
Abstract
Subsets of high-grade gliomas, including glioblastoma (GBM), are known to utilize the alternative lengthening of telomeres (ALT) pathway for telomere length maintenance. However, the telomere maintenance profile of one subtype of GBM—giant cell GBM—has not been extensively studied. Here, we investigated the prevalence of ALT, as well as ATRX and SMARCAL1 protein loss, in a cohort of classic giant cell GBM and GBM with giant cell features. To determine the presence of ALT, a telomere-specific fluorescence in situ hybridization assay was performed on 15 cases of classic giant cell GBM, 28 additional GBMs found to have giant cell features, and 1 anaplastic astrocytoma with giant cell features. ATRX, SMARCAL1, and IDH1 protein status were assessed in a proportion of cases by immunohistochemistry and were compared to clinical-pathologic and molecular characteristics. In the overall cohort of 44 cases, 19 (43%) showed evidence of ALT. Intriguingly, of the ALT-positive cases, only 9 (47.4%) displayed loss of the ALT suppressor ATRX by immunohistochemistry. Since inactivating mutations in SMARCAL1 have been identified in ATRX wild-type ALT-positive gliomas, we developed an immunohistochemistry assay for SMARCAL1 protein expression using genetically validated controls. Of the 19 ALT-positive cases, 6 (31.5%) showed loss or mis-localization of SMARCAL1 by immunohistochemistry. Of these cases, four retained ATRX protein expression, while two cases also displayed ATRX loss. Additionally, we assessed five cases from which multiple temporal samples were available and ALT status was concordant between both tumor biopsies. In summary, we have identified a subset of giant cell GBM that utilize the ALT telomere maintenance mechanism. Importantly, in addition to ATRX loss, ALT-positive tumors harboring SMARCAL1 alterations are prevalent in giant cell GBM.
Similar content being viewed by others

Introduction
Glioblastoma (GBM) is the most commonly diagnosed primary brain tumor, with an extremely poor 5-year survival rate of about five percent [1]. The giant cell GBM variant (IDH wild type) is characterized histologically by the presence of large, bizarre multinucleated cells and a dense reticulin network [2], making up roughly five percent or less of all GBM cases [3, 4]. Giant cell GBM tends to be more circumscribed than conventional GBM, which makes it more amenable to gross total resection, and may be associated therefore with a better prognosis [5]. These tumors also have a high frequency of TP53 mutations [6]. A subset of GBMs may also contain pleomorphic giant cells, although this may not be the dominant feature.
All cancers must activate a mechanism to maintain telomere lengths above a critical threshold in order to maintain an unlimited replicative potential [7]. While most cancers upregulate the telomerase reverse transcriptase to lengthen telomeres, subsets of high-grade glial tumors, including GBM, have shown a propensity toward the alternative lengthening of telomeres (ALT) telomere maintenance mechanism, which has been associated with a more favorable prognosis [8,9,10,11,12,13]. ALT is a telomerase-independent telomere maintenance mechanism that is thought to involve a recombination-mediated process similar to homology-directed DNA repair [14]. ALT can be readily identified in fixed cancer specimens through the presence of large, ultrabright telomeric DNA foci measured by telomere-specific fluorescence in situ hybridization (FISH) [12, 15].
ALT-positive tumors display a distinct genetic profile. Most commonly, ALT-positive gliomas harbor inactivating mutations in the gene ATRX leading to loss of protein expression. ATRX is a chromatin remodeling protein [12, 16,17,18], which forms a histone chaperone complex with DAXX that normally is responsible for the replication-independent deposition of the histone variant H3.3 in repetitive heterochromatin, particularly at the pericentromeric and telomeric regions [19,20,21,22]. Given the near perfect concordance between ALT-positivity and ATRX loss in gliomas, it has been suggested that ATRX acts as an ALT suppressor [23, 24]. Importantly, these ALT-positive tumors lack mutations in the promoter of the gene TERT, which encodes the telomerase reverse transcriptase, and leads to the more common mechanism of telomere maintenance via telomerase activation [25]. Furthermore, ALT-positive gliomas commonly harbor mutations in TP53 and IDH1 [8, 11], as well as in histone H3 (K27M or G34) [26, 27] or NF1 [28,29,30] in subsets of cases. Interestingly, genetic profiling of giant cell GBMs revealed IDH1 mutations in 5.3% and TERT promoter mutations in 25% of cases analyzed, while 19% showed loss of ATRX immunoreactivity [31, 32]. In addition, inactivating mutations in SMARCAL1, an ATP-dependent helicase involved in resolving stalled replication forks, were identified in a subset of ALT-positive ATRX wild-type gliomas, suggesting that SMARCAL1, like ATRX and DAXX, may function as an ALT-suppressor [33].
Despite the known importance of ALT in gliomas, as a whole, ALT has not been investigated in the limited numbers of giant cell GBMs analyzed, to date. Herein, we have investigated the prevalence of ALT, as well as alterations in ATRX and SMARCAL1, in giant cell GBM.
Materials and methods
Case selection
Cases of giant cell GBM or GBMs with giant cells were retrieved from the pathology files at Johns Hopkins Hospital in the years 1989–2017. All histologic slides and available immunostains were systematically re-reviewed by a neuropathologist (FJR). Tumors were classified as giant cell GBM using current WHO criteria (2016 WHO). In brief, tumors were designated giant cell GBM if they had a predominance of bizarre multinucleated glial cells (>50% of tumors areas). Cases with a focal giant cell pattern (>20%) but that was not dominant were classified as having giant cell features (Fig. 1).

Classic giant cell glioblastomas (GC-GBM) were characterized by bizarre pleomorphic cells encompassing the majority of the specimen, had strong p53 positivity, retained ATRX and lacked IDH1 mutations. Glioblastomas with giant cell features (GBM-GC Fx) were characterized by the presence of large pleomorphic cells (arrows), but encompassing only a subset of tumor. A small proportion of tumors with ALT had IDH1 mutations (GBM-GC IDH1). For all panels, original magnification ×400. For the inset highlighting ATRX loss, original magnification ×600.
Telomere-specific fluorescence in situ hybridization (FISH)
ALT status was interpreted using previously published criteria and was characterized by the presence of distinct large telomeric DNA signals using a telomere-specific FISH assay [12, 13, 28]. Briefly, slides of whole sections were heated and flooded with xylenes to remove paraffin, and then subjected to an ethanol gradient to hydrate the tissue. Antigen retrieval was performed using citrate, pH 6 (Cat# H-3300, Vector Laboratories, Burlington, CA). Slides were then re-hydrated and exposed to a Cy3-labeled peptide nucleic acid (PNA) probe complementary to telomeric DNA (Cat# F1002, Panagene, South Korea). As a positive control for hybridization efficiency, an Alexa Fluor 488–labeled PNA probe with specificity for centromeric DNA repeats (Cat# F3012, Panagene, South Korea) was included in the hybridization solution. Slides were counterstained with 4′,6-diamidino-2-phenylindole (Sigma-Aldrich, St. Louis, MO) for labeling total nuclear DNA and mounted with Prolong anti-fade mounting medium (Invitrogen, Carlsbad, CA). ALT positivity was determined using criteria previously published, based on the presence of large, ultrabright telomeric signals by FISH. In brief, ALT-positive cases were defined as those with foci >10-fold brighter compared with the average signal of telomeres in benign cells, and present in >1% of cancer nuclei [12, 13, 28].
Immunohistochemistry
Formalin-fixed, paraffin-embedded tissue sections were deparaffinized and hydrated prior to antigen retrieval. Immunohistochemistry was performed in automated instruments using antibodies directed against ATRX (Rabbit polyclonal, 1:200 dilution, catalog# HPA001906, Sigma-Aldrich, St. Louis, MO) and mutant IDH1 protein (Clone H09, 1:100 dilution, catalog# DIA-H09, Dianova, Hamburg, Germany). DAXX and SMARCAL1 immunohistochemistry were performed manually. For DAXX, slides were steamed in citrate-based antigen unmasking solution (Cat# H-3300-250, Vector Laboratories, Burlingame, CA) for 30 min, blocked twice (Cat# S2003 and X0909, Dako, Denmark), then were incubated for 90 min at room temperature in primary antibody against DAXX (1:100 dilution, Cat# HPA008736, Atlas Antibodies, Bromma, Sweden). For SMARCAL1, slides were steamed in EDTA buffer (Cat# AM9849, Invitrogen, Carlsbad, CA) for 45 min, blocked twice (Cat# S2003 and X0909, Dako, Denmark), then were incubated for 2 h at room temperature in primary antibody against SMARCAL1 (1:100 dilution, Cat# HPA020337, Atlas Antibodies, Bromma, Sweden). For both DAXX and SMARCAL1, antibody signal was detected using HRP-conjugated Anti-Rabbit IgG (Cat# PV6119, Leica, Buffalo Grove, IL) and visualized by 3,3′ diamino-benzidine (Cat# D1468, Millipore-Sigma) prior to hematoxylin counterstaining (Cat# S2009, Dako, Denmark), dehydration, and mounting. For ATRX, DAXX, and SMARCAL1 scoring, retained protein expression was defined as nuclear staining within the tumor cells, and loss of protein expression was defined as the lack of immunolabeling in the tumor cells. Importantly, to confirm ATRX, DAXX, and SMARCAL1 immunoreactivity, positive nuclear staining in internal nonneoplastic cell components was required in all cases scored. Of note, one case was categorized as mis-localized for SMARCAL1 because the tumor cells, but none of the nonneoplastic cells, displayed abnormal nuclear foci instead of pan-nuclear immunolabeling.
Fluorescence microscopy and image analysis
High-resolution fluorescent images were obtained using a Nikon Ti deconvolution wide-field epifluorescence microscope equipped with a Nikon Intensilight C-HGFI illuminator and appropriate fluorescence excitation/emission filters. Grayscale images were captured at ×400 magnification using Nikon NIS-Elements software and an attached Andor Zyla sCMOS camera, pseudo-colored, and merged. For the ALT-associated telomere focus size quantification analysis, fluorescent images were obtained using a Nikon 50i epifluorescence microscope equipped with X-Cite series 120 illuminator (EXFO Photonics Solutions Inc., Mississauga, Ontario, Canada) and appropriate fluorescence excitation/emission filters. Grayscale images were captured using Nikon NIS-Elements software and an attached Photometrics CoolsnapEZ digital camera, pseudo-colored, and merged. Brightfield images were obtained using a Nikon 50i epifluorescence microscope, Nikon NIS-Elements software, and an attached Nikon Digital Sight DS-Fi1 camera. Telomere foci from ALT-positive cases were imaged at 100 ms exposure at ×200 magnification. Telomere foci were detected using algorithms available through Image J. Ultrabright telomere foci were defined as signals above a predetermined threshold of 150 using Renyi Entropy settings. The total area of each resulting particle was measured.
Statistical analysis
All statistical tests were performed using GraphPad Prism software (version 8). P values less than 0.05 were considered significant.
Results
The prevalence of ALT in giant cell GBM
ALT is a prevalent telomere maintenance mechanism in gliomas. In prior studies of ALT in glioma, in which giant cell GBM cases were included, ALT has not been identified [16, 34]. However, a recent study identified ATRX loss in 53% (9/17) of giant cell GBM cases with more than 30% of giant cells [32]. Thus, to better understand the potential importance of ALT in giant cell GBM, we performed telomere-specific FISH on 15 cases of classic giant cell GBM. ALT-positive cases were defined as those harboring large, ultrabright telomere-specific foci after telomere-specific FISH. ALT was identified in 8 (53%) of 15 cases, suggesting that the ALT telomere maintenance mechanism is relevant to the biology of many giant cell GBMs.
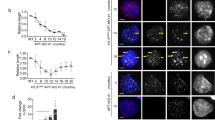
Most ALT-positive gliomas display loss of the ATRX protein [12, 16,17,18]. Therefore, we assessed the ATRX status of the giant cell GBM cases in our cohort, and the results are summarized in Table 1. Surprisingly, of the 8 ALT-positive cases, only 3 (37.5%) showed ATRX loss by immunohistochemistry. Inactivating mutations in SMARCAL1 were recently identified in ATRX wild type, ALT-positive gliomas [33]. Therefore, we sought to assess SMARCAL1 status in our giant cell GBM cohort. Conditions for SMARCAL1 immunohistochemistry were optimized using U87 glioma cells wild type for SMARCAL1 (Fig. 2A) or in which SMARCAL1 had been knocked out via CRISPR (Fig. 2B; [33]). Upon antibody validation and optimization of staining conditions, immunohistochemistry for SMARCAL1 was performed in the giant cell GBM cohort (Fig. 2C). Of the 8 ALT-positive cases, 3 (37.5%) showed loss or mis-localization of SMARCAL1; notably, these 3 cases retained ATRX protein expression. Finally, mutant IDH1 (R132H) protein was assessed by immunohistochemistry and 14 cases (93.3%) were negative.

IHC for SMARCAL1 was performed on U87 cell line variants, either SMARCAL1 wild-type (A) or SMARCAL1 knockout (B). C Example of giant cell GBM lacking SMARCAL1 protein expression in the cancer cells as revealed by IHC. For all panels, original magnification ×400.
Given the prevalence of ALT in classic giant cell GBM, we sought to expand our cohort to GBMs found to have giant cell features (e.g. cases with a focal giant cell pattern >20% but not dominant). We performed telomere-specific FISH on 28 GBM cases with giant cell features. Of this additional cohort, 10 (36%) displayed ALT. In these additional cases, ATRX and SMARCAL1 were each assessed by immunohistochemistry. Of the 10 ALT-positive cases, 7 (70%) displayed ATRX loss, while 3 (30%) displayed loss of SMARCAL1. Furthermore, 6 (60%) of the ALT-positive cases were negative for IDH1 R132H IHC. Interestingly, 2 ALT-positive cases showed simultaneous loss of ATRX and SMARCAL1, while in contrast, 2 ALT-positive cases retained both ATRX and SMARCAL1. In addition, we identified one anaplastic astrocytoma (WHO grade III) with giant cell features. This case was ALT-positive; however, ATRX and SMARCAL1 protein expression remained intact. In total, we identified 5 ALT-positive cases that retained both ATRX and SMARCAL1 protein expression. Since loss of DAXX, an ATRX binding partner, has been previously strongly associated with ALT in other cancer types, we evaluated the status of DAXX protein expression in these cases. However, we observed that all five cases retained DAXX nuclear protein expression via immunohistochemistry.
In total, we assessed 44 cases of giant cell GBM, GBM with giant cell features, or anaplastic astrocytoma with giant cell features. Of this combined cohort, 19 (43%) showed evidence of ALT. Of the ALT-positive cases, only 9 (47.4%) displayed loss of ATRX by immunohistochemistry, which has typically been associated with ALT positivity in gliomas. Based on work by Diplas et al., we assessed SMARCAL1 expression in our cohort, as well. Of the ALT-positive cases, 6 (31.5%) showed loss or mis-localization of SMARCAL1. In the four cases with SMARCAL1 loss or mis-localization, ATRX was retained, while the remaining two cases did not display loss of either ATRX or SMARCAL1. While SMARCAL1 alterations were more prevalent in the context of ATRX retention, there was no significant relationship found between ATRX and SMARCAL1 status (p = 0.35; Fisher’s exact test).
Temporal analysis of ALT in giant cell GBM
In our combined cohort, we assessed five cases from which two temporal samples were available, and the time interval between the biopsies ranged from 2 months to 29 months. ALT was identified in two of these five cases. For each ALT-positive case, ALT was present in both samples; conversely, for each ALT-negative case, ALT was not identified in either sample. These results suggest that the telomere maintenance program utilized by a giant cell GBM is determined relatively early in the clinical progression of the tumor and is maintained over time, although given the lack of lower grade precursors for these tumors, the role of these alterations in tumor initiation remains unclear. One ALT-positive giant cell GBM showed SMARCAL1 retention and ATRX loss in both tumor biopsies taken 23 months apart. Another ALT-positive GBM with giant cell features showed loss of ATRX in both tumor biopsies, but loss of SMARCAL1 only on the tumor biopsy taken at a timepoint 29 months later.
ALT features in giant cell GBM with SMARCAL1 or ATRX loss
SMARCAL1 loss in ALT-positive cells has been shown to increase the size of ALT-associated ultrabright telomeric foci [35]. In order to assess the impact of ATRX and SMARCAL1 status on telomere focus size in our giant cell GBM cohort, image analysis was performed. All ALT-positive cases were imaged at a constant exposure time. Representative images from an ALT-negative case (Fig. 3A), as well as ALT-positive cases lacking ATRX (Fig. 3B) or SMARCAL1 (Fig. 3C) protein expression are shown. For all cases, telomere foci were identified by threshold analysis in Image J software and the size of telomere foci were calculated; ALT-positive cases were stratified by ATRX and SMARCAL1 status (Fig. 3D). Analysis of focus size revealed no significant differences in median telomere focus size based on SMARCAL1 status alone (p = 0.5908; Mann–Whitney test; data not shown). In contrast, cases with ATRX retention showed significantly higher median telomere focus size (p = 0.0117; Mann–Whitney test; data not shown). Further analysis was performed to determine the relative frequency of extremely large telomere foci—defined herein as being larger than 75 pixels2 by image analysis—based on ATRX and SMARCAL1 status (Fig. 3D). The frequency of these extremely large foci was significantly increased in cases with SMARCAL1 loss and ATRX retention (p = 0.0497; Kruskal–Wallis test). Therefore, in the context of ATRX retention, SMARCAL1 loss appears to promote the development of these abnormally large ALT-associated telomere DNA foci.

Representative telomere (red) and centromere (green) FISH images from A an ALT-negative giant cell GBM case, and from ALT-positive giant cell GBM cases with either B ATRX loss or C SMARCAL1 loss (original magnification ×400). Scale bar equals 50 microns and the arrows point to ultrabright telomeric FISH signals present in the ALT-positive cases. D Focus size was measured for all ALT-positive giant cell GBM cases under constant exposure, and focus sizes were compared. A cutoff of 75 pixels2 (red line) was chosen for comparative analyses.
Finally, we tested whether there were any differences in overall survival based on the assessed molecular markers in the combined cohort of classic giant cell GBMs and GBMs with giant cell features. As shown in Fig. 4A, B, a Kaplan–Meier curve and log rank test demonstrates that, while there may be a trend, ATRX and SMARCAL1 status do not display significant differences in overall survival (p = 0.08 and p = 0.09, respectively). However, as shown in Fig. 4C, patients with ALT-positive tumors showed significantly improved overall survival compared to patients with ALT-negative tumors (p < 0.03). Of note, while we did not observe an association between patient age as dichotomized groups (median cut-off: <55 years; p = 0.4), we did observe a positive correlation between ALT and older age (p < 0.05; Wilcoxon rank-sum test).

Kaplan–Meier curves and log rank tests demonstrate that there is not significant difference in overall survival based on A ATRX status (p = 0.08) or B SMARCAL1 status (p = 0.09); however, C there was a significantly better overall survival in patients with ALT-positive tumors compared to patients with ALT-negative tumors (p < 0.03).
Discussion
Subsets of high-grade gliomas, including GBM, are known to utilize the ALT telomere maintenance mechanism [8,9,10,11,12,13]. However, the telomere maintenance profile of one subtype of GBM—giant cell GBM—has not been extensively studied. We have sought to examine the presence of ALT in a cohort of giant cell GBM, as well as GBM with giant cell features that do not quite satisfy criteria for giant cell GBM.
In our cohort, we have identified ALT in 53% of giant cell GBM cases and 36% of cases of GBM with giant cell features. While prior studies have not shown ALT in giant cell GBM, numbers of giant cell GBM cases were limiting [16, 34]. By expanding the number of giant cell GBM cases examined, we have demonstrated that ALT occurs at a high frequency in both giant cell GBM and GBM with giant cell features. Of the 19 total ALT-positive cases examined, only five (26.3%) showed evidence of an IDH1 mutation. Therefore, compared to classic GBM, in which ALT-positive cases tend to harbor mutant IDH1 [11], the genetic profile of ALT-positive giant cell GBM is distinct.
In addition, we assessed the degree to which ALT was associated with loss of ATRX, a chromatin remodeling protein predicted to suppress ALT in gliomas. Interestingly, in both of our cohorts, ATRX loss did not perfectly associate with ALT status. Specifically, in our combined cohort, only 47.4% displayed loss of ATRX. While isolated ALT-positive gliomas seemingly retaining ATRX have been previously identified, we were struck by the large percentage of cases in our cohort displaying this behavior. As such, we developed an immunohistochemistry assay to detect expression of SMARCAL1, which was recently identified as being mutated in a subset of ALT-positive gliomas [33]. Loss or mis-localization of SMARCAL1 was identified in 31.5% of ALT-positive cases examined herein. Four ALT-positive cases showed aberrant SMARCAL1 expression and retained ATRX expression, while two cases seemingly lost both ATRX and SMARCAL1. While this result may seem counterintuitive, Diplas et al. did identify cases in which both ATRX and SMARCAL1 showed inactivating mutations, indicating that these two ALT-associated genetic events can co-occur [33]. Interestingly, five ALT-positive cases retained both ATRX, SMARCAL1, and DAXX protein expression, potentially indicating loss of function mutations in these ALT suppressors that did not yield a complete loss of protein. Alternatively, additional heretofore unidentified ALT suppressor(s) may contribute to the development of ALT in giant cell GBM.
Loss of SMARCAL1 in vitro was previously shown to induce the formation of extrachromosomal, circular telomeric sequences (c-circles), which have been associated with ALT activity [36, 37]. In addition, in cancer cell lines already utilizing the ALT mechanism, SMARCAL1 loss was associated with a dramatic increase in the size of ALT-associated telomeric DNA foci [35]. Thus, in our ALT-positive cohort, we assessed the contribution of the status of ATRX and SMARCAL1 to telomere focus size. Overall, cases with ATRX retention, irrespective of SMARCAL1 expression, were more likely to have a higher median telomere focus size. While we did not observe a significant relationship between SMARCAL1 status alone and median telomere focus size, cases with SMARCAL1 loss and ATRX retention showed a greater propensity toward the presence of extremely large ALT-associated ultrabright telomeric foci. These results suggest that the manifestation of ALT may differ depending on the ALT suppressor that is lost—ATRX or SMARCAL1.
There are a number of strengths of this study. We assessed the presence of ALT using a robust telomere-specific FISH assay and identified a subset of giant cell GBM that utilize the ALT telomere maintenance mechanism. Additionally, we optimized a SMARCAL1 immunohistochemistry assay using genetically defined controls. Using this assay, we identified that, in addition to ATRX loss, ALT-positive tumors harboring SMARCAL1 alterations are prevalent in giant cell GBM. However, despite these strengths, there are also limitations to our study. Due to the scant availability of archived tissue, we were unable to sequence the SMARCAL1 gene to confirm the presence of somatic mutations. In addition, since this is a rare disease, the conclusions rely on a relatively small cohort of samples. However, our findings warrant future investigations that include larger cohorts collected across multiple institutions.
In summary, we have identified a substantial subset of giant cell GBM tumors that utilize the ALT telomere maintenance mechanism. Furthermore, in addition to ALT-positive tumors with ATRX loss, ALT-positive tumors harboring SMARCAL1 loss or mis-localization are prevalent in giant cell GBM. Further study will illuminate the existence of ALT-specific therapies that may prove useful in this tumor type.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Ostrom QT, Cote DJ, Ascha M, Kruchko C, J.S B-S. Adult Glioma Incidence and Survival by Race or Ethnicity in the United States From 2000 to 2014. JAMA Oncol. 2018;4:1254–62.
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131:803–20.
Kozak KR, Moody JS. Giant cell glioblastoma: a glioblastoma subtype with distinct epidemiology and superior prognosis. Neuro Oncol. 2009;11:833–41.
Oh T, Rutkowski MJ, Safaee M, Sun MZ, Sayegh ET, Bloch O, et al. Survival outcomes of giant cell glioblastoma: institutional experience in the management of 20 patients. J Clin Neurosci. 2014;21:2129–34.
Jin MC, Wu A, Xiang M, Azad TD, Soltys SG, Li G, et al. Prognostic factors and treatment patterns in the management of giant cell glioblastoma. World Neurosurg. 2019;128:e217–e224.
Meyer-Puttlitz B, Hayashi Y, Waha A, Rollbrocker B, Bostrom J, Wiestler OD, et al. Molecular genetic analysis of giant cell glioblastomas. Am J Pathol. 1997;151:853–857.
Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70.
Chen YJ, Hakin-Smith V, Teo M, Xinarianos GE, Jellinek DA, Carroll T, et al. Association of mutant TP53 with alternative lengthening of telomeres and favorable prognosis in glioma. Cancer Res. 2006;66:6473–6476.
Hakin-Smith V, Jellinek DA, Levy D, Carroll T, Teo M, Timperley WR, et al. Alternative lengthening of telomeres and survival in patients with glioblastoma multiforme. Lancet. 2003;361:836–838.
Henson JD, Hannay JA, McCarthy SW, Royds JA, Yeager TR, Robinson RA, et al. A robust assay for alternative lengthening of telomeres in tumors shows the significance of alternative lengthening of telomeres in sarcomas and astrocytomas. Clin Cancer Res. 2005;11:217–25.
McDonald KL, McDonnell J, Muntoni A, Henson JD, Hegi ME, von Deimling A, et al. Presence of alternative lengthening of telomeres mechanism in patients with glioblastoma identifies a less aggressive tumor type with longer survival. J Neuropathol Exp Neurol. 2010;69:729–36.
Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C, et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science. 2011;333:425.
Heaphy CM, Subhawong AP, Hong SM, Goggins MG, Montgomery EA, Gabrielson E, et al. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am J Pathol. 2011;179:1608–15.
Cesare AJ, Reddel RR. Alternative lengthening of telomeres: models, mechanisms and implications. Nat Rev Genet. 2010;11:319–30.
Meeker AK, Gage WR, Hicks JL, Simon I, Coffman JR, Platz EA, et al. Telomere length assessment in human archival tissues: combined telomere fluorescence in situ hybridization and immunostaining. Am J Pathol. 2002;160:1259–68.
Nguyen DN, Heaphy CM, de Wilde RF, Orr BA, Odia Y, Eberhart CG, et al. Molecular and morphologic correlates of the alternative lengthening of telomeres phenotype in high-grade astrocytomas. Brain Pathol. 2013;23:237–43.
Jiao Y, Killela PJ, Reitman ZJ, Rasheed AB, Heaphy CM, de Wilde RF, et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget. 2012;3:709–22.
Abedalthagafi M, Phillips JJ, Kim GE, Mueller S, Haas-Kogen DA, Marshall RE, et al. The alternative lengthening of telomere phenotype is significantly associated with loss of ATRX expression in high-grade pediatric and adult astrocytomas: a multi-institutional study of 214 astrocytomas. Mod Pathol. 2013;26:1425–32.
Xue Y, Gibbons R, Yan Z, Yang D, McDowell TL, Sechi S, et al. The ATRX syndrome protein forms a chromatin-remodeling complex with Daxx and localizes in promyelocytic leukemia nuclear bodies. Proc Natl Acad Sci USA. 2003;100:10635–40.
Drane P, Ouararhni K, Depaux A, Shuaib M, Hamiche A. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 2010;24:1253–65.
Goldberg AD, Banaszynski LA, Noh KM, Lewis PW, Elsaesser SJ, Stadler S, et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010;140:678–91.
Lewis PW, Elsaesser SJ, Noh KM, Stadler SC, Allis CD. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc Natl Acad Sci USA. 2010;107:14075–80.
Napier CE, Huschtscha LI, Harvey A, Bower K, Noble JR, Hendrickson EA, et al. ATRX represses alternative lengthening of telomeres. Oncotarget. 2015;6:16543–58.
Clynes D, Jelinska C, Xella B, Ayyub H, Scott C, Mitson M, et al. Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat Commun. 2015;6:7538.
Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA Jr., et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci USA. 2013;110:6021–6.
Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet. 2014;46:444–50.
Korshunov A, Capper D, Reuss D, Schrimpf D, Ryzhova M, Hovestadt V, et al. Histologically distinct neuroepithelial tumors with histone 3 G34 mutation are molecularly similar and comprise a single nosologic entity. Acta Neuropathol. 2016;131:137–46.
Rodriguez FJ, Vizcaino MA, Blakeley J, Heaphy CM. Frequent alternative lengthening of telomeres and ATRX loss in adult NF1-associated diffuse and high-grade astrocytomas. Acta Neuropathol. 2016;132:761–3.
D’Angelo F, Ceccarelli M, Tala, Garofano L, Zhang J, Frattini V, et al. The molecular landscape of glioma in patients with Neurofibromatosis 1. Nat Med. 2019;25:176–87.
Rodriguez FJ, Graham MK, Brosnan-Cashman JA, Barber JR, Davis C, Vizcaino MA, et al. Telomere alterations in neurofibromatosis type 1-associated solid tumors. Acta Neuropathol Commun. 2019;7:139.
Oh JE, Ohta T, Nonoguchi N, Satomi K, Capper D, Pierscianek D, et al. Genetic alterations in gliosarcoma and giant cell glioblastoma. Brain Pathol. 2016;26:517–22.
Cantero D, Mollejo M, Sepulveda JM, D’Haene N, Gutierrez-Guaman MJ, Rodriguez de Lope A, et al. TP53, ATRX alterations, and low tumor mutation load feature IDH-wildtype giant cell glioblastoma despite exceptional ultra-mutated tumors. Neurooncol Adv. 2020;2:vdz059.
Diplas BH, He X, Brosnan-Cashman JA, Liu H, Chen LH, Wang Z, et al. The genomic landscape of TERT promoter wildtype-IDH wildtype glioblastoma. Nat Commun. 2018;9:2087.
Silvestre DC, Pineda JR, Hoffschir F, Studler JM, Mouthon MA, Pflumio F, et al. Alternative lengthening of telomeres in human glioma stem cells. Stem Cells. 2011;29:440–51.
Cox KE, Marechal A, Flynn RL. SMARCAL1 resolves replication stress at ALT telomeres. Cell Rep. 2016;14:1032–40.
Poole LA, Zhao R, Glick GG, Lovejoy CA, Eischen CM, Cortez D. SMARCAL1 maintains telomere integrity during DNA replication. Proc Natl Acad Sci USA. 2015;112:14864–9.
Cesare AJ, Griffith JD. Telomeric DNA in ALT cells is characterized by free telomeric circles and heterogeneous t-loops. Mol Cell Biol. 2004;24:9948–57.
Acknowledgements
We would like to acknowledge the assistance of the Boston University Cellular Imaging Core.
Funding
This study was supported by the Department of Defense (W81XWH-18-1-0496), a National Cancer Institute training grant (2T32CA009110-39A1), and a postdoctoral fellowship from the Rally Foundation for Childhood Cancer Research, The Truth 365, and Open Hands Overflowing Hearts. Core facilities were funded through a National Cancer Institute Cancer Center Support grant (P30 CA006973). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
JAB, CMD, AKM, FJR, and CMH planned the experiments; JAB, CMD, BHD performed the experiments; JAB, FJR, and CMH analyzed the data; BHD contributed essential reagents; JAB, FJR, and CMH drafted the initial manuscript. All authors read and approved the final paper.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethics approval/Consent to participate
The study was performed under approval by the Johns Hopkins Hospital Institutional Review Board and in accordance with the Declaration of Helsinki.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Brosnan-Cashman, J.A., Davis, C.M., Diplas, B.H. et al. SMARCAL1 loss and alternative lengthening of telomeres (ALT) are enriched in giant cell glioblastoma. Mod Pathol 34, 1810–1819 (2021). https://doi.org/10.1038/s41379-021-00841-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-021-00841-7
This article is cited by
-
Genome maintenance meets mechanobiology
Chromosoma (2024)
