
Abstract
Classic adenoid cystic carcinomas (C-AdCCs) of the breast are rare, relatively indolent forms of triple negative cancers, characterized by recurrent MYB or MYBL1 genetic alterations. Solid and basaloid adenoid cystic carcinoma (SB-AdCC) is considered a rare variant of AdCC yet to be fully characterized. Here, we sought to determine the clinical behavior and repertoire of genetic alterations of SB-AdCCs. Clinicopathologic data were collected on a cohort of 104 breast AdCCs (75 C-AdCCs and 29 SB-AdCCs). MYB expression was assessed by immunohistochemistry and MYB-NFIB and MYBL1 gene rearrangements were investigated by fluorescent in-situ hybridization. AdCCs lacking MYB/MYBL1 rearrangements were subjected to RNA-sequencing. Targeted sequencing data were available for 9 cases. The invasive disease-free survival (IDFS) and overall survival (OS) were assessed in C-AdCC and SB-AdCC. SB-AdCCs have higher histologic grade, and more frequent nodal and distant metastases than C-AdCCs. MYB/MYBL1 rearrangements were significantly less frequent in SB-AdCC than C-AdCC (3/14, 21% vs 17/20, 85% P < 0.05), despite the frequent MYB expression (9/14, 64%). In SB-AdCCs lacking MYB rearrangements, CREBBP, KMT2C, and NOTCH1 alterations were observed in 2 of 4 cases. SB-AdCCs displayed a shorter IDFS than C-AdCCs (46.5 vs 151.8 months, respectively, P < 0.001), independent of stage. In summary, SB-AdCCs are a molecularly heterogeneous but clinically aggressive group of tumors. Less than 25% of SB-AdCCs display the genomic features of C-AdCC. Defining whether these tumors represent a single entity or a collection of different cancer types with a similar basaloid histologic appearance is warranted.
Similar content being viewed by others

Introduction
Classic adenoid cystic carcinoma of the breast (C-AdCC) is characterized by t (6;9) MYB-NFIB rearrangements, or less commonly by MYBL1 rearrangements or MYB gene amplification1,2,3,4. Despite its triple-negative (TN) phenotype, the classic form of AdCC (C-AdCC) displays an indolent clinical behavior5,6,7,8.
The solid and basaloid variant of breast adenoid cystic carcinoma (SB-AdCC) is currently perceived as an exceedingly rare histologic subgroup of AdCCs, with <100 published cases9,10,11,12,13. SBAdCCs are characterized histologically by a basaloid cytologic appearance, consisting of rounded nests and trabeculae, typically embedded in a fibromyxoid stroma9.
Owing to the rarity of SB-AdCCs, there is a paucity of data on the clinical behavior and repertoire of somatic genetic alterations these cancers harbor. Albeit reported to harbor a greater propensity for lymph node metastasis than C-AdCCs9,10, the clinical behavior of SB-AdCCs remain to be fully characterized. Retrospective analyses have suggested that SB-AdCCs would be more aggressive than C-AdCCs12,13. Whether SB-AdCCs have a clinical behavior akin to that of C-AdCC, a low-grade TN breast cancer (TNBC) associated with good outcome14, remains unclear.
Here, we sought to determine the clinicopathologic characteristics and genetic repertoire of SB-AdCCs relative to C-AdCCs. We found that, at variance with C-AdCCs, SB-AdCCs are clinically aggressive tumors whose molecular features differ from those of the classic variant. Only a minority of SB-AdCCs were found to harbor MYB rearrangements, the hallmark molecular features of C-AdCCs, raising the possibility that only a subset of these cancers may be related to C-AdCCs.
Methods
Case selection
Following approval by the Memorial Sloan Kettering Cancer Center (MSK) Institutional Review Board, in accordance with the Helsinki Declaration of 1975 as revised in 1983, we conducted a retrospective review of our institution’s database using key words “breast” and “adenoid cystic carcinoma” diagnosed from 1991-2019 at MSK. Clinical features, including patient age, tumor size, treatment, and clinical follow-up data, were retrieved from the electronic medical records (EMR).
We reviewed pathology reports and performed a central pathology review of the cases using all the available hematoxylin-and-eosin (H&E) and/or digital slides for each case. The central histopathologic review and Nottingham grading were conducted independently for each case by three breast pathologists (C.J.S., F.P., and H.W.) according to criteria put forward by the World Health Organization (WHO)15. SB-AdCCs were defined as having a solid-basaloid growth pattern in at least 90% of the tumor area, as previously described1. Information on estrogen receptor (ER), progesterone receptor (PR), and HER2 expression, assessed by immunohistochemical stains (IHC) according to current American Society of Clinical Oncology/College of American Pathologists (ASCO/CAP) guidelines, was collected16,17. Information on P63 IHC (monoclonal antibody clone 4A4, Roche, Basel, Switzerland) performed at the time of diagnosis, scored as positive nuclear expression in at least 10% of cells, was collected according to methods previously described18.
MYB immunohistochemistry and fluorescence in situ hybridization
Immunohistochemical assessment of MYB was performed using BenchMark ULTRA platform (Roche), following antigen retrieval with a cell conditioning solution (CC1, Roche). Tissue sections were incubated with the monoclonal antibody clone EP76769Y (Abcam, Cambridge, MA) at a 1:200 dilution and detected using the OptiView DAB detection system (Roche). Tumors with strong MYB immunoexpression in <5% of tumor cells were classified as positive, as previously described19, and further categorized as focal (5 to 50%) or diffuse (>50%). All cases were reviewed together with two pathologists (CJS and HW).
Fluorescence in situ hybridization (FISH) was performed on paraffin embedded tumors using a 3-color probe designed to detect MYB-NFIB fusion or other MYB rearrangements (n = 34) and a 2-color MYBL1 break-apart probe (n = 13), as previously described4. The bacterial artificial chromosome clones (Roswell Park Cancer Institute, Buffalo New York) used in the probe-mix were as follows: 5′ MYB (RP11-614H6, RP11-104D9; Green), 3′ MYB (RP11-323N12, RP11-1060C14; Orange), 3′ NFIB (RP11-413D24, RP11-589C16; Red), 3′ MYBL1 (RP11-766A10, RP11-346I3; Orange), and 5′ MYBL1 (RP11-271O1, RP11-315E14; Green). Probe labeling, hybridization, post-hybridization washing, and fluorescence detection were performed according to procedures established at the Molecular Cytogenetics Core Facility. Slides were scanned using a Zeiss Axioplan 2i epifluorescence microscope (Carl Zeiss Microscopy, Thornwood, NY) equipped with Isis imaging software
(MetaSystems Group Inc, Waltham, MA). FISH signals were imaged through the depth of the tissue (compressed stack of 12 z-sections at 0.5-micron intervals) and for each case, at least 50 discrete nuclei analyzed as previously described4. A case was accordingly considered positive if >15% cells showed at least one 5′MYB-3′NFIB (RG) fusion signal or MYB break-apart signal or MYBL1 break-apart signal, as previously described4.
RNA sequencing and fusion gene discovery
All samples were anonymized prior to tissue processing. C-AdCC (n = 3) and SB-AdCCs (n = 11) lacking MYB-NFIB or MYBL1 rearrangements were microdissected from 8-μ-thick histologic sections under a stereomicroscope (Olympus SZ61) to ensure a tumor cell content >80%, as previously described20. RNA was extracted using the RNeasy FFPE kit following manufacturer’s instructions. RNA was subjected to paired-end RNA sequencing using validated protocols at MSKCC’s Integrated Genomics Operation, as previously described20,21. Read pairs supporting fusion transcripts were identified using INTEGRATE22, Defuse23, and FusionCatcher24, as previously described20. Candidate fusion genes and read-throughs detected in a set of 297 normal samples from The Cancer Genome Atlas (TCGA)25 were excluded, as previously described20,21. The remaining candidate fusion transcripts were annotated using OncoFuse [v1.0.9b226], to define their oncogenic potential. In addition, the presence of candidate fusion genes was assessed across 33 cancer types in the tumor fusion gene data portal27, which comprises a list of 20,731 fusion genes across 33 cancer types (n = 9950).
MSK-IMPACT targeted sequencing
Five C-AdCC (n = 5) and four SB-AdCC had been previously subjected to targeted sequencing using the FDA-cleared MSK-Integrated Mutation Profiling of Actionable Targets Assay (MSK-IMPACT)28, targeting all coding regions of 410 (n = 2) or 468 (n = 7) cancer related genes. Non-synonymous somatic mutations, amplifications, and homozygous deletions were detected using the MSKCC clinical genomics pipeline and sequencing results were retrieved from the final diagnostic molecular pathology reports and cBioportal29.
Statistical analysis
Statistical analyses were performed using GraphPad Prism (v8.4.3) and RStudio (v3.1.2). Fisher’s exact test and Mann–Whitney U test were used for comparisons between categorical variables and continuous variables, respectively. For survival analysis, the primary endpoint was invasive disease-free survival [IDFS, 30]. Secondary survival endpoints were overall survival (OS) and disease-specific survival (DSS). IDFS was calculated from the date of primary diagnosis to the date of first occurrence of ipsilateral invasive breast tumor recurrence, local/regional invasive breast cancer recurrence, distant recurrence, death attributable to any cause, contralateral invasive breast cancer, or second primary non-breast invasive cancer (whichever came first)30. OS was defined as time from the primary diagnosis to the date of death or last follow-up. DSS was defined as time from the primary diagnosis to the date of death related to breast cancer or last follow-up. Patients who died from causes other than breast cancer (assessed through the EMR) were excluded from the DSS survival analysis. Patients without events were censored at the last date of follow-up. Survival rates were computed using the Kaplan–Meier method with log-rank test and the Cox proportional-hazards model.
Hazard ratio (HR) along with 95% confidence interval (CI) were provided for Cox proportional-hazards regression analyses. Median follow-up was estimated using the reverse Kaplan–Meier method31. Variables with a P value of <0.1 at univariate analysis were entered into the multivariable models. An interaction term was included in the statistical models when subgroup analyses were performed, as previously described32. All tests were two-sided, as p values of <0.05 were considered statistically significant.
Results
Clinicopathologic characteristics
Our study cohort consisted of 104 primary breast AdCCs which upon histopathologic review were classified as C-AdCC (n = 75; 72%) or SB-AdCC (n = 29; 28%; Table 1). The patient age at diagnosis was significantly older for SB-AdCCs relative to C-AdCCs (median 63, range 44–88 vs. median 55.5, range 46–78, P value < 0.05, Mann–Whitney U test, Table 1). No differences in T stage or N stage were observed between the two groups (Table 1). The C-AdCCs included in this study displayed a cribriform or tubular growth pattern with the characteristic dual population of cells (Fig. 1A), whereas the SB-AdCCs in this study were characterized by solid and rounded nests/trabeculae of basaloid-appearing tumor cells, often with distinctive myxoid stroma (Fig. 1B, C). There was no histologic evidence of high-grade transformation in C-AdCC, nor examples of C-AdCC with a prominent, but not satisfying the 90% threshold, solid and/or basaloid component.

Representative hematoxylin-and-eosin (H&E) and MYB expression micrographs of A classic adenoid cystic carcinoma (Case 15) displaying cribriform growth with two-cell population and abundant basement membrane material (inset) and diffuse MYB expression (B). C Solid-basaloid adenoid cystic carcinoma (Case 29), displaying Grade 3 nuclei, necrosis and readily identifiable mitotic figures (inset), and diffuse MYB expression (D). E Solid-basaloid adenoid cystic carcinoma (Case 27), showing fibromyxoid stroma and focal MYB expression (F). Scale bars in A-C-E, 500 μm; B-D-F, 200 μm. Scale bars in A-C-E, 500 μm; B-D-F, 200 μm.
The median tumor size between groups was comparable (C-AdCC, 2.13 cm vs. SB-AdCC 1.94 cm, P value = 0.29, Mann–Whitney U test, Table 1). As compared to C-AdCCs, SB-AdCCs demonstrated aggressive features and were associated with a higher histologic grade (P value < 0.05, Fisher’s exact test, Table 1), higher rates of lymphovascular invasion (8/29, 28% in SB-ADCCs vs 2/63, 3% in C-AdCCs, P value < 0.05, Fisher’s exact test) and lymph node metastasis in cases where sentinel lymph node biopsy or axillary lymph node dissection was performed (3/21, 14% in SB-AdCCs vs 0/56, 0% in C-AdCCs, P value < 0.05, Fisher’s exact test; Table 1). Most C-AdCCs (46/54, 85%) and SB-AdCCs (23/24, 96%) lacked ER, PR, and HER2 (0–1 + ) expression. Eight C-AdCCs (15%) and one SB-AdCCs (4%) demonstrated weak ER expression in <10% of cells (P value = 0.27, Fisher’s exact test; Table 1). The myoepithelial component showed strong p63 nuclear expression in all C-AdCCs tested (20/20, 100%), whereas few SB-AdCCs (5/14, 36%) showed positivity for p63 (P value < 0.05, Fisher’s exact test; Table 1).
Assessment of MYB status in AdCC
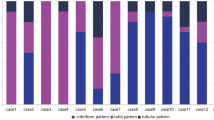
Given that rearrangements involving MYB have been reported at a lower frequency in SB-AdCCs than C-AdCCs (10,13) and that MYB immunohistochemistry (IHC) has been considered a surrogate for the presence of MYB rearrangements (18), we sought to evaluate MYB status in cases with available material using IHC and FISH (experimental workflow detailed in Fig. 2; Supplementary Table 1). Our analyses revealed that nearly all C-AdCCs displayed MYB protein expression by IHC (19/20, 95%, Fig. 2A; Table 2) in either a focal (9/19, 47%) or diffuse pattern (10/19, 53%, Fig. 1D). Immunohistochemical analysis also revealed MYB protein expression in 64% (9/14) in SBAdCCs, a prevalence numerically lower than that observed in C-AdCCs (P value = 0.07, Fisher’s exact test, Table 2). Of the SB-AdCCs expressing MYB, most did so in a diffuse pattern 66% (6/9; Figs. 2A and 1E), whereas focal positive MYB expression was observed in the 33% (3/9) of cases (Fig. 1F). By comparison, no MYB expression was detected in a series of 232 consecutive TNBCs in tumor tissue microarrays utilizing the same immunohistochemistry protocol (data not shown).

Depiction of analysis workflow and various techniques (fluorescence in situ hybridization, immunohistochemistry, and RNA sequencing) employed for the study of solid-basaloid (SB-AdCC, n = 14) and classic breast adenoid cystic carcinoma (C-AdCC, n = 20).
We next sought to assess the presence of MYB-NFIB fusion gene in the AdCCs analyzed here. Thirty-four cases with blocks available were interrogated using a 3 color MYB-NFIB break-apart probe, including 20 C-AdCCs (primary, n = 17 and metastatic, n = 3) and 14 SB-AdCCs (primary, n = 12, recurrent, n = 1 and metastatic, n = 1; Fig. 2). In agreement with prior studies (10, 13), most C-AdCCs (16/20, 80%) were found to harbor the MYB-NFIB fusion gene. In contrast, only one of 14 (7%) SB-AdCCs interrogated displayed the MYB-NFIB fusion gene (P < 0.05, Fisher’s exact test, Fig. 3A, B; Table 2). In two of the 14 remaining SB-AdCCs, we observed a complex signal pattern with loss of the 3′ MYB probe, suggestive of a rearrangement involving MYB (Table 2). The partner gene, however, could not be determined. Notably, SB-AdCCs with MYB rearrangements did not display any areas of C-AdCC nor were distinctive in appearance relative to tumors lacking MYB rearrangements. Given the prior observation of MYBL1 rearrangements in AdCC (4), we evaluated 15 AdCCs (C-AdCC = 4; SB-AdCC = 11) lacking MYB rearrangements using a two-color MYBL1 break-apart FISH probe. MYBL1 rearrangements were detected in one C-AdCC (1/4, 25%) and in none of the SB-AdCCs studied (0/11). These observations are consistent with those of prior studies10,13, and demonstrate a statistically significant lower frequency of MYB-NFIB, MYB rearrangements or MYBL1 rearrangements in SB-AdCCs than in C-AdCCs (P < 0.05; Fisher’s exact test, Fig. 3A; Table 2). These findings support the contention that SB-AdCCs differ not only histologically but also genetically from C-AdCCs.

A MYB and MYBL1 rearrangement status as analyzed by fluorescence in situ hybridization (FISH) and immunohistochemistry (IHC) of classic adenoid cystic carcinomas (C-AdCC; n = 20) and solid-basaloid adenoid cystic carcinomas (SB-AdCC; n = 14). B Representative hematoxylin and eosin (H&E) micrograph (top left panel) and corresponding FISH image using a 3 color probe demonstrating MYB-NFIB rearrangement (green, 5′MYB; orange, 3′ MYB; red 3′ NFIB) of C-AdCC Case 2 (top right panel). Representative H&E micrograph (bottom left panel) and corresponding FISH image of SB-AdCC Case 11 showing the lack of MYB rearrangement (bottom right panel).
We next sought to define whether tumors lacking MYB/MYBL1 rearrangements would be underpinned by alternative fusion genes. We performed an RNA-sequencing of the remaining AdCCs lacking rearrangements in MYB/MYBL1 (C-AdCC, n = 3 and SB-AdCC, n = 11) utilizing a previously validated pipeline optimized for the detection of high-confidence, novel oncogenic fusion genes18.
Our analysis revealed a novel oncogenic/likely oncogenic KMT2C-WEE2 fusion gene in one SB-AdCC (1/11). The KMT2C-WEE2 fusion gene identified was an in-frame chimeric transcript composed of the first 5 exons of KMT2C on chromosome 7q with exons 3-12 of WEE2 (Fig. 4A, F). The predicted consequence of this event is a promoter swapping between KMT2C and WEE2 and disruption of the KMT2C beyond exon 5. No other fusion genes were identified in the remaining SB-AdCCs or C-AdCCs, including fusion transcripts involving MYB paralogs or other genes in the MYB signaling pathway.

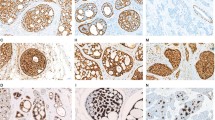
A Schematic diagram of a KMT2C-WEE2 fusion transcript identified in SB-AdCC Case 27 using RNA sequencing, depicting the exons and protein domains involved. The breakpoints of KMT2C and WEE2 are represented by vertical lines. The spanning reads crossing the genomic breakpoint are depicted aligned to the predicted junction sequence. Scale bars in B, 100 μm. B Oncoprint summarizing the non-synonymous somatic mutations and gene copy number alterations identified by MSK-IMPACT targeted sequencing in breast classic adenoid cystic carcinomas (C-AdCC; n = 5) and solid-basaloid adenoid cystic carcinomas (SB-AdCC; n = 4). Cases are shown in columns and genes in rows. MYB-NFIB rearrangement status and current disease status are shown in phenobars (top). Histologic features of solid-basaloid adenoid cystic carcinomas by genetic alteration. Representative hematoxylin and eosin (H&E) of C solid-basaloid adenoid cystic carcinoma (SB-AdCC with MYB-NFIB rearrangement (case 29). D SB-AdCC with MYB rearrangement (case 25). E SB-AdCC lacking MYB rearrangement (case 30). F SB-AdCC with KMT2C-WEE2 rearrangement (case 27). G, H SB-AdCC lacking MYB rearrangement with NOTCH1/KMT2C alterations (case 23 and 24). Scale bars, 200 μm.
Targeted sequencing of AdCCs
We retrieved the targeted sequencing data of five C-AdCCs (primary, n = 2; metastatic, n = 3) and four SB-AdCCs (primary, n = 2; metastatic, n = 2) subjected to MSK-IMPACT sequencing in the clinical setting (Fig. 4B). All SB-AdCCs lacked MYB rearrangements (despite 2 of 4 tumors showing positive MYB expression by IHC), whereas MYB-NFIB rearrangement and MYB expression were present in the 5 C-AdCCs sequenced.
Unlike TNBCs of no special type33, the tumors analyzed lacked TP53 and PIK3CA mutations, with few copy number alterations and a low tumor mutational burden (Supplementary Table 2). CREBBP was the most frequently altered gene (33%, 3/9) across AdCCs, including a truncating and a missense mutation in two metastatic SB-AdCCs (n = 2), and a rearrangement in a metastatic C-AdCC (n = 1). Recurrent alterations in KDM6A, NOTCH1, and KMT2C were also observed (2/9, 22% each; Fig. 4B). Notably, 2/4 SB-AdCCs harbored likely oncogenic mutations in NOTCH1 (50%, 2/4), and in the chromatin remodeling gene, KMT2C (50%, 2/4), in the form of truncating mutation and deep deletion (Fig. 4B, E, F). The findings agree with a prior study reporting on genetic alterations affecting NOTCH and chromatin remodeling genes in SB-AdCCs13. In summary, SB-AdCCs display a heterogeneous repertoire of somatic genetic alterations that is not dissimilar to that of C-AdCCs (Fig. 4C–H).
Treatment and clinical outcomes
No statistical differences in terms of surgical approach or adjuvant radiotherapy between C-AdCC and SB-AdCC groups were observed (Table 1). No patients in this study received neoadjuvant therapy. Follow-up were available for 69% of C-AdCCs (n = 52) and 76% of SB-AdCCs patients (n = 20; Table 1). The rates of adjuvant chemotherapy were significantly higher in SB-AdCCs (13/20, 68.4%) compared to C-AdCCs (9/52, 17.3%; P value < 0.05, Fisher’s exact test, Table 1). At a median follow-up of 92.3 months (interquartile range, 27.7–136.1), 32 (44%), 17 (24%), and 12 (17%) IDFS, OS, and DSS disease events were observed, respectively. As compared to patients with C-AdCCs, patients with SB-AdCCs were more likely to show local recurrences (6/20, 30% vs. 2/52, 4%), display metastases to distant organs (13/20, 65% vs. 10/52, 19%) and die of disease (8/20, 40% vs. 4/52, 8%).
SB-AdCCs were associated with a lower OS as compared to C-AdCCs (median OS 142 vs 204 months; HR 4.2, 95% CI 1.6–11.2, P = 0.01; Fig. 5A), although this difference (including adjuvant chemotherapy and radiotherapy) did not meet significance in the multivariate model (Supplementary Table 3 and Supplementary Fig. 1A). Similarly, DSS was significantly lower for SB-AdCCs when compared to C-AdCCs (median DSS 110 vs 305 months, HR 8.4, 95% CI 2.1–33.1, P < 0.001, Fig. 5B). After adjusting for other variables that were statistically significant at univariable analysis in our cohort (adjuvant chemotherapy and radiotherapy), SB-AdCCs were not associated with a reduced DSS in the multivariable model though a trend was observed (Supplementary Table 3 and Supplementary Fig. 1B). SB-AdCCs were associated with a statistically significant reduction of IDFS when compared to C-AdCCs (median IDFS 46.5 vs 151.8 months: HR 5.7, 95% CI 2.8–12, P < 0.001; Fig. 5C). On multivariate analysis, SB-AdCCs remained independently associated with a reduced IDFS (HR 4.0, 95% CI 1.69–9.3), independent of tumor stage, radiotherapy, and adjuvant chemotherapy (Supplementary Table 3 and Supplementary Fig. 1C).

Kaplan–Meier curves for overall survival (A), disease-specific survival (B), and invasive disease-free survival (IDFS) (C) and of classic adenoid cystic carcinoma (C-AdCC, n = 54) and solid-basaloid morphology (SB-AdCC, n = 20). Kaplan–Meier curves for IDFS and in patients with C-AdCC and SB-AdCC stratified by tumor size (D) and adjuvant chemotherapy (E). CI confidence interval, CT chemotherapy, HR hazard ratio, NR not reached.
Neither tumor stage (P for interaction = 0.375; Fig. 5D and Supplementary Table 4) nor adjuvant chemotherapy (P for interaction = 0.120; Fig. 5E and Supplementary Table 4) retained statistical significance in the analysis of IDFS according to the type of AdCC (i.e., C-AdCCs vs SB-AdCCs). The size of the study, however, precluded sufficient statistical power to further analyze the subgroups of patients with chemotherapy-treated C-AdCC and chemotherapy-untreated SB-AdCC.
Discussion
SB-AdCCs are vanishingly rare forms of TNBC, with a paucity of data on their clinical behavior and genetic underpinning. We detected the canonical MYB-NFIB fusion gene in only one of the SB-AdCCs analyzed here (7%), at variance with the prevalence of this fusion gene in >70% of breast AdCCs1,2,3,4. Two additional SB-AdCCs displayed rearrangements of MYB suggestive but not conclusive for the presence of MYB rearrangements with a yet to be identified partner. Our findings agree with those from prior studies indicating a significantly lower frequency of the MYB-NFIB fusion genes in SB-AdCCs than in C-AdCCs10, despite frequent immunohistochemical expression of MYB13.
Based on these observations, it would be plausible that SB-AdCCs may represent a heterogenous group of tumors in which some may be etiologically linked to AdCCs, whereas others may represent a distinct tumor type that shares similar histologic features, such as an adnexal-like carcinoma or other basaloid-appearing carcinoma. In fact, none of the SB-AdCCs included in this study displayed areas of C-AdCC admixed with or at the vicinity of the SB-AdCC areas. SB-AdCCs were either negative or focally positive for p63, a marker often expressed in adnexal neoplasms34. The detection of MYB expression in SB-AdCCs has been interpreted as supportive of an etiologic link between these tumors and C-AdCCs. It should be noted, however, that SB-AdCCs did express MYB protein but at a lower frequency than C-AdCCs and, at variance with C-AdCCs, where the expression of MYB is driven by the MYB-NFIB fusion gene, MYB or MYBL1 rearrangements or MYB gene amplification4. These rearrangements and fusion genes were found only in a minority (21%) of SB-AdCCs studied here, suggesting that the mechanisms underpinning MYB expression in these cancers may differ from those driving MYB expression in C-AdCCs. In fact, MYB expression has also been reported in up to 14% of adnexal tumors18. In the latter group, MYB expression is, in fact, driven by mechanisms other than those observed in C-AdCCs. For instance, a subset of sporadic dermal cylindromas overexpress MYB protein via MYB gene duplication, in the absence of canonical MYB-NFIB rearrangements35, highlighting that from an etiologic standpoint, expression of MYB protein is not necessarily a surrogate for MYB rearrangements and that tumors may express MYB through different molecular mechanisms. In a limited survey of SB-AdCCs lacking MYB rearrangement subjected to NGS, alterations were identified in NOTCH1 and chromatin remodeling genes (i.e., KMT2C and CREBBP), supporting a previous study13. In that study13, SB-AdCCs and C-AdCCs were found to have profoundly distinct expression profiles at the whole transcriptomic level, despite the overlap in their repertoire of somatic mutations13. Taken together, it is unclear as to whether SB-AdCCs are bona fide variants of C-AdCCs, as currently accepted, or if these cancers represent either a distinct entity or a heterogeneous group of tumors that share similar histologic features. Further whole transcriptomic studies (i.e., comparing SB-AdCCs, C-AdCCs and adnexal tumors of the same spectrum) are required to determine the taxonomy of these rare cancers.
SB-AdCCs were found to be aggressive tumors in comparison to C-AdCCs, with markedly reduced IDFS in patients, irrespective of tumor size. Our findings are consistent with those of Slodkowska et al.12, who reported that SB-AdCCs are clinically more aggressive than C-AdCCs. In that study12, margin status and Nottingham grade were found to be associated with decreased recurrence free survival. Masse et al.13 found higher rates of local recurrence or distant metastasis in a comparison of 17 SB-AdCCs with 16 C-AdCCs. Our findings support the notion that SB-AdCCs should not be included in the low-grade spectrum of TNBC nor be grouped with C-AdCCs14, as these tumors have a high propensity for tumor relapses after primary surgery and adjuvant systemic treatment. While lymph node involvement is low in AdCCs5 and absent in the C-AdCCs analyzed here, 15% of SB-AdCCs displayed lymph node metastases at the time of surgery. These findings support the notion that axillary staging should be routinely considered in SB-AdCCs.
Currently, the optimal adjuvant chemotherapy regimen in SB-AdCC is unknown. Prior studies have shown a limited or marginal role for chemotherapy in AdCC36,37, apart from anthracyclines38. Our study does not allow for definitive conclusions on chemotherapy based on the small numbers of patients presented here. Hence, additional studies to define the clinical management of SB-AdCC more accurately are warranted. Given that only three patients had SB-AdCCs harboring MYB rearrangements (two of whom were lost to follow-up, and one is alive with disease at the time of study), we were unable to determine if these would have a clinical outcome similar to those of patients with C-AdCC.
Our study has important limitations, such as the small size of the cohort, owing to the rarity of this entity. Furthermore, targeted sequencing data was available for only a subset of cases and was limited to the genes included in the MSK-IMPACT panel. Despite these limitations, our series is the largest in the literature with complete clinical data and molecular characterization of this rare but problematic form of TNBC. Although C-AdCCs and SB-AdCCs are phenotypically similar, our data show that the repertoire of genetic alterations of SB-AdCC is heterogeneous, neither resembling that of high-grade TNBCs nor entirely recapitulating that of C-AdCCs. Our findings do not support the notion that the majority of SB-AdCCs would be part of the spectrum of breast AdCCs; further studies to define how to best position these cancers in the context of breast neoplasms are warranted.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Wetterskog, D. et al. Adenoid cystic carcinomas constitute a genomically distinct subgroup of triple-negative and basal-like breast cancers. J. Pathol. 226, 84–96 (2012).
Martelotto, L. G. et al. Genomic landscape of adenoid cystic carcinoma of the breast. J. Pathol. 237, 179–189 (2015).
Andreasen, S. et al. Adenoid cystic carcinomas of the salivary gland, lacrimal gland, and breast are morphologically and genetically similar but have distinct microRNA expression profiles. Mod. Pathol. 31, 1211–1225 (2018).
Kim, J. et al. MYBL1 rearrangements and MYB amplification in breast adenoid cystic carcinomas lacking the MYB-NFIB fusion gene. J Pathol 244, 143–150 (2018).
Kulkarni, N. et al. Rare breast cancer: 933 adenoid cystic carcinomas from the National Cancer Data Base. Ann. Surg. Oncol. 20, 2236–2241 (2013).
Liao, H. Y. et al. The clinicopathological features and survival outcomes of different histological subtypes in triple-negative breast cancer. J Cancer 9, 296–303 (2018).
Treitl, D. et al. Adenoid cystic carcinoma of the breast, 20 years of experience in a single center with review of literature. Breast Cancer 25, 28–33 (2018).
Wang, S. et al. 36 cases adenoid cystic carcinoma of the breast in China: Comparison with matched grade one invasive ductal carcinoma-not otherwise specified. Pathol. Res. Pract. 213, 310–315 (2017).
Shin, S. J. & Rosen, P. P. Solid variant of mammary adenoid cystic carcinoma with basaloid features: a study of nine cases. Am. J. Surg. Pathol. 26, 413–420 (2002).
D’alfonso, T. M. et al. MYB-NFIB gene fusion in adenoid cystic carcinoma of the breast with special focus paid to the solid variant with basaloid features. Hum. Pathol. 45, 2270–2280 (2014).
Foschini, M. P. et al. Solid variant of adenoid cystic carcinoma of the breast: a case series with proposal of a new grading system. Int. J. Surg. Pathol. 24, 97–102 (2016).
Slodkowska, E. et al. Predictors of outcome in mammary adenoid cystic carcinoma: a multi-institutional study. Am. J. Surg. Pathol. 44, 214–223 (2020).
Massé, J. et al. Solid-type adenoid cystic carcinoma of the breast, a distinct molecular entity enriched in NOTCH and CREBBP mutations. Mod. Pathol. 33, 1041–1055 (2020).
Pareja, F., Weigelt, B. & Reis-Filho, J. S. Problematic breast tumors reassessed in light of novel molecular data. Mod. Pathol. 34, 38–47 (2021).
Lokuhetty, D., White, V. A., Watanabe, R. & Cree, I. A. WHO Classification of Tumours Editorial Board. Breast tumours. WHO classification of tumours of the breast, 5th ed. Lyon: International Agency for Research on Cancer (WHO classifiction of tumours series, 5th ed). 2 (2019).
Allison, K. H. et al. Estrogen and progesterone receptor testing in breast cancer: ASCO/CAP guideline update. J. Clin. Oncol. 38, 1346–1366 (2020).
Wolff, A. C. et al. Human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Focused Update. J. Clin. Oncol. 36, 2105–2122 (2018).
Rakha, E. A. et al. Morphological and immunophenotypic analysis of breast carcinomas with basal and myoepithelial differentiation. J. Pathol. 208, 495–506 (2006).
Brill, L. B. et al. Analysis of MYB expression and MYB-NFIB gene fusions in adenoid cystic carcinoma and other salivary neoplasms. Mod. Pathol. 24, 1169–1176 (2011).
Pareja, F. et al. Assessment of HMGA2 and PLAG1 rearrangements in breast adenomyoepitheliomas. NPJ Breast Cancer 5, 6 (2019).
Kim, S. H. et al. Identification of recurrent FHL2-GLI2 oncogenic fusion in sclerosing stromal tumors of the ovary. Nat. Commun. 11, 44 (2020).
Zhang, J. et al. INTEGRATE: gene fusion discovery using whole genome and transcriptome data. Genome Res. 26, 108–118 (2016).
McPherson, A. et al. Defuse: an algorithm for gene fusion discovery in tumor rna-seq data. PLoS Comput. Biol. 7, e1001138 (2011).
Edgren, H. et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome Biol. 12, R6 (2011).
Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012).
Shugay, M., Ortiz de Mendíbil, I., Vizmanos, J. L. & Novo, F. J. Oncofuse: a computational framework for the prediction of the oncogenic potential of gene fusions. Bioinformatics 29, 2539–2546 (2013).
TUMOR FUSION GENE DATA PORTAL. http://www.tumorfusions.org. Accessed on March 2019.
Cheng, D. T. et al. Memorial sloan kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J. Mol. Diagn. 17, 251–264 (2015).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 (2013).
Hudis, C. A. et al. Proposal for standardized definitions for efficacy end points in adjuvant breast cancer trials: the STEEP system. J. Clin. Oncol. 25, 2127–2132 (2007).
Schemper, M. & Smith, T. L. A note on quantifying follow-up in studies of failure time. Control Clin Trials 17, 343–346 (1996).
Zhang, Z. Survival analysis in the presence of competing risks. Ann. Transl. Med. 5, 47 (2017).
Nik-Zainal, S. et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534, 47–54 (2016).
Ivan, D. et al. Use of p63 expression in distinguishing primary and metastatic cutaneous adnexal neoplasms from metastatic adenocarcinoma to skin. J Cutan. Pathol. 34, 474–480 (2007).
Rajan, N. et al. Overexpression of MYB drives proliferation of CYLD-defective cylindroma cells. J. Pathol. 239, 197–205 (2016).
Trapani, D. et al. Benefit of adjuvant chemotherapy in patients with special histology subtypes of triple-negative breast cancer: a systematic review. Breast Cancer Res. Treat. 187, 323–337 (2021).
Chae, Y. K. et al. Adenoid cystic carcinoma: current therapy and potential therapeutic advances based on genomic profiling. Oncotarget 6, 37117–37134 (2015).
Laurie, S. A., Ho, A. L., Fury, M. G., Sherman, E. & Pfister, D. G. Systemic therapy in the management of metastatic or locally recurrent adenoid cystic carcinoma of the salivary glands: a systematic review. Lancet Oncol. 12, 815–824 (2011).
Acknowledgements
The authors would like to acknowledge Center Core grant (P30-CA008748), the Molecular Diagnostics Service in the Department of Pathology and the Marie-Josee and Henry R. Kravis Center for Molecular Oncology.
Funding
This work was primarily funded by the Breast Cancer Research Foundation and in part by the Marie-Josée and Henry R. Kravis Center for Molecular Oncology and the National Cancer Institute Cancer Center Core Grant No. P30-CA008748. F.P. is partially funded by an NIH K12 CA184746 grant. F.P., B.W., and J.S.R.-F. are funded in part by the NIH/NCI P50 CA247749 01 grant. B.W. is funded in part by a Cycle for Survival grant. E.M.dS. is partially funded by the MSK-MIND grant.
Author information
Authors and Affiliations
Contributions
C.J.S.: conceptualization, data curation, histologic review, IHC analysis, validation, writing– original draft, and writing–review and editing; E.B.: conceptualization, histologic review, resources, writing–review, and editing; A.M.: survival analysis, validation, writing–review and editing; A.D.C.P.: RNA seq analysis, bioinformatics, and validation; G.N.: FISH analysis and validation; E.M.dS.: formal analysis and validation; S.P.: survival analysis and validation; S.S.: data curation; K.V.: data curation; P.R.: conceptualization and writing—review and editing; L.N.: conceptualization and writing–review and editing; T.D.‘A.: writing–review and editing; B.W.: writing–review and editing; F.P.: conceptualization, histologic review, formal analysis, validation, and writing–review and editing; J.S. R.-F.: conceptualization, resources, supervision, writing–review and editing; H.Y.W.: conceptualization, histologic review, IHC analysis, supervision, writing–review and editing.
Corresponding authors
Ethics declarations
Conflict of interest
J.S.R.-F. reports receiving personal/consultancy fees from Goldman Sachs, REPARE Therapeutics, Paige.AI and Eli Lilly, membership of the scientific advisory boards of VolitionRx, REPARE Therapeutics and Paige.AI, membership of the Board of Directors of Grupo Oncoclinicas, and ad hoc membership of the scientific advisory boards of Roche Tissue Diagnostics, Ventana Medical Systems, Novartis, Genentech, and InVicro, outside the scope of this study. J.S.R.-F. also report ownership of Paige.AI stock options and REPARE Therapeutics stocks. P.R. received institutional grant/funding from Grail, Illumina, Novartis, Epic Sciences, ArcherDx, and consultation fees or honoraria from Novartis, Foundation Medicine, AstraZeneca, Epic Sciences, Inivata, Natera, Tempus. The other authors reported no conflicts of interest.
Ethics approval
The retrospective data for the paper was approved by the Memorial Sloan Kettering Cancer Center (MSK) Institutional Review Board and 12-245 Data and Tissue Utilization Committee, in accordance with the Helsinki Declaration of 1975 as revised in 1983.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Schwartz, C.J., Brogi, E., Marra, A. et al. The clinical behavior and genomic features of the so-called adenoid cystic carcinomas of the solid variant with basaloid features. Mod Pathol 35, 193–201 (2022). https://doi.org/10.1038/s41379-021-00931-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-021-00931-6
This article is cited by
-
Molecular pathology in breast disease: diagnostic, prognostic, and therapeutic tools
Virchows Archiv (2024)
-
Rare subtypes of triple negative breast cancer: Current understanding and future directions
npj Breast Cancer (2023)
-
A novel EWSR1-MYB fusion in an aggressive advanced breast adenoid cystic carcinoma with mixed classical and solid-basaloid components
Virchows Archiv (2023)
-
Solid-basaloid variant of adenoid cystic carcinoma of the breast with near complete response to neoadjuvant chemotherapy
npj Breast Cancer (2022)
-
Distinct clinicopathological and genomic features in solid and basaloid adenoid cystic carcinoma of the breast
Scientific Reports (2022)
