors by pregnane derivatives. J Neurosci. 2016;36:2161–75.
Park-Chung M, Wu FS, Purdy RH, Malayev AA, Gibbs TT, Farb DH. Distinct sites for inverse modulation of N-methyl-D-aspartate receptors by sulfated steroids. Mol Pharm. 1997;52:1113–23.
Mennerick S, Zeng CM, Benz A, Shen W, Izumi Y, Evers AS, et al. Effects on γ-aminobutyric acid GABAA receptors of a neuroactive steroid that negatively modulates glutamate neurotransmission and augments GABA neurotransmission. Mol Pharm. 2001;60:732–41.
Ziolkowski L, Mordukhovich I, Chen DM, Chisari M, Shu HJ, Lambert PM, et al. A neuroactive steroid with a therapeutically interesting constellation of actions at GABAA and NMDA receptors. Neuropharmacology. 2021;183:108358.
Todorovic SM, Pathirathna S, Brimelow BC, Jagodic MM, Ko SH, Jiang X, et al. Reduced neuroactive steroids are novel voltage-dependent blockers of T-Type Ca2+ channels in rat sensory neurons in vitro and potent peripheral analgesics in vivo. Mol Pharm. 2004;66:1223–35.
Atluri N, Joksimovic SM, Oklopcic A, Milanovic D, Klawitter J, Eggan P, et al. A neurosteroid analogue with T-type calcium channel blocking properties is an effective hypnotic, but is not harmful to neonatal rat brain. Br J Anaesth. 2018;120:768–78.
Manzella FM, Covey DF, Jevtovic-Todorovic V, Todorovic SM. Synthetic neuroactive steroids as new sedatives and anaesthetics: back to the future. J Neuroendocrinol. 2022;34:e13086.
Boero G, Porcu P, Morrow AL. Pleiotropic actions of allopregnanolone underlie therapeutic benefits in stress-related disease. Neurobiol Stress. 2019;12:100203.
Uzunova V, Sheline Y, Davis JM, Rasmusson A, Uzunov DP, Costa E, et al. Increase in the cerebrospinal fluid content of neurosteroids in patients with unipolar major depression who are receiving fluoxetine or fluvoxamine. Proc Natl Acad Sci USA. 1998;95:3239–44.
Dornellas A, Macedo GC, McFarland MH, Gómez-A A, O'Buckley TK, Da Cunha C, et al. Allopregnanolone decreases evoked dopamine release differently in rats by sex and estrous stage. Front Pharm. 2021;11:608887.
Jiang X, Shu HJ, Krishnan K, Qian M, Taylor AA, Covey DF, et al. A clickable neurosteroid photolabel reveals selective Golgi compartmentalization with preferential impact on proximal inhibition. Neuropharmacology. 2016;108:193–206.
Chen ZW, Chen LH, Akentieva N, Lichti CF, Darbandi R, Hastings R, et al. A neurosteroid analogue photolabeling reagent labels the colchicine-binding site on tubulin: a mass spectrometric analysis. Electrophoresis. 2012;33:666–74.
Kim HN, Lee S-J, Koh J-Y. The neurosteroids, allopregnanolone and progesterone, induce autophagy in cultured astrocytes. Neurochem Int. 2012;60:125–33.
Ishikawa M, Takaseki S, Yoshitomi T, Covey DF, Zorumski CF, Izumi Y. The neurosteroid allopregnanolone protects retinal neurons by effects on autophagy and GABRs/GABAA receptors in rat glaucoma models. Autophagy. 2020;17:1–18. https://doi.org/10.1080/15548627.2020.1731270.
Ishikawa M, Nakazawa T, Kunikata H, Sato K, Yoshitomi T, Krishnan K, et al. The enantiomer of allopregnanolone prevents pressure-mediated retinal degeneration via autophagy. Front Pharm. 2022;0:717.
Balan I, Beattie MC, O’Buckley TK, Aurelian L, Morrow AL. Endogenous neurosteroid (3α,5α)3-hydroxypregnan-20-one inhibits toll-like-4 receptor activation and pro-inflammatory signaling in macrophages and brain. Sci Rep. 2019;9:1220.
Balan I, Aurelian L, Schleicher R, Boero G, O'Buckley T, Morrow AL. Neurosteroid allopregnanolone (3α,5α-THP) inhibits inflammatory signals induced by activated MyD88-dependent toll-like receptors. Transl Psychiatry. 2021;11:145.
Balan I, Patterson R, Boero G, Krohn H, O'Buckley TK, Meltzer-Brody S, et al. Brexanolone therapeutics in post-partum depression involves inhibition of systemic inflammatory pathways. EBioMedicine. 2023;89:104473.
Zorumski CF, Wittmer LL, Isenberg KE, Hu YF, Covey DF. Effects of neurosteroid and benz[e]indene enantiomers on GABAA receptors in cultured hippocampal neurons and transfected HEK-293 cells. Neuropharmacology. 1996;35:1161–8.
Wittmer LL, Hu Y, Kalkbrenner M, Evers AS, Zorumski CF, Covey DF. Enantioselectivity of steroid-induced gamma-aminobutyric acidA receptor modulation and anesthesia. Mol Pharm. 1996;50:1581–6.
Hu Y, Wittmer LL, Kalkbrenner M, Evers AS, Zorumski CF, Covey DF. Neurosteroid analogues. Part 5. Enantiomers of neuroactive steroids and benz[e]indenes: total synthesis, electrophysiological effects on GABAA receptor function and anesthetic actions in tadpoles. J Chem Soc Perkin Trans. 1997;1:3665–71.
VanLandingham JW, Cutler SM, Virmani S, Hoffman SW, Covey DF, Krishnan K, et al. The enantiomer of progesterone acts as a molecular neuroprotectant after traumatic brain injury. Neuropharmacology. 2006;51:1078–85.
Covey DF, Nathan D, Kalkbrenner M, Nilsson KR, Hu Y, Zorumski CF, et al. Enantioselectivity of pregnanolone-induced gamma-aminobutyric acidA receptor modulation and anesthesia. J Pharmacol Exp Ther. 2000;293:1009–16.
Langmade SJ, Gale SE, Frolov A, Mohri I, Suzuki K, Mellon SH, et al. Pregnane X receptor (PXR) activation: a mechanism for neuroprotection in a mouse model of Niemann-Pick C disease. Proc Natl Acad Sci USA. 2006;103:13807–12.
Frank P, Jokela M, Batty GD, Cadar D, Steptoe A, Kivimäki M. Association between systemic inflammation and individual symptoms of depression: a pooled analysis of 15 population-based cohort studies. Am J Psychiatry. 2021;178:1107–18.
Kiecolt-Glaser JK, Derry HM, Fagundes CP. Inflammation: depression fans the flames and feasts on the heat. Am J Psychiatry. 2015;172:1075–91.
Kohler O, Krogh J, Mors O, Benros ME. Inflammation in depression and the potential for anti-inflammatory treatment. Curr Neuropharmacol. 2016;14:732–42.
He J, Evans CO, Hoffman SW, Oyesiku NM, Stein DG. Progesterone and allopregnanolone reduce inflammatory cytokines after traumatic brain injury. Exp Neurol. 2004;189:404–12.
Murugan S, Jakka P, Namani S, Mujumdar V, Radhakrishnan G. The neurosteroid pregnenolone promotes degradation of key proteins in the innate immune signaling to suppress inflammation. J Biol Chem. 2019;294:4596–607.
Bianchi M, Baulieu EE. 3β-Methoxy-pregnenolone (MAP4343) as an innovative therapeutic approach for depressive disorders. Proc Natl Acad Sci USA. 2012;109:1713–8.
Abramian AM, Comenencia-Ortiz E, Vithlani M, Tretter EV, Sieghart W, Davies PA, et al. Protein kinase C phosphorylation regulates membrane insertion of GABAA receptor subtypes that mediate tonic inhibition. J Biol Chem. 2010;285:41795–805.
Modgil A, Parakala ML, Ackley MA, Doherty JJ, Moss SJ, Davies PA. Endogenous and synthetic neuroactive steroids evoke sustained increases in the efficacy of GABAergic inhibition via a protein kinase C-dependent mechanism. Neuropharmacology. 2017;113:314–22.
Thomas P, Pang Y. Membrane progesterone receptors: evidence for neuroprotective, neurosteroid signaling and neuroendocrine functions in neuronal cells. Neuroendocrinology. 2012;96:162–71.
Parakala ML, Zhang Y, Modgil A, Chadchankar J, Vien TN, Ackley MA, et al. Metabotropic, but not allosteric, effects of neurosteroids on GABAergic inhibition depend on the phosphorylation of GABAA receptors. J Biol Chem. 2019;294:12220–30.
Irwin RW, Solinsky CM, Brinton RD. Frontiers in therapeutic development of allopregnanolone for Alzheimer’s disease and other neurological disorders. Front Cell Neurosci. 2014;8:203.
Frye CA, Paris JJ, Walf AA, Rusconi JC. Effects and mechanisms of 3α,5α,-THP on emotion, motivation, and reward functions involving pregnane xenobiotic receptor. Front Neurosci. 2012;5:136.
Fan J, Shimizu Y, Chan J, Wilkinson A, Ito A, Tontonoz P, et al. Hormonal modulators of glial ABCA1 and apoE levels. J Lipid Res. 2013;54:3139–50.
Lamba V, Yasuda K, Lamba JK, Assem M, Davila J, Strom S, et al. PXR (NR1I2): splice variants in human tissues, including brain, and identification of neurosteroids and nicotine as PXR activators. Toxicol Appl Pharm. 2004;199:251–65.
Rong X, Albert CJ, Hong C, Duerr MA, Chamberlain BT, Tarling EJ, et al. LXRs regulate ER stress and inflammation through dynamic modulation of membrane phospholipid composition. Cell Metab. 2013;18:685–97.
Almeida FB, Nin MS, Barros HMT. The role of allopregnanolone in depressive-like behaviors: focus on neurotrophic proteins. Neurobiol Stress. 2020;12:100218.
Serrao JM, Goodchild CS. Alfaxalone anaesthesia increases brain derived neurotrophic factor levels and preserves postoperative cognition by activating pregnane-X receptors: an in vitro study and a double blind randomised controlled trial. BMC Anesthesiol. 2022;22:401.
Shirayama Y, Fujita Y, Oda Y, Iwata M, Muneoka K, Hashimoto K. Allopregnanolone induces antidepressant-like effects through BDNF-TrkB signaling independent from AMPA receptor activation in a rat learned helplessness model of depression. Behav Brain Res. 2020;390:112670.
Casarotto PC, Girych M, Fred SM, Kovaleva V, Moliner R, Enkavi G, et al. Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell. 2021;184:1299–1313.e19.
Zanos P, Gould TD. Mechanisms of ketamine action as an antidepressant. Mol Psychiatry. 2018;23:801–11.
Yang T, Nie Z, Shu H, Kuang Y, Chen X, Cheng J, et al. The role of BDNF on neural plasticity in depression. Front Cell Neurosci. 2020;14:82. https://doi.org/10.3389/fncel.2020.00082.
Schüle C, Nothdurfter C, Rupprecht R. The role of allopregnanolone in depression and anxiety. Prog Neurobiol. 2014;113:79–87.
Chen S, Gao L, Li X, Ye Y. Allopregnanolone in mood disorders: mechanism and therapeutic development. Pharm Res. 2021;169:105682.
Agis-Balboa RC, Guidotti A, Pinna G. 5α-reductase type 1 expression is downregulated in the prefrontal cortex/Brodmann’s area 9 (BA9) of depressed patients. Psychopharmacology. 2014;231:3569–80.
Liang JJ, Rasmusson AM. Overview of the molecular steps in steroidogenesis of the GABAergic neurosteroids allopregnanolone and pregnanolone. Chronic Stress. 2018;2:2470547018818555.
Rupprecht R, Wetzel CH, Dorostkar M, Herms J, Albert NL, Schwarzbach J, et al. Translocator protein (18kDa) TSPO: a new diagnostic or therapeutic target for stress-related disorders? Mol Psychiatry. 2022;27:2918–26.
Shang C, Yao RM, Guo Y, Ding ZC, Sun LJ, Ran YH, et al. Translocator protein–mediated fast-onset antidepressant-like and memory-enhancing effects in chronically stressed mice. J Psychopharmacol. 2020;34:441–51.
Diviccaro S, Melcangi RC, Giatti S. Post-finasteride syndrome: an emerging clinical problem. Neurobiol Stress. 2019;12:100209.
Melcangi RC, Caruso D, Abbiati F, Giatti S, Calabrese D, Piazza F, et al. Neuroactive steroid levels are modified in cerebrospinal fluid and plasma of post-finasteride patients showing persistent sexual side effects and anxious/depressive symptomatology. J Sex Med. 2013;10:2598–603.
Walton NL, Antonoudiou P, Barros L, Dargan T, DiLeo A, Evans-Strong A, et al. Impaired endogenous neurosteroid signaling contributes to behavioral deficits associated with chronic stress. Biol Psychiatry. 2023. https://doi.org/10.1016/J.BIOPSYCH.2023.01.022.
Meltzer-Brody S, Kanes SJ. Allopregnanolone in postpartum depression: role in pathophysiology and treatment. Neurobiol Stress. 2020;12:100212.
Serra M, Mostallino MC, Talani G, Pisu MG, Carta M, Mura ML, et al. Social isolation-induced increase in α4 and δ subunit gene expression is associated with a greater efficacy of ethanol on steroidogenesis and GABAA receptor function. J Neurochem. 2006;98:122–33.
Dong E, Matsumoto K, Uzunova V, Sugaya I, Takahata H, Nomura H, et al. Brain 5α-dihydroprogesterone and allopregnanolone synthesis in a mouse model of protracted social isolation. Proc Natl Acad Sci USA. 2001;98:2849–54.
Serra M, Pisu MG, Littera M, Papi G, Sanna E, Tuveri F, et al. Social isolation-induced decreases in both the abundance of neuroactive steroids and GABA(A) receptor function in rat brain. J Neurochem. 2000;75:732–40.
Agís-Balboa RC, Pinna G, Pibiri F, Kadriu B, Costa E, Guidotti A. Down-regulation of neurosteroid biosynthesis in corticolimbic circuits mediates social isolation-induced behavior in mice. Proc Natl Acad Sci USA. 2007;104:18736–41.
Pinna G. Allopregnanolone, the neuromodulator turned therapeutic agent: thank you, next? Front Endocrinol. 2020;11:236.
Okonogi T, Sasaki T. Theta-range oscillations in stress-induced mental disorders as an oscillotherapeutic target. Front Behav Neurosci. 2021;15:698753.
Headley DB, Paré D. In sync: gamma oscillations and emotional memory. Front Behav Neurosci. 2013;7:170.
Sejnowski TJ, Paulsen O. Network oscillations: emerging computational principles. J Neurosci. 2006;26:1673–6.
Buzsáki G, Draguhn A. Neuronal oscillations in cortical networks. Science. 2004;304:1926–9.
Durstewitz D, Huys QJM, Koppe G. Psychiatric illnesses as disorders of network dynamics. Biol Psychiatry Cogn Neurosci Neuroimaging. 2021;6:865–76.
Bassett DS, Xia CH, Satterthwaite TD. Understanding the emergence of neuropsychiatric disorders with network neuroscience. Biol Psychiatry Cogn Neurosci Neuroimaging. 2018;3:742–53.
Shor O, Yaniv-Rosenfeld A, Valevski A, Weizman A, Khrennikov A, Benninger F. EEG-based spatio-temporal relation signatures for the diagnosis of depression and schizophrenia. Sci Rep. 2023;13:776.
Nandrino JL, Pezard L, Martinerie J, el Massioui F, Renault B, Jouvent R, et al. Decrease of complexity in EEG as a symptom of depression. Neuroreport. 1994;5:528–30.
Wu CT, Huang HC, Huang S, Chen IM, Liao SC, Chen CK, et al. Resting-state EEG signal for major depressive disorder detection: a systematic validation on a large and diverse dataset. Biosensors. 2021;11:499.
Avots E, Jermakovs K, Bachmann M, Päeske L, Ozcinar C, Anbarjafari G. Ensemble approach for detection of depression using EEG features. Entropy. 2022;24:211.
Wu W, Zhang Y, Jiang J, Lucas MV, Fonzo GA, Rolle CE, et al. An electroencephalographic signature predicts antidepressant response in major depression. Nat Biotechnol. 2020;38:439–47.
Zhdanov A, Atluri S, Wong W, Vaghei Y, Daskalakis ZJ, Blumberger DM, et al. Use of machine learning for predicting escitalopram treatment outcome from electroencephalography recordings in adult patients with depression. JAMA Netw Open. 2020;3:e1918377.
Jaworska N, De La Salle S, Ibrahim MH, Blier P, Knott V. Leveraging machine learning approaches for predicting antidepressant treatment response using electroencephalography (EEG) and clinical data. Front Psychiatry. 2019;9:768.
Knott VJ, Telner JI, Lapierre YD, Browne M, Horn ER. Quantitative EEG in the prediction of antidepressant response to imipramine. J Affect Disord. 1996;39:175–84.
Li BJ, Friston K, Mody M, Wang HN, Lu HB, Hu DW. A brain network model for depression: from symptom understanding to disease intervention. CNS Neurosci Ther. 2018;24:1004–19.
Price ME, McCool BA. Structural, functional, and behavioral significance of sex and gonadal hormones in the basolateral amygdala: a review of preclinical literature. Alcohol. 2022;98:25–41.
Daviu N, Bruchas MR, Moghaddam B, Sandi C, Beyeler A. Neurobiological links between stress and anxiety. Neurobiol Stress. 2019;11:100191.
Boyle LM. A neuroplasticity hypothesis of chronic stress in the basolateral amygdala. Yale J Biol Med. 2013;86:117–25.
Antonoudiou P, Colmers P, Walton NL, Weiss GL, Smith AC, Nguyen DP, et al. Allopregnanolone mediates affective switching through modulation of oscillatory states in the basolateral amygdala. Biol Psychiatry. 2022;91:283–93.
Akwa Y, Purdy RH, Koob GF, Britton KT. The amygdala mediates the anxiolytic-like effect of the neurosteroid allopregnanolone in rat. Behav Brain Res. 1999;106:119–25.
Shirayama Y, Muneoka K, Fukumoto M, Tadokoro S, Fukami G, Hashimoto K, et al. Infusions of allopregnanolone into the hippocampus and amygdala, but not into the nucleus accumbens and medial prefrontal cortex, produce antidepressant effects on the learned helplessness rats. Hippocampus. 2011;21:1105–13.
Engin E, Treit D. The anxiolytic-like effects of allopregnanolone vary as a function of intracerebral microinfusion site: the amygdala, medial prefrontal cortex, or hippocampus. Behav Pharmacol. 2007;18:461–70.
Berry SJ, Ewing LL, Coffey DS, Strandberg JD. Effect of age, castration, and testosterone replacement on the development and restoration of canine benign prostatic hyperplasia. Prostate. 1986;9:295–302.
Lambert PM, Ni R, Benz A, Rensing NR, Wong M, Zorumski CF, et al. Non-sedative cortical EEG signatures of allopregnanolone and functional comparators. Neuropsychopharmacology. 2023;48:371–9.
Lambert PM, Lu X, Zorumski CF, Mennerick S. Physiological markers of rapid antidepressant effects of allopregnanolone. J Neuroendocrinol. 2022;34:e13023.
Acknowledgements
The authors thank members of their laboratories and collaborators for input.
Funding
Work in the authors’ laboratories is supported by MH123748 (SM), MH122379 (SM and JLM), R01AA026256 (JLM), R21NS120868 (JLM), R01NS105628 (JLM), R01NS102937 (JLM), R01MH128235 (JLM).
Author information
Authors and Affiliations
Contributions
SM and JLM equally contributed to the conceptualization, organization, and writing of the article.
Corresponding author
Ethics declarations
Competing interests
JLM serves on the Scientific Advisory Board for Sage Therapeutics. SM has no competing interests to disclose.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Maguire, J.L., Mennerick, S. Neurosteroids: mechanistic considerations and clinical prospects. Neuropsychopharmacol. 49, 73–82 (2024). https://doi.org/10.1038/s41386-023-01626-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-023-01626-z
This article is cited by
-
Antidepressants enter cells, organelles, and membranes
Neuropsychopharmacology (2024)
