
Abstract
The pathophysiology of major depressive disorder (MDD) is thought to result from impaired connectivity between key brain networks. Gamma-aminobutyric acid (GABA) is the key inhibitory neurotransmitter in the brain, working primarily via GABAA receptors, with an important role in virtually all physiologic functions in the brain. Some neuroactive steroids (NASs) are positive allosteric modulators (PAMs) of GABAA receptors and potentiate phasic and tonic inhibitory responses via activation of synaptic and extrasynaptic GABAA receptors, respectively. This review first discusses preclinical and clinical data that support the association of depression with diverse defects in the GABAergic system of neurotransmission. Decreased levels of GABA and NASs have been observed in adults with depression compared with healthy controls, while treatment with antidepressants normalized the altered levels of GABA and NASs. Second, as there has been intense interest in treatment approaches for depression that target dysregulated GABAergic neurotransmission, we discuss NASs approved or currently in clinical development for the treatment of depression. Brexanolone, an intravenous NAS and a GABAA receptor PAM, is approved by the U.S. Food and Drug Administration for the treatment of postpartum depression (PPD) in patients 15 years and older. Other NASs include zuranolone, an investigational oral GABAA receptor PAM, and PH10, which acts on nasal chemosensory receptors; clinical data to date have shown improvement in depressive symptoms with these investigational NASs in adults with MDD or PPD. Finally, the review discusses how NAS GABAA receptor PAMs may potentially address the unmet need for novel and effective treatments with rapid and sustained antidepressant effects in patients with MDD.
Similar content being viewed by others


Introduction
Depression is a common and debilitating mental health disorder that negatively impacts a person’s health and functioning and is a leading cause of disability globally [1]. According to the Diagnostic and Statistical Manual of Mental Disorders, 5th edition, text revision, major depressive disorder (MDD) is characterized by ≥2 weeks of at least 5 of the following symptoms (at least 1 of which is depressed mood or anhedonia): depressed mood, anhedonia (loss of interest or pleasure in daily activities), feelings of guilt/low self-esteem, changes in sleep, weight loss or loss of appetite, psychomotor retardation or agitation, fatigue, poor concentration, and suicidal thought that represents a change from previous functioning. The major depressive episode must not be due to another disorder [2]. The global annual age-standardized prevalence of depressive disorders (MDD and dysthymia) in 2019 was estimated to be 3440.1 per 100,000 individuals [3]. Based on the 2021 National Survey on Drug Use and Health (NSDUH) in the United States, the annual prevalence of major depressive episodes in adults was 8.3% and that of major depressive episodes with severe impairment was 5.7% [4].
The etiology of depression has not yet been fully established, but considering the heterogeneity of symptoms, underlying genetics, and treatment responses, it is generally believed that the cause of MDD may be multifactorial. Various genetic and environmental factors (eg, first-degree family members with MDD, adverse childhood experiences, stressful life events) can influence the development of depression [2]. A genome-wide association study of genetic and health records of 1.2 million individuals from 4 separate data banks identified variations in 178 genes that were linked to MDD [5]. Stressful environmental signals can be integrated into the genome via epigenetic mechanisms, such as DNA methylation and histone modifications [6, 7]. Evidence also shows that modified DNA methylation patterns due to stress can affect brain plasticity and emotion in patients with depression [8]. Furthermore, brain imaging studies have shown structural and functional changes associated with depression [9,10,11]. Structural changes include a loss of glial cells, morphologic changes in neurons, and decreased volume in the cingulate cortex, prefrontal cortex (PFC), hippocampus, and amygdala [12]. Functional changes in MDD involve abnormal connectivity in the central executive, default mode, and salience networks, the key neuronal networks controlling mood, arousal, behavior, and cognition [13, 14]. Elevated activity of the hypothalamic-pituitary-adrenal (HPA) axis, neurotrophic deficit, and neuroinflammation are other potential mechanisms proposed for the development of depression [15, 16].
More recent data implicate alterations in the sensorimotor network as being the most consistent factor in depression [17, 18]. Evidence also suggests an association of altered connectivity in the default mode network with postpartum depression (PPD) [19]. These functional networks communicate using several neurotransmitters, including amino acids such as glutamate and gamma-aminobutyric acid (GABA), the primary excitatory and inhibitory neurotransmitters in the brain, respectively, and monoamines such as norepinephrine, dopamine, and serotonin [12, 20].
Hypotheses of depression
Several hypotheses exist for the pathophysiology of depression as it relates to altered neurotransmitter levels. The early monoamine hypothesis, which posits that a core pathophysiologic feature of depression is depletion of brain monoamine neurotransmitters (eg, norepinephrine, dopamine, and serotonin), originated from the observation that most standard-of-care antidepressant therapies (ADTs) can increase extracellular concentrations of these neurotransmitters [15, 16, 21, 22].
The glutamatergic hypothesis of depression suggests an association between elevated glutamate levels and depression [16, 22]. This hypothesis is based on preclinical evidence of the antidepressant effects of N-methyl-D-aspartate (NMDA)-receptor antagonists [23]. Glutamate binds to NMDA receptors, resulting in excitatory neurotransmission [24]. Elevated glutamate levels lead to overactivation of NMDA receptors and induce calcium ion (Ca2+) influx, which in turn may lead to long-term potentiation and long-term depression [25, 26]. However, the evidence for elevated glutamate levels in depression is inconsistent. A postmortem study of adults with MDD reported increased glutamate levels in the frontal cortex of patients with MDD [27], and a proton magnetic resonance spectroscopy study showed increased glutamate levels in the occipital cortex of patients with MDD [28]. Conversely, a meta-analysis of proton magnetic resonance spectroscopy studies examining levels of glutamatergic neurometabolites reported significant decreases in the combined glutamine-plus-glutamate level within the medial PFC in patients with depression compared with healthy volunteers but not in the dorsolateral PFC or medial temporal cortex; differences in glutamate levels between the two groups were not significant in any of these areas [29]. Another meta-analysis showed that glutamate levels were lower within the anterior cingulate cortex of patients with depression compared with healthy volunteers [30].
The GABAergic deficit hypothesis proposes that defects in GABAergic neural inhibition causally contribute to the common phenotypes of MDD, and, conversely, the efficacy of an ADT may be linked to its ability to restore GABAergic neurotransmission [31]. It is based on findings of reduced levels of GABA in the plasma, cerebral cortex, and cerebrospinal fluid (CSF), altered expression and subunit composition of GABAA receptors, and reduced levels of neuroactive steroids (NASs) in CSF among individuals with depression [22, 32]. The hypothesis is also supported by results from multidisciplinary contemporary approaches that combined large-scale genome-wide association studies, postmortem cytology, and functional and structural imaging studies to clarify the shared origins of otherwise biologically heterogeneous MDD [33, 34]. A consistent association was noted between principal neuroimaging findings in individuals with depression and downregulated genetic markers for cortical somatostatin-expressing GABAergic interneurons and astrocytes [33]. Polygenic somatostatin interneuron markers were most expressed in the subgenual anterior cingulate, medial PFC, anterior insula, and temporal lobes, coinciding with brain areas where imaging studies confirmed cortical thinning and aberrant connectivity in individuals with depression [33]. The expression of the MDD-associated somatostatin gene marker SST was found to be significantly negatively correlated with structural differences in cortical regions of individuals with MDD relative to healthy controls [34]. Impaired GABAergic signaling is also thought to be implicated in PPD [35, 36] and bipolar disorder [37, 38].
This narrative review provides preclinical and clinical data supporting the role of GABAergic signaling in the brain and the GABAergic deficit hypothesis of depression, and how modulation of GABA signaling by GABAergic compounds and NASs could potentially be employed to treat depression. We also examine currently approved and investigational NAS therapies and their hypothesized mechanisms of action in depression, supporting the potential link of science and practice for physicians and clinical researchers. The mechanisms and novel therapies reviewed may impact the approach to rapid treatment of MDD with improved long-term outcomes. Publications were selected from the literature based on their relevance to the covered topics (ie, the role of GABA signaling and the potential role of GABAergic compounds and NASs in MDD) and author experience and preference.
GABAergic signaling and normal brain functioning
The complex interplay between excitatory glutamatergic neurons and inhibitory GABAergic neurons is essential to achieving balanced cortical neural activity [39, 40]. The glutamatergic-GABAergic balance is tightly regulated by the biosynthesis, transport, and signaling of the respective neurotransmitters (ie, glutamate and GABA) in the central nervous system (CNS) [39, 40]. The biosynthesis of glutamate and GABA are interrelated via the glutamate/GABA-glutamine cycle [41]. Briefly, glutamatergic neurons release glutamate via synaptic vesicles into the synaptic cleft, where it is taken up by astrocytes and converted to glutamine. Glutamine is transported back to glutamatergic neurons, hydrolyzed to glutamate, and repackaged into synaptic vesicles [41]. GABAergic neurons release GABA into the synapse, where it is taken up by astrocytes and ultimately converted to glutamine. Glutamine is then transported to GABAergic neurons, where it is converted to glutamate by glutaminase and then to GABA by glutamate decarboxylase [42].
Physiologic role of GABA and GABAergic neurons
Excitatory glutamatergic and inhibitory GABAergic neurons predominantly communicate through synaptic interactions [43]. GABA is present primarily in local interneurons, but also in long projection neurons in the PFC, anterior cingulate cortex, amygdala, nucleus accumbens, ventral tegmental area, and the hippocampus—the regions functionally associated with decision-making, cognition, intelligence, memory, sleep, emotions, motivation, and pleasure [12, 44,45,46,47]. GABA plays an important role in neuronal proliferation, migration, differentiation, and preliminary circuit-building during brain development [44, 48] and is implicated in the development of interstitial neurons in white matter and oligodendrocytes [44]. GABA also regulates connectivity between the major brain functional networks (eg, default mode and executive control networks) [49].
GABAergic projection neurons are widely distributed throughout the brain and make dense connections between brain regions involved in mood regulation and reward learning (Fig. 1) [12]; GABAergic interneurons play a vital role in local neural circuitry and activity [50]. Altogether, GABAergic neurons play an important role in regulating various physiologic brain functions, such as learning and memory, sensorimotor processing, and neuroplasticity [51]. GABAergic neurons also terminate the innate physiologic stress response by regulating the HPA axis and restoring homeostasis, suggesting the critical role of GABAergic signaling in normal brain function [45].

Glutamatergic projections illustrated here include those from the frontal cortex to the anterior cingulate cortex (ACC), thalamus (TH), ventral tegmental area (VTA), hippocampus (HPC) and nucleus accumbens (NAc); from hippocampus to hypothalamus (HT), VTA, NAc and PFC; and from amygdala to HT, ACC and NAc. Major GABAergic projections are from HT to the occipital and parietal cortex, HPC to PFC, and from NAc to TH and VTA. Only a subset of known interconnections is shown here. Depression is associated with reduced brain volume and decreased glial cell density in various brain regions, including ACC, PFC, hippocampus, and amygdala. (Figure is reproduced from Sarawagi et al. 2021 [12] according to the terms of the Creative Commons Attribution License [CC BY]).
GABA receptors
GABA mediates neural inhibition in the brain by activating the 2 major GABA receptors: (1) GABAA, ionotropic ligand-gated ion channels that signal via direct ligand-mediated opening; and (2) GABAB, metabotropic G protein-coupled receptors that act indirectly via intracellular signaling cascades [32, 52, 53]. GABAB receptor-mediated signaling relies on the activation of G protein signaling pathways to inhibit neurotransmitter release and modulate action potential propagation [52, 54]. GABAB receptor expression or activity does not appear to be consistently altered in individuals with depression and has therefore attracted less research interest compared with GABAA receptors in this disease [55]. However, GABAB receptor activation has been reported to increase membrane trafficking of GABAA receptors in dentate gyrus granule cells, resulting in enhanced GABAA receptor current [56, 57].
GABAA receptors are widely distributed in the brain and play an important role in many brain functions [58, 59]. In addition to GABA, other endogenous ligands include zinc, NASs, and certain amino acids [60,61,62]. GABAA receptors are encoded by 19 subunit genes, six α (α1-α6), three β (β1-β3), three γ (γ1-γ3), three ρ (ρ1-ρ3), and one each of the δ, ε, π, and θ subunits [63]. GABAA receptors belong to a large heterogeneous class of pentameric chloride channels comprising 2 α, 2 β, and 1 γ, δ, ρ, θ, or ε subunits, with the α1β2γ2 GABAA receptors being the most abundant [32, 53]. The complex and heterogenous nature of GABAA receptors results in considerable diversity in their physiology, location, and pharmacologic profile [53]. Upon GABA binding and activation of the GABAA receptor, chloride ions flow into the cell, leading to rapid membrane hyperpolarization and inhibition of action potentials in the postsynaptic neuron [52, 64]. A subclass of GABAA receptors, termed GABAA-ρ (previously GABAC) receptors, is a group of receptors composed exclusively of ρ subunits, which are typically insensitive to GABAA allosteric modulators (eg, benzodiazepines, barbiturates, and most NASs) [65]. However, there are some GABAA receptor modulators that can also engage GABAA-ρ receptors, such as pregnanolone, allopregnanolone, and some synthetic NASs [65,66,67].
The subunit composition of the GABAA receptor defines its biophysical and pharmacologic properties and whether it localizes to a synaptic or extrasynaptic site. The widely expressed α1–3β1–3γ2 GABAA receptors are predominantly localized to the synapses, while the α4–6β2–3δ GABAA receptors are largely present extrasynaptically [53, 68, 69]. Activation of low-affinity synaptic γ subunit-containing receptors is transient and mediates rapid phasic inhibition, while extrasynaptic δ subunit-containing receptors mediate tonic inhibition through persistent activation by low concentrations of ambient extracellular GABA [64, 70]. In neurons with both synaptic and extrasynaptic conductance, the tonic currents may produce a larger net inhibitory effect than do the phasic currents [71].
Neuroactive steroids
Neuroactive steroids are a class of steroids that are synthesized de novo in neurons and glia of the central and peripheral nervous systems following transport of cholesterol into the mitochondria (Fig. 2). Additionally, some circulating sterols (eg, progesterone, dehydroepiandrosterone [DHEA]) can cross the blood-brain barrier to be used as precursor molecules [64, 72,73,74,75]. Endogenous NASs are generally categorized as: (1) pregnane-derived (eg, allopregnanolone, allotetrahydrodeoxycorticosterone [allo-THDOC]); (2) androstane-derived (eg, androstanediol, etiocholanolone); or (3) sulfated (eg, pregnenolone sulfate, dehydroepiandrosterone sulfate) [64, 72].

There it is metabolized by P450scc into pregnenolone, the precursor of all endogenous NASs. Biosynthetic enzymes are denoted in green; neuroactive steroids and substrates are denoted in red. *Allotetrahydrodeoxycorticosterone is also known as tetrahydrodeoxycorticosterone (same Chemical Abstract Services number). Allo-THDOC allotetrahydrodeoxycorticosterone, DHEAS dehydroepiandrosterone sulfate, DHT 5α-dihydrotestosterone, HSD hydroxysteroid dehydrogenase, NAS neuroactive steroid, P450aro cytochrome P450-aromatase, P450c11β cytochrome P450 11β-hydroxylase, P450c17 cytochrome P450 17α-hydroxylase, P450c21 cytochrome P450 21-hydroxylase, P450scc cytochrome P450 side chain cleavage, PREGS pregnenolone sulfate, SULT sulfotransferase.
Neuroactive steroids regulate neuronal excitability via rapid non-genomic action [64], primarily through interaction with neuronal membrane receptors and ion channels like the ionotropic GABAA receptors [64, 72]. Activity of NASs at neuronal GABAA receptors occurs within minutes, compared with the slow-onset (delayed by hours) and prolonged duration of action of steroid hormones, which act via intracellular steroid hormone receptors [76]. In general, NASs bind to GABA, NMDA, serotonin, and σ-1 receptors to modulate neurotransmitter signaling [72]. They modulate excitatory-inhibitory balance and homeostatic mechanisms, thus regulating brain functions that control mood, aggression, cognition, memory, and pain [77]. Neuroactive steroids can function as positive allosteric modulators (PAMs) of both synaptic and extrasynaptic GABAA receptors to activate and potentiate phasic and tonic currents, respectively [64, 78,79,80], or as negative allosteric modulators (NAMs) to dampen the response to neurotransmitter ligands such as glutamate and GABA [81]. NAS GABAA receptor NAMs are activation-dependent, non-competitive inhibitors of GABAA receptors and can also inhibit the effects of NAS GABAA receptor PAMs [80]. NAS GABAA receptor PAMs also regulate neuroplasticity, neuroinflammation, and HPA axis function and may play an important role in neurogenesis [36, 82,83,84]. Allopregnanolone, pregnanolone, and allo-THDOC are among the more potent endogenous NAS PAMs of GABAergic neurotransmission [85].
GABAA receptor activation by NASs occurs via 2 discrete sites in the α and β subunit transmembrane domains, one at the α-β subunit interface for activation and the other exclusively on α subunits for potentiation of response to NASs [62, 79]. Binding of nanomolar concentrations of NAS GABAA receptor PAMs increases the mean open time and decreases the mean closed time of the GABAA receptor chloride channel in the presence of sub-saturating concentrations of GABA, thereby increasing the chloride current through the channel and reducing neuronal excitability [64, 86]. In the absence of GABA, micromolar concentrations of PAMs can directly open GABAA receptor chloride channels [87]. Interestingly, NAS GABAA receptor PAMs have also been shown to increase phosphorylation of certain GABAA receptor subunits, leading to increased cell surface expression of those GABAA receptors [88,89,90]
The sensitivity of GABAA receptors to NASs is determined by the receptor subunit composition; at normal extracellular GABA concentrations, extrasynaptic δ subunit-containing GABAA receptors are more sensitive to NAS modulation compared with synaptic γ subunit-containing GABAA receptors [91], allowing for greater enhancement in GABAA receptor currents [92]. This preferential interaction of NASs with extrasynaptic δ subunit-containing receptors is secondary to GABA acting as a partial agonist at these receptors [92]. However, allopregnanolone modulates both γ- and δ subunit-containing GABAA receptors within a similar potency range and may therefore enhance both phasic and tonic currents, respectively [93].
Dysregulated GABAergic signaling
Major depressive disorder has been linked to dysregulation of the excitatory-inhibitory balance within the brain and the reduced ability to maintain homeostasis in response to internal or external stimuli [12, 45, 94, 95]. Both preclinical and clinical data support an association of depression with diverse defects in GABAergic neurotransmission. Epigenetic changes of the GABAergic system have been shown to be responsible for adult hippocampus neurogenesis and depression-like behaviors in prenatal-stressed mice [96]. In addition, alterations in DNA methyltransferase mRNA expression have been observed in the brains of individuals with MDD who died by suicide compared with the brains of non-MDD/suicide individuals, and this change in expression was associated with gene-specific aberrations in DNA methylation in the GABAA receptor α1 subunit promoter region within the frontopolar cortex [97]. Alterations in the DNA methylation signatures of GABA-related genes have also been reported in other psychiatric disorders, including autism spectrum disorder [98], schizophrenia [99, 100], psychosis with a history of chronic alcohol abuse [101], and bipolar disorder [102].
A review of postmortem studies found variable expression of various GABAA receptor subunit mRNAs of suicide victims with depressive disorders and patients with MDD [32, 103]. A large human gene expression analysis of cortical and subcortical regions from the brains of depression-related suicides found that the expression levels of genes involved in GABAergic transmission were among the most consistently changed [104]. Genetic alteration of the γ-subunit of the GABAA receptor disrupted the regulatory response of GABAergic neurons and led to depressive and anxiogenic behaviors in rodents [105,106,107]. Genetic studies in mice have shown that deletion of the GABAA receptor γ subunit led to impaired GABAergic signaling and behavioral and cognitive deficits that could be reversed by chronic desipramine or acute ketamine [105, 108], while deletion of the α2 subunit led to depressive- or anxiety-like behaviors [109, 110]. Mice with decreased GABAA receptor δ subunit expression displayed anxiety-like behavior and maternal neglect postpartum, and administration of the δ subunit selective agonist 4,5,6,7-tetrahydroisoxazolo[5,4-c]pyridin-3-ol (THIP) reduced the abnormal behaviors [111]. Similarly, administration of SGE-516, a NAS GABAA receptor PAM, rescued these abnormal behaviors in this same model [112].
GABA levels and GABAA receptor function were found to be diminished in several brain regions in rodent models of acute and chronic stress [113,114,115,116]. GABAA receptor agonists or GABAA receptor PAMs prevented and reversed rodent behavioral models of depression [117]; conversely, administration of GABAA receptor antagonists to normal rodents caused behaviors that mimicked these models of depression [118]. Individuals with depression exhibit reduced functioning of GABAergic interneurons and defects in GABAergic neural inhibition compared with healthy controls [28, 119, 120]. Calbindin-D28K, a calcium-binding and buffering protein critical for preventing neuronal death as well as maintaining calcium homeostasis, is expressed ubiquitously across multiple brain regions that are intimately involved in regulating emotional behaviors, particularly in GABAergic interneurons in the PFC, amygdala, and hippocampus [121]. A postmortem study showed that the density and size of GABAergic interneurons immunoreactive for calbindin-D28K were significantly decreased in the PFC of individuals with MDD versus those without MDD [122]. Positron emission tomography imaging showed reduced GABAA receptor binding of [11C]-flumazenil in the limbic parahippocampal temporal gyrus and right lateral superior temporal gyrus of individuals with MDD versus healthy controls, suggesting a decreased number of GABAA receptors and/or reduced affinity to benzodiazepine-site ligands [123]. GABAergic inhibitory neurotransmission in cerebral cortex, as assessed using transcranial magnetic stimulation, has been shown to be reduced significantly in individuals with MDD [124].
Individuals with depression, compared with healthy controls, also exhibited diminished GABA levels in the brain, plasma, and CSF [38, 125, 126] that are most pronounced in melancholic and treatment-resistant depression [119, 127], and remission from MDD was accompanied by normalization of GABA levels in the brain [125]. Additionally, severity of anhedonia is inversely correlated with GABA levels in the anterior cingulate cortex as shown in adolescents with MDD [128, 129], further supporting the correlation between dysregulated GABAergic neurotransmission and depression. Findings of reduced levels of glutamate decarboxylase in postmortem PFC of individuals with untreated MDD compared with healthy controls provide additional evidence for a link between GABAergic dysfunction and depression [130]. ADTs that affect monoaminergic neurotransmission may also show downstream effects on GABA- and glutamatergic neurotransmission [22, 131]. In animal models, studies showed that selective serotonin reuptake inhibitors (SSRIs), serotonin and norepinephrine reuptake inhibitors (SNRIs), tricyclic antidepressants (TCAs), and monoamine oxidase inhibitors (MAOIs) decrease glutamatergic signaling [132,133,134]. Treatment with SSRIs or electroconvulsive therapy in individuals with depression has resulted in normalization of decreased plasma GABA levels in the brain and plasma [135,136,137]. Due to limited study group sizes, no significant correlation was found between measures of clinical response and the change in cortical GABA concentrations in 2 of these studies [136, 137]. However, in a study of inpatients with MDD treated with SSRIs, 70% of responders had increased GABA levels and 64% had decreased glutamate levels [135].
Dysregulated GABA neurotransmission is also linked to anxiety and insomnia, 2 common comorbidities in individuals with depression. An association between anxiety and GABAergic signaling is supported by the preclinical findings of anxiety behaviors related to chronic inhibition of GABA synthesis [138] and the disruption of the anxiolytic-like effect of diazepam due to diminished levels of glutamate decarboxylase [139]. Additionally, review of the therapeutic mechanism of action for different anxiolytics found that these drugs may share a final common pathway involving enhancement of GABAergic neurotransmission [131]. GABA is also believed to be involved in the regulation of sleep [140]. Time awake after sleep onset has been found to be inversely correlated with GABA levels in individuals with primary insomnia, although results regarding changes in GABA levels of individuals with primary insomnia versus healthy controls are inconsistent [141, 142]. Furthermore, drugs targeting GABAA receptors, such as benzodiazepines and Z-drugs, exhibit sedative and hypnotic effects [143, 144].
GABAergic and monoaminergic neurons are interconnected, and, consequently, GABAA receptor deficits can also alter dopaminergic, serotonergic, and noradrenergic activity [32] (Fig. 3). Additionally, inadequate signaling in somatostatin-positive GABAergic interneurons in prefrontal microcircuits (established as one of the key substrates in MDD) can potentially produce attenuated pyramidal neuron output from the PFC and subsequent downstream regulation of threat and danger circuits (amygdala and bed nucleus of the stria terminalis) and sensory and motor processing in the thalamus, mimicking monoamine insufficiency in the brainstem [145, 146].

Serotonergic neurons originating in the dorsal raphe nucleus and projecting to the prefrontal cortex (PFC) regulate excitability of GABAergic and glutamatergic neurons, which in turn, modulate the excitability of serotonergic neurons in the dorsal raphe nucleus by the GABA-glutamate balance (left). Chronic stress affects local networks regulating activity within the medial PFC (mPFC), leading to changes in local excitatory-inhibitory balance. In a proposed mechanistic model (right), somatostatin-expressing GABAergic neurons provide reduced dendritic inhibition of glutamatergic pyramidal neurons in the infralimbic mPFC under chronic stress, reducing filtering of information flow into the PFC [145]. An altered glutamate and GABA neurotransmission might appear as a disturbance in monoamine signaling. (Part of this figure is adapted from McKlveen et al. 2019 [145], with permission from Elsevier).
Role of neuroactive steroids in depression and other brain disorders
The downregulated biosynthesis of NAS GABAA receptor PAMs has been implicated in various psychiatric disorders (eg, MDD, PPD, premenstrual dysphoric disorder, and posttraumatic stress disorder [PTSD]) [31, 147]. Changes in NAS GABAA receptor PAMs synthesis pathways have been linked to the pathologies of neurodegenerative and inflammatory brain diseases (eg, Alzheimer’s disease, Parkinson’s disease, multiple sclerosis) based on postmortem studies [148], as well as to self-reported pain symptoms (eg, chest pain, muscle soreness) [149] in humans. Levels of allopregnanolone have been shown to be significantly decreased in individuals with PTSD, a condition that is highly comorbid with MDD [150].
Reduced levels of allopregnanolone in the CSF and plasma have also been reported in individuals with mood disorders such as MDD, in addition to decreased GABA levels [151, 152] and decreased PFC expression of 5α-reductase, the enzyme catalyzing the rate-limiting step in allopregnanolone biosynthesis [153]. Decreased levels of allopregnanolone in the plasma or serum were also found in individuals with postpartum “blues” or pharmacologically induced panic attacks [154, 155], in contrast to the increased level of the 3β isomer of allopregnanolone, which antagonizes GABAA receptor function in panic attacks [148]. Significant fluctuations in the blood and brain levels of allopregnanolone were shown to be strongly correlated with alterations in function and plasticity of GABAA receptors in rodents [156, 157]. The failure to upregulate GABAA receptors in response to the rapid drop in levels of allopregnanolone postpartum is likely involved in the development of PPD [35].
While acute stressors can lead to increased levels of NAS GABAA receptor PAMs (ie, allopreg-nanolone and allo-THDOC) in animal models [36, 158], chronic stress, a major predictor of MDD, was shown to result in altered GABAergic signaling and decreased production of endogenous GABAA receptor PAMs (ie, allopregnanolone) in rodent stress models [80, 159, 160]. Chronic stress-induced reduction in allopregnanolone levels was associated with abnormal behaviors such as aggression, enhanced fear, depressive- or anxiety-like behaviors, and impaired adult hippocampal neurogenesis in animal models [161,162,163,164].
Selective serotonin reuptake inhibitors such as fluoxetine and norfluoxetine can normalize decreased levels of allopregnanolone in the brain while decreasing behavioral abnormalities associated with mood disorders, as demonstrated in socially isolated mice [165, 166]. Studies in individuals with depression also showed that treatment with fluoxetine could increase allopregnanolone levels in the CSF [151, 152], and these changes were correlated with improvements in depressive symptoms [151]. The important role of NAS GABAA receptor PAMs in depression is further supported by the findings that allopregnanolone administration prevented or normalized depressive- or anxiety-like behaviors in a social isolation rodent model [163].
GABAA receptor positive allosteric modulators
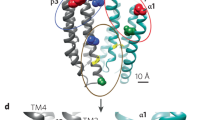
Most GABAA receptor-targeting drugs (ie, barbiturates, benzodiazepines, and NASs) function via allosteric binding to the receptor at sites distinct from the GABA binding sites (Fig. 4) [58, 62, 79, 91, 167, 168]. GABA binding sites are located at the α-β subunit interface on both synaptic and extrasynaptic receptors [58]. Barbiturates, benzodiazepines, and NASs bind the GABAA receptor at allosteric sites and increase the GABAA receptor current by increasing chloride conductance [169]. The presence of GABA is necessary for benzodiazepine response. Binding of benzodiazepines to the synaptic GABAA receptor locks the receptor into a conformation for which GABA has much higher affinity, thus increasing the frequency of the chloride channel opening, with minimal effect on the duration of bursts [170, 171]. Barbiturates, on the other hand, bind in the presence of GABA to both synaptic and extrasynaptic GABAA receptors and increase the duration of chloride channel opening without altering the frequency of bursts [169, 171]. Only at high doses can barbiturates directly stimulate GABAA receptors in the absence of GABA [172].

NAS GABAA receptor PAMs, such as allopregnanolone, bind to GABAA receptors at sites distinctive from those for benzodiazepines (BZDs). NAS GABAA receptor PAMs bind to both synaptic γ subunit-containing and extrasynaptic δ subunit-containing GABAA receptors, potentiating phasic and tonic currents, respectively. In contrast, benzodiazepines bind to γ subunit-containing GABAA receptors only and primarily augment phasic inhibition. Extrasynaptic GABAA receptors containing δ subunits are insensitive to benzodiazepines [32, 53, 62, 64, 78, 79, 91, 92, 167].
Barbiturates bind to the α-β subunit interface on synaptic and extrasynaptic receptors and to γ-β subunit interfaces on synaptic receptors [58]. Barbiturates also non-selectively bind to the entire superfamily of ligand-gated ion channels [173]. Barbiturates act as antagonists of ionotropic glutamate receptors, such as α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and kainate receptors, thus inhibiting the bulk of fast excitatory synaptic transmission and glutamate release throughout the CNS [173]. These actions may account for the antidepressant, anxiolytic, hypnotic, and anticonvulsant activities of barbiturates, but may also account for the physical and psychological addiction potential and high rates of tolerance and dependence associated with this class of drugs. Barbiturates also have a high overdose potential due to a very narrow dosage margin [174]. Barbiturates were used to treat depression, anxiety, and insomnia in the early part of the 20th century but were generally replaced by benzodiazepines in the 1960s because the many potential drawbacks outweighed their usefulness [175].
Benzodiazepines are sometimes used to treat specific symptoms that are frequently associated with depression (eg, anxiety and insomnia). However, while a meta-analysis showed that treatment with the benzodiazepine alprazolam led to a higher percentage of individuals with MDD achieving response on the 17-item Hamilton Rating Scale for Depression (HAMD-17; ≥50% reduction in total score) or Clinical Global Impression-Improvement (CGI-I) scale (much improved or very much improved) compared with placebo, other benzodiazepines such as chlordiazepoxide and diazepam did not exhibit clear antidepressant activity [176]. In addition, chronic use of benzodiazepines more than 2–4 weeks is not recommended as it may result in decreased GABAergic and monoaminergic function, cognitive and psychomotor impairment, and interference with neurogenesis [177, 178]. These concerns, in addition to the risk of dependence and abuse [179] and an overall increase in the risk of attempting or completing suicide [180] may further limit the potential use of benzodiazepines in the treatment of depression [178].
Given that benzodiazepines appear to have questionable antidepressant activity, it is speculated that lack of antidepressant activity may be associated with the subunit composition of the GABAA receptors with which benzodiazepines interact. Benzodiazepines bind at the α-γ subunit interface on synaptic GABAA receptors (primarily augmenting phasic inhibition) [58]. Preclinical data suggest that α1 subunit-containing GABAA receptors play a major role in sedation and addiction [181, 182], positive modulation of α2 subunit-containing receptors may have more consistent antidepressant effects [109], and activation of α3 subunit-containing receptors have pro-depressant actions [183]. Benzodiazepines show a higher affinity for α1 subunit-containing receptors [53] and concomitantly activate α2- or α3 subunit-containing receptors [184], the net effect of which is the promotion of sedation and addiction and a null effect on depression. In addition, benzodiazepines may only enhance phasic inhibitory currents through their binding to synaptic (γ subunit-containing) GABAA receptors [185]. GABAA receptor PAMs that bind to both synaptic and extrasynaptic GABAA receptors and enhance both phasic and tonic inhibitory currents, respectively, may have greater therapeutic utility than benzodiazepines in treating depression.
The ability of NASs to target both synaptic and extrasynaptic GABAA receptors is especially important in conditions where synaptic GABAA receptors are downregulated, a condition which could lead to benzodiazepine tolerance [185]. NASs can interact with most GABAA receptors, including the benzodiazepine-insensitive receptors containing α4 and α6 subunits or lacking the γ subunit [91]. In addition to allosteric modulation of GABAA receptors, NAS GABAA receptor PAMs can also exert metabotropic effects on GABAergic inhibition via activation of the G protein-coupled membrane progesterone receptors (mPRs); mPR-dependent modulation of GABAA receptor phosphorylation results in increased cell surface expression of GABAA receptors and thus a sustained elevation in tonic current [90], further differentiating them from benzodiazepines, which are associated with a downregulation of GABAA receptors [186]. The tonic current is resistant to the competitive GABAA receptor antagonist gabazine, confirming that it is not generated from GABA binding to these receptors [187].
Current treatment options for mdd in clinical practice
The 8 general groups of approved drugs for MDD are: SSRIs, SNRIs, TCAs, tetracyclic antidepressants (TeCAs), MAOIs, atypical and multimodal antidepressants, NMDA receptor antagonists, and GABAA receptor modulators (Table 1) [188,189,190,191,192,193,194].
Standard-of-care ADTs used in the current pharmacologic management of MDD primarily target monoamine neurotransmitter systems. Compared with first-generation ADTs (TCAs and MAOIs), SSRIs and SNRIs have generally been shown to cause relatively fewer adverse effects and therefore appear to be more widely used [195, 196]. A meta-analysis of the efficacy and tolerability of SSRIs against TCAs in patients with MDD showed that although TCAs demonstrated similar efficacy with SSRIs (with superior efficacy in hospitalized patients), they are associated with significantly more adverse effects due to their inhibition of cholinergic, α-1 adrenergic, and histaminergic receptors [197]. TCAs are also more likely to induce toxicity and can be fatal if overdosed [198]. Although efficacious, MAOIs are not commonly prescribed because of potentially fatal reactions including increased blood pressure, heart attack, stroke, or serotonin syndrome when used together with foods containing high levels of tyramine (eg, aged cheese, spoiled meat, soy sauce) or other drugs [199, 200]. Monoaminergic ADTs often require 4 to 6 weeks or longer to take effect [201,202,203]. In addition, the STAR*D Study has shown that as many as approximately 50% of patients may not respond adequately [203]. Relapse rates can be high in patients taking standard-of-care ADTs, especially in those who require multiple treatment steps, as demonstrated in the STAR*D Study among patients with MDD (relapse rates ranged from 40%–70% during a 12-month naturalistic follow-up) [202].
Novel ADTs with targets that have been implicated in the neurobiology of depression beyond monoamines (eg, glutamate and GABA), are being investigated. For example, while the mechanism of action for the antidepressant effects of ketamine is not fully understood, it is thought that it may block NMDA receptors on GABAergic interneurons, thereby preventing their activation [204]. Subsequently, downstream disinhibition of glutamatergic neurons causes a glutamate surge. Increased extracellular glutamate initiates activation of postsynaptic AMPA receptors, leading to potentiation of BDNF and mTORC1 synaptogenic signaling pathways (Table 1) [190, 204, 205]. In addition, one study found that the antidepressant effects of ketamine were blocked when naltrexone, an opioid antagonist, was administered prior to ketamine [206], suggesting that the antidepressant effect of ketamine may be dependent on opioid receptor activation and not necessarily due to neurological actions mediated by NMDA receptors. Ketamine’s S-enantiomer, esketamine, was recently approved by the U.S. Food and Drug Administration (FDA) for treatment-resistant depression and MDD with acute suicidal ideation or behavior [207]. In contrast to standard monoaminergic ADTs, ketamine has demonstrated rapid antidepressant effects that peak at approximately 24 h and are sustained for approximately 1 week after administration in adults with MDD or bipolar depression [208]. However, the long-term use of ketamine may induce urologic toxicity [209], and chronic abuse of ketamine can negatively affect brain structure and functioning and cause cognitive impairment [210].
Newer ADTs targeting dysregulated GABA neurotransmission are also being developed. These include GABAA and GABAB receptor modulators (allosteric modulators, NASs, agonists, and antagonists), and GABAergic interneuron-targeting neuropeptides [45].
Neuroactive steroids for treatment of depression
Among the more recent additions to the treatment landscape, brexanolone, a NAS GABAA receptor PAM that is chemically identical to endogenous allopregnanolone (Table 1), was approved in 2019 by the FDA to treat adults with PPD (Table 2) [211]. This indication was expanded in 2022 to include patients ≥15 years of age [212]. Prior to the approval of brexanolone, the standard of care for PPD was psychotherapy, psychotropics, or combination treatment. Medications adapted from MDD treatment but not specifically approved for PPD included SSRIs, SNRIs, and TCAs [213]. In pivotal phase 2 and 3 clinical trials, adult women with PPD who received brexanolone demonstrated significant improvement in depressive symptoms compared with those who received placebo; improvement was rapid (at Hour 60) and sustained (through day 30) [214,215,216]. Brexanolone was generally well tolerated in these trials [214, 216]. An intravenous preparation of brexanolone was used because of low oral bioavailability and high in vivo clearance of endogenous allopregnanolone [216]. While the use of brexanolone can be limited by the relatively long, continuous infusion time (60 h), these data have led to an increased interest in the therapeutic potential of GABAA receptor-modulating NASs.
Another NAS, PRAX-114 is a primarily extrasynaptic GABAA receptor PAM in oral formulation that was being investigated for the treatment of MDD [217, 218] (Table 2). Interim results from a non–placebo-controlled, 3-arm, fixed-dose, phase 2 safety and tolerability study conducted in Australia showed improvements from baseline in depression severity as assessed by HAMD-17 total score reductions following a 14-day treatment course with PRAX-114 [219]. Changes from baseline (CFB) in the HAMD-17 total score (reductions of 15–19 points) were observed in all 3 arms over an 8-day period. The safety and efficacy of a 28-day treatment course with PRAX-114 as monotherapy for severe MDD were also assessed in the phase 2/3, randomized, double-blind, placebo-controlled Aria trial (N = 216) [217]. However, this study failed to meet its primary endpoint of CFB in HAMD-17 total score on day 15, nor did it meet any of the secondary endpoints [220]. The sponsor has closed screening in its randomized, double-blind, placebo-controlled phase 2 trial as adjunctive and monotherapy treatment for patients with MDD and inadequate response to antidepressant treatment (N = 110), has stopped enrollment in a PTSD phase 2 trial, and has discontinued an essential tremor trial. The sponsor has no plans to pursue further development of PRAX-114 for psychiatric disorders.
PH10 is an investigational, synthetic NAS from the family of pherines, formulated as a nasal spray, currently under clinical development for treatment of MDD [221] (Table 2). PH10 acts on nasal chemosensory receptors to modulate neural circuits in the brain, including connections to the limbic amygdala and other basal forebrain structures, leading to antidepressant effects [221]. In a 3-arm (high-dose, low-dose, and placebo) phase 2a pilot study in patients with MDD (N = 30), treatment with PH10 led to a greater improvement in depressive symptoms as assessed by CFB (reductions) in HAMD-17 total score compared with placebo after 8 weeks of treatment, with minimal side effects and potentially a rapid (week 1) onset of effects [221, 222]. Mean CFB in HAMD-17 total score at week 8 were 17.8 (high dose), 16.3 (low dose), and 10.9 for placebo (overall p = 0.07; high dose p = 0.02; low dose p = 0.10). HAMD-17 responder rates of the 3 doses at week 8 were 80% (high dose; p > 0.05), 90% (low dose; p > 0.05), and 60% (placebo), and remission rates were 60% (p > 0.05), 80% (p < 0.05), and 20%, respectively. Adverse events that were more common with PH10 compared with placebo included increased appetite, daytime sleepiness, nasal dryness, headache, and bitter taste. A phase 2b trial of PH10 nasal spray for the treatment of MDD has been planned [223]. In addition, future development as a treatment for PPD, treatment-resistant depression, and suicidal ideation is under consideration [224].
Zuranolone is an oral, investigational, synthetic NAS and PAM of both synaptic and extrasynaptic GABAA receptors that upregulates GABAA receptor expression and enhances inhibitory GABAergic signaling [225]. It is currently in clinical development and being investigated as an oral, 14-day treatment for adults with MDD or PPD (Table 2). Zuranolone has a pharmacokinetic profile that enables oral once-daily dosing with increased bioavailability [226, 227]. In two phase 3 trials in adults with PPD assessing zuranolone 30 mg (N = 150) or zuranolone 50 mg (N = 195), those who received a once-daily, 14-day treatment course of zuranolone demonstrated significant improvements in depressive symptoms as assessed by CFB (reductions) in HAMD-17 total score at day 15 compared with those who received placebo [228, 229]. Mean CFB in HAMD-17 total score at day 15 were 17.8 (vs placebo 13.6; p < 0.05) with zuranolone 30 mg and 15.6 (vs placebo −11.6; p < 0.05) with zuranolone 50 mg. Rapid (day 3) and sustained (day 45) improvements in depressive symptoms were significantly greater with zuranolone than with placebo (p < 0.05) in both studies. HAMD-17 response rates at day 45 (end of study) were 75.3% (vs placebo 56.5%; nominal p > 0.05) and 61.9% (vs placebo 54.1%; nominal p > 0.05), and remission rates were 53.4% (vs placebo 30.4%; nominal p < 0.01) and 44.0% (vs placebo 29.4%; nominal p > 0.05) in the 2 studies [228, 229]. In a phase 2 trial (zuranolone 30 mg, N = 89) and a phase 3 trial (zuranolone 50 mg, N = 534) in adults with MDD, those who received treatment with zuranolone demonstrated significantly greater improvements in depressive symptoms as assessed by CFB (reductions) in HAMD-17 total score at day 15 compared with those who received placebo [230, 231]; mean CFB in HAMD-17 total score at day 15 were 17.4 (vs placebo 10.3; p < 0.05) and 14.1 (vs placebo −12.3; p < 0.05), respectively. Another phase 3 trial assessing zuranolone 20 mg (N = 194) and 30 mg (N = 194) in patients with MDD did not meet its primary endpoint [232]; mean CFB in HAMD-17 total score at day 15 was 12.5 with zuranolone 30 mg compared with 11.1 with placebo (N = 193) (p > 0.05). Rapid (by day 2 or 3) improvement in depressive symptoms were observed in these 3 trials in MDD (nominal p < 0.05 vs placebo) [233]. HAMD-17 response rates at day 42 (end of study) were 61.9% (vs placebo 56.4%; nominal p > 0.05) in the phase 2 trial, 52.9% (vs placebo 45.9%; nominal p > 0.05) in the phase 3 zuranolone 50 mg trial, and 43.4% (vs placebo 41.5%; nominal p > 0.05) in the phase 3 zuranolone 20 or 30 mg trial; HAMD-17 remission rates were 45.2% (vs placebo 33.3%; nominal p > 0.05), 30.8% (vs placebo 29.6%; nominal p > 0.05), and 24.3% (vs placebo 25.9%; nominal p > 0.05), respectively [230,231,232]. Use of standard ADTs at baseline was allowed in these trials, providing the patient was on a stable dose. Zuranolone as a co-initiation therapy was evaluated in a phase 3 trial; rapid and significantly greater improvement from baseline in HAMD-17 total score was observed at day 3 with zuranolone versus placebo when co-initiated with standard-of-care ADTs in adults with MDD (p < 0.001) [234]. Moreover, in an ongoing open-label study that includes assessment of the need for repeat treatment courses with zuranolone over 1 year, of enrolled adults with MDD who responded at day 15 to treatment (≥50% reduction from baseline in HAMD-17 score) with an initial 14-day treatment course of zuranolone 50 mg and continued beyond day 28, 79.5% received a total of 1 or 2 treatment courses during their time of up to 1 year in the study [235, 236]. Zuranolone was generally well tolerated. In clinical trials, adverse events that were more common (>5% in zuranolone) with zuranolone compared with placebo included somnolence, dizziness, sedation, and fatigue. The overall incidence of serious adverse events was low, reported in <2% of zuranolone-treated patients across the trials. No patient enrolled in any clinical trial to date (February 2023) has reported developing withdrawal syndrome after discontinuation of zuranolone.
Conclusions
Treatment responses to standard-of-care oral antidepressants have been suboptimal in many individuals with MDD, potentially due to slow onset of effects, low response rates, adverse effects, and the need for chronic treatment. There remains an unmet need for novel and effective treatments with rapid, robust, and sustained antidepressant effects; with better safety and tolerability than standard-of-care ADTs, and ideally without the need for chronic treatment. The development of novel therapeutics for MDD relies on a deep, comprehensive, and evolving understanding of the pathophysiology of depression.
There has been increased interest in GABAA receptor-based treatment approaches for MDD. Recent research on the proposed mechanism of action of NASs for PPD and MDD underscores the potential role of GABAergic signaling in the pathophysiology of depression. Although the placebo effect in depression may be a factor associated with failure to establish efficacy of novel treatments in clinical trials [237], data reviewed here indicate that NAS GABAA receptor PAMs may potentially offer rapid and sustained antidepressant benefits for individuals with MDD. Further research is necessary to better understand the role of NAS GABAA receptor PAMs in MDD.
References
Depression. World Health Organization. Available at https://www.who.int/news-room/fact-sheets/detail/depression. Accessed: March 18.
American Psychiatric Association. Diagnostic and statistical manual of mental disorders, 5th edition, text revision (DSM-5-TR) (American Psychiatric Association Publishing, Washington, DC, 2022).
GBD Mental Disorders Collaborators. Global, regional, and national burden of 12 mental disorders in 204 countries and territories, 1990-2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet Psychiatry. 2022;9:137–50.
National Survey on Drug Use and Health 2021. Available at https://www.samhsa.gov/data/sites/default/files/reports/rpt39441/NSDUHDetailedTabs2021/NSDUHDetailedTabs2021/NSDUHDetTabsSect6pe2021.htm. Accessed: February 22.
Levey DF, Stein MB, Wendt FR, Pathak GA, Zhou H, Aslan M, et al. Bi-ancestral depression GWAS in the Million Veteran Program and meta-analysis in >1.2 million individuals highlight new therapeutic directions. Nat Neurosci. 2021;24:954–63.
Lax E. DNA methylation as a therapeutic and diagnostic target in major depressive disorder. Front Behav Neurosci. 2022;16:759052.
Cheng Z, Su J, Zhang K, Jiang H, Li B. Epigenetic mechanism of early life stress-induced depression: focus on the neurotransmitter systems. Front Cell Dev Biol. 2022;10:929732.
Reszka E, Jablonska E, Lesicka M, Wieczorek E, Kapelski P, Szczepankiewicz A, et al. An altered global DNA methylation status in women with depression. J Psychiatr Res. 2021;137:283–9.
Evans JW, Szczepanik J, Brutsche N, Park LT, Nugent AC, Zarate CA Jr. Default mode connectivity in major depressive disorder measured up to 10 days after ketamine administration. Biol Psychiatry. 2018;84:582–90.
MacQueen G, Frodl T. The hippocampus in major depression: evidence for the convergence of the bench and bedside in psychiatric research? Mol Psychiatry. 2011;16:252–64.
Savitz J, Drevets WC. Bipolar and major depressive disorder: neuroimaging the developmental-degenerative divide. Neurosci Biobehav Rev. 2009;33:699–771.
Sarawagi A, Soni ND, Patel AB. Glutamate and GABA homeostasis and neurometabolism in major depressive disorder. Front Psychiatry. 2021;12:637863.
Fischer AS, Keller CJ, Etkin A. The clinical applicability of functional connectivity in depression: pathways toward more targeted intervention. Biol Psychiatry Cogn Neurosci Neuroimaging. 2016;1:262–70.
Dai L, Zhou H, Xu X, Zuo Z. Brain structural and functional changes in patients with major depressive disorder: a literature review. PeerJ. 2019;7:e8170.
Jesulola E, Micalos P, Baguley IJ. Understanding the pathophysiology of depression: from monoamines to the neurogenesis hypothesis model - are we there yet? Behav Brain Res. 2018;341:79–90.
Li Z, Ruan M, Chen J, Fang Y. Major depressive disorder: advances in neuroscience research and translational applications. Neurosci Bull. 2021;37:863–80.
Javaheripour N, Li M, Chand T, Krug A, Kircher T, Dannlowski U, et al. Altered resting-state functional connectome in major depressive disorder: a mega-analysis from the PsyMRI consortium. Transl Psychiatry. 2021;11:511.
Martino M, Magioncalda P, Huang Z, Conio B, Piaggio N, Duncan NW, et al. Contrasting variability patterns in the default mode and sensorimotor networks balance in bipolar depression and mania. Proc Natl Acad Sci USA. 2016;113:4824–9.
Deligiannidis KM, Fales CL, Kroll-Desrosiers AR, Shaffer SA, Villamarin V, Tan Y, et al. Resting-state functional connectivity, cortical GABA, and neuroactive steroids in peripartum and peripartum depressed women: a functional magnetic resonance imaging and spectroscopy study. Neuropsychopharmacology. 2019;44:546–54.
Petroff OA. GABA and glutamate in the human brain. Neuroscientist. 2002;8:562–73.
McEwen BS, Chattarji S, Diamond DM, Jay TM, Reagan LP, Svenningsson P, et al. The neurobiological properties of tianeptine (Stablon): from monoamine hypothesis to glutamatergic modulation. Mol Psychiatry. 2010;15:237–49.
Luscher B, Fuchs T. GABAergic control of depression-related brain states. Adv Pharm. 2015;73:97–144.
Trullas R, Skolnick P. Functional antagonists at the NMDA receptor complex exhibit antidepressant actions. Eur J Pharm. 1990;185:1–10.
Adell A. Brain NMDA receptors in schizophrenia and depression. Biomolecules. 2020;10:947.
Wang S, Bian L, Yin Y, Guo J. Targeting NMDA receptors in emotional disorders: their role in neuroprotection. Brain Sci. 2022;12:1329.
Wang J, Wang F, Mai D, Qu S. Molecular mechanisms of glutamate toxicity in Parkinson’s disease. Front Neurosci. 2020;14:585584.
Hashimoto K, Sawa A, Iyo M. Increased levels of glutamate in brains from patients with mood disorders. Biol Psychiatry. 2007;62:1310–6.
Sanacora G, Mason GF, Rothman DL, Behar KL, Hyder F, Petroff OA, et al. Reduced cortical gamma-aminobutyric acid levels in depressed patients determined by proton magnetic resonance spectroscopy. Arch Gen Psychiatry. 1999;56:1043–7.
Moriguchi S, Takamiya A, Noda Y, Horita N, Wada M, Tsugawa S, et al. Glutamatergic neurometabolite levels in major depressive disorder: a systematic review and meta-analysis of proton magnetic resonance spectroscopy studies. Mol Psychiatry. 2019;24:952–64.
Luykx JJ, Laban KG, van den Heuvel MP, Boks MP, Mandl RC, Kahn RS, et al. Region and state specific glutamate downregulation in major depressive disorder: a meta-analysis of (1)H-MRS findings. Neurosci Biobehav Rev. 2012;36:198–205.
Lüscher B, Möhler H. Brexanolone, a neurosteroid antidepressant, vindicates the GABAergic deficit hypothesis of depression and may foster resilience. F1000Res. 2019;8:F1000 Faculty Rev-751. Published 2019 May 29.
Luscher B, Shen Q, Sahir N. The GABAergic deficit hypothesis of major depressive disorder. Mol Psychiatry. 2011;16:383–406.
Anderson KM, Collins MA, Kong R, Fang K, Li J, He T, et al. Convergent molecular, cellular, and cortical neuroimaging signatures of major depressive disorder. Proc Natl Acad Sci USA. 2020;117:25138–49.
Li J, Seidlitz J, Suckling J, Fan F, Ji GJ, Meng Y, et al. Cortical structural differences in major depressive disorder correlate with cell type-specific transcriptional signatures. Nat Commun. 2021;12:1647.
Gunduz-Bruce H, Takahashi K, Huang MY. Development of neuroactive steroids for the treatment of postpartum depression. J Neuroendocrinol. 2022;34:e13019.
Maguire J. Neuroactive steroids and GABAergic involvement in the neuroendocrine dysfunction associated with major depressive disorder and postpartum depression. Front Cell Neurosci. 2019;13:83.
Fee C, Banasr M, Sibille E. Somatostatin-positive gamma-aminobutyric acid interneuron deficits in depression: cortical microcircuit and therapeutic perspectives. Biol Psychiatry. 2017;82:549–59.
Romeo B, Choucha W, Fossati P, Rotge JY. Meta-analysis of central and peripheral gamma-aminobutyric acid levels in patients with unipolar and bipolar depression. J Psychiatry Neurosci. 2018;43:58–66.
Xu JC, Fan J, Wang X, Eacker SM, Kam TI, Chen L, et al. Cultured networks of excitatory projection neurons and inhibitory interneurons for studying human cortical neurotoxicity. Sci Transl Med. 2016;8:333ra348.
Nadadhur AG, Emperador Melero J, Meijer M, Schut D, Jacobs G, Li KW, et al. Multi-level characterization of balanced inhibitory-excitatory cortical neuron network derived from human pluripotent stem cells. PLoS One. 2017;12:e0178533.
Bak LK, Schousboe A, Waagepetersen HS. The glutamate/GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J Neurochem. 2006;98:641–53.
Tiwari V, Ambadipudi S, Patel AB. Glutamatergic and GABAergic TCA cycle and neurotransmitter cycling fluxes in different regions of mouse brain. J Cereb Blood Flow Metab. 2013;33:1523–31.
Chang CL, Trimbuch T, Chao HT, Jordan JC, Herman MA, Rosenmund C. Investigation of synapse formation and function in a glutamatergic-GABAergic two-neuron microcircuit. J Neurosci. 2014;34:855–68.
Wu C, Sun D. GABA receptors in brain development, function, and injury. Metab Brain Dis. 2015;30:367–79.
Fogaca MV, Duman RS. Cortical GABAergic dysfunction in stress and depression: new insights for therapeutic interventions. Front Cell Neurosci. 2019;13:87.
Bird CM, Burgess N. The hippocampus and memory: insights from spatial processing. Nat Rev Neurosci. 2008;9:182–94.
Eban-Rothschild A, Appelbaum L, de Lecea L. Neuronal mechanisms for sleep/wake regulation and modulatory drive. Neuropsychopharmacology. 2018;43:937–52.
Cellot G, Cherubini E. Functional role of ambient GABA in refining neuronal circuits early in postnatal development. Front Neural Circuits. 2013;7:136.
Chen X, Fan X, Hu Y, Zuo C, Whitfield-Gabrieli S, Holt D, et al. Regional GABA concentrations modulate inter-network resting-state functional connectivity. Cereb Cortex. 2019;29:1607–18.
Tremblay R, Lee S, Rudy B. GABAergic interneurons in the neocortex: from cellular properties to circuits. Neuron. 2016;91:260–92.
Mohler H. Molecular regulation of cognitive functions and developmental plasticity: impact of GABAA receptors. J Neurochem. 2007;102:1–12.
Jewett BE, Sharma S. Physiology, GABA. (StatPearls Publishing, 2021).
Olsen RW, Sieghart W. GABAA receptors: subtypes provide diversity of function and pharmacology. Neuropharmacology. 2009;56:141–8.
Bettler B, Kaupmann K, Mosbacher J, Gassmann M. Molecular structure and physiological functions of GABAB receptors. Physiol Rev. 2004;84:835–67.
Pehrson AL, Sanchez C. Altered gamma-aminobutyric acid neurotransmission in major depressive disorder: a critical review of the supporting evidence and the influence of serotonergic antidepressants. Drug Des Devel Ther. 2015;9:603–24.
Li N, Tao W, Yang L, Spain WJ, Ransom CB. GABA-B receptors enhance GABA-A receptor currents by modulation of membrane trafficking in dentate gyrus granule cells. Neurosci Lett. 2022;773:136481.
Tao W, Higgs MH, Spain WJ, Ransom CB. Postsynaptic GABAB receptors enhance extrasynaptic GABAA receptor function in dentate gyrus granule cells. J Neurosci. 2013;33:3738–43.
Ghit A, Assal D, Al-Shami AS, Hussein DEE. GABAA receptors: structure, function, pharmacology, and related disorders. J Genet Eng Biotechnol. 2021;19:123.
Young AB, Chu D. Distribution of GABAA and GABAB receptors in mammalian brain: potential targets for drug development. Drug Dev Res. 1990;21:161–7.
Hosie AM, Dunne EL, Harvey RJ, Smart TG. Zinc-mediated inhibition of GABA(A) receptors: discrete binding sites underlie subtype specificity. Nat Neurosci. 2003;6:362–9.
Mori M, Gähwiler BH, Gerber U. Beta-alanine and taurine as endogenous agonists at glycine receptors in rat hippocampus in vitro. J Physiol. 2002;539:191–200.
Hosie AM, Wilkins ME, Smart TG. Neurosteroid binding sites on GABAA receptors. Pharm Ther. 2007;116:7–19.
Sieghart W, Fuchs K, Tretter V, Ebert V, Jechlinger M, Hoger H, et al. Structure and subunit composition of GABA(A) receptors. Neurochem Int. 1999;34:379–85.
Reddy DS. Neurosteroids: endogenous role in the human brain and therapeutic potentials. Prog Brain Res. 2010;186:113–37.
Naffaa MM, Hung S, Chebib M, Johnston GAR, Hanrahan JR. GABA-rho receptors: distinctive functions and molecular pharmacology. Br J Pharm. 2017;174:1881–94.
Morris KD, Moorefield CN, Amin J. Differential modulation of the gamma-aminobutyric acid type C receptor by neuroactive steroids. Mol Pharm. 1999;56:752–9.
Li W, Jin X, Covey DF, Steinbach JH. Neuroactive steroids and human recombinant rho1 GABAC receptors. J Pharm Exp Ther. 2007;323:236–47.
Belelli D, Harrison NL, Maguire J, Macdonald RL, Walker MC, Cope DW. Extrasynaptic GABAA receptors: form, pharmacology, and function. J Neurosci. 2009;29:12757–63.
Maguire JL, Stell BM, Rafizadeh M, Mody I. Ovarian cycle–linked changes in GABAA receptors mediating tonic inhibition alter seizure susceptibility and anxiety. Nat Neurosci. 2005;8:797–804.
Engin E, Benham RS, Rudolph U. An emerging circuit pharmacology of GABAA receptors. Trends Pharm Sci. 2018;39:710–32.
Nusser Z, Mody I. Selective modulation of tonic and phasic inhibitions in dentate gyrus granule cells. J Neurophysiol. 2002;87:2624–8.
Tuem KB, Atey TM. Neuroactive steroids: receptor interactions and responses. Front Neurol. 2017;8:442.
Lloyd-Evans E, Waller-Evans H. Biosynthesis and signalling functions of central and peripheral nervous system neurosteroids in health and disease. Essays Biochem. 2020;64:591–606.
Tsutsui K, Ukena K, Usui M, Sakamoto H, Takase M. Novel brain function: biosynthesis and actions of neurosteroids in neurons. Neurosci Res. 2000;36:261–73.
Baulieu EE. Neurosteroids: a novel function of the brain. Psychoneuroendocrinology. 1998;23:963–87.
Joels M. Steroid hormones and excitability in the mammalian brain. Front Neuroendocrinol. 1997;18:2–48.
Joksimovic SL, Covey DF, Jevtovic-Todorovic V, Todorovic SM. Neurosteroids in pain management: a new perspective on an old player. Front Pharm. 2018;9:1127.
Rudolph U, Knoflach F. Beyond classical benzodiazepines: novel therapeutic potential of GABAA receptor subtypes. Nat Rev Drug Discov. 2011;10:685–97.
Hosie AM, Wilkins ME, da Silva HM, Smart TG. Endogenous neurosteroids regulate GABAA receptors through two discrete transmembrane sites. Nature. 2006;444:486–9.
Zorumski CF, Paul SM, Covey DF, Mennerick S. Neurosteroids as novel antidepressants and anxiolytics: GABA-A receptors and beyond. Neurobiol Stress. 2019;11:100196.
Dubrovsky BO. Steroids, neuroactive steroids and neurosteroids in psychopathology. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:169–92.
Borowicz KK, Piskorska B, Banach M, Czuczwar SJ. Neuroprotective actions of neurosteroids. Front Endocrinol (Lausanne). 2011;2:50.
Schverer M, Lanfumey L, Baulieu EE, Froger N, Villey I. Neurosteroids: non-genomic pathways in neuroplasticity and involvement in neurological diseases. Pharm Ther. 2018;191:190–206.
Maguire J, Mody I. Neurosteroid synthesis-mediated regulation of GABAA receptors: relevance to the ovarian cycle and stress. J Neurosci. 2007;27:2155–62.
Ratner MH, Kumaresan V, Farb DH. Neurosteroid actions in memory and neurologic/neuropsychiatric disorders. Front Endocrinol (Lausanne). 2019;10:169.
Puia G, Santi M, Vicini S, Pritchett DB, Purdy RH, Paul SM, et al. Neurosteroids act on recombinant human GABAA receptors. Neuron. 1990;4:759–65.
Alvarez LD, Pecci A, Estrin DA. In search of GABAA receptor’s neurosteroid binding sites. J Med Chem. 2019;62:5250–60.
Abramian AM, Comenencia-Ortiz E, Modgil A, Vien TN, Nakamura Y, Moore YE, et al. Neurosteroids promote phosphorylation and membrane insertion of extrasynaptic GABAA receptors. Proc Natl Acad Sci USA. 2014;111:7132–7.
Modgil A, Parakala ML, Ackley MA, Doherty JJ, Moss SJ, Davies PA. Endogenous and synthetic neuroactive steroids evoke sustained increases in the efficacy of GABAergic inhibition via a protein kinase C-dependent mechanism. Neuropharmacology. 2017;113:314–22.
Parakala ML, Zhang Y, Modgil A, Chadchankar J, Vien TN, Ackley MA, et al. Metabotropic, but not allosteric, effects of neurosteroids on GABAergic inhibition depend on the phosphorylation of GABAA receptors. J Biol Chem. 2019;294:12220–30.
Reddy DS, Estes WA. Clinical potential of neurosteroids for CNS disorders. Trends Pharm Sci. 2016;37:543–61.
Bianchi MT, Macdonald RL. Neurosteroids shift partial agonist activation of GABAA receptor channels from low- to high-efficacy gating patterns. J Neurosci. 2003;23:10934–43.
Antonoudiou P, Colmers PLW, Walton NL, Weiss GL, Smith AC, Nguyen DP, et al. Allopregnanolone mediates affective switching through modulation of oscillatory states in the basolateral amygdala. Biol Psychiatry. 2022;91:283–93.
Northoff G, Sibille E. Why are cortical GABA neurons relevant to internal focus in depression? A cross-level model linking cellular, biochemical and neural network findings. Mol Psychiatry. 2014;19:966–77.
Lener MS, Niciu MJ, Ballard ED, Park M, Park LT, Nugent AC, et al. Glutamate and gamma-aminobutyric acid systems in the pathophysiology of major depression and antidepressant response to ketamine. Biol Psychiatry. 2017;81:886–97.
Zhong H, Rong J, Zhu C, Liang M, Li Y, Zhou R. Epigenetic modifications of GABAergic interneurons contribute to deficits in adult hippocampus neurogenesis and depression-like behavior in prenatally stressed mice. Int J Neuropsychopharmacol. 2020;23:274–85.
Poulter MO, Du L, Weaver ICG, Palkovits M, Faludi G, Merali Z, et al. GABAA receptor promoter hypermethylation in suicide brain: implications for the involvement of epigenetic processes. Biol Psychiatry. 2008;64:645–52.
Nardone S, Sams DS, Zito A, Reuveni E, Elliott E. Dysregulation of cortical meuron DNA methylation profile in autism spectrum disorder. Cereb Cortex. 2017;27:5739–54.
Costa E, Grayson DR, Guidotti A. Epigenetic downregulation of GABAergic function in schizophrenia: potential for pharmacological intervention? Mol Inter. 2003;3:220–9.
Ruzicka WB, Zhubi A, Veldic M, Grayson DR, Costa E, Guidotti A. Selective epigenetic alteration of layer I GABAergic neurons isolated from prefrontal cortex of schizophrenia patients using laser-assisted microdissection. Mol Psychiatry. 2007;12:385–97.
Guidotti A, Dong E, Gavin DP, Veldic M, Zhao W, Bhaumik DK, et al. DNA methylation/demethylation network expression in psychotic patients with a history of alcohol abuse. Alcohol Clin Exp Res. 2013;37:417–24.
Ho AM, Winham SJ, Armasu SM, Blacker CJ, Millischer V, Lavebratt C, et al. Genome-wide DNA methylomic differences between dorsolateral prefrontal and temporal pole cortices of bipolar disorder. J Psychiatr Res. 2019;117:45–54.
Klempan TA, Sequeira A, Canetti L, Lalovic A, Ernst C, ffrench-Mullen J, et al. Altered expression of genes involved in ATP biosynthesis and GABAergic neurotransmission in the ventral prefrontal cortex of suicides with and without major depression. Mol Psychiatry. 2009;14:175–89.
Sequeira A, Mamdani F, Ernst C, Vawter MP, Bunney WE, Lebel V, et al. Global brain gene expression analysis links glutamatergic and GABAergic alterations to suicide and major depression. PLoS One. 2009;4:e6585.
Shen Q, Lal R, Luellen BA, Earnheart JC, Andrews AM, Luscher B. gamma-Aminobutyric acid-type A receptor deficits cause hypothalamic-pituitary-adrenal axis hyperactivity and antidepressant drug sensitivity reminiscent of melancholic forms of depression. Biol Psychiatry. 2010;68:512–20.
Smith KS, Rudolph U. Anxiety and depression: mouse genetics and pharmacological approaches to the role of GABAA receptor subtypes. Neuropharmacology. 2012;62:54–62.
Hewitt SA, Wamsteeker JI, Kurz EU, Bains JS. Altered chloride homeostasis removes synaptic inhibitory constraint of the stress axis. Nat Neurosci. 2009;12:438–43.
Ren Z, Pribiag H, Jefferson SJ, Shorey M, Fuchs T, Stellwagen D, et al. Bidirectional homeostatic regulation of a depression-related brain state by gamma-aminobutyric acidergic deficits and ketamine treatment. Biol Psychiatry. 2016;80:457–68.
Vollenweider I, Smith KS, Keist R, Rudolph U. Antidepressant-like properties of alpha2-containing GABAA receptors. Behav Brain Res. 2011;217:77–80.
Koester C, Rudolph U, Haenggi T, Papilloud A, Fritschy JM, Crestani F. Dissecting the role of diazepam-sensitive gamma-aminobutyric acid type A receptors in defensive behavioral reactivity to mild threat. Pharm Biochem Behav. 2013;103:541–9.
Maguire J, Mody I. GABAAR plasticity during pregnancy: relevance to postpartum depression. Neuron. 2008;59:207–13.
Melon L, Hammond R, Lewis M, Maguire J. A novel, synthetic, neuroactive steroid Is effective at decreasing depression-like behaviors and improving maternal care in preclinical models of postpartum depression. Front Endocrinol (Lausanne). 2018;9:703.
Acosta GB, Rubio MC. GABAA receptors mediate the changes produced by stress on GABA function and locomotor activity. Neurosci Lett. 1994;176:29–31.
Otero Losada ME. Changes in central GABAergic function following acute and repeated stress. Br J Pharm. 1988;93:483–90.
Czeh B, Vardya I, Varga Z, Febbraro F, Csabai D, Martis LS, et al. Long-term stress disrupts the structural and functional integrity of GABAergic neuronal networks in the medial prefrontal cortex of rats. Front Cell Neurosci. 2018;12:148.
Shalaby A, Kamal S. Effect of Escitalopram on GABA level and anti-oxidant markers in prefrontal cortex and nucleus accumbens of chronic mild stress-exposed albino rats. Int J Physiol Pathophysiol Pharm. 2009;1:154–61.
Sherman AD, Petty F. Neurochemical basis of the action of antidepressants on learned helplessness. Behav Neural Biol. 1980;30:119–34.
Petty F, Sherman AD. GABAergic modulation of learned helplessness. Pharm Biochem Behav. 1981;15:567–70.
Sanacora G, Gueorguieva R, Epperson CN, Wu YT, Appel M, Rothman DL, et al. Subtype-specific alterations of gamma-aminobutyric acid and glutamate in patients with major depression. Arch Gen Psychiatry. 2004;61:705–13.
Bhagwagar Z, Wylezinska M, Jezzard P, Evans J, Ashworth F, Sule A, et al. Reduction in occipital cortex gamma-aminobutyric acid concentrations in medication-free recovered unipolar depressed and bipolar subjects. Biol Psychiatry. 2007;61:806–12.
DeFelipe J. Types of neurons, synaptic connections and chemical characteristics of cells immunoreactive for calbindin-D28K, parvalbumin and calretinin in the neocortex. J Chem Neuroanat. 1997;14:1–19.
Rajkowska G, O’Dwyer G, Teleki Z, Stockmeier CA, Miguel-Hidalgo JJ. GABAergic neurons immunoreactive for calcium binding proteins are reduced in the prefrontal cortex in major depression. Neuropsychopharmacology. 2007;32:471–82.
Klumpers UM, Veltman DJ, Drent ML, Boellaard R, Comans EF, Meynen G, et al. Reduced parahippocampal and lateral temporal GABAA-[11C]flumazenil binding in major depression: preliminary results. Eur J Nucl Med Mol Imaging. 2010;37:565–74.
Levinson AJ, Fitzgerald PB, Favalli G, Blumberger DM, Daigle M, Daskalakis ZJ. Evidence of cortical inhibitory deficits in major depressive disorder. Biol Psychiatry. 2010;67:458–64.
Schur RR, Draisma LW, Wijnen JP, Boks MP, Koevoets MG, Joels M, et al. Brain GABA levels across psychiatric disorders: a systematic literature review and meta-analysis of 1 H-MRS studies. Hum Brain Mapp. 2016;37:3337–52.
Godfrey KEM, Gardner AC, Kwon S, Chea W, Muthukumaraswamy SD. Differences in excitatory and inhibitory neurotransmitter levels between depressed patients and healthy controls: a systematic review and meta-analysis. J Psychiatr Res. 2018;105:33–44.
Price RB, Shungu DC, Mao X, Nestadt P, Kelly C, Collins KA, et al. Amino acid neurotransmitters assessed by proton magnetic resonance spectroscopy: relationship to treatment resistance in major depressive disorder. Biol Psychiatry. 2009;65:792–800.
Gabbay V, Mao X, Klein RG, Ely BA, Babb JS, Panzer AM, et al. Anterior cingulate cortex gamma-aminobutyric acid in depressed adolescents: relationship to anhedonia. Arch Gen Psychiatry. 2012;69:139–49.
Gabbay V, Bradley KA, Mao X, Ostrover R, Kang G, Shungu DC. Anterior cingulate cortex gamma-aminobutyric acid deficits in youth with depression. Transl Psychiatry. 2017;7:e1216.
Karolewicz B, Maciag D, O’Dwyer G, Stockmeier CA, Feyissa AM, Rajkowska G. Reduced level of glutamic acid decarboxylase-67 kDa in the prefrontal cortex in major depression. Int J Neuropsychopharmacol. 2010;13:411–20.
Goddard AW. Cortical and subcortical gamma amino acid butyric acid deficits in anxiety and stress disorders: clinical implications. World J Psychiatry. 2016;6:43–53.
Sanacora G, Treccani G, Popoli M. Towards a glutamate hypothesis of depression: an emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology. 2012;62:63–77.
Kim YK, Na KS. Role of glutamate receptors and glial cells in the pathophysiology of treatment-resistant depression. Prog Neuropsychopharmacol Biol Psychiatry. 2016;70:117–26.
Todd KG, Baker GB. Neurochemical effects of the monoamine oxidase inhibitor phenelzine on brain GABA and alanine: a comparison with vigabatrin. J Pharm Pharm Sci. 2008;11:14s–21s.
Kucukibrahimoglu E, Saygin MZ, Caliskan M, Kaplan OK, Unsal C, Goren MZ. The change in plasma GABA, glutamine and glutamate levels in fluoxetine- or S-citalopram-treated female patients with major depression. Eur J Clin Pharm. 2009;65:571–7.
Sanacora G, Mason GF, Rothman DL, Krystal JH. Increased occipital cortex GABA concentrations in depressed patients after therapy with selective serotonin reuptake inhibitors. Am J Psychiatry. 2002;159:663–5.
Sanacora G, Mason GF, Rothman DL, Hyder F, Ciarcia JJ, Ostroff RB, et al. Increased cortical GABA concentrations in depressed patients receiving ECT. Am J Psychiatry. 2003;160:577–9.
Sajdyk T, Johnson P, Fitz S, Shekhar A. Chronic inhibition of GABA synthesis in the bed nucleus of the stria terminalis elicits anxiety-like behavior. J Psychopharmacol. 2008;22:633–41.
Heldt SA, Mou L, Ressler KJ. In vivo knockdown of GAD67 in the amygdala disrupts fear extinction and the anxiolytic-like effect of diazepam in mice. Transl Psychiatry. 2012;2:e181.
Siegel JM. The neurotransmitters of sleep. J Clin Psychiatry. 2004;65:4–7.
Winkelman JW, Buxton OM, Jensen JE, Benson KL, O’Connor SP, Wang W, et al. Reduced brain GABA in primary insomnia: preliminary data from 4T proton magnetic resonance spectroscopy (1H-MRS). Sleep. 2008;31:1499–506.
Morgan PT, Pace-Schott EF, Mason GF, Forselius E, Fasula M, Valentine GW, et al. Cortical GABA levels in primary insomnia. Sleep. 2012;35:807–14.
Wang L, Pan Y, Ye C, Guo L, Luo S, Dai S, et al. A network meta-analysis of the long- and short-term efficacy of sleep medicines in adults and older adults. Neurosci Biobehav Rev. 2021;131:489–96.
Gunja N. The clinical and forensic toxicology of Z-drugs. J Med Toxicol. 2013;9:155–62.
McKlveen JM, Moloney RD, Scheimann JR, Myers B, Herman JP. “Braking” the prefrontal cortex: the role of glucocorticoids and interneurons in stress adaptation and pathology. Biol Psychiatry. 2019;86:669–81.
Kamigaki T. Dissecting executive control circuits with neuron types. Neurosci Res. 2019;141:13–22.
Hantsoo L, Epperson CN. Premenstrual dysphoric disorder: epidemiology and treatment. Curr Psychiatry Rep. 2015;17:87.
Luchetti S, Huitinga I, Swaab DF. Neurosteroid and GABA-A receptor alterations in Alzheimer’s disease, Parkinson’s disease and multiple sclerosis. Neuroscience. 2011;191:6–21.
Naylor JC, Kilts JD, Szabo ST, Dunn CE, Keefe FJ, Tupler LA, et al. Allopregnanolone levels are inversely associated with self-reported pain symptoms in U.S. Iraq and Afghanistan-era veterans: implications for biomarkers and therapeutics. Pain Med. 2016;17:25–32.
Rasmusson AM, King MW, Valovski I, Gregor K, Scioli-Salter E, Pineles SL, et al. Relationships between cerebrospinal fluid GABAergic neurosteroid levels and symptom severity in men with PTSD. Psychoneuroendocrinology. 2019;102:95–104.
Uzunova V, Sheline Y, Davis JM, Rasmusson A, Uzunov DP, Costa E, et al. Increase in the cerebrospinal fluid content of neurosteroids in patients with unipolar major depression who are receiving fluoxetine or fluvoxamine. Proc Natl Acad Sci USA. 1998;95:3239–44.
Romeo E, Strohle A, Spalletta G, di Michele F, Hermann B, Holsboer F, et al. Effects of antidepressant treatment on neuroactive steroids in major depression. Am J Psychiatry. 1998;155:910–3.
Agis-Balboa RC, Guidotti A, Pinna G. 5alpha-reductase type I expression is downregulated in the prefrontal cortex/Brodmann’s area 9 (BA9) of depressed patients. Psychopharmacol (Berl). 2014;231:3569–80.
Nappi RE, Petraglia F, Luisi S, Polatti F, Farina C, Genazzani AR. Serum allopregnanolone in women with postpartum “blues”. Obstet Gynecol. 2001;97:77–80.
Strohle A, Romeo E, di Michele F, Pasini A, Hermann B, Gajewsky G, et al. Induced panic attacks shift gamma-aminobutyric acid type A receptor modulatory neuroactive steroid composition in patients with panic disorder: preliminary results. Arch Gen Psychiatry. 2003;60:161–8.
Concas A, Mostallino MC, Porcu P, Follesa P, Barbaccia ML, Trabucchi M, et al. Role of brain allopregnanolone in the plasticity of gamma-aminobutyric acid type A receptor in rat brain during pregnancy and after delivery. Proc Natl Acad Sci USA. 1998;95:13284–9.
Grobin AC, Morrow AL. 3Alpha-hydroxy-5alpha-pregnan-20-one levels and GABA(A) receptor-mediated 36Cl(−) flux across development in rat cerebral cortex. Brain Res Dev Brain Res. 2001;131:31–39.
Purdy RH, Morrow AL, Moore PH Jr., Paul SM. Stress-induced elevations of gamma-aminobutyric acid type A receptor-active steroids in the rat brain. Proc Natl Acad Sci USA. 1991;88:4553–7.
Maguire J. Stress-induced plasticity of GABAergic inhibition. Front Cell Neurosci. 2014;8:157.
Dong E, Matsumoto K, Uzunova V, Sugaya I, Takahata H, Nomura H, et al. Brain 5alpha-dihydroprogesterone and allopregnanolone synthesis in a mouse model of protracted social isolation. Proc Natl Acad Sci USA. 2001;98:2849–54.
Matsumoto K, Pinna G, Puia G, Guidotti A, Costa E. Social isolation stress-induced aggression in mice: a model to study the pharmacology of neurosteroidogenesis. Stress. 2005;8:85–93.
Pibiri F, Nelson M, Guidotti A, Costa E, Pinna G. Decreased corticolimbic allopregnanolone expression during social isolation enhances contextual fear: a model relevant for posttraumatic stress disorder. Proc Natl Acad Sci USA. 2008;105:5567–72.
Evans J, Sun Y, McGregor A, Connor B. Allopregnanolone regulates neurogenesis and depressive/anxiety-like behaviour in a social isolation rodent model of chronic stress. Neuropharmacology. 2012;63:1315–26.
Serra M, Pisu MG, Littera M, Papi G, Sanna E, Tuveri F, et al. Social isolation-induced decreases in both the abundance of neuroactive steroids and GABAA receptor function in rat brain. J Neurochem. 2000;75:732–40.
Pinna G, Costa E, Guidotti A. Fluoxetine and norfluoxetine stereospecifically and selectively increase brain neurosteroid content at doses that are inactive on 5-HT reuptake. Psychopharmacol (Berl). 2006;186:362–72.
Pinna G, Dong E, Matsumoto K, Costa E, Guidotti A. In socially isolated mice, the reversal of brain allopregnanolone down-regulation mediates the anti-aggressive action of fluoxetine. Proc Natl Acad Sci USA. 2003;100:2035–40.
Locci A, Pinna G. Neurosteroid biosynthesis down-regulation and changes in GABAA receptor subunit composition: a biomarker axis in stress-induced cognitive and emotional impairment. Br J Pharm. 2017;174:3226–41.
Vega Alanis BA, Iorio MT, Silva LL, Bampali K, Ernst M, Schnurch M, et al. Allosteric GABAA receptor modulators-a review on the most recent heterocyclic chemotypes and their synthetic accessibility. Molecules. 2020;25:999.
Twyman RE, Rogers CJ, Macdonald RL. Differential regulation of gamma-aminobutyric acid receptor channels by diazepam and phenobarbital. Ann Neurol. 1989;25:213–20.
Hanson SM, Czajkowski C. Structural mechanisms underlying benzodiazepine modulation of the GABA(A) receptor. J Neurosci. 2008;28:3490–9.
Bianchi MT, Botzolakis EJ, Lagrange AH, Macdonald RL. Benzodiazepine modulation of GABA(A) receptor opening frequency depends on activation context: a patch clamp and simulation study. Epilepsy Res. 2009;85:212–20.
Baer AB & Holstege CP Barbiturates, long-acting. in Encyclopedia of Toxicology (Second Edition) (ed. Wexler, P) 209-10 (Elsevier, New York, 2005).
Löscher W, Rogawski MA. How theories evolved concerning the mechanism of action of barbiturates. Epilepsia. 2012;53:12–25.
Ciraulo DA & Oldham M Chapter Sixteen - Sedative hypnotics. in The Effects of Drug Abuse on the Human Nervous System (eds. Madras, B & Kuhar, M) 499-532 (Academic Press, Boston, 2014).
Balon R, Starcevic V, Silberman E, Cosci F, Dubovsky S, Fava GA, et al. The rise and fall and rise of benzodiazepines: a return of the stigmatized and repressed. Braz J Psychiatry. 2020;42:243–4.
Petty F, Trivedi MH, Fulton M, Rush AJ. Benzodiazepines as antidepressants: does GABA play a role in depression? Biol Psychiatry. 1995;38:578–91.
Lim B, Sproule BA, Zahra Z, Sunderji N, Kennedy SH, Rizvi SJ. Understanding the effects of chronic benzodiazepine use in depression: a focus on neuropharmacology. Int Clin Psychopharmacol. 2020;35:243–53.
Lader M. Benzodiazepines revisited–will we ever learn? Addiction. 2011;106:2086–109.
Cheng T, Wallace DM, Ponteri B, Tuli M. Valium without dependence? Individual GABA(A) receptor subtype contribution toward benzodiazepine addiction, tolerance, and therapeutic effects. Neuropsychiatr Dis Treat. 2018;14:1351–61.
Dodds TJ. Prescribed benzodiazepines and suicide risk: a review of the literature. Prim Care Companion CNS Disord. 2017;19(2). https://doi.org/10.4088/PCC.16r02037.
McKernan RM, Rosahl TW, Reynolds DS, Sur C, Wafford KA, Atack JR, et al. Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABA(A) receptor alpha1 subtype. Nat Neurosci. 2000;3:587–92.
Crestani F, Martin JR, Mohler H, Rudolph U. Mechanism of action of the hypnotic zolpidem in vivo. Br J Pharm. 2000;131:1251–4.
Fiorelli R, Rudolph U, Straub CJ, Feldon J, Yee BK. Affective and cognitive effects of global deletion of alpha3-containing gamma-aminobutyric acid-A receptors. Behav Pharm. 2008;19:582–96.
Engin E, Liu J, Rudolph U. α2-containing GABA(A) receptors: a target for the development of novel treatment strategies for CNS disorders. Pharm Ther. 2012;136:142–52.
Martinez Botella G, Salituro FG, Harrison BL, Beresis RT, Bai Z, Blanco MJ, et al. Neuroactive steroids. 2. 3alpha-hydroxy-3beta-methyl-21-(4-cyano-1H-pyrazol-1’-yl)-19-nor-5beta-pregnan-20 -one (SAGE-217): a clinical next generation neuroactive steroid positive allosteric modulator of the (gamma-Aminobutyric Acid)A receptor. J Med Chem. 2017;60:7810–9.
Jacob TC, Michels G, Silayeva L, Haydon J, Succol F, Moss SJ. Benzodiazepine treatment induces subtype-specific changes in GABAA receptor trafficking and decreases synaptic inhibition. Proc Natl Acad Sci USA. 2012;109:18595–600.
Wlodarczyk AI, Sylantyev S, Herd MB, Kersante F, Lambert JJ, Rusakov DA, et al. GABA-independent GABAA receptor openings maintain tonic currents. J Neurosci. 2013;33:3905–14.
Santarsieri D, Schwartz TL. Antidepressant efficacy and side-effect burden: a quick guide for clinicians. Drugs Context. 2015;4:212290.
Mantas I, Saarinen M, Xu ZD, Svenningsson P. Update on GPCR-based targets for the development of novel antidepressants. Mol Psychiatry. 2022;27:534–58.
Aleksandrova LR, Phillips AG, Wang YT. Antidepressant effects of ketamine and the roles of AMPA glutamate receptors and other mechanisms beyond NMDA receptor antagonism. J Psychiatry Neurosci. 2017;42:222–9.
Depression Medicines. U. S. Food and Drug Administration Office of Women’s Health. Available at https://www.fda.gov/consumers/free-publications-women/depression-medicines. Accessed: November 13, 2022.
Protti M, Mandrioli R, Marasca C, Cavalli A, Serretti A, Mercolini L. New-generation, non-SSRI antidepressants: Drug-drug interactions and therapeutic drug monitoring. Part 2: NaSSAs, NRIs, SNDRIs, MASSAs, NDRIs, and others. Med Res Rev. 2020;40:1794–832.
Mandrioli R, Protti M, Mercolini L. New-generation, non-SSRI antidepressants: therapeutic drug monitoring and pharmacological interactions. Part 1: SNRIs, SMSs, SARIs. Curr Med Chem. 2018;25:772–92.
Sheffler ZM, Patel P & Abdijadid S. Antidepressants. in StatPearls (Treasure Island (FL), 2022).
Chu A & Wadhwa R. Selective serotonin reuptake inhibitors. in StatPearls (Treasure Island (FL), 2021).
Lambert O, Bourin M. SNRIs: mechanism of action and clinical features. Expert Rev Neurother. 2002;2:849–58.
Anderson IM. Selective serotonin reuptake inhibitors versus tricyclic antidepressants: a meta-analysis of efficacy and tolerability. J Affect Disord. 2000;58:19–36.
White N, Litovitz T, Clancy C. Suicidal antidepressant overdoses: a comparative analysis by antidepressant type. J Med Toxicol. 2008;4:238–50.
Gerhard DM, Wohleb ES, Duman RS. Emerging treatment mechanisms for depression: focus on glutamate and synaptic plasticity. Drug Discov Today. 2016;21:454–64.
Fiedorowicz JG, Swartz KL. The role of monoamine oxidase inhibitors in current psychiatric practice. J Psychiatr Pr. 2004;10:239–48.
De Crescenzo F, Perelli F, Armando M, Vicari S. Selective serotonin reuptake inhibitors (SSRIs) for post-partum depression (PPD): a systematic review of randomized clinical trials. J Affect Disord. 2014;152–154:39–44.
Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, Stewart JW, Warden D, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163:1905–17.
Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry. 2006;163:28–40.
Matveychuk D, Thomas RK, Swainson J, Khullar A, MacKay MA, Baker GB, et al. Ketamine as an antidepressant: overview of its mechanisms of action and potential predictive biomarkers. Ther Adv Psychopharmacol. 2020;10:2045125320916657.
Royo M, Escolano BA, Madrigal MP, Jurado S. AMPA receptor function in hypothalamic synapses. Front Synaptic Neurosci. 2022;14:833449.
Williams NR, Heifets BD, Blasey C, Sudheimer K, Pannu J, Pankow H, et al. Attenuation of antidepressant effects of ketamine by opioid receptor antagonism. Am J Psychiatry. 2018;175:1205–15.
Janssen Pharmaceuticals Inc. SPRAVATO® (esketamine) nasal spray, CIII [prescribing information]. Titusville, NJ: Janssen Pharmaceuticals, Inc. (2020).
Kishimoto T, Chawla JM, Hagi K, Zarate CA, Kane JM, Bauer M, et al. Single-dose infusion ketamine and non-ketamine N-methyl-d-aspartate receptor antagonists for unipolar and bipolar depression: a meta-analysis of efficacy, safety and time trajectories. Psychol Med. 2016;46:1459–72.
Winstock AR, Mitcheson L, Gillatt DA, Cottrell AM. The prevalence and natural history of urinary symptoms among recreational ketamine users. BJU Int. 2012;110:1762–6.
Strous JFM, Weeland CJ, van der Draai FA, Daams JG, Denys D, Lok A, et al. Brain changes associated with long-term ketamine abuse, a systematic review. Front Neuroanat. 2022;16:795231.
SAGE Therapeutics Inc. ZULRESSO (brexanolone) injection for intravenous use [prescribing information]. Cambridge, MA: SAGE Therapeutics, Inc. 2019;
Sage Therapeutics Inc. ZULRESSO (brexanolone) injection for intravenous use [prescribing information]. Cambridge, MA: Sage Therapeutics Inc. 2022;
Frieder A, Fersh M, Hainline R, Deligiannidis KM. Pharmacotherapy of postpartum depression: current approaches and novel drug development. CNS drugs. 2019;33:265–82.
Meltzer-Brody S, Colquhoun H, Riesenberg R, Epperson CN, Deligiannidis KM, Rubinow DR, et al. Brexanolone injection in post-partum depression: two multicentre, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet. 2018;392:1058–70.
Gerbasi ME, Meltzer-Brody S, Acaster S, Fridman M, Bonthapally V, Hodgkins P, et al. Brexanolone in postpartum depression: post hoc analyses to help inform clinical decision-making. J Women’s Health (Larchmt). 2021;30:385–92.
Kanes S, Colquhoun H, Gunduz-Bruce H, Raines S, Arnold R, Schacterle A, et al. Brexanolone (SAGE-547 injection) in post-partum depression: a randomised controlled trial. Lancet. 2017;390:480–9.
A clinical trial of PRAX-114 in participants with major depressive disorder. ClinicalTrials.gov identifier: NCT04832425. Available at https://clinicaltrials.gov/ct2/show/NCT04832425?term=Prax+114&draw=2&rank=2. Accessed: June 10.
A clinical trial of adjunctive and monotherapy PRAX-114 in participants with major depressive disorder. ClinicalTrials.gov identifier: NCT04969510. Available at https://clinicaltrials.gov/ct2/show/NCT04969510?term=Prax+114&draw=2&rank=3. Accessed: June 10.
Hecking J, Davoudian PA, Wilkinson ST. Emerging therapeutics based on the amino acid neurotransmitter system: an update on the pharmaceutical pipeline for mood disorders. Chronic Stress (Thousand Oaks). 2021;5:24705470211020446.
Praxis Precision Medicines reports negative results from PRAX-114 phase 2/3 monotherapy Aria Study in patients with major depressive disorder. News release. Praxis Precision Medicines. June 6, 2022. Accessed March 14, 2023. https://investors.praxismedicines.com/news-releases/news-release-details/praxis-precision-medicines-reports-negative-results-prax-114.
Monti L, Nicolini H, Liebowitz MR, Hanover R. A placebo controlled trial of PH10: test of a rapidly acting intranasally administered antidepressant. Br J Pharm Med Res. 2019;4:12.
VistaGen announces positive results of newly published exploratory phase 2a study of PH10 for rapid-onset treatment of major depressive disorder. News release. VistaGen Therapeutics. Februrary 20, 2020. Accessed March 14, 2023. https://www.vistagen.com/news-releases/news-release-details/vistagen-announces-positive-results-newly-published-exploratory.
VistaGen’s PH10 nasal spray demonstrates different mechanism of action from benzodiazepines in preclinical study. News release. VistaGen Therapeutics. March 11, 2021. Accessed March 14, 2023. https://www.vistagen.com/news-releases/news-release-details/vistagens-ph10-nasal-spray-demonstrates-different-mechanism
Pipeline overview. Available at https://www.vistagen.com/pipeline/overview. Accessed: June 24, 2022.
Althaus AL, Ackley MA, Belfort GM, Gee SM, Dai J, Nguyen DP, et al. Preclinical characterization of zuranolone (SAGE-217), a selective neuroactive steroid GABAA receptor positive allosteric modulator. Neuropharmacology. 2020;181:108333.
Hoffmann E, Nomikos GG, Kaul I, Raines S, Wald J, Bullock A, et al. SAGE-217, a novel GABAA receptor positive allosteric modulator: clinical pharmacology and tolerability in randomized phase I dose-finding studies. Clin Pharmacokinet. 2020;59:111–20.
Smith DA, Beaumont K, Maurer TS, Di L. Relevance of half-life in drug design. J Med Chem. 2018;61:4273–82.
Deligiannidis KM, Meltzer-Brody S, Gunduz-Bruce H, Doherty J, Jonas J, Li S, et al. Effect of zuranolone vs placebo in postpartum depression: a randomized clinical trial. JAMA Psychiatry. 2021;78:951–9.
Deligiannidis K, Meltzer-Brody S, Maximos B, et al. Zuranolone for the treatment of adults with postpartum depression. Am J Psychiatry. 2023. In press.
Gunduz-Bruce H, Silber C, Kaul I, Rothschild AJ, Riesenberg R, Sankoh AJ, et al. Trial of SAGE-217 in patients with major depressive disorder. N. Engl J Med. 2019;381:903–11.
Clayton AH, Lasser R, Parikh SV, Iosifescu DV, Jung J, Kotecha M, et al. Zuranolone for the treatment of adults with major depressive disorder: a phase 3, randomized, placebo‐controlled trial. Am J Psychiatry. 2023;appiajp20220459. https://doi.org/10.1176/appi.ajp.20220459.
Clayton AH, Lasser R, Nandy I, Sankoh AJ, Jonas J, Kanes SJ. Zuranolone in major depressive disorder: results from MOUNTAIN—a phase 3, multicenter, double-blind, randomized, placebo-controlled trial. J Clin Psychiatry. 2023;84:22m14445.
Clayton AH, Deligiannidis KM, Kotecha M, Doherty J. Overview of the rapid antidepressant effects observed in the zuranolone clinical development program [A164]. Obstet Gynecol. 2022;139:47S–48S.
Sage Therapeutics and Biogen announce the phase 3 CORAL Study met its primary and key secondary endpoints - comparing zuranolone 50 mg co-initiated with standard of sare antidepressant vs. standard of care co-initiated with placebo in people with MDD. News release. Sage Therapeutics Inc. Februrary 16, 2022. Accessed March 14, 2023. https://investor.sagerx.com/news-releases/news-release-details/sage-therapeutics-and-biogen-announce-phase-3-coral-study-met.
Sage Therapeutics announces continued positive zuranolone data for both 30 mg and 50 mg doses in open-label SHORELINE Study in patients with MDD. News release. Sage Therapeutics Inc. March 17, 2021. Accessed March 14, 2023. https://investor.sagerx.com/news-releases/news-release-details/sage-therapeutics-announces-continued-positive-zuranolone-data.
Sage Therapeutics and Biogen announce positive, one-year zuranolone 50 mg data in the ongoing open-label SHORELINE Study in patients with MDD. News release. Biogen Inc. December 1, 2021. Accessed March 14, 2023. https://investors.biogen.com/news-releases/news-release-details/sage-therapeutics-and-biogen-announce-positive-one-year.
Jones BDM, Razza LB, Weissman CR, Karbi J, Vine T, Mulsant LS, et al. Magnitude of the placebo response across treatment modalities used for treatment-resistant depression in adults: a systematic review and meta-analysis. JAMA Netw Open. 2021;4:e2125531.
Dell’Osso B, Palazzo MC, Oldani L, Altamura AC. The noradrenergic action in antidepressant treatments: pharmacological and clinical aspects. CNS Neurosci Ther. 2011;17:723–32.
Moraczewski J & Aedma KK. Tricyclic antidepressants. in StatPearls (Treasure Island (FL), 2021).
Sub Laban T & Saadabadi A. Monoamine oxidase inhibitors (MAOI). in StatPearls (Treasure Island (FL), 2021).
Sabri MA & Saber-Ayad MM. MAO Inhibitors. in StatPearls (Treasure Island (FL), 2022).
Nguyen L, Robson MJ, Healy JR, Scandinaro AL, Matsumoto RR. Involvement of sigma-1 receptors in the antidepressant-like effects of dextromethorphan. PLoS One. 2014;9:e89985.
Iosifescu DV, Jones A, O’Gorman C, Streicher C, Feliz S, Fava M, et al. Efficacy and safety of AXS-05 (dextromethorphan-bupropion) in patients with major depressive disorder: a phase 3 randomized clinical trial (GEMINI). J Clin Psychiatry. 2022;83:21m14345.
Schwartz J, Murrough JW, Iosifescu DV. Ketamine for treatment-resistant depression: recent developments and clinical applications. Evid Based Ment Health. 2016;19:35–38.
Acknowledgements
Medical writing and editorial assistance were provided by Symbiotix, LLC (Linda M Ritter, PhD, Medical Director) and funded by Sage Therapeutics, Inc., and Biogen Inc.
Author information
Authors and Affiliations
Contributions
AJC, GWM, and VM were involved in the conceptualization, writing, and editing of the paper.
Corresponding author
Ethics declarations
Competing interests
AJC serves as a consultant to AbbVie, Acadia, AiCure, Alfasigma, Alkermes, Axsome, BioXcel, Boehringer Ingelheim, Cognitive Research, Corium, Intra-Cellular Therapies, Janssen, Jazz Pharmaceuticals, Lundbeck, MedAvante-Prophase, Neurocrine, Noven, Otsuka, Relmada, Sage Therapeutics, Inc., Sunovion, Supernus, Takeda, and Teva. Dr. Cutler serves as a speaker for/receives promotional honoraria from AbbVie, Acadia, Alfasigma, Alkermes, Axsome, Corium, Intra-Cellular Therapies, Janssen, Lundbeck, Neurocrine, Noven, Otsuka, Sunovion, Supernus, Takeda, and Teva. He has received research grants from Acadia, Alkermes, Allergan, Axsome, Biohaven, Intra-Cellular Therapies, Janssen, Lilly, Lundbeck, Novartis, Otsuka, Sage Therapeutics, Inc., Sunovion, and Takeda. Dr. Cutler is also an employee and board member of the Neuroscience Education Institute. GWM serves as a researcher for AbbVie, Akili, Alkermes, Axsome, Boehringer Ingelheim, Genentech, Janssen, Lundbeck, Medgenics, NLS Pharma, Otsuka, Reckitt Benckiser, Roche, Sage Therapeutics, Inc., Sunovion, Supernus, Takeda, Taisho, and Teva. Dr. Mattingly serves as a consultant for AbbVie, Alkermes, Alfasigma, Ironshore, Janssen, Lundbeck, Major League Baseball, Otsuka, National Football League, Neos, NLS Pharma, Purdue, Rhodes, Sage Therapeutics, Inc., Sunovion, Supernus, Takeda, Teva, and Vanda. Additionally, he serves as a speaker for AbbVie, Alkermes, Ironshore, Janssen, Lundbeck, Otsuka, Neos, Shire, Sunovion, Takeda, and Teva. VM serves as a consultant for AbbVie/Allergan, Acadia Pharmaceuticals, Inc., Alfasigma, USA, Inc., Alkermes, Inc., Eisai, Intra-Cellular Therapies, Ironshore, Janssen, Lundbeck A/S, Jazz Pharmaceuticals, Noven Pharmaceuticals Inc, Otsuka America Pharmaceutical, Inc., Sage Therapeutics, Inc., Sunovion, Supernus, and Takeda. Dr. Maletic also serves as a speaker for AbbVie, Acadia, Alfasigma, Alkermes, Allergan, Eisai, Ironshore, Intra-Cellular, Janssen, H. Lundbeck A/S, Otsuka America Pharmaceutical, Inc., Sunovion, Supernus, and Takeda.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cutler, A.J., Mattingly, G.W. & Maletic, V. Understanding the mechanism of action and clinical effects of neuroactive steroids and GABAergic compounds in major depressive disorder. Transl Psychiatry 13, 228 (2023). https://doi.org/10.1038/s41398-023-02514-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-023-02514-2
