
Abstract
Squamous cell carcinomas of the lower female genital tract may be human papillomavirus-associated or independent. We studied the HPV status, mutational repertoire, histology, and clinical data of 28 samples from 26 patients, 65% with a vulvar primary and 35% with a vaginal primary. These represented invasive vulvovaginal squamous cell carcinomas that underwent clinical tumor-normal targeted massively parallel sequencing analysis. HPV status was determined using the HPV high-risk RNA ISH assay and/or by MSK-IMPACT. Eleven patients had HPV-associated squamous cell carcinoma (four vulvar and seven vaginal) and 15 patients had HPV-independent SqCC (13 vulvar and 2 vaginal). Well-differentiated squamous cell carcinomas were always HPV-independent. HPV-independent moderately and poorly differentiated carcinomas frequently had alterations in the NOTCH signaling pathway (6/7), which were also associated with increased tumor budding (P: 0.002). HPV-associated vulvovaginal squamous cell carcinoma had PIK3CA activating mutations (7/11, 64%) as the most common genomic event, while TERT gene alterations, mainly TERT promoter mutations (14/15 cases, 93%) featured significantly in HPV-independent carcinomas. Other common abnormalities in HPV-independent tumors were TP53 mutations (13/15, 87%), CDKN2A alterations (10/15, 67%), and NOTCH1 and FAT1 mutations (7/15, 47% each). A subset of both HPV-associated and -independent tumors had NOTCH pathway alterations (6/11, 55% and 10/15, 67% respectively), but different genes in this pathway were altered in these tumors. In summary, TERT, TP53, CDKN2A, and NOTCH1 gene alterations strongly point away from an HPV-driven process (odds ratios: 0.01, 0.07, 0, and 0, respectively with p values < 0.02 for all four genes), while PIK3CA activating mutations without the other mutations strongly favors an HPV-driven tumor (odds ratio: 10.12, p value: 0.016). HPV-independent carcinomas are more likely to be moderately-poorly differentiated with intermediate to high tumor cell budding. Cancer cell fraction analysis of HPV-independent squamous carcinomas suggests that TERT and/or NOTCH1 alterations along with TP53 alterations can be the initiating event in these tumors.
Similar content being viewed by others

Introduction
The incidence of invasive vulvovaginal squamous cell carcinoma nearly doubled over the last 15 years and is expected to rise because of an aging population. It has been estimated that ~6120 new cases of vulvar cancer will be diagnosed in the US in 2020, with roughly 1350 attributable deaths, accounting for 5–6% of all gynecologic cancers1.
There are at least two mechanisms that lead to the development of Vulvovaginal squamous cell carcinomas: HPV-associated squamous cell carcinoma, typically vaginal, lacking TP53 mutation, with high grade squamous intraepithelial lesion in form of usual-type vulvar intraepithelial neoplasia (uVIN) as a precursor; and HPV-independent vulvar SqCC, typically vulvar, associated with TP53 mutation, with differentiated vulvar intraepithelial neoplasia (dVIN) as a precursor2,3,4,5,6. HPV-independent vulvovaginal squamous cell carcinoma is mostly a disease of postmenopausal women and is frequently associated with chronic inflammatory skin diseases such as lichen sclerosus and/or lichen simplex chronicus, lesions thought to be non-obligate precursors of such vulvovaginal squamous cell carcinomas. The mechanism of the progression of differentiated VIN or, as suggested by some authors, of lichen sclerosus to vulvovaginal squamous cell carcinoma is under investigation3. Approximately one-third of all vulvovaginal squamous cell carcinoma patients suffer from recurrence, for which therapeutic options are limited7. Recently, an additional type of HPV-independent pathogenetic pathway has been described, which involves mutations in the PIK3CA gene and progression from differentiated exophytic vulvar intraepithelial lesion/vulvar acanthosis with altered differentiation (DEVIL/VAAD) to verrucous carcinoma8,9 and HPV-independent squamous cell carcinoma.
The aim of this study was to characterize the molecular landscape of vulvovaginal squamous cell carcinoma and identify the molecular underpinnings of HPV-associated and HPV-independent tumors.
Methods and methods
This study, including review and analysis of data, was approved by the Institutional Review Board at Memorial Sloan Kettering Cancer Center (MSKCC).
Study cohort
All vulvovaginal squamous cell carcinoma that underwent clinical tumor-normal targeted massively parallel sequencing analysis of 410–468 cancer-related genes (Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets; MSK-IMPACT) were selected for this study. We studied 28 samples of vulvovaginal squamous cell carcinoma from 26 patients; 17 primary tumors, 4 local recurrences and 7 metastatic tumors (lung, pelvic soft tissue, bone, lymph nodes, and others) were sequenced. Clinical sequencing was performed per institutional guidelines and as required by the clinical team, as such the cohort is highly selected for tumors with recurrence/disease persistence or otherwise adverse clinical presentation.
All cases were reviewed by a gynecologic pathologist at MSKCC. Tumors were graded as well-, moderately-, or poorly- differentiated squamous cell carcinoma based on a combination of cytologic atypia and keratinization. Briefly, a well-differentiated vulvovaginal squamous cell carcinoma has no to minimal cytologic atypia, abundant keratinization, and/or keratin pearls. Moderately differentiated VSqCC showed moderate cytologic atypia with less notable areas of keratinization. Poorly differentiated carcinomas showed marked cytologic atypia with no or minimal keratinization10,11. The evaluation of tumor budding was based on criteria from International Tumor Budding Consensus Conference 2016 recommendations. Tumor budding was defined as a single cancer cell or a cluster of <5 cancer cells in the stroma found at the periphery of the tumor. If multiple areas where different grades were found in one specimen, the highest grade was taken (Hot spot method). Tumor budding was categorized into low (<5 buds/field), intermediate (≥5 to <10 buds/field), and high (≥10 buds/field). Budding was counted using ×20 objective with an adjusted standard field size of 0.785 mm2. Sections with no tumor buds were included in the low category group12,13. Patients’ demographic characteristics and relevant clinical data were extracted from the electronic medical records.
DNA extraction and targeted capture massively parallel sequencing
Genomic DNA from surgically resected tumor and patient‐matched normal samples were extracted from formalin‐fixed paraffin‐embedded specimens. Massively parallel sequencing was performed using Memorial Sloan Kettering‐Integrated Mutation Profiling of Actionable Cancer Targets (MSK‐IMPACT™) platform designed for targeted sequencing of exons and select introns of 410–468 cancer‐related genes as previously reported. Allelic-specific copy number alterations and allelic-specific loss of heterozygosity were defined using FACETS14. Cancer cell fractions of all mutations were inferred using ABSOLUTE (v1.0.6) and tumor clonal evolution was evaluated using PyClone (v0.13.1), as described previously15.
HPV status determination
The high-risk human papillomavirus (HR-HPV) status in each case was determined based on either positivity of P16 immunohistochemistry as diffuse, strong and continuous, nuclear and cytoplasmic staining16 and/or HPV-HR RNA ISH17 as presence of dark-brown, dot-like cytoplasmic and/or nuclear positivity. Furthermore, sequencing reads from MSK-IMPACT test were filtered to analyze only reads that do not align to the human genome. The non-human reads were then analyzed with “blastn” algorithm utilizing the NCBI NT database. Reads that mapped with greater than 90% identity to an entry in the database were labeled as a single read present for the database entry. The database entry was cross walked to the NCBI Taxonomy and the read quantity was calculated as the number of reads that mapped to the HPV subtype. This method has been validated and shown to have similar performance to high-risk HPV ISH testing18.
Statistical analysis
Statistical analyses were performed using the R.22. Clinical characteristics, and molecular alterations were compared using Chi-squared Fisher’s exact test for qualitative variables, and Mann-Whitney’s U test for nonparametric continuous variables. p values < 0.05 were considered statistically significant.
Results
Clinicopathologic characteristics and prognosis relative to HPV status
28 samples from 26 patients were evaluated in this study. These included 17 (65%) vulvar carcinoma patients and 9 (35%) with a vaginal primary.
HPV testing was performed in 23 of the 26 patients (25 samples) with 12 specimens tested using p16 assay and 15 specimens tested using the HPV high-risk RNA ISH assay. HPV status was further investigated using the MSK-IMPACT assay in all samples. The summative results of HPV testing from all assays showed that 11 patients had HPV-associated Squamous cell carcinoma and 15 patients had HPV-independent SqCC. The HPV-associated and HPV-independent tumors showed site differences with the majority of vulvar squamous cell carcinoma being HPV-independent (13/17) in contrast to the vaginal tumors where the majority were HPV associated (7/9) (X2 P: 0.008).
The median age of the patients at initial diagnosis was 56.5 years (range: 26–92 years) with no statistically significant difference observed between the HPV-associated and HPV-independent tumors (Mann-Whitney U P: 0.889). Clinical staging information was available for 24 patients which showed that 50% of the patients presented with stage I disease (n = 12), followed by stage III, stage IV, and stage II disease (n = 8, 3, 2, respectively). No difference in stage distribution was observed based on the HPV status (X2 P: 0.871).
Evaluation of the background squamous epithelium (when possible) showed that lichen sclerosus was only seen in association with the HPV-independent tumors (X2 P: 0.013) with 8 of the 15 tumors showing background LS. dVIN was also mainly seen in HPV-independent tumors (X2 P: 0.004) with 11 (73.3%) of the HPV-independent vulvovaginal squamous cell carcinoma showing associated dVIN in the pathologic review of the specimen. In contrast, only one vulvovaginal squamous cell carcinoma with HPV positivity on ISH evaluation had associated dVIN (see below for further discussion of this case).
The median tumor size, width and depth of invasion were 3.3 cm (range: 0.7–9.8 cm), 2.0 cm (range: 0.5–3.5 cm), and 0.67 cm (range: 0.25–2.2 cm), respectively. The depth of invasion showed significant difference between the HPV-associated (median: 0.5 cm) and HPV-independent (median: 0.8 cm) tumors (Mann-Whitney U P: 0.023).
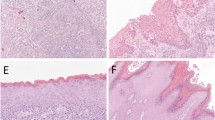
The vulvovaginal squamous cell carcinoma in our cohort consisted of eight (30.8%) well-differentiated tumors with the remainder being moderately or poorly differentiated tumors (n = 9 (34.6%) each). Well-differentiated tumors were only seen in HPV-independent tumors; all HPV-associated tumors were either moderately or poorly differentiated (X2 P: 0.013) (Fig. 1A, B). In HPV-independent tumors, the majority of the moderately/poorly differentiated tumors were associated with alterations in the NOTCH signaling pathway (6/7). Furthermore, NOTCH signaling pathway alterations were associated with increased tumor budding (Fig. 1C). Budding was able to be assessed in 17 cases, 14/15 HPV-independent and 3/9 HPV-associated. Low tumor budding was present in six HPV-independent cases (five NOTCH mutations and six TERT mutations) and three HPV-associated cases. Intermediate (n = 5; five TERT and one NOTCH) and high budding (n = 3; three TERT and NOTCH) were only found in HPV-independent cases.

A HPV-associated vulvovaginal squamous cell carcinomas are often moderately/poorly differentiated and are associated with a usual vulvar/vaginal intraepithelial neoplasm. B HPV-independent vulvovaginal squamous cell carcinomas can be well/moderately/poorly differentiated and are associated with a differentiated vulvar intraepithelial neoplasm. C Frequent tumor budding was seen in vulvovaginal squamous cell carcinomas with NOTCH signaling pathway alterations. D A case of vulvar squamous cell carcinoma is shown, which was positive for HPV infection (tested by high-risk HPV ISH) (E); further investigation showed this sample to harbor TERT and TP53 mutations and presence of a low-risk HPV (HPV 10) infection. The positive ISH result is likely a false-positive finding.
With a median follow-up of 20 months overall and 28 months among living patients, all 26 patients (100%) exhibited disease progression, including 2 patients with local recurrence, 3 with distant metastasis and 21 with both. The median disease-free survival was 8 months (interquartile range [IQR]: 4–21 months). 18 patients had disease progression with the median time to progression of 11 months (IQR: 6–27 months). 16 patients died of disease with median disease-specific survival of 42 months (IQR: 19–122 months). It must be noted that the clinical tumor sequencing was mainly performed on tumors with disease persistence or recurrence and as such we have avoided subgroup analysis for prognostic markers due to possible selection bias.
The clinical and pathologic features of the study cohort are summarized in Table 1.
One of our cases (Sample 15) consisted of a poorly differentiated vulvar squamous cell carcinoma with a dVIN precursor lesion (Fig. 1D). HPV high-risk ISH showed diffusely distributed signals (Fig. 1E). P16 immunostaining showed diffuse staining. Off-site capture by NGS showed the presence of Alphapapilloma virus DNA, more specifically human papillomavirus 10, which has been associated with anogenital warts but not malignant or premalignant lesions. Sequencing also showed that the tumor harbored TERT promoter and TP53 gene mutations, while PIK3CA alterations were not found. This leads us to believe that the tumor is an HPV-independent squamous cell carcinoma with incidental non-high-risk HPV infection. The capture of HPV-10 by the HPV ISH assay is problematic as it raises concern for cross-reactivity of HPV HR ISH with some of the rare yet non-high-risk HPV strains.
Genomic landscape of vulvovaginal squamous cell carcinoma
NGS results for 26 samples were evaluated. Overall, the most common genomic alterations in vulvovaginal squamous cell carcinomas are mutations in TP53 (62%), upstream promoter region of TERT (58%), PIK3CA (35%), and deletions/truncating mutations of CDKN2A (38%). Other common genomic events include alterations of FAT1 (31%), NOTCH1 (27%), JAK3 (15%), and KMT2D (15%). These findings are summarized in Fig. 2A.

A Oncoprint depicting the most recurrent genomic alterations in vulvovaginal squamous cell carcinomas. Each column represents a tumor with the bar graph at the top depicting the number/distribution of alterations per sample, and the Oncoprint rows showing alterations for each gene. The bottom part of the graph shows the summary of histopathologic and clinical information for each case. The bar graph on the right of the panel shows the number and distribution of alterations for each gene. Mutation types and clinicopathologic features are color-coded according to the legend. Samples without alteration in any of the genes included in the Oncoprint are not shown. B Bar plot highlighting the differences in the genomic landscape of HPV-positive versus HPV-negative vulvovaginal squamous cell carcinomas. Note a single sample of HPV-positive vulvovaginal squamous cell carcinoma was noted to harbor TERT promoter mutation, further investigation showed the detected HPV to be a low-risk variant. OR: odds ratio.
The overall mutational profile of HPV-associated vulvovaginal squamous cell carcinoma is considerably different from HPV-independent tumors (Fig. 2B). HPV-associated vulvovaginal squamous cell carcinoma commonly has PIK3CA activating mutations (7/11, 64%) involving either codon 545 (p.E545K/G/Q) (six cases) or codon 542 (p.E542K) (one case) (Fig. 3A). Other common repetitive alterations in HPV-associated vulvovaginal squamous cell carcinoma include FBXW7 and BAP1 mutations (3/11, 27% each) followed by CREBBP mutations (2/11, 18%).

A Lollipop plot showing that all PIK3CA mutations in vulvovaginal squamous cell carcinomas in our cohort are activating mutations involving codons 542 and 545. B Oncoprint showing the NOTCH pathway alterations in vulvovaginal squamous cell carcinomas. C Interaction matrix plot showing the mutual exclusivity/co-occurrence of the common genomic alterations in vulvovaginal squamous cell carcinomas.
The HPV-independent tumors are dominated by the presence of TERT gene alterations (mainly TERT promoter mutations) that were present in 14/15 cases (93%). Conversely, only one HPV-positive case (a spurious result, sample 15, as discussed above) was shown to harbor a TERT promoter mutation (1/11, 9%). Other common alterations in HPV-independent tumors include TP53 mutations (13/15, 87%), CDKN2A alterations (10/15, 67%), NOTCH1 and FAT1 mutations (7/15, 47% each). In fact, the presence of alterations in any of the TERT, TP53, CDKN2A, and NOTCH1 genes strongly points away from an HPV-driven process (odds ratios: 0.01, 0.07, 0, and 0, respectively with P < 0.02 for all four genes). Conversely, the presence of PIK3CA activating mutations in the absence of the above-mentioned genes strongly favors an HPV-driven tumor (odds ratio: 10.12, P: 0.016) (Fig. 2B). It must be noted that subclonal PIK3CA mutations were observed in two of the HPV-independent tumors.
Evaluation of co-occurrence/mutual exclusivity in genomic alterations reveals two distinct molecular profiles of vulvovaginal squamous cell carcinomas with PIK3CA showing mutual exclusivity with TP53 and TERT. Other significant associations include CDKN2A, NOTCH1, and FAT1 co-occurrence. These findings are shown in Fig. 3C.
In terms of signaling pathways, HPV-associated tumors often have PI3K pathway activation (7/11, 64%), while HPV-independent tumors have P53 pathway alterations (13/15, 87%), and cell-cycle pathway alterations (11/15, 73%). A subset of both HPV-driven and HPV-independent tumors have NOTCH pathway alterations (6/11, 55% and 10/15, 67%, respectively), however, different genes in the NOTCH signaling pathway are altered in these tumors: In HPV-independent tumors NOTCH1, NOTCH2, and NOTCH3 mutations predominate while in HPV-associated tumors FBXW7, SPEN, and NCOR1 mutations are seen (Fig. 3B).
Clonal evolution of HPV-independent vulvovaginal squamous cell carcinoma
Evaluation of cancer cell fractions of mutations in the HPV-independent tumors was performed in 15 samples, which showed that the mutations with highest cancer cell fraction (likely being the initial genomic alteration) were TERT alterations (n = 6/15) followed by NOTCH alterations (both NOTCH1 and NOTCH2) (n = 5/15) and TP53 alterations (n = 4/15). Interestingly, TP53 mutations were more likely to have the second-highest cancer cell fraction (n = 10/15), i.e., they are more likely to be the second step in the tumor evolution of at least some HPV-independent vulvovaginal squamous cell carcinoma (Fig. 4A).

A Alluvial graph showing the distribution of the three genomic alterations in vulvovaginal squamous cell carcinomas with the highest cancer cell fractions. All HPV-independent vulvovaginal squamous cell carcinomas have truncal alterations in a combination of TP53, TERT and NOTCH1 genes and any of these genes can have the highest cancer cell fraction in the tumors. B, C Phylogenetic reconstruction of the tumor evolution in two cases of metastatic/recurrent vulvovaginal squamous cell carcinomas.
Two patients with HPV-independent vulvovaginal squamous cell carcinoma had both primary and recurrence samples sequenced. The first patient had a well-differentiated vulvar primary squamous cell carcinoma that presented as clinical-stage Ib disease, which was excised. 6 months later, she developed lung metastasis and died of disease 19 months after the initial diagnosis. Sequencing of the primary tumor showed alterations in TERT, TP53, FAT1, and CDKN2A genes among others. Sequencing of the metastatic tumor showed mostly similar mutational landscape except for absence of the CDKN2A mutation and gain of a PDGFRB mutation (Fig. 4B).
The second patient had a moderately differentiated vulvar primary squamous cell carcinoma which presented as clinical stage Ib disease that was excised. She had disease recurrence after 54 months with recurrent tumor in the vagina and involvement of lymph nodes. She died after 121 months. Sequencing of the primary tumor showed mutations in TERT, TP53, FAT1, NOTCH1, and CDKN2A genes among others. At recurrence in addition to these mutations, the tumor showed alterations of ASXL1, KMT2D, HIST1H3D, and EPHB1 genes (Fig. 4C).
Discussion
The pathogenesis of vulvovaginal squamous cell carcinoma is linked to three distinct mechanisms with three different precursor lesions: uVIN (HSIL); dVIN, and DEVIL/VAAD3,9. The latter precursor lesion is believed to be associated with verrucous carcinoma which generally tends to have favorable outcomes9 and similar to HPV-associated squamous cell carcinomas19 are often associated with PIK3CA alterations20. None of the cases in our cohort was verrucous carcinoma nor were they preceded or accompanied by DEVIL/VAAD.
Vulvovaginal squamous cell carcinoma can be generalized into two distinct etiologic categories: HPV-associated tumors, as the name implies, are due to pathogenic HPV infections and often arise in the setting of high-grade usual vulvar or vaginal intraepithelial lesions (uVIN/HSIL). In contrast, HPV-independent tumors occur in elderly post-menopausal patients and are often associated with differentiated vulvar intraepithelial lesions (dVIN)21. The etiology and pathogenesis of HPV-associated vulvovaginal squamous cell carcinoma are very similar to cervical squamous cell carcinoma with a large body of evidence dedicated to understanding of this disease22,23. In contrast, the molecular mechanisms involved in the development of HPV-independent vulvovaginal squamous cell carcinoma are yet to be fully elucidated, however, there is evidence that these tumors represent clonal evolution and mutational progression of dVIN lesions24,25. These lesions and their precursor (dVIN) have a tendency for multifocality and in such cases evidence for a common clonal origin exists in a substantial proportion of the tumors24. Several studies have described the genomic profiles of HPV-associated and HPV-independent vulvovaginal squamous cell carcinoma;2,20,26,27,28,29,30 HPV-independent vulvovaginal squamous cell carcinoma have been previously shown to harbor mutations in TP53, CDKN2A, HRAS, KRAS, PIK3CA, PPP2R1A, and PTEN genes, with TP53 mutations being the most commonly reported alteration5,24,25,31. In contrast, TP53 alterations are uncommon in HPV-associated vulvovaginal squamous cell carcinomas which often show PIK3CA mutations in a higher proportion of tumors compared to HPV-independent carcinomas2,19,25,27,32. In this study we have described the genomic findings of a cohort of HPV-independent and HPV-associated vulvovaginal squamous cell carcinomas; it must be noted that the HPV-independent and HPV-associated tumors were primarily seen in vulva and vagina respectively, which may at least partially explain the distinct molecular underpinnings of these tumors.
The prognostic significance of HPV in vulvar squamous cell carcinoma is controversial, some studies have suggested that the HPV status is not an independent prognostic factor33,34, while others have shown that HPV-independent vulvar squamous cell carcinoma is associated with significantly worse outcomes35,36. TP53 alterations in vulvovaginal squamous cell carcinomas have been shown to be associated with worse overall survival25,37 and recurrence-free survival37. Although the influence of HPV infection on the prognosis of vulvovaginal squamous cell carcinoma remains unclear33, HPV infection appears to be integral to the induction of precancerous vulvar/vaginal intraepithelial neoplasia (VIN) and its evolution to HPV-associated vulvovaginal squamous cell carcinoma22.
High-risk HPV through the action of viral oncoproteins—E6 and E7—disrupts the normal function of several important tumor suppressors including p53, Rb, and p16 in the cells leading to cell cycle progression and replication38,39,40,41. The effects of the E7 viral protein also lead to strong overexpression of the p16 protein42, which has been shown to be an adequate surrogate of HPV infection status in vulvovaginal squamous cell carcinomas43.
There is an inverse relationship between HPV infection and TP53 mutation; in HPV-associated tumors the inactivation of the p53 protein occurs via the degradation induced by the viral E6 protein44, rendering gene mutations redundant. Degradation of the p53 protein by the viral oncoprotein leads to wild-type staining in most HPV-associated tumors. Conversely, somatic inactivating missense mutations of the TP53 gene are often associated with increased stability of the mutant p53 protein in HPV-independent vulvovaginal squamous cell carcinoma with resultant overexpression45.
Furthermore, the HPV E6 protein also interacts with the Myc protein which in turn activates the TERT promoter, inducing TERT mRNA transcription in genital keratinocytes, thereby increasing the cellular telomerase activity46,47. There is experimental evidence suggesting that the neoplastic transformation of the cells infected with HR HPV is at least partially mediated by the viral E6 protein-induced telomerase activation48.
TERT gene mutations and truncating/deleterious alterations of the TP53 gene appear to be present in most HPV-independent tumors (87%) which is in line with prior studies19. Other common alterations in this tumor subtype involve CDKN2A, and NOTCH1 genes. These alterations are absent/uncommon in HPV-associated vulvovaginal squamous cell carcinoma (P < 0.05 for all), suggesting that HPV exerts a carcinogenic effect in cells that is similar to the effects of the alterations of these genes. Furthermore, we have shown that TERT, TP53, and NOTCH1 alterations can all be the initiating events in HPV-independent vulvovaginal squamous cell carcinoma, which is contrary to the belief that TP53 alterations are the sole initiating event49,50. It has been suggested previously that a combination of HPV and p53 status can adequately separate vulvar squamous cell carcinomas into three distinct subgroups (HPV-positive, HPV-negative/p53-mutant, and HPV-negative/p53-wild type) with distinct clinical outcomes37. We argue that TERT mutational status, rather than TP53, may be the main defining factor for differentiating between HPV-independent vulvovaginal squamous cell carcinoma and HPV-associated vulvovaginal squamous cell carcinoma as mutations in TP53 can be observed in both subtypes. However, it remains to be seen whether TERT status determination is any more informative than TP53 status in terms of clinical outcomes, especially since surrogate immunohistochemical markers for TERT alterations do not yet exist and the current methodologies such as TERT mRNA ISH can only show overexpression, failing to distinguish between the underlying molecular mechanisms leading to overexpression51,52.
Similar to our observation in vulvovaginal squamous cell carcinoma, NOTCH1 alterations in head and neck squamous cell carcinomas are more commonly seen in HPV-independent tumors53 and it is believed that the loss of function of canonical Notch signaling drives squamous cell carcinogenesis in these sites54. We believe that NOTCH1 loss of function alterations may have a similar effect in vulvovaginal squamous cell carcinoma and act as an early initiating event in HPV-independent vulvovaginal squamous cell carcinoma. Other common alterations in HPV-independent vulvovaginal squamous cell carcinoma include mutation/deletion of FAT1 gene. Previously, it was shown that in head and neck squamous cell carcinomas FAT1 alterations are also more common in HPV-independent tumors compared to HPV-associated tumors. In that cohort, FAT1 mutations were associated better with overall survival55, it remains to be seen whether FAT1 alterations confer any survival advantage in HPV-independent vulvovaginal squamous cell carcinoma. We also observed that NOTCH-mutated, HPV-independent carcinomas are more likely to be moderately-poorly differentiated with a high rate of tumor cell budding.
Previous studies in head and neck squamous cell carcinomas12 and colorectal adenocarcinoma13 revealed that tumor budding is an independent prognostic factor that may be associated with high tumor grade, presence of lymphovascular invasion, and consequently lymph node metastasis. Similarly, recent studies have shown that in patients with locally advanced gynecological cancers tumor budding is an independent prognostic biomarker in predicting tumor recurrence and survival outcomes56,57,58. Herein, we report that only HPV-independent tumors had intermediate to high budding, but the analysis was limited by a paucity of HPV-associated tumors for study.
Cancers mediated by HPV infection are enriched in alterations activating the PI3K/AKT/mTOR pathway with many of these tumors harboring hotspot mutations in the PIK3CA gene59. In our cohort, we showed that PIK3CA mutations commonly occur in HPV-associated tumors and rarely in HPV-independent tumors. While an HPV-independent pathogenetic pathway involving mutations in the PIK3CA gene and progression from DEVIL/VAAD to HPV-independent squamous cell carcinoma has been described, our two cases of HPV-independent tumors with PIK3CA mutation, only had subclonal PIK3CA mutations, i.e., PIK3CA mutation was likely acquired after TERT and TP53 mutations, rendering a transformed DEVIL/VAAD unlikely. The dearth of corresponding PI3K/AKT/mTOR pathway alterations in HPV-independent tumors is interesting and may suggest a yet unknown genomic or epigenetic alteration in this subtype. Furthermore, We showed that significant statistical difference exists in frequency of alterations in FBXW7, and BAP1 genes which, in our limited cohort, were only observed in the HPV-associated vulvovaginal squamous cell carcinoma.
As we used a clinical-grade NGS assay, our study is limited to cases with requests for clinical sequencing, which limited our cohort to patients with advanced or progressing tumors. As a result, patients with verrucous vulvar squamous cell carcinoma, which is much more indolent, were not included in our study. Further studies, on larger cohorts, are needed to determine the prognostic significance of the genomic alterations in vulvovaginal squamous cell carcinoma.
In summary, we have shown that the altered function of p53 and TERT (activation) is pivotal in vulvovaginal squamous cell carcinoma. In HPV-independent tumors, these altered functions occur through genomic alterations of the involved genes, while in HPV-associated tumors the functional alterations are likely achieved via the effects of the viral proteins. Furthermore, we have shown that NOTCH1 loss of function is another driver in the carcinogenesis of HPV-independent vulvovaginal squamous cell carcinoma and may be associated with increased tumor cell budding. TERT, NOTCH, and TP53 alterations may alI be initiating events in HPV-independent squamous cell carcinomas.
Data availability
The data that support the findings of this study are available from the corresponding author on reasonable request.
References
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 70, 7–30 (2020).
Han, M. R. et al. Mutational signatures and chromosome alteration profiles of squamous cell carcinomas of the vulva. Exp. Mol. Med. 50, e442 (2018).
Del Pino, M., Rodriguez-Carunchio, L. & Ordi, J. Pathways of vulvar intraepithelial neoplasia and squamous cell carcinoma. Histopathology 62, 161–175 (2013).
Clancy, A. A., Spaans, J. N. & Weberpals, J. I. The forgotten woman’s cancer: Vulvar Squamous Cell Carcinoma (VSCC) and a targeted approach to therapy. Ann. Oncol. 27, 1696–1705 (2016).
Trietsch, M. D., Nooij, L. S., Gaarenstroom, K. N. & van Poelgeest, M. I. Genetic and epigenetic changes in vulvar squamous cell carcinoma and its precursor lesions: a review of the current literature. Gynecol. Oncol. 136, 143–157 (2015).
Hoang, L. N., Park, K. J., Soslow, R. A. & Murali, R. Squamous precursor lesions of the vulva: current classification and diagnostic challenges. Pathology 48, 291–302 (2016).
Cormio, G. et al. Groin recurrence in carcinoma of the vulva: management and outcome. Eur. J. Cancer Care 19, 302–307 (2010).
Akbari, A. et al. Differentiated exophytic vulvar intraepithelial lesion: clinicopathologic and molecular analysis documenting its relationship with verrucous carcinoma of the vulva. Mod Pathol 33, 2011–2018 (2020).
Nascimento, A. F. et al. Vulvar acanthosis with altered differentiation: a precursor to verrucous carcinoma? Am. J. Surg. Pathol. 28, 638–643 (2004).
Kurman R. J., C. M, Herrington C. S. WHO Classification of Tumours of Female Reproductive Organs. 4th edn, Vol. 6th (Inter-national Agency for Research on Cancer: Lyon, 2014).
Zaino, R. J. et al. Histopathologic predictors of the behavior of surgically treated stage IB squamous cell carcinoma of the cervix. A Gynecologic Oncology Group study. Cancer 69, 1750–1758 (1992).
Xu, B. et al. The prognostic role of histologic grade, worst pattern of invasion, and tumor budding in early oral tongue squamous cell carcinoma: a comparative study. Virchows Arch. 479, 597–606 (2021).
Lugli, A. et al. Recommendations for reporting tumor budding in colorectal cancer based on the International Tumor Budding Consensus Conference (ITBCC) 2016. Mod. Pathol. 30, 1299–1311 (2017).
Shen, R. & Seshan, V. E. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 44, e131 (2016).
Roth, A. et al. PyClone: statistical inference of clonal population structure in cancer. Nat. Methods 11, 396–398 (2014).
Darragh, T. M. et al. The Lower Anogenital Squamous Terminology Standardization Project for HPV-associated Lesions: background and consensus recommendations from the College of American Pathologists and the American Society for Colposcopy and Cervical Pathology. Arch. Pathol. Lab. Med. 136, 1266–1297 (2012).
Mills, A. M., Dirks, D. C., Poulter, M. D., Mills, S. E. & Stoler, M. H. HR-HPV E6/E7 mRNA in situ hybridization: validation against PCR, DNA in situ hybridization, and p16 immunohistochemistry in 102 samples of cervical, vulvar, anal, and head and neck neoplasia. Am. J. Surg. Pathol. 41, 607–615 (2017).
Bowman, A. et al. Identification of viral integration sites in cancer genomes using unmapped reads in targeted next-generation sequencing data. J. Mol. Diagn. 963-964 (Elsevier Science Inc, New York, NY, USA).
Williams, E. A. et al. Vulvar squamous cell carcinoma: comprehensive genomic profiling of HPV+ versus HPV- forms reveals distinct sets of potentially actionable molecular targets. JCO Precis. Oncol. 4, 647–661 (2020).
Watkins, J. C. et al. Differentiated exophytic vulvar intraepithelial lesions are genetically distinct from keratinizing squamous cell carcinomas and contain mutations in PIK3CA. Mod. Pathol. 30, 448–458 (2017).
Mulvany, N. J. & Allen, D. G. Differentiated intraepithelial neoplasia of the vulva. Int. J. Gynecol. Pathol. 27, 125–135 (2008).
van de Nieuwenhof, H. P. et al. The etiologic role of HPV in vulvar squamous cell carcinoma fine tuned. Cancer Epidemiol. Biomark. Prev. 18, 2061–2067 (2009).
van der Avoort, I. A. et al. Vulvar squamous cell carcinoma is a multifactorial disease following two separate and independent pathways. Int. J. Gynecol. Pathol. 25, 22–29 (2006).
Pors, J. et al. Targeted molecular sequencing of recurrent and multifocal non-HPV-associated squamous cell carcinoma of the vulva. Int. J. Gynecol. Pathol. 40, 391–399 (2021).
Tessier-Cloutier, B. et al. Molecular characterization of invasive and in situ squamous neoplasia of the vulva and implications for morphologic diagnosis and outcome. Mod. Pathol. 34, 508–518 (2021).
Zieba, S. et al. Somatic mutation profiling of vulvar cancer: exploring therapeutic targets. Gynecol. Oncol. 150, 552–561 (2018).
Nooij, L. S. et al. Genomic characterization of vulvar (pre)cancers identifies distinct molecular subtypes with prognostic significance. Clin. Cancer Res. 23, 6781–6789 (2017).
Weberpals, J. I. et al. Vulvar squamous cell carcinoma (VSCC) as two diseases: hpv status identifies distinct mutational profiles including oncogenic fibroblast growth factor receptor 3. Clin. Cancer Res. 23, 4501–4510 (2017).
Zieba, S. et al. Somatic mutation profiling in premalignant lesions of vulvar squamous cell carcinoma. Int. J. Mol. Sci. 21, 4880 (2020).
Kunjoonju, J. P., Raitanen, M., Grenman, S., Tiwari, N. & Worsham, M. J. Identification of individual genes altered in squamous cell carcinoma of the vulva. Genes Chromosomes Cancer 44, 185–193 (2005).
Trietsch, M. D. et al. CDKN2A(p16) and HRAS are frequently mutated in vulvar squamous cell carcinoma. Gynecol. Oncol. 135, 149–155 (2014).
Carreras-Dieguez, N. et al. Molecular landscape of vulvar squamous cell carcinoma. Int. J. Mol. Sci. 22, 7069 (2021).
Pinto, A. P. et al. Prognostic significance of lymph node variables and human papillomavirus DNA in invasive vulvar carcinoma. Gynecol. Oncol. 92, 856–865 (2004).
Alonso, I. et al. Does human papillomavirus infection imply a different prognosis in vulvar squamous cell carcinoma? Gynecol. Oncol. 122, 509–514 (2011).
McAlpine, J. N. et al. Human papillomavirus (HPV)-independent vulvar squamous cell carcinoma has a worse prognosis than HPV-associated disease: a retrospective cohort study. Histopathology 71, 238–246 (2017).
Allo, G. et al. HPV-independent vulvar squamous cell carcinoma is associated with significantly worse prognosis compared with HPV-associated tumors. Int. J. Gynecol. Pathol. 39, 391–399 (2020).
Kortekaas, K. E. et al. Vulvar cancer subclassification by HPV and p53 status results in three clinically distinct subtypes. Gynecol. Oncol. 159, 649–656 (2020).
Crook, T., Vousden, K. H. & Tidy, J. A. Degradation of p53 can be targeted by HPV E6 sequences distinct from those required for p53 binding and trans-activation. Cell 67, 547–556 (1991).
Yim, E. K. & Park, J. S. The role of HPV E6 and E7 oncoproteins in HPV-associated cervical carcinogenesis. Cancer Res. Treat. 37, 319–324 (2005).
Agoff, S. N. et al. p16(INK4a) expression correlates with degree of cervical neoplasia: a comparison with Ki-67 expression and detection of high-risk HPV types. Mod. Pathol. 16, 665–673 (2003).
Santos, M. et al. p16 overexpression identifies HPV-positive vulvar squamous cell carcinomas. Am. J. Surg. Pathol. 30, 1347–1356 (2006).
GEIßLER, C. et al. The role of p16 expression as a predictive marker in HPV-positive oral SCCHN–a retrospective single-center study. Anticancer Res. 33, 913–916 (2013).
Rakislova, N. et al. Histological characteristics of HPV-associated and -independent squamous cell carcinomas of the vulva: a study of 1594 cases. Int. J. Cancer 141, 2517–2527 (2017).
Pilotti, S. et al. Papillomavirus, p53 alteration, and primary carcinoma of the vulva. Diagn Mol Pathol 4, 239–248 (1995).
Soufir, N. et al. Inactivation of the CDKN2A and the p53 tumour suppressor genes in external genital carcinomas and their precursors. Br. J. Dermatol. 156, 448–453 (2007).
Panczyszyn, A., Boniewska-Bernacka, E. & Glab, G. Telomeres and telomerase during human papillomavirus-induced carcinogenesis. Mol. Diagn. Ther. 22, 421–430 (2018).
Branca, M. et al. Upregulation of telomerase (hTERT) is related to the grade of cervical intraepithelial neoplasia, but is not an independent predictor of high-risk human papillomavirus, virus persistence, or disease outcome in cervical cancer. Diagn. Cytopathol. 34, 739–748 (2006).
Miller, J. et al. HPV16 E7 protein and hTERT proteins defective for telomere maintenance cooperate to immortalize human keratinocytes. PLoS Pathog. 9, e1003284 (2013).
Pinto, A. P. et al. Differentiated vulvar intraepithelial neoplasia contains Tp53 mutations and is genetically linked to vulvar squamous cell carcinoma. Mod. Pathol. 23, 404–412 (2010).
Choschzick, M. et al. Role of TP53 mutations in vulvar carcinomas. Int. J. Gynecol. Pathol. 30, 497–504 (2011).
Fan, Y. et al. Telomerase expression by aberrant methylation of the TERT promoter in melanoma arising in giant congenital Nevi. J. Invest. Dermatol. 136, 339–342 (2016).
Hellgren, L. S. et al. Nuclear-specific accumulation of telomerase reverse transcriptase (TERT) mRNA in TERT promoter mutated follicular thyroid tumours visualised by in situ hybridisation: a possible clinical screening tool? J. Clin. Pathol. https://doi.org/10.1136/jclinpath-2021-207631 (2021).
Morris, L. G. T. et al. The molecular landscape of recurrent and metastatic head and neck cancers: insights from a precision oncology sequencing platform. JAMA Oncol. 3, 244–255 (2017).
Nyman, P. E., Buehler, D. & Lambert, P. F. Loss of function of canonical notch signaling drives head and neck carcinogenesis. Clin. Cancer Res. 24, 6308–6318 (2018).
Kim, K. T., Kim, B. S. & Kim, J. H. Association between FAT1 mutation and overall survival in patients with human papillomavirus-negative head and neck squamous cell carcinoma. Head Neck 38, E2021–E2029 (2016). Suppl 1.
Park, J. Y. et al. Tumor budding in cervical cancer as a prognostic factor and its possible role as an additional intermediate-risk factor. Gynecol. Oncol. 159, 157–163 (2020).
Park, J. Y., Hong, D. G., Chong, G. O. & Park, J. Y. Tumor budding is a valuable diagnostic parameter in prediction of disease progression of endometrial endometrioid carcinoma. Pathol. Oncol. Res. 25, 723–730 (2019).
Huang, B., Cai, J., Xu, X., Guo, S. & Wang, Z. High-grade tumor budding stratifies early-stage cervical cancer with recurrence risk. PLoS One 11, e0166311 (2016).
Zhang, L., Wu, J., Ling, M. T., Zhao, L. & Zhao, K. N. The role of the PI3K/Akt/mTOR signalling pathway in human cancers induced by infection with human papillomaviruses. Mol. Cancer 14, 87 (2015).
Funding
Research reported in this publication was supported in part by the Cancer Center Support Grant of the National Institutes of Health/National Cancer Institute under award number P30CA008748.
Author information
Authors and Affiliations
Contributions
R.A.S. conceived the study and supervised the work. R.A.S., A.S. performed pathologic review. A.M.B., A.S., and C.V. performed the data extraction and electronic health records review. A.M.B., A.S., M.L., and C.V. performed data analysis and data interpretation. A.S. and A.M.B. wrote the first draft of the manuscript. All authors edited and approved the final draft.
Corresponding author
Ethics declarations
Competing interests
No competing financial interests exist for all contributory authors.
Ethics approval
This study was performed with the approval of the Institutional Review Board of Memorial Sloan-Kettering Cancer Center (New York, NY).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Salama, A.M., Momeni-Boroujeni, A., Vanderbilt, C. et al. Molecular landscape of vulvovaginal squamous cell carcinoma: new insights into molecular mechanisms of HPV-associated and HPV-independent squamous cell carcinoma. Mod Pathol 35, 274–282 (2022). https://doi.org/10.1038/s41379-021-00942-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-021-00942-3
