
Abstract
Adenomyoepitheliomas (AMEs) of the breast are uncommon and span the morphologic spectrum of benign, atypical, in situ, and invasive forms. In exceptionally rare cases, these tumors metastasize to regional lymph nodes or distant sites. In the era of genomic characterization, data is limited regarding AMEs. The aim of this study was to provide insight into the molecular underpinnings of a spectrum of AMEs. Seven cases of AMEs of the breast (benign-1, atypical-2, in situ-1, invasive-3) were identified in our files. The seven samples were interrogated using the Oncomine Comprehensive Assay v3 (ThermoFisher). Two atypical AMEs and the malignant in situ AME harbored the same gain-of-function PIK3CA mutation. The malignant in situ AME also showed EGFR amplification, not described previously. Both a benign AME and a malignant invasive AME shared the same gain-of-function AKT1 variant. The benign AME also showed a GNAS mutation. Moreover, the same gain-of-function HRAS mutation was present in an atypical AME and a malignant invasive AME. We also identified co-occurring HRAS and PIK3CA mutations in an ER-positive atypical AME, which has not been previously described. No fusion drivers were detected. We describe the molecular characteristics of the spectrum of AME tumors of the breast, which harbor alterations in the PI3K/AKT pathway. Our findings are clinically relevant with respect to the current options of targeted therapy in the rare instances where malignant AME tumors of the breast progress.
Similar content being viewed by others
Introduction
Adenomyoepitheliomas (AMEs) of the breast are rare lesions first described by Hamperl in 1970 [1]. These biphasic tumors are composed of both epithelial and myoepithelial components and are thought to be variants of intraductal papillomata [2]. Based on architecture and/or cytomorphology, AMEs can be classified into spindle, tubular, lobulated, papillary, and mixed histologic patterns [3, 4].
A majority of AMEs have a benign clinical course, however, distant metastases from cases lacking atypia or a proliferative component have been reported [5, 6]. Therefore, according to the most recent edition of the World Health Organization Classification of Tumors, AMEs may be regarded as neoplasms with low malignant potential [7]. Overt malignant transformation of AMEs has been documented and is considered to be an extremely rare occurrence [8,9,10,11,12,13,14]. Atypical AMEs demonstrate cytological atypia without tumoral necrosis, high mitotic rates, or overgrowth of the epithelial or myoepithelial components. Malignant AMEs can be classified as either in situ malignant AME or invasive malignant AME. In malignant AMEs, the malignant component may be solely epithelial, solely myoepithelial, or both epithelial and myoepithelial [7]. Metastases from malignant AMEs have been documented in lymph nodes, soft tissue, lung, liver, thyroid, kidney, and brain [6, 8,9,10, 15,16,17,18].
Given the rarity of AMEs, the literature regarding genetic alterations is limited. Studies of both benign AMEs and malignant AMEs have employed DNA ploidy analysis, comparative genomic hybridization (cGH) analysis, cytogenetics, targeted next-generation sequencing (NGS) and whole exome sequencing (WES) [10, 16, 19,20,21,22,23,24,25]. Early genetic studies of benign AMEs showed a variety of molecular alterations including microsatellite instability and reciprocal translocations involving chromosomes 8 and 16 [22, 26]. Jones et al. reported a case of malignant AME with losses at chromosomes 11q and 16q in the primary tumor and an additional loss at 12q in the liver metastasis [10].
Sequencing analyses have demonstrated recurrent mutations in PIK3CA, AKT1, HRAS, and PIK3R1 genes [16, 20, 23]. Furthermore, a recent study by Geyer et al. highlighted different recurrent alterations in AMEs based on estrogen receptor (ER) status, with ER-positive tumors more frequently harboring PIK3CA and AKT1 activating mutations and ER-negative tumors harboring HRAS mutations [23]. The aim of this study was to describe a complete spectrum of AMEs and provide further insight into the molecular underpinnings of AMEs of the breast using a target NGS-sequencing platform.
Materials and methods
Case selection
Seven cases of AMEs of the breast were identified in our files including four cases of malignant AMEs (three invasive and one in situ), two atypical AMEs, and one benign AME. All available pathological material for each case were reviewed. All clinical data were retrieved from the medical records and dates of recurrence or last contact were recorded. Institutional review board approval was obtained for all aspects of this study.
Pathologic examination
All available hematoxylin and eosin (H&E) slides from each case were reviewed by four pathologists (SJS/SH/PSG/PJM). For benign and atypical AMEs, the following characteristics were evaluated: nuclear atypia, mitoses, and prominent component (in atypical AME). For cases with carcinoma, the following characteristics were evaluated: uniphasic versus biphasic malignant proliferation, necrosis, nuclear grade, and presence of non-glandular differentiation. The block with the best preserved and highest volume of tumor was selected for further analysis. In cases with carcinoma, the block with the greatest volume of neoplastic component was chosen for further analysis.
Immunohistochemistry
ER immunohistochemistry was performed on slides prepared from representative tumor blocks. Immunostained slides were examined by routine light microscopy by a single pathologist (PSG) and scored. Positivity for ER was defined as ≥1% nuclear positivity.
Nucleic acid extraction
Tumor DNA and RNA were extracted from 5 μm-thick formalin-fixed paraffin-embedded (FFPE) tissue sections. Tumor tissue was macrodissected based on annotation of a corresponding H&E slide. Extraction was performed by using the Maxwell® 16 FFPE Plus LEV DNA & RNA Purification Kits on an automated Maxwell 16 Research extraction system (Promega, Madison, USA). The Maxwell RNase A solution and DNase I were used to, respectively, digest RNA and DNA during the two different procedures of nucleic acids’ isolation. The Qubit® 3.0 Fluorometer (Thermo Fisher Scientific) was employed for sample quantitation by means of the highly sensitive and accurate fluorescence-based Qubit® quantitation assays.
DNA and RNA NGS and data analysis
NGS library preparation for the Oncomine Comprehensive Assay v3 (OCAv3, ThermoFisher Scientific) using extracted DNA and RNA was performed using the Ion AmpliSeq™ Library Preparation on the IonChef™ System protocol (ThermoFisher Scientific). Sequencing was performed on the IonTorrent™ S5 XL platform, following manufacturer protocols and using positive control cell line mixtures (Horizon Discovery). OCAv3 is an amplicon-based, targeted assay that enables the detection of relevant SNVs (gene hotspots and full coding regions, see Supplementary Table 1), amplifications, gene fusions, and indels from 161 unique genes. Genomic data were analyzed, and alterations were detected using the IonReporter™ software, version 5.6 (ThermoFisher Scientific).
Sanger sequencing
For genes of interest (PIK3CA, AKT1, and HRAS), PCR was performed using custom PCR primers designed to amplify short (~200–400 bp) regions in FFPE samples. A human gDNA control sample was run in parallel with the FFPE samples to confirm a successful PCR and end-sequencing was performed using PCR primers. After enzymatic purification, sequencing was achieved through BigDye Terminator Cycle Sequencing. Data analysis was performed with DNASTAR Lasergene12 software and the threshold for SNP detection was set to 10%. Mutations from the reference sequence were called whenever sequence quality and coverage allowed.
Results
Patient characteristics and follow-up
All patients were female with a median age of 56 years (range: 42–78 years; mean: 58 years). The median tumor size was 1.5 cm (range: 1.2–4.8 cm; mean: 2.1 cm). Five patients underwent breast-conserving surgery and two patients had mastectomies. Additional clinicopathologic characteristics of the cohort are summarized in Table 1. Follow up was available for six patients with a median of 22 months (range: 11–37; mean: 22 months). Of the four patients with malignant AME, 1 patient (AME1) presented with lung metastases 8 months following surgery and died of disease 24 months following surgery. The other three cases of malignant AME (AME3, AME4, and AME7) were all alive without disease at 23, 37, and 16 months following surgery, respectively. The two cases of atypical AME were alive without disease at last follow-up.
Histopathology
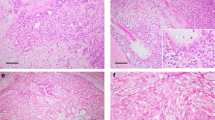
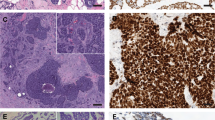
All seven AME cases were biphasic in nature with a nodular proliferation of epithelial and myoepithelial cells. The case of benign AME was a well-circumscribed and nodular tumor lacking a complete fibrous capsule (AME2) (Fig. 1a and b). No overt cytologic atypia was present and there were equal ratios of epithelial and myoepithelial components. One atypical AME was multinodular and displayed moderate cytological atypia with a prominent myoepithelial component. Mitoses were identified within myoepithelial and epithelial cells (AME5). The second atypical AME was well circumscribed with equal proportions of the epithelial and myoepithelial components and displayed cytologic atypia predominantly in the epithelial component (Fig. 1c and d) (AME6). Mitoses were identified within the epithelial component. The distinction of atypical AME and malignant in situ AME was based on the presence of severe cytologic atypia, necrosis, and brisk mitotic activity which would be beyond that allowable for the diagnosis of an atypical AME. The malignant in situ AME was multinodular and demonstrated a neoplastic proliferation of epithelial cells with moderate to severe cytologic atypia, focal necrosis, and brisk mitotic activity. Focal sebaceous and squamous differentiation was also present (Fig. 2a and b) (AME3). No stromal invasion was identified. Beyond the borders of the malignant in situ AME, was an adjacent 5 mm focus of ductal carcinoma in situ (DCIS) comprised of solid and cribriform architectural types with low nuclear grade and calcifications (not shown). In this case (AME3), the malignant component comprised a vast proportion of the tumor (~90%). Of the three malignant invasive AMEs, one had metastasis to a regional lymph node. The primary tumor was infiltrative with large areas of necrosis. The tumor was biphasic and composed of malignant glandular and focal squamous epithelial cells and malignant myoepithelial cells with spindle and clear cell morphology (Fig. 2c and d) (AME1). The associated lymph node metastasis also demonstrated focal squamous differentiation (Fig. 2e and f). In this case (AME1), the malignant component represented the majority of the tumor (~80%). The second invasive malignant AME was multinodular with invasive tumor borders. The tumor was biphasic and composed of predominately malignant myoepithelial cells with spindle and clear cell morphology and also malignant glandular epithelial cells (AME4). In this case (AME4), the malignant component comprised a large part of the tumor (~60%). The third invasive malignant AME was diffusely infiltrative and associated with necrosis. The biphasic tumor was comprised of malignant glandular epithelial cells (AME7). All of the malignant AME (one in situ and three invasive) demonstrated high-grade nuclei and mitoses were easily identified. In this case (AME7), the malignant component comprised a broad proportion of the tumor (~80%). Additional clinicopathologic characteristics of the cohort are summarized in Table 2. No variables demonstrated a clear association with specific genomic alterations.

a Example of benign AME that is well-circumscribed with nodular growth and a papillary configuration. Sanger sequencing confirmed the presence of an AKT1 variant (E17K) (inset). B This benign AME demonstrates equal proportions of epithelial and myoepithelial elements with bland cytologic features. c Example of atypical AME that is well circumscribed with focal papillary architecture (upper right). d This atypical AME shows myoepithelial prominence with cytologic atypia.

a Example of malignant in situ AME with a nodular configuration. b High magnification of this malignant in situ AME shows a monotonous population of epithelial cells with intermediate grade nuclei and focal squamous features. Sanger sequencing confirmed the presence of a PIK3CA mutation (H1047P) (inset). c Example of malignant AME with invasive growth and necrosis. d This malignant invasive AME showed biphasic proliferation with glandular, clear cell, and spindle morphology. Sanger sequencing showed an HRAS mutation (G12D) (inset). e The same malignant invasive AME was associated with metastases to a lymph node. f The metastatic AME in lymph node showed both glandular and myoepithelial components and squamous differentiation (left).
Immunohistochemistry
The benign AME, the two atypical AMEs (Fig. 3a and b), and one malignant invasive AME (AME4) were ER-positive. ER positivity was present in both the epithelial and myoepithelial cells for all four of these tumors, however, the epithelial positivity was predominately more diffuse and strong than that of the myoepithelial cell staining. The malignant in situ AME and two other malignant invasive AME (AME1 and AME7) were negative for ER (Table 1).

a Atypical AME with multinodular growth and papillary configuration. b This atypical AME shows cytologic atypia and mitosis. The tumor was positive for estrogen receptor (inset).
Mutations detected by oncomine v3
The two atypical AMEs and the malignant in situ AME both harbored the same gain-of-function PIK3CA mutation (H1047P; COSM249874), with a similar variant allele frequency of 31–35%. The malignant in situ AME also showed EGFR amplification. The EGFR amplification showed a 45.83-fold change (CNV confidence interval: 5%: 40.55; 95%: 51.8). The benign AME and one malignant invasive AME (AME4) both shared the same gain-of-function AKT1 variant (E17K; COSM33765) with variant allele frequencies of 31% and 16%, respectively. The benign AME also demonstrated the gain-of-function GNAS mutation (R201C; COSM27887), with an allele frequency of 28%. Moreover, the same gain-of-function HRAS mutation (G12D; COSM484) was present in one atypical AME (AME5) and in a malignant invasive AME (AME1). The variant allele frequencies for HRAS were 45% and 21%, respectively, and average read depth was 1082×. The Oncomine Comprehensive Assay v3 (OCAv3) interrogates HRAS hotspots that include both codon 61 and codon 12 (Supplementary Table 1). No fusions were detected in any of our cases. The sequencing results of the cohort are summarized in Fig. 4. Since microdissection was not performed, the aforementioned detected alterations were not stratified based on epithelial versus myoepithelial components of the tumors. While copy number alterations are evaluable for a specific set of genes, evaluation of broad copy number alterations are beyond the scope of the OCAv3 test.

Somatic mutations and amplifications in adenomyoepitheliomas of the breast identified using the Oncomine Comprehensive Assay v3 are plotted. Allelic frequency of mutations is noted. Cases are shown in rows (Benign cases at the top; Malignant cases at the bottom), and estrogen receptor status and genes in columns.
Sanger sequencing
Sanger sequencing confirmed a selection of mutations in four representative cases: PIK3CA on AME3, AKT1 on AME4 and AME2, and HRAS on AME1.
Discussion
We present the molecular features of the spectrum of AMEs of the breast including benign, atypical, malignant in situ, and malignant invasive cases. Recurrent molecular alterations include activating mutations in PIK3CA, AKT1, and HRAS.
The presence of alterations in the PI3K/AKT/mTOR pathway is common in breast cancer. Of the mutated genes, PIK3CA is the most common (36%) with somatic alterations with the other genes including PIK3R1 (3%), PTEN (3%), and AKT1 (2%) being very uncommon [27, 28]. In the reported literature, 48% of AMEs harbor PIK3CA mutations and 17% AMEs harbor AKT1 mutations [16, 20, 23, 29, 30]. We too found that PIK3CA and AKT1 mutations are common in AMEs (42% and 29%, respectively). We reaffirm that PIK3CA and AKT1 mutations are mutually exclusive and that AKT1 mutations are common in ER-positive tumors [23, 29]. These findings suggest that similar to breast cancer, the PI3K/AKT/mTOR pathway is important in the pathogenesis of AMEs, however, the frequency of AKT1 gene mutations is higher in AMEs compared to breast carcinomas, which is in keeping with recent studies. While malignant AMEs and metastatic disease are rare, treatment options for patients with advanced disease are often limited to conventional chemotherapies for which there are little data available regarding efficacy [17]. Given the advent of PI3K, AKT, and mTOR inhibitors, the possibility of targeted therapy exists for patients with malignant AMEs that harbor mutations in the PI3K/AKT/mTOR pathway [31, 32].
Contrary to prior studies, we report co-occurring HRAS and PIK3CA mutations in an ER-positive AME [23, 29]. While others have suggested that HRAS mutation is possibly instrumental in the acquisition of an ER-negative aggressive phenotype, our finding suggests that this may not be the only mechanism involved. We note that while most of the HRAS mutations in AMEs involve Q61, we identified G12D mutations in our tumors. Both of these are hotspot mutations, and G12D mutations have also been observed in other benign and malignant mammary lesions [33, 34]. Additionally, MAPK/MEK inhibitors may be considered in patients with tumors harboring HRAS mutations and data suggests that patients with concurrent HRAS and PIK3CA may benefit from mTOR inhibitors [35].
To our knowledge, this is the first report of an EGFR gene amplification in an AME. EGFR gene mutations have been previously reported, however, no prior cases have shown amplification [29]. EGFR gene amplifications are present in a subset of lung adenocarcinomas and squamous cell carcinomas and in approximately half of glioblastomas [36]. While anti-EGFR therapy has shown little efficacy in the treatment of EGFR gene-amplified glioblastoma, EGFR gene amplification has been implicated as a mechanism of drug resistance in other cancer types [37, 38]. As such, its role as a therapeutic target in AMEs is unclear.
In the benign AME, we found co-occurring AKT1 and GNAS mutations, a finding which has been previously reported [29]. Mutant GNAS results in constitutively increased cAMP formation. The GNAS R201C mutation detected in our benign AME tumor has been reported in other tumor types including ovarian Leydig cell tumors [39, 40], precursor and malignant lesions of the gastrointestinal tract [41], fibrous dysplasia [42], melanomas [41], and in at least one invasive mammary carcinoma [43]. Distinct functionally relevant GNAS mutations (i.e. R201H) have also been reported in usual ductal hyperplasia arising in mammary papillomas [44]. The presence of oncogenic GNAS mutations suggests a yet unrecognized role of altered G-protein signaling in a subset of benign and malignant mammary lesions, including AMEs.
Finally, we failed to detect any TP53 mutations, including in our ER-negative malignant AMEs, similar to previous findings [16, 20, 23, 29, 30].
We acknowledge that small sample size, targeted sequencing approach, and limited clinical follow-up are limitations of our study. Despite these, we report new findings in adenomyoepithelial tumors of the breast: the first case of an ER-positive AME with HRAS G12D and PIK3CA mutations and an EGFR gene amplification in an AME.
Conclusion
Herein, we add to the limited data regarding the molecular characteristics of AMEs of the breast. While we confirm prior findings that these tumors frequently harbor mutations in the PI3K/AKT/mTOR pathway, that PIK3CA and AKT1 mutations are mutually exclusive, and that AKT1 mutations are common in ER-positive tumors, we report the first cases of co-existing HRAS G12D and PIK3CA mutations in an ER-positive AME. These findings suggest that our understanding of the molecular underpinnings of these rare tumors will further evolve as additional studies are available. Additionally, we report the first EGFR-amplified AME. Our findings further suggest that therapy targeting PIK3CA, AKT1, and HRAS may be options for patients with progressive malignant AMEs of the breast.
References
Hamperl H. The myothelia (myoepithelial cells). Normal state; regressive changes; hyperplasia; tumors. Curr Top Pathol. 1970;53:161–220.
Rosen PP. Adenomyoepithelioma of the breast. Hum Pathol. 1987;18:1232–7.
Schmitt D, Tan PH, Dabbs D, Jones L. Myoepithelial and epithelial–myoepithelial lesions. In: Lakhani SR, editor. WHO classification of tumours of the breast. 4th ed. International Agency for Research on Cancer: Lyon; 2012. p. 120–3.
Tavassoli FA. Myoepithelial lesions of the breast. Myoepitheliosis, adenomyoepithelioma, and myoepithelial carcinoma. Am J Surg Pathol. 1991;15:554–68.
Nadelman CM, Leslie KO, Fishbein MC. “Benign,” metastasizing adenomyoepithelioma of the breast: a report of 2 cases. Arch Pathol Lab Med. 2006;130:1349–53.
Korolczuk A, Amarowicz M, Bak K, Korobowicz E, Koncewicz T. Adenomyoepithelioma of the breast with late pulmonary metastases—case report and review of the literature. J Cardiothorac Surg. 2016;11:121.
World Health Organization Classification of Tumours. Breast tumours. 5th ed. International Agency for Research on Cancer: Lyon; 2019.
Bult P, Verwiel JM, Wobbes T, Kooy-Smits MM, Biert J, Holland R. Malignant adenomyoepithelioma of the breast with metastasis in the thyroid gland 12 years after excision of the primary tumor. Case report and review of the literature. Virchows Arch. 2000;436:158–66.
Honda Y, Iyama K. Malignant adenomyoepithelioma of the breast combined with invasive lobular carcinoma. Pathol Int. 2009;59:179–84.
Jones C, Tooze R, Lakhani SR. Malignant adenomyoepithelioma of the breast metastasizing to the liver. Virchows Arch. 2003;442:504–6.
Moritz AW, Wiedenhoefer JF, Profit AP, Jagirdar J. Breast adenomyoepithelioma and adenomyoepithelioma with carcinoma (malignant adenomyoepithelioma) with associated breast malignancies: a case series emphasizing histologic, radiologic, and clinical correlation. Breast. 2016;29:132–9.
Qureshi A, Kayani N, Gulzar R. Malignant adenomyoepithelioma of the breast: a case report with review of literature. BMJ Case Rep. 2009;2009 pii: bcr01.2009.1442.
Xu J, Tang X, Iida Y, Fuchinoue F, Kusumi T, Yagihashi N, et al. Adenomyoepithelioma with carcinoma of the breast: a report of two cases and a review of the literature. Pathol Res Pr. 2016;212:130–4.
Yang Y, Wang Y, He J, Pan G, Tuo X, Jiang A, et al. Malignant adenomyoepithelioma combined with adenoid cystic carcinoma of the breast: a case report and literature review. Diagn Pathol. 2014;9:148.
Rasbridge SA, Millis RR. Adenomyoepithelioma of the breast with malignant features. Virchows Arch. 1998;432:123–30.
Ericson-Lindquist K, Johansson A, Leveen P, Elmberger G, Jonsson G, Staaf J, et al. Targeted sequencing may facilitate differential diagnostics of pulmonary tumours: a case series. Diagn Pathol. 2017;12:31.
Lee S, Oh SY, Kim SH, Lee JH, Kim DC, Cho SH, et al. Malignant adenomyoepithelioma of the breast and responsiveness to Eribulin. J Breast Cancer. 2015;18:400–3.
Logie N, Hugh J, Paulson K, Pearcey R, King KM. Radiotherapy in the multidisciplinary management of adenomyoepithelioma of the breast with an axillary lymph node metastasis: a case report and review of the literature. Cureus. 2017;9:e1380.
Baraban E, Zhang PJ, Jaffer S, Lubin D, Feldman M, Bleiweiss IJ, et al. MYB rearrangement and immunohistochemical expression in adenomyoepithelioma of the breast: a comparison with adenoid cystic carcinoma. Histopathology. 2018;73:897–903.
Baum JE, Sung KJ, Tran H, Song W, Ginter PS. Mammary epithelial-myoepithelial carcinoma: report of a case with HRAS and PIK3CA mutations by next-generation sequencing. Int J Surg Pathol. 2018;4:441–5.
Da Silva L, Buck L, Simpson PT, Reid L, McCallum N, Madigan BJ, et al. Molecular and morphological analysis of adenoid cystic carcinoma of the breast with synchronous tubular adenosis. Virchows Arch. 2009;454:107–14.
Gatalica Z, Velagaleti G, Kuivaniemi H, Tromp G, Palazzo J, Graves KM, et al. Gene expression profile of an adenomyoepithelioma of the breast with a reciprocal translocation involving chromosomes 8 and 16. Cancer Genet Cytogenet. 2005;156:14–22.
Geyer FC, Li A, Papanastasiou AD, Smith A, Selenica P, Burke KA, et al. Recurrent hotspot mutations in HRAS Q61 and PI3K-AKT pathway genes as drivers of breast adenomyoepitheliomas. Nat Commun. 2018;9:1816.
Nomura K, Fukunaga M, Uchida K, Aizawa S. Adenomyoepithelioma of the breast with exaggerated proliferation of epithelial cells: report of a case. Pathol Int. 1996;46:1011–4.
Trojani M, Guiu M, Trouette H, De Mascarel I, Cocquet M. Malignant adenomyoepithelioma of the breast. An immunohistochemical, cytophotometric, and ultrastructural study of a case with lung metastases. Am J Clin Pathol. 1992;98:598–602.
Salto-Tellez M, Putti TC, Lee CK, Chiu LL, Koay ES. Adenomyoepithelioma of the breast: description of allelic imbalance and microsatellite instability. Histopathology. 2005;46:230–1.
Yang SX, Polley E, Lipkowitz S. New insights on PI3K/AKT pathway alterations and clinical outcomes in breast cancer. Cancer Treat Rev. 2016;45:87–96.
Ng CK, Schultheis AM, Bidard FC, Weigelt B, Reis-Filho JS. Breast cancer genomics from microarrays to massively parallel sequencing: paradigms and new insights. J Natl Cancer Inst. 2015;107:djv015.
Lubin D, Toorens E, Zhang PJ, Jaffer S, Baraban E, Bleiweiss IJ, et al. Adenomyoepitheliomas of the breast frequently harbor recurrent hotspot mutations in PIK3-AKT pathway-related genes and a subset show genetic similarity to salivary gland epithelial–myoepithelial carcinoma. Am J Surg Pathol. 2019;43:1005–13.
Watanabe S, Otani T, Iwasa T, Takahama T, Takeda M, Sakai K, et al. A case of metastatic malignant breast adenomyoepithelioma with a codon-61 mutation of HRAS. Clin Breast Cancer. 2019;19:e589–e92.
Hyman DM, Smyth LM, Donoghue MTA, Westin SN, Bedard PL, Dean EJ, et al. AKT inhibition in solid tumors with AKT1 mutations. J Clin Oncol. 2017;35:2251–9.
LoRusso PM. Inhibition of the PI3K/AKT/mTOR pathway in solid tumors. J Clin Oncol. 2016;34:3803–15.
Myers MB, Banda M, McKim KL, Wang Y, Powell MJ, Parsons BL. Breast cancer heterogeneity examined by high-sensitivity quantification of PIK3CA, KRAS, HRAS, and BRAF mutations in normal breast and ductal carcinomas. Neoplasia. 2016;18:253–63.
Troxell ML, Levine J, Beadling C, Warrick A, Dunlap J, Presnell A, et al. High prevalence of PIK3CA/AKT pathway mutations in papillary neoplasms of the breast. Mod Pathol. 2010;23:27–37.
Kiessling MK, Curioni-Fontecedro A, Samaras P, Atrott K, Cosin-Roger J, Lang S, et al. Mutant HRAS as novel target for MEK and mTOR inhibitors. Oncotarget. 2015;6:42183–96.
Roskoski R Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharm Res. 2014;79:34–74.
Sigismund S, Avanzato D, Lanzetti L. Emerging functions of the EGFR in cancer. Mol Oncol. 2018;12:3–20.
An Z, Aksoy O, Zheng T, Fan QW, Weiss WA. Epidermal growth factor receptor and EGFRvIII in glioblastoma: signaling pathways and targeted therapies. Oncogene. 2018;37:1561–75.
Bieler BM, Gerber SM, Perez de Nanclares G, Hardisson D, Lecumberri B. Intratumoral activating GNAS (R201C) mutation in two unrelated patients with virilizing ovarian Leydig cell tumors. Endocrinol Diabetes Nutr. 2017;64:335–7.
Fragoso MC, Latronico AC, Carvalho FM, Zerbini MC, Marcondes JA, Araujo LM, et al. Activating mutation of the stimulatory G protein (gsp) as a putative cause of ovarian and testicular human stromal Leydig cell tumors. J Clin Endocrinol Metab. 1998;83:2074–8.
Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703–13.
Jour G, Oultache A, Sadowska J, Mitchell T, Healey J, Nafa K, et al. GNAS mutations in fibrous dysplasia: a comparative study of standard sequencing and locked nucleic acid PCR sequencing on decalcified and nondecalcified formalin-fixed paraffin-embedded tissues. Appl Immunohistochem Mol Morphol. 2016;24:660–7.
Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, et al. Comprehensive molecular portraits of invasive lobular breast. Cancer Cell. 2015;163:506–19.
Jahn SW, Kashofer K, Thuringer A, Abete L, Winter E, Eidenhammer S, et al. Mutation profiling of usual ductal hyperplasia of the breast reveals activating mutations predominantly at different levels of the PI3K/AKT/mTOR pathway. Am J Pathol. 2016;186:15–23.
Acknowledgements
This work was supported in part by the Translational Research Program of the Department of Pathology and Laboratory Medicine at Weill Cornell Medicine. Furthermore, we would like to acknowledge Dr. F. Menken, Dr. D. Gingell, Dr. N.T. Wongchaowart, Dr. T.J. Lillemoe, M. Soaita, and Dr. B. Lee for providing clinical information and/or histologic material for this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This work was presented in part at the 107th Annual Meeting of the United States and Canadian Academy of Pathology in March 2018, Vancouver, BC
Supplementary information
Rights and permissions
About this article

Cite this article
Ginter, P.S., McIntire, P.J., Kurtis, B. et al. Adenomyoepithelial tumors of the breast: molecular underpinnings of a rare entity. Mod Pathol 33, 1764–1772 (2020). https://doi.org/10.1038/s41379-020-0552-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-020-0552-x
This article is cited by
-
Triple-negative breast carcinomas of low malignant potential: review on diagnostic criteria and differential diagnoses
Virchows Archiv (2022)
-
Papillary lesions of the breast
Virchows Archiv (2022)
-
HRAS is a therapeutic target in malignant chemo-resistant adenomyoepithelioma of the breast
Journal of Hematology & Oncology (2021)
-
Papillary neoplasms of the breast—reviewing the spectrum
Modern Pathology (2021)
-
Uncommon Tumors and Uncommon Presentations of Cancer in the Breast
Current Breast Cancer Reports (2021)
