
Abstract
Adenoid cystic carcinoma (ACC) of the breast with a predominant solid pattern is difficult to diagnose with certainty and differentiate from more common triple-negative breast cancers (TNBCs) of basal-phenotype. To better characterize solid ACC, we performed a clinical, morphological, immunohistochemical, and molecular comparative analysis of 33 ACCs of the breast comprising 17 solid variant ACCs and 16 conventional ACCs. Solid ACCs displayed basaloid morphology with an exclusive or predominant epithelial cell population associated with decreased myoepithelial differentiation, while demonstrating MYB protein overexpression similar to the more common type of ACC. Strong and diffuse MYB expression by immunochemistry was observed in 14/17 (82%) of solid ACCs while MYB rearrangements were detected by break apart fluorescence in situ hybridization (FISH) in only 3/16 (19%) of solid ACCs. Conversely, weak MYB immunohistochemical expression was observed in only 7/204 (3%) of TNBC. Solid ACCs displayed a transcriptomic profile distinct from conventional ACCs with 549 genes showing a highly significant differential expression between conventional and solid ACC [false discovery rate (FDR) < 0.01; log2FC > |1|]. EnrichR and Kegg Pathway analyses identified PI3K-Akt and focal adhesion signaling pathways as significantly overexpressed in conventional ACCs compared with solid ACCs which significantly overexpressed the nitrogen metabolism pathway. CREBBP mutations and NOTCH activating gene mutations were only present in solid ACCs, concerning 5/16 (31%) of cases for each gene. Tumors with NOTCH activating mutations displayed a strong diffuse nuclear NICD1 staining, an established marker of Notch pathway activation. Solid ACCs also differed from basal-type TNBC, with fewer TP53 mutations and a more stable genomic profile on array comparative genomic hybridization (CGH). In summary, solid-type ACC of the breast is a distinct molecular entity within the ACC family and is different from common basal-type TNBC. MYB is a diagnostically useful biomarker of solid ACC and NOTCH could be a novel potential therapeutic target in 30% of cases.
Similar content being viewed by others

Introduction
Adenoid cystic carcinoma (ACC) of the breast is a very rare neoplasm that represents <0.1% of invasive breast carcinomas and shares histological characteristics and cytogenetic anomalies with salivary gland-type tumors of the head and neck, lacrimal gland, lung, prostate, cervix, and vulva [1,2,3,4,5,6,7,8]. Breast ACC is a subtype of triple-negative breast cancer (TNBC) defined by the absence of estrogen receptor (ER), progesterone receptor (PR), and a human epidermal growth factor receptor 2 (HER2) negative status. They share many features with the more frequent salivary ACC, although the later has poorer prognosis, with a 10-year overall survival rate of 60% compared with 75–93.8% for breast ACC [9,10,11,12,13,14]. The conventional forms of breast and salivary ACC are characterized morphologically and immunohistochemically by a dual population of cells, a basal/myoepithelial component which expresses basal markers, such as p63 and a luminal/epithelial component, which displays consistent positivity for CK7 and CKIT. Breast and salivary gland ACC are also characterized by the recurrent translocation, t(6;9)(q22-23;p23-24), resulting in the fusion of MYB oncogene with the NFIB transcription factor in 33–100% of breast ACC [2, 15,16,17]. MYB encodes a transcription factor that functions as a regulator of progenitor cells and is implicated in tumorigenesis. The MYB–NFIB gene fusion, discovered in 2009, results in increased MYB protein expression that can be detected using immunohistochemistry (IHC) [18]. The canonical MYB-NFIB translocation is not found in a number of breast and salivary gland ACCs, despite a typical morphology. Alternative genetic alterations resulting in MYB or MYBL1 overexpression, a structurally related protein of the same c-MYB transcription factor family, have also been described in salivary gland and breast tumors [19,20,21]. Breast ACCs with a conventional morphology are usually treated only by surgery, without systemic treatment. A subgroup of ACCs with a solid pattern and predominance of epithelial cells, also termed solid variant ACC or grade III according to Ro, are more difficult to diagnose with certainty [15, 22, 23]. Indeed, in this group, MYB rearrangement is uncommon, and clinical and genetic characteristics differ from conventional ACCs. In everyday practice, the pathologist is confronted with the question of whether these solid variants constitute a true subgroup of the breast ACC family or if they resemble the more common TNBC of basal-type phenotype. The latter is a potential diagnostic pitfall, of worse prognosis, with frequent early metastatic relapses and the need for more aggressive treatment with chemotherapy [24].
The aim of our study was therefore to compare the clinical, morphological, immunohistochemical, and molecular characteristics of breast ACCs solid variant, conventional breast ACCs, and TNBCs basal-type.
Materials and methods
Cases
This retrospective analysis includes 33 cases of conventional and solid variant breast ACCs diagnosed at Institute Bergonié, Bordeaux between 2000 and 2017. These cases were treated at our institution or were sent to us for second opinion. The clinical features, treatment, and follow-up data for each patient were obtained either directly from the Bergonié Institute patients’ files or by mailed questionnaire to referring pathologists, physicians, and surgeons for patients treated outside the center.
In addition, a previously described tissue microarray (TMA) containing 207 cases of TNBCs from patients treated at Institute Bergonié between 2007 and 2012 was used to perform a comparative immunohistochemical analysis [25]. These TMAs included three cases of ACC.
Morphology
All cases were reviewed by two pathologists (JM and GM). We determined for each tumor the growth pattern, Elston & Ellis (E&E) histological grade, Ro histological grade, presence of a characteristic dual epithelial–myoepithelial cell population, necrosis, lymphatic tumor emboli, and perineural invasion. The tumors were classified into two groups: Conventional-type ACC comprising 16 cases (cases 1–16), characterized by a combination of epithelial and myoepithelial cells, glandular and pseudoglandular cavities. Glandular cavities contained an eosinophilic material and were lined by luminal cells with eosinophilic cytoplasms and small nonatypical nuclei. Pseudoglandular cavities contained a myxoid or hyalinized material and were lined by basal-type cells with scant cytoplasms and monomorphic low-grade nuclei. The mitotic count was <4 mitoses/mm2. Nine tumors (cases 1–9) displayed the typical MYB rearrangement, and seven tumors (cases 10–16) did not have MYB rearrangement, as determined by the fluorescence in situ hybridization (FISH) assay. The second group, solid-type ACC, comprising 17 cases (cases 17–33) was defined by the presence of infiltrative trabecular, solid, or polyadenoid structures made of relatively monomorphic basal-type cells, within a hyalinized, myxoid, or desmoplastic stroma. The characteristic dual cell population, as described in conventional-type ACC, was absent. Solid-type ACC tumor cells displayed intermediate grade nuclei, with oval or angulated contours, with dense chromatin and sometimes prominent nucleoli. The mitotic count was variable, ranging from 1 to 8 mitoses/mm2.
Immunohistochemistry (IHC)
The immunohistochemical study was performed using blocks of representative formalin-fixed and paraffin-embedded (FFPE) tumor material. These blocks were the subject of serial sections of 3 µm thick, which were mounted on positively charged glass slides. The immunohistochemical study was carried out on a BenchMark® ULTRA (ROCHE-VENTANA) automat with ultraView Universal DAB (760-500) or OptiView DAB IHC (760-700) detection kits according to the protocol of the different primary antibodies used. Details of antibody clones, manufacturers, dilutions used, incubation times, pretreatment buffers, and staining kits are summarized in Supplementary Table 1. Nuclear staining was assessed for ER, PR, MYB, NICD1, p63, and Ki-67. Membranous staining was assessed for HER2/neu and cytoplasmic staining was assessed for CK5, CK14, CK7, and CKIT. A negative ER and/or PR status was defined by the presence of <1% positive tumor cells. HER2 status was determined according to the 2018 American Society of Clinical Oncology/College of American Pathologists recommendations [26]. Ki-67 index was determined according to the international recommendations on the assessment of Ki-67 in breast cancer [27]. The H-score was used for the interpretation of the immunohistochemical stainings of MYB, NICD1, CK5, CK7, and CK14. The H-score is obtained by multiplying the percentage of positive tumor cells by the staining intensity (0: no staining, 1: weak staining, 2: moderate staining, and 3: strong staining). The resulting score ranges from 0 to 300. Staining of p63 was assessed as present or absent. If present, it was qualified as (a) diffuse (all the tumor cells in the solid structures were positive), (b) peripheral (the periphery but not the center of the solid tumor structures was positive), or (c) peripheral and central (it surrounded the tumor structures and the cavities within the tumor structures).
Fluorescence in situ hybridization
FISH assay for the MYB gene was performed using the DAKO Histology FISH Accessory Kit (Agilent Technologies) according to the manufacturer’s instructions. Interphase molecular cytogenetic studies using a commercially available MYB break-apart (BA) probe (ZytoLight® SPEC MYB Dual Color BA Probe; Zytovision) were performed on a 4-µm paraffin-embedded section cut from whole section blocks. Nuclei were scored for nonrearranged patterns (red and green fusion signals) and rearranged and unbalanced patterns (split of red and green signals or extra single green signals) using a Nikon Eclipse 80i fluorescent microscope with appropriate filters. For each case, 100 nuclei were counted, and a case was declared positive if red and green signals were separated by a distance of >2 hybridization signals (split-apart signals) or if there were extra single green signals in more than 30% of the nuclei.
Array-CGH analysis
The areas of high tumor cellularity (>30%) were marked on a hematoxylin and eosin–stained histologic section and were bored on the FFPE tumor block. DNA was extracted using the QIAamp DSP DNA FFPE Tissue Kit (Qiagen) according to the manufacturer’s instructions. Genomic DNA was then quantified using a Nanodrop 1000 spectrophotometer (Thermo Fisher Scientific) and a Qubit fluorometer (Thermo Fisher Scientific) using the Qubit® dsDNA BR Assay Kit or the Qubit® dsDNA HS Assay Kit according to the manufacturer’s recommendations (Thermo Fisher Scientific). The tumors’ genomic profiles were obtained using the OncoScan™ CNV Assay (Thermo Fisher Scientific) according to the manufacturer’s instructions. Profiles were visualized and analyzed with the Chromosome Analysis Suite Software (Thermo Fisher Scientific).
To take into account the number and types of change in the copies of the tumor DNA, we calculated for each case the genomic index (GI), such that GI = A2/C, with A corresponding to the total number of gains and losses and C corresponding to the number of chromosomes involved [28].
Next-generation sequencing analysis
The Oncomine Comprehensive Assay v3 (Thermo Fisher Scientific) was used to perform the next-generation sequencing analysis (Supplementary Methods). The panel content covers 161 of the most relevant cancer driver genes, including increased kinase domain coverage and representation of genes involved in DNA repair (Supplementary Table 2).
RNA sequencing analysis
RNA extraction
FFPE tumors were analyzed by a pathologist to determine the tumor cell content. Samples were macrodissected to obtain a tumor cell content of at least 30%. RNA was extracted using the Maxwell-16 LEV RNA FFPE Purification kit (Promega) according to the manufacturer’s protocol. RNA quality and quantity were assessed by spectrophotometry with absorbance at 230, 260, and 280 nm.
RNA sequencing
rRNA-depleted RNA was used for the library preparation with a TruSeq RNA library Prep Kit (Illumina) according to the manufacturer’s instructions as described previously [29]. RNA sequencing was performed on a NextSeq500 device (Illumina). The libraries were sequenced with paired-end 76-bp “reads”.
Statistical analysis
RNA-seq expression analysis
Abundance of transcripts from RNA-seq data was quantified using the kallisto software. It is based on pseudoalignment for rapidly determining the compatibility of reads with targets, without the need for alignment [30]. The pseudoalignment was based on the reference human genome hg19. The transcript abundances are quantified by kallisto [30] in transcripts per million units. Quantification data were then imported using the Bioconductor tximport package [31] and transformed in a gene-level count matrix for use with DESeq2 [32]. Only protein coding genes were kept for further analysis. Calculations were performed using high-performance computing resources from Pôle des Systèmes d’Information et des Usages du Numérique—Centre de Calcul de l’Université de Bourgogne. Statistical analysis was performed using the open-source software R (https://cran.r-project.org/). Principal component analysis (PCA), an unsupervised method that allows the user to visualize the variability contained in the data, was performed on the RNA expression of coding genes for the 33 patients. Further, a differential analysis to select genes that have different expression profiles between the two groups was conducted using the DESeq2 R package. Genes with p values < 0.01 after false discovery rate (FDR) correction (Benjamini–Hochberg) and with log2 fold changes (log2FC) above 1 were kept, resulting in the selection of 549 genes. Genes are clustered by Pearson correlation, and log-expression values are mean-corrected by genes. Clustering was performed using the Ward method. To perform gene set enrichment analysis we used Enrichr tool [33, 34]. More precisely, we focused on the KEGG pathway databases (Kyoto Encyclopedia of Genes and Genomes), which integrates genomic, chemical, and systemic functional information. This gene set enrichment analysis allows to select KEGG pathways that are overrepresented in genes overexpressed, respectively, in conventional ACCs and solid ACCs.
Fusion gene detection
To identify candidate fusion transcripts from RNA-seq data, we used the STAR-Fusion tool [35], based on the STAR aligner. STAR-Fusion performs a fast mapping of fusion evidence to reference transcript structure annotations and filters likely artifacts to report accurate fusion predictions. The reference genome was hg19.
Results
Clinical characteristics
Clinical features, therapeutic, and follow-up data are summarized in Table 1. All cases were women, and the mean age at diagnosis was 62 years (48–83 years) in conventional ACCs and 68 years (33–84 years) in solid ACCs. The average tumor size was 27 mm (15–70 mm) in conventional ACCs and 25 mm (12–70 mm) in solid ACCs. The majority of patients were treated by lumpectomy: 11/15 (73.3%) of conventional ACCs and 11/17 (64.7%) of solid ACCs, associated with axillary lymph node dissection or sentinel lymph node mostly in solid ACCs [15/16 (93%) of patients] compared with conventional ACCs [10/15 (67%) of patients]. One patient with conventional ACC had neoadjuvant chemotherapy due to the large size of the tumor (case 10), and three patients with solid ACCs had adjuvant chemotherapy. Mean follow-up time was higher in conventional ACCs [70,6 months (12–348 months)] than in solid ACCs [63.8 months (12–228 months)]. Two patients with conventional-type ACCs had one local recurrence (cases 3 and 5). One patient with solid-type ACC had a lymph node metastasis (case 26), and three others had one or two local recurrences (cases 18, 19, and 31).
Morphologic features and correlation by immunohistochemistry
Morphologic and immunohistochemical characteristics are illustrated in Fig. 1 and Table 2. By definition, all conventional-type ACCs displayed a dual epithelial/myoepithelial cell population. These tumors were grade 1 or 2 according to the E&E and Ro grading systems, whereas solid-type ACCs were E&E grade 2 or 3 and Ro grade III, except for one tumor (case 27) which was Ro grade I, displaying a cribriform pattern but without a dual population (Supplementary Fig. 1). Tumor cell necrosis was present in two conventional-type ACCs and six solid-type ACCs (Supplementary Fig. 1). Only one tumor (case 30, in the solid-type ACC group) had perineural invasion (Supplementary Fig. 1).

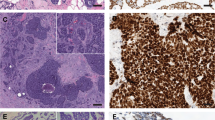
Conventional-type ACC with MYB rearrangement (case 8) (a–e), without MYB rearrangement (case 11) (f–j), and solid-type ACC (case 30) (k–o). Morphology (a, f, k) (hematoxylin & eosin stains) showing a cribriform growth pattern in conventional-type ACCs and a solid growth pattern in solid-type ACC. p63 immunostaining (b, g, l) is positive in the myoepithelial cells in conventional-type ACCs and negative in this solid-type ACC. MYB immunostaining (c, h, m) is strongly positive in myoepithelial cells in conventional-type ACCs and diffusely positive in this solid-type ACC. CK7 immunostaining (d, i, n) is positive in the epithelial cells of conventional-type ACCs and highlights intercalated duct-like structures embedded in the tumor islands in this solid-type ACC. NICD1 nuclear immunostaining (e, j, o) is positive in the epithelial cells of conventional-type ACCs, and diffuse in this solid-type ACCs.
None of the 33 ACCs-expressed ER or PR and all cases were HER2 negative. p63 was expressed in a peripheral and central pattern in conventional-type ACCs. p63 was totally negative in eight solid-type ACCs and positive in the remaining nine. Of these remaining nine, six tumors displayed a peripheral pattern, two tumors expressed a peripheral and central pattern, and one tumor displayed a diffuse staining pattern (case 29) (Supplementary Fig. 1). CK7-positive epithelial cells were observed in conventional-type ACCs (median H-score: 210), admixed with the p63-positive myoepithelial cells. CK7-positive epithelial cells were diffusely positive in ten solid-type ACCs (median H-score: 275) and highlighted intercalated duct-like structures embedded in the tumor islands within seven tumors (median H-score: 150). MYB expression was observed in the two groups with a median H-score of 165 (0–240) in conventional-type ACCs and 210 (0–240) in solid-type ACCs. The expression pattern was stronger in the myoepithelial cells than in the luminal cells in conventional-type ACCs, whereas solid-type ACCs exhibited a more diffuse MYB staining pattern with an H-score > 150 in 14/17 (82%) of cases (Fig. 1). Nine tumors in the two groups showed only peripheral staining (Supplementary Fig. 1). Focal and weak MYB expression was observed in only 7/204 (3%) of non-ACC TNBCs.
Fluorescence in situ hybridization MYB break apart
MYB FISH status was known at the start of the study for the conventional-type ACCs and was present in 9/16 cases. MYB rearrangement was detected in 3/16 (19%) of solid-type ACCs (cases 20, 25, and 27).
Whole transcriptome analysis
RNA-seq results were generated for all tumors. Figure 2a shows the result of the PCA performed on the RNA expression of coding genes for the 33 patients. Different gene expression profiles were observed between the two groups. Furthermore, we performed a differential analysis to select protein coding genes that have different expression profiles between conventional and solid-type ACCs. By using strict, conservative criteria (FDR < 0.01; log2FC > |1|), we selected 549 genes with a highly significant differential expression (Supplementary Table 3), including 324 genes (62%) upregulated in conventional ACCs and 225 genes (38%) upregulated in solid ACCs. Results are presented in a heatmap (Fig. 2b). Using Enrichr and Kegg Pathway computational analysis, we identified PI3K-Akt and focal adhesion signaling pathways as significantly overexpressed in conventional-type ACCs compared with solid-type ACCs, whereas these last significantly overexpressed the nitrogen metabolism pathway (Fig. 2c, d).

a Principal component analysis of coding gene RNA expression in our series shows that conventional-type ACCs (blue) and solid-type ACCs (green) exhibit different gene expression profiles. b Heatmap representing the 549 differentially selected genes in rows and patients in columns. Under-expressed genes are in green and overexpressed genes in red. Conventional and solid-type ACCs were relatively uniformly separated, except cases 16 and 29. EnrichR Kegg pathways representation based on the 549 differential genes (c) 324 genes were overexpressed in conventional-type ACCs with PI3K-Akt signaling pathway as the first overexpressed Kegg pathway (d) 225 genes were overexpressed in solid-type ACCs with Nitrogen metabolism as the first overexpressed Kegg pathway.
Fusion gene detection
Fusion gene detection was interpretable in 28 cases by Oncomine and 28 cases by transcriptome analysis. Candidate fusion transcripts are listed in Table 2. The nine conventional-type ACCs that featured a rearrangement of MYB by FISH BA had a MYB–NFIB fusion transcript with Oncomine. Not all MYB–NFIB fusion transcripts were found in conventional ACCs by RNA-seq (cases 4, 5, and 7) because the altered RNA quality of these samples did not allow sufficient depth of reading for the detection of fusions. Oncomine was more sensitive to detection of MYB–NFIB because it is a targeted technique by PCR with specific primers. RNA-seq analysis did not confirm FISH BA MYB rearrangement in three cases (cases 20, 25, and 27). Case 20 was not interpretable by Oncomine or RNA-seq. Cases 25 and 27 were interpretable by at least one technique, therefore we concluded there to be an absence of a MYB–NFIB fusion gene in these cases. Furthermore, we performed a FISH dual fusion MYB–NFIB analysis on these two cases to check if MYB’s partner was NFIB. The result was negative for both cases.
Five cases had a RN7SKP203-EIF4G2 fusion transcript, concomitant with a MYB–NFIB fusion transcript for three conventional-type ACCs (cases 5, 6, and 9) and concomitant with a MYB rearrangement by FISH in one solid-type ACC (case 27).
Array-CGH analysis
Array comparative genomic hybridization (a-CGH) was interpretable in all cases. Profiles with the GI are available in the Supplementary Fig. 2. The main recurrent alterations were observed in 15 tumors (Fig. 3). They included loss on 12q (6/33, 18%) seen in five conventional-type ACCs and only one solid-type ACC (case 29), loss on 14p (7/33, 21%) observed observed more frequently in solid-type ACCs, and loss on 17p (6/33, 18%) followed by a gain on 17p (5/33, 15%) in the two groups. Eight tumor profiles were totally flat or showed only one alteration (cases 1, 4, 7, 11, 12, 13, 25, and 27) (Supplementary Fig. 2). Case 31, a solid-type ACC, had the greatest instability with a GI at 28.5. The remaining tumors showed rearranged profiles without any recurrent alteration with a GI ranging from 2 to 15.12 (Supplementary Fig. 2). Case 29 showed a CCND1 locus amplification (on chromosome 11) (Fig. 3) associated with strong cyclin D1 overexpression by IHC (Supplementary Fig. 1).

The main alterations involved chromosomes 12, 14, and 17. Losses are represented in blue and gains in red. On chromosome 12, minimal common area deleted in cases 1, 2, 5, 7, 8, and 29 concerned 4.1 Mb and involved 110 genes. On chromosome 14, minimal common area deleted in cases 10, 16, 18, 21, 22, 31, and 32 concerned 6.3 Mb and involved 22 genes. On chromosome 17, minimal common area deleted in cases 5, 8, 9, 29, 30 and 31, concerned 10.8 Mb and involved 291 genes. On this same chromosome, cases 5, 8, 9, 29, and 30 also had a gain. Case 29 was the only case having amplification of the CCND1 locus seen on chromosome 11.
Single nucleotide variant analysis
SNV analysis was interpretable in 31 cases, and all mutations are summarized in Supplementary Table 4. Cases 1 and 17 were not interpretable, and cases 12 and 18 had no mutation. The most frequently mutated genes in the series were CDK12 in 11/29 (38%) of cases, NOTCH1 in 8/29 (28%) of cases, CREBBP in 5/29 (17%) of cases and ARID1A in 4/29 (14%) of cases (Fig. 4a). The comparative oncoplot (Fig. 4b) showed that the NOTCH genes were more frequently mutated in solid-type ACCs than in conventional-type ACCs. In conventional ACC, there were two NOTCH1 missense mutations in a single tumor (case 2), whereas in solid ACC, 14 NOTCH somatic mutations were identified in eight tumors (8/16, 50%). NOTCH1 had four truncating mutations [p. (Val2476*) case 23, p. (Leu2419Hisfs* 10) case 28, p.(Ser2467*) and p.(Asn914Thrfs*189) in case 30]. NOTCH2 had two truncating mutations [p.(Gln2325*) case 20 and p.(Glu1148*) case 27] (Table 2). Four of these mutations were in exon 34, a mutational hotspot identified in human cancers, leading to deletion of the C terminal NOTCH PEST domain and predicted to induce NOTCH gain of function. The seven other NOTCH1 mutations and the only NOTCH3 mutation were missense mutations (cases 19, 20, 22, 27, and 32) (Table 2). These mutations were located in the EGF-like extracellular domain of the NOTCH receptor that is not subjected to cleavage activation by γ-secretase, nor to degradation of the protein regulated by the PEST domain. In order to correlate NOTCH mutations and their impact on Notch pathway activation, we performed NICD1 immunostaining, nuclear NICD1 localization being an established marker of Notch pathway activation [36] (Table 2 and Supplementary Fig. 3). Nuclear NICD1 expression was present in 30 tumors (30/33, 90%) with a median H-score of 80 (30–120) in conventional-type ACCs, where expression was confined to the epithelial cells (Fig. 1) and with a median H-score of 60 (0–280) in solid-type ACCs with a diffuse expression when it was positive (Fig. 1). The three NICD1-negative cases were in solid-type ACCs (cases 18, 19, and 33) and were true negative with an internal positive control (endothelial cells). The NICD1 staining pattern was more diffuse and stronger in cases with NOTCH1 gain of function mutations in exon 34 (cases 23, 28, and 30; Table 2 and Supplementary Fig. 3) and in cases with NOTCH2 gain of function mutation in exons 34 and 21 (cases 20 and 27, respectively; Table 2 and Supplementary Fig. 3). These five tumors had a median NICD1 H-score of 150 (110–280). Case 32 had a missense mutation of NOTCH3 in exon 2 with a NICD1 H-score of 160. Cases 2 and 19 which had a NOTCH1 mutation in exon 9, had a weak NICD1 expression, suggesting that this mutation was not activating. In the TNBC TMA, NICD1 was expressed by 11/204 (5%) of cases. Regarding other SNVs, CREBBP mutations involved in chromatin state regulation were exclusively found in solid-type ACCs [5/16 (31%) of cases]. Three cases had a CREBBP nonsense mutation [p.(Gln1292*); p.(Gln357*); p.(Gln1910*)], one case had a frameshift mutation [p.(Glu1149Alafs*21)], and the last case had a missense mutation [p.(Pro2363Leu)]. Only one TP53 mutation was observed in the series, and it concerned a splice site mutation (intron 3) in a solid-type ACC (Case 31). Somatic mutation genes were classified by five main pathways, represented in Fig. 5: DNA repair, receptor tyrosine kinase, chromatin modifications, Notch pathway, and Other. The Notch pathway was significantly more represented in solid ACCs than in conventional ACCs, according to Fischer test (p value = 0.04). Genes with molecular aberration classified by their involved pathway are listed in Supplementary Table 5.

Oncoplots representing recurrent mutations observed in 24 out of 29 cases of breast ACC with mutations (a) in all samples and (b) in a separate oncoplot for each tumor group. Each line corresponds to a gene presenting a mutation (classified into frameshift deletions or insertions, nonsense, missense, and multihit mutations) and each column corresponds to a patient presenting this mutated gene. Side and top barplots on a represent the number of mutations per type, respectively, by gene and by patient.

Each column corresponds to a pathway and each line to a patient (cases 1 to 33). The color gradient indicates the number of genes of this pathway presenting a mutation.
Discussion
The aim of this study was to better characterize, through a comprehensive pathological and molecular analysis, the solid-type of ACC, a rare tumor initially morphologically described by Shin and Rosen [22]. To the best of our knowledge, our series of solid-type breast ACCs is the largest reported to date.
We demonstrated through this study that these tumors represent a different subgroup in the ACC family. As previously described in cases of salivary gland and breast ACC [37, 38], solid-type ACCs in our series displayed an exclusive or predominant epithelial cell population with decreased myoepithelial differentiation, while also demonstrating MYB protein overexpression similar to the more common type of ACC. FISH MYB rearrangements were present in 3/16 (19%) of our solid-type breast ACCs, similar to the results described by d’Alfonso et al. [15]. However, MYB immunohistochemical expression was present in most solid breast ACCs, underlining alternative activating mechanisms of the MYB pathway. Poling et al. have described MYB immunohistochemical detection as a relatively sensitive and specific marker of ACCs if it is strong and diffuse [18]. We found weak MYB expression in only 3% of the tumors of our TNBC TMA, confirming the idea that MYB immunostaining in TNBC is ACC specific. The peripheral staining of MYB observed in some tumors in the two groups could be explained by the short half-life of MYB as described by Kim [20] (Supplementary Fig. 1). In our series, we did not observe other MYB family rearrangements, in particular MYBL1, as previously described by Kim et al. in breast [20] and by Mitani et al. in salivary gland ACCs [19]. A new fusion gene RN7SKP203-EIF4G2 was found in five tumors and has not been previously described. The aberrant editing of EIF4G2 could contribute to the potent oncogenesis [39]. Transcriptional analysis showed that solid-type ACC constitutes a distinct entity, as seen by PCA, the heatmap and the expression of signaling pathways (Fig. 2). Genome profiling showed an overall low level of genomic rearrangements in conventional ACCs and increased heterogeneous genomic rearrangements in solid ACCs, but not to the extent of what is commonly observed in TNBC [40]. We notably observed recurrent alterations on chromosomes 12, 14, and 17, as seen by Wetterskog et al. [16] and Martelotto et al. [2]. By SNV analysis, solid-type ACCs, significantly differed from conventional-type ACCs, by a higher mutation rate in the Notch pathway. This signaling pathway is known to be mutated in many human diseases. Three tumors had a NOTCH1 truncating mutation in exon 34, already described in T-cell acute lymphoblastic leukemia as a mutational hotspot [41]. NOTCH1 p.(Ser2467*) mutation has also been described in salivary solid-type ACCs by Ferrarotto et al. [42]. Case 27 showed a missense NOTCH1 mutation p.(Ala2044Thr), previously described in squamous cell carcinoma [43] and case 20 had a NOTCH2 truncating mutation [p.(Gln2325*)], observed in splenic marginal zone lymphoma [44]. NOTCH activation downregulates p63 and promotes differentiation toward the luminal epithelial fate which could explain loss of p63 immunostaining in some tumors and the progression towards a solid histology [21, 45, 46]. Furthermore, diffuse nuclear NICD1 typically correlated with areas of solid epithelial growth pattern in our series. Solid-type ACCs also differed from conventional-type ACCs by the presence of inactivating CREBBP mutations, a chromatin remodeling gene, paralog of EP300 [47]. NOTCH1 and EP300 mutations have been previously described in a case of ACC with a high-grade TNBC component [37]. CREBBP inactivation is also known to be a marker of aggressiveness in bladder carcinoma [48].
Conversely to TNBC NST, where 80% of tumors harbor a TP53 mutation [49] only one solid-type ACC tumor in our series had a TP53 mutation (1/16; 6%), and it was the same tumor that had the most rearranged profile by a-CGH (case 31) (Supplementary Fig. 2). We still think that this case is a genuine solid-type ACC because it displayed the characteristic basaloid morphological features and overexpressed MYB protein (Supplementary Fig. 1). Clinically, solid-type ACC have already been described as more aggressive than conventional ACC, with more frequent axillary and visceral metastases, especially to the lung [15, 22, 50,51,52,53]. In this series, we only observed one lymph node metastasis out of the 15 axillary lymph node explorations that were documented, three local relapses and no visceral metastasis; however, follow-up may not have been long enough to confirm previously published data. However, the vast majority of TNBC NST recurrences occur within the first five years pointing to the fact that even if solid-type ACC appear to be more aggressive than conventional ACC, they are not as aggressive as TNBC NST.
There are some limitations to this study. Due to the rarity of ACCs and even more of solid-type ACC in the breast, the size of our series is relatively small. Furthermore, we did not have any frozen tumor material, so the extent of our molecular analyses, in particular for the search of fusion genes, did not allow sufficient depth of reading for the detection of fusions due to the altered RNA quality of some samples. Given the small numbers and the variations in treatment any relationship of the outcomes to group categories should be interpreted with caution. Almost all patients with TNBC NST are offered chemotherapy which raises the question if the same should be done for solid-type ACC of the breast. The data in our study is insufficient for making a recommendation for or against chemotherapy in this subtype. However, salivary gland ACC is known not to be very chemo sensitive [14] so we wonder if this would be an effective treatment in solid-type breast ACC. Given the molecular alterations observed, enrolling patients in clinical trials exploring novel targeted agents would be the best alternative.
To conclude, we demonstrate through a comprehensive pathological and molecular analysis that solid-type ACC is different from both conventional-type ACC and the more common basal-like TNBC NST. Solid-type ACC display a basaloid morphology with an exclusive or predominant epithelial cell population associated with frequent MYB protein overexpression. Solid-type ACC is a distinct entity, with morphological and molecular criteria of aggressiveness, and is enriched in activating NOTCH and CREBBP gene mutations. In the era of targeted therapies, Notch signaling inhibitors are currently under investigation in clinical trials and may be effective in patients with NOTCH gene activating mutations [54].
References
Sapino A, Sneige N, Eusebi V. Special subtypes. In: Lakhani S, Ellis I, Schnitt S, Tan PH, van de Vijver MJ, editors. WHO classification of tumours of the breast. 4th ed. Lyon, France: International Agency for Research on Cancer; 2012. p. 56–57.
Martelotto LG, De Filippo MR, Ng CKY, Natrajan R, Fuhrmann L, Cyrta J, et al. Genomic landscape of adenoid cystic carcinoma of the breast. J Pathol. 2015;237:179–89.
Moskaluk CA. Adenoid cystic carcinoma: clinical and molecular features. Head Neck Pathol. 2013;7:17–22.
Dillon PM, Chakraborty S, Moskaluk CA, Joshi PJ, Thomas CY. Adenoid cystic carcinoma: a review of recent advances, molecular targets, and clinical trials. Head Neck. 2016;38:620–7.
Di Palma S, Fehr A, Danford M, Smith C, Stenman G. Primary sinonasal adenoid cystic carcinoma presenting with skin metastases genomic profile and expression of the MYB-NFIB fusion biomarker. Histopathology. 2014;64:453–5.
Roden AC, Greipp PT, Knutson DL, Kloft-Nelson SM, Jenkins SM, Marks RS, et al. Histopathologic and cytogenetic features of pulmonary adenoid cystic carcinoma. J Thorac Oncol. 2015;10:1570–5.
Halat SK, MacLennan GT. Adenoid cystic/basal cell carcinoma of the prostate. J Urol. 2008;179:1576.
von Holstein SL, Fehr A, Persson M, Therkildsen MH, Prause JU, Heegaard S, et al. Adenoid cystic carcinoma of the lacrimal gland: MYB gene activation, genomic imbalances, and clinical characteristics. Ophthalmology. 2013;120:2130–8.
Lloyd S, Yu JB, Wilson LD, Decker RH. Determinants and patterns of survival in adenoid cystic carcinoma of the head and neck, including an analysis of adjuvant radiation therapy. Am J Clin Oncol. 2011;34:76–81.
Arpino G, Clark GM, Mohsin S, Bardou VJ, Elledge RM. Adenoid cystic carcinoma of the breast: molecular markers, treatment, and clinical outcome. Cancer. 2002;94:2119–27.
Millar B-AM, Kerba M, Youngson B, Lockwood GA, Liu F-F. The potential role of breast conservation surgery and adjuvant breast radiation for adenoid cystic carcinoma of the breast. Breast Cancer Res Treat. 2004;87:225–32.
Xu B, Drill E, Ho A, Ho A, Dunn L, Prieto-Granada CN, et al. Predictors of outcome in adenoid cystic carcinoma of salivary glands: a clinicopathologic study with correlation between MYB fusion and protein expression. Am J Surg Pathol. 2017;41:1422–32.
Marchio C, Weigelt B, Reis-Filho JS. Adenoid cystic carcinomas of the breast and salivary glands (or “The strange case of Dr Jekyll and Mr Hyde” of exocrine gland carcinomas). J Clin Pathol. 2010;63:220–8.
Chae YK, Chung SY, Davis AA, Carneiro BA, Chandra S, Kaplan J, et al. Adenoid cystic carcinoma: current therapy and potential therapeutic advances based on genomic profiling. Oncotarget. 2015;6:37117–34.
D’Alfonso TM, Mosquera JM, MacDonald TY, Padilla J, Liu Y-F, Rubin MA, et al. MYB-NFIB gene fusion in adenoid cystic carcinoma of the breast with special focus paid to the solid variant with basaloid features. Hum Pathol. 2014;45:2270–80.
Wetterskog D, Lopez-Garcia MA, Lambros MB, A’Hern R, Geyer FC, Milanezi F, et al. Adenoid cystic carcinomas constitute a genomically distinct subgroup of triple-negative and basal-like breast cancers. J Pathol. 2012;226:84–96.
Persson M, Andren Y, Mark J, Horlings HM, Persson F, Stenman G. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc Natl Acad Sci USA. 2009;106:18740–4.
Poling JS, Yonescu R, Subhawong AP, Sharma R, Argani P, Ning Y, et al. MYB labeling by immunohistochemistry is more sensitive and specific for breast adenoid cystic carcinoma than MYB labeling by FISH. Am J Surg Pathol. 2017;41:973–9.
Mitani Y, Liu B, Rao PH, Borra VJ, Zafereo M, Weber RS, et al. Novel MYBL1 gene rearrangements with recurrent MYBL1-NFIB fusions in salivary adenoid cystic carcinomas lacking t(6;9) translocations. Clin Cancer Res. 2016;22:725–33.
Kim J, Geyer FC, Martelotto LG, Ng CK, Lim RS, Selenica P, et al. MYBL1 rearrangements and MYB amplification in breast adenoid cystic carcinomas lacking the MYB-NFIB fusion gene. J Pathol. 2018;244:143–50.
Drier Y, Cotton MJ, Williamson KE, Gillespie SM, Ryan RJH, Kluk MJ, et al. An oncogenic MYB feedback loop drives alternate cell fates in adenoid cystic carcinoma. Nat Genet. 2016;48:265–72.
Shin SJ, Rosen PP. Solid variant of mammary adenoid cystic carcinoma with basaloid features: a study of nine cases. Am J Surg Pathol. 2002;26:413–20.
Ro JY, Silva EG, Gallager HS. Adenoid cystic carcinoma of the breast. Hum Pathol. 1987;18:1276–81.
Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363:1938–48.
Laurent E, Begueret H, Bonhomme B, Veillon R, Thumerel M, Velasco V, et al. SOX10, GATA3, GCDFP15, androgen receptor, and mammaglobin for the differential diagnosis between triple-negative breast cancer and TTF1-negative lung adenocarcinoma. Am J Surg Pathol. 2019;43:293–302.
Wolff AC, Hammond MEH, Allison KH, Harvey BE, Mangu PB, Bartlett JMS, et al. Human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Focused Update. J Clin Oncol. 2018;36:2105–22.
Dowsett M, Nielsen TO, A’Hern R, Bartlett J, Coombes RC, Cuzick J, et al. Assessment of Ki67 in breast cancer: recommendations from the International Ki67 in Breast Cancer working group. J Natl Cancer Inst. 2011;103:1656–64.
Lagarde P, Perot G, Kauffmann A, Brulard C, Dapremont V, Hostein I, et al. Mitotic checkpoints and chromosome instability are strong predictors of clinical outcome in gastrointestinal stromal tumors. Clin Cancer Res. 2012;18:826–38.
Alsadoun N, MacGrogan G, Truntzer C, Lacroix-Triki M, Bedgedjian I, Koeb M-H, et al. Solid papillary carcinoma with reverse polarity of the breast harbors specific morphologic, immunohistochemical and molecular profile in comparison with other benign or malignant papillary lesions of the breast: a comparative study of 9 additional cases. Mod Pathol. 2018;31:1367–80.
Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34:525–7.
Soneson C, Love MI, Robinson MD. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 2015;4:1521.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Burstein MD, Tsimelzon A, Poage GM, Covington KR, Contreras A, Fuqua SAW, et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin Cancer Res. 2015;21:1688–98.
Franks JM, Cai G, Whitfield ML. Feature specific quantile normalization enables cross-platform classification of molecular subtypes using gene expression data. Bioinformatics. 2018;34:1868–74.
Haas B, Dobin A, Stransky N, Li B, Yang X, Tickle T, et al. STAR-fusion: fast and accurate fusion transcript detection from RNA-Seq. bioRxiv. 2017. http://biorxiv.org/lookup/doi/10.1101/120295. Accessed 15 Jun 2019.
Kluk MJ, Ashworth T, Wang H, Knoechel B, Mason EF, Morgan EA, et al. Gauging NOTCH1 activation in cancer using immunohistochemistry. PLoS ONE. 2013;8:e67306.
Fusco N, Geyer FC, De Filippo MR, Martelotto LG, Ng CKY, Piscuoglio S, et al. Genetic events in the progression of adenoid cystic carcinoma of the breast to high-grade triple-negative breast cancer. Mod Pathol. 2016;29:1292–305.
Bell D, Bell AH, Bondaruk J, Hanna EY, Weber RS. In-depth characterization of the salivary adenoid cystic carcinoma transcriptome with emphasis on dominant cell type. Cancer. 2016;122:1513–22.
Lee T, Pelletier J. Eukaryotic initiation factor 4F: a vulnerability of tumor cells. Future Med Chem. 2012;4:19–31.
Curtis C, Shah SP, Chin S-F, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–52.
Weng AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–71.
Ferrarotto R, Mitani Y, Diao L, Guijarro I, Wang J, Zweidler-McKay P, et al. Activating NOTCH1 mutations define a distinct subgroup of patients with adenoid cystic carcinoma who have poor prognosis, propensity to bone and liver metastasis, and potential responsiveness to Notch1 inhibitors. J Clin Oncol. 2017;35:352–60.
South AP, Purdie KJ, Watt SA, Haldenby S, den Breems N, Dimon M, et al. NOTCH1 mutations occur early during cutaneous squamous cell carcinogenesis. J Investig Dermatol. 2014;134:2630–8.
Kiel MJ, Velusamy T, Betz BL, Zhao L, Weigelin HG, Chiang MY, et al. Whole-genome sequencing identifies recurrent somatic NOTCH2 mutations in splenic marginal zone lymphoma. J Exp Med. 2012;209:1553–65.
Nguyen B-C. Cross-regulation between Notch and p63 in keratinocyte commitment to differentiation. Genes Dev. 2006;20:1028–42.
Yalcin-Ozuysal Ö, Fiche M, Guitierrez M, Wagner K-U, Raffoul W, Brisken C. Antagonistic roles of Notch and p63 in controlling mammary epithelial cell fates. Cell Death Differ. 2010;17:1600–12.
Attar N, Kurdistani SK. Exploitation of EP300 and CREBBP lysine acetyltransferases by cancer. Cold Spring Harb Perspect Med. 2017;7:a026534.
Duex JE, Swain KE, Dancik GM, Paucek RD, Owens C, Churchill MEA, et al. Functional impact of chromatin remodeling gene mutations and predictive signature for therapeutic response in bladder cancer. Mol Cancer Res. 2018;16:69–77.
Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70.
Defaud-Hénon F, Tunon-de-Lara C, Fournier M, Marty M, Velasco V, de Mascarel I, et al. [Adenoid cystic carcinoma of the breast: clinical, histological and immunohistochemical characterization]. Ann Pathol. 2010;30:7–16.
Fukuoka K, Hirokawa M, Shimizu M, Sadahira Y, Manabe T, Kurebayashi J, et al. Basaloid type adenoid cystic carcinoma of the breast. APMIS. 1999;107:762–6.
Mastropasqua MG, Maiorano E, Pruneri G, Orvieto E, Mazzarol G, Vento AR, et al. Immunoreactivity for c-kit and p63 as an adjunct in the diagnosis of adenoid cystic carcinoma of the breast. Mod Pathol. 2005;18:1277–82.
Foschini MP, Rizzo A, De Leo A, Laurino L, Sironi M, Rucco V. Solid variant of adenoid cystic carcinoma of the breast: a case series with proposal of a new grading system. Int J Surg Pathol. 2016;24:97–102.
Massard C, Azaro A, Soria J-C, Lassen U, Le Tourneau C, Sarker D, et al. First-in-human study of LY3039478, an oral Notch signaling inhibitor in advanced or metastatic cancer. Ann Oncol. 2018;29:1911–7.
Acknowledgements
We wish to thank patients for their support in the realization of this study. The authors thank Pippa McKelvie-Sebileau for medical editing in English, Virginie Duvignau for art editing, and Dorothée Quincy for manuscript submission.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics
This study was approved by the Institut Bergonié institutional review board and data collection was in compliance with the French “Jardé” law—reference methodology 004.
Informed consent
Informed consent was obtained from all study participants, including permission to use their tumor material.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Massé, J., Truntzer, C., Boidot, R. et al. Solid-type adenoid cystic carcinoma of the breast, a distinct molecular entity enriched in NOTCH and CREBBP mutations. Mod Pathol 33, 1041–1055 (2020). https://doi.org/10.1038/s41379-019-0425-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-019-0425-3
This article is cited by
-
A novel EWSR1-MYB fusion in an aggressive advanced breast adenoid cystic carcinoma with mixed classical and solid-basaloid components
Virchows Archiv (2023)
-
Current understanding of adenoid cystic carcinoma in the gene expression and targeted therapy
Holistic Integrative Oncology (2023)
-
OncoSNIPE® Study Protocol, a study of molecular profiles associated with development of resistance in solid cancer patients
BMC Cancer (2022)
-
Comparison of the mutational profiles of neuroendocrine breast tumours, invasive ductal carcinomas and pancreatic neuroendocrine carcinomas
Oncogenesis (2022)
-
Distinct clinicopathological and genomic features in solid and basaloid adenoid cystic carcinoma of the breast
Scientific Reports (2022)
