
Abstract
Glioblastomas are the most common IDH-wildtype adult high-grade gliomas, frequently harboring mutations in the TERT gene promoter (pTERT) and utilizing the subsequent telomerase overexpression for telomere length maintenance. However, some rare cases show loss of ATRX and use alternative mechanisms of telomere lengthening. In this study, we performed the first complex genomic analysis specifically concentrating on the latter subgroup. Comprehensive genomic profiling of 12 ATRX-deficient and 13 ATRX-intact IDH-wildtype adult high-grade gliomas revealed that ATRX and pTERT mutations are mutually exclusive. DNMT3A alterations were confined to ATRX-deficient, while PTEN mutations to ATRX-intact cases. RAS–MAPK pathway alterations, including NF1 mutations, were more characteristic in the ATRX-deficient group. Variants of genes related to homologous recombination repair showed different patterns of affected genes. Two ATRX-deficient tumors with high tumor mutational burden and mismatch repair deficiency were found. One of these contained a novel fusion involving the NTRK2 and LRRFIP2 genes, while the other showed loss of MSH2 and MSH6 without genetic alterations in the encoding genes suggesting an epigenetic background. Genetic characteristics of ATRX-deficient IDH-wildtype adult high-grade gliomas suggest that these tumors are particularly intriguing targets of potential future therapeutic interventions including immunotherapies combined with MAPK pathway inhibition and DNA repair inhibitors.
Similar content being viewed by others

Introduction
Glioblastoma [GB; World Health Organization (WHO) grade 4] is the most common adult-type high-grade glioma, currently defined as an IDH-wildtype and H3-wildtype diffuse astrocytic tumor1. The prognosis is still extremely poor and effective therapeutic interventions are lacking. Most molecularly targeted and immunotherapeutic approaches failed to show significant effects in clinical trials2. Therefore, detailed characterization of GB subgroups and the search for novel predictive biomarkers and actionable targets are crucial in order to develop future personalized treatment options.
Approximately 80% of GBs harbors hotspot mutations in the promoter region of the TERT gene (pTERT) currently considered as entity-defining molecular markers3. The subsequent overexpression of the encoded catalytic subunit of the telomerase enzyme is the main mechanism of telomere length maintenance in these tumors. In contrast, IDH-mutant astrocytomas and H3-altered high-grade pediatric-type diffuse gliomas are characterized by the loss of ATRX (α thalassemia/mental retardation syndrome X-linked), a chromatin-remodeling factor and histone chaperone4. Loss of ATRX protein induces alternative lengthening of telomeres (ALT), a telomerase-independent mechanism of telomere maintenance being dependent on homologous recombination (HR) between telomeric sequences5. Besides the abovementioned entities, ALT is also involved in the pathogenesis of a small subset of pTERT-wildtype GBs. This latter subgroup frequently harbors loss-of-function mutations of either ATRX or SMARCAL1 genes as it was recently described6.
ATRX-deficient IDH-wildtype and H3-wildtype adult gliomas are rare and include mostly GBs and some recently described new entities, such as high-grade astrocytoma with piloid features (HGAP)7 and glioneuronal tumor with ATRX alteration, kinase fusion and anaplastic features (GTAKA)8. Many cases of these latter entities previously fell into the GB category. Due to the rarity of these tumors, only limited data are available regarding their genetic characteristics, particularly the clinically relevant molecular patterns. Therefore, further investigation is needed to elucidate their potential unique biological, genomic and clinical properties. In order to address this issue, we performed comprehensive genomic profiling (CGP) of an IDH-wildtype and H3-wildtype adult high-grade astrocytic glioma cohort showing ATRX loss (n = 12) in parallel with the analysis of an ATRX-intact GB cohort being similar in size (n = 13), as a clinically meaningful comparison. This was implemented using the Illumina TruSight Oncology 500 (TSO-500) targeted next-generation sequencing (NGS) panel. For the list of genes tested for small variants [single nucleotide variants (SNVs), small insertions and deletions (InDels), copy number variations (CNVs)] and fusions, see Supplementary Table S1.
Results
Clinical and histopathological characteristics of ATRX-deficient and ATRX-intact tumors
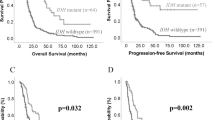
Twenty five adult IDH-wildtype and H3-wildtype gliomas showing obvious high-grade histological features were included in this study. Among these, 12 showed ATRX loss and 13 showed retained ATRX expression according to the immunohistochemical (IHC) analysis (Table 1). There was no significant difference in terms of patient age between the ATRX-deficient and ATRX-intact groups (p = 0.1241, Mann–Whitney test), but more patients above 60 years of age were included into the ATRX-deficient group (Supplementary Fig. S1a). Kaplan–Meier analysis did not reveal significant difference between the overall survival (OS) of ATRX-deficient and ATRX-intact cases (hazard ratio = 1.829, 95% confidence interval 0.7085–4.722, log-rank test: χ2 (1) = 1.98, p = 0.1593) (Supplementary Fig. S1b). However, median OS of the ATRX-deficient group is still conspicuously lower (Table 1) being probably related to the higher proportion of elderly patients and not to real biological difference. There are 3 cases with OS above 36 months and therefore considered to be long-term survivors9, 1 in the ATRX-deficient and 2 in the ATRX-intact groups. The majority of patients received radio-chemotherapy according to the standard guidelines which was complemented with bevacizumab in 13 cases. Oncological treatment could not be used in 3 cases due to the poor general status of the patients. Recurrent tumor samples were available for molecular analysis in 7 cases (Table 1). In these cases, only recurrent tumor samples were subjected to genetic testing.
In the ATRX-deficient group, 10 cases were diagnosed as GB (WHO grade 4) based on the morphological, IHC and available molecular data. One spinal tumor (case #2) in this group contained areas reminiscent of pilocytic astrocytoma and harbored KIAA1549::BRAF fusion (Supplementary Fig. S2b) together with ATRX loss being highly suggestive of HGAP7. Another tumor (case #3) was diagnosed as astrocytoma, not elsewhere classified (NEC) (WHO grade 3) from a stereotaxic biopsy sample due to the IDH-wildtype and H3-wildtype state and lack of criteria defining GB. In this case, the patient had received the clinical diagnosis of neurofibromatosis type I. Case #17 showed an oligodendroglioma-like morphology (see later), diffuse OLIG2 nuclear positivity, but only focal expression of GFAP. Fluorescent in situ hybridization (FISH) analysis revealed relative deletions of 1p36 and 19q13 chromosomal regions in 50% and 75% of the polysomic tumor cells, respectively. However, the IDH-wildtype state excluded the diagnosis of oligodendroglioma. These latter cases demonstrate some degree of heterogeneity of the ATRX-deficient group, but it is still in accordance with the originally defined group of interest. Each case in the ATRX-intact group was morphologically and molecularly consistent with GB (WHO grade 4). Readily identifiable multinucleated giant cells were present in 7 (58.3%) and 3 (23.1%) cases within the ATRX-deficient and ATRX-intact groups, respectively (p = 0.1107, Fisher’s exact test). However, giant cells were usually scattered and exceeded the proportion of 30% in only two ATRX-deficient and one ATRX-intact tumors.
Genetic landscape of ATRX-deficient gliomas
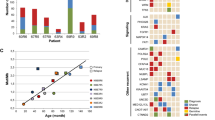
NGS analysis revealed pathogenic (P) or likely pathogenic (LP) variants of ATRX in all ATRX-deficient, but in none of ATRX-intact tumors. pTERT mutations (namely c.-124C > T) were detected in 12 of 13 ATRX-intact, but none of ATRX-deficient gliomas (Fig. 1a,b). Mutual exclusivity of ATRX and pTERT mutations were confirmed by the statistical analysis (p = 2.692e−6; padj = 1.345e−5, Fisher’s exact test).

(a) Summary of the main clinical characteristics and alterations affecting genes well documented to be involved in GB pathogenesis in ATRX-deficient and ATRX-intact glioma cases (divided cells denote multiple variants in the same gene). (b) Comparison of ATRX, pTERT, PTEN, DNMT3A and NF1 mutational as well as EGFR amplification frequencies in the ATRX-deficient and ATRX-intact groups, respectively.
As expected, the most frequently affected gene among receptor tyrosine kinases (RTK) was EGFR. The occurrence of EGFR amplification was apparently, but not significantly lower in ATRX-deficient cases (p = 0.2016, padj = 0.2016, Fisher’s exact test) (Fig. 1a,b). Other RTKs, namely PDGFRA, KIT and MET were affected less frequently, with no obvious differences between the analyzed groups. Amplification of the PDGFRA gene was detected in 3 cases and accompanied by complex alterations including fusions, mutations and deletions as well as concurrent amplification of the KIT gene (Fig. 1a). Fusions of RTK genes were detected in 3 ATRX-deficient and 3 ATRX-intact cases, respectively (Supplementary Fig. S2a)10. These rearrangements included the SEPTIN14::EGFR (case #8), PDGFRA::ADGRL3 (case #12), KDR::PDGFRA, PTPRZ1::MET, WASL::MET (case #15) and DNAJB6::MET (case #18) fusions. In addition, one GB (case #16) harboring FGFR3::TACC3 fusion was found in the ATRX-intact group, but the previously reported characteristic morphological features11 frequently accompanying this fusion were not evident (Supplementary Fig. S2c). Notably, fusions affecting the NTRK2 gene, namely NTRK2::LRRFIP2 and LRRFIP2::NTRK2 were detected in case #17 (Fig. 3i,j).
Among members of the PI3K-AKT pathway, PTEN was the most frequently affected gene in ATRX-intact tumors (53.9%). In contrast, none of the ATRX-deficient tumors harbored variants in PTEN fulfilling the inclusion criteria (p = 0.0052, padj = 0.013, Fisher’s exact test) (Fig. 1a,b). Regarding RAS–MAPK pathway, NF1 mutations occurred in 42% of ATRX-deficient, while only 15% of ATRX-intact tumors, although the difference was not statistically significant (p = 0.2016, padj = 0.2016, Fisher’s exact test) (Fig. 1a, b). In addition, besides the one case harboring a KIAA1549::BRAF fusion, another contained BRAF p.V600E mutation in the ATRX-deficient group. PTPN11 mutations occurred in 3 cases, among which the two P variants were detected in the ATRX-intact group. TP53 mutations were frequent in both groups (66.7% and 53.9% in ATRX-deficient and ATRX-intact groups, respectively). PPM1D, an oncogene related to p53 signaling, was altered in 2 additional ATRX-deficient cases, namely the 2 cases characterized by high tumor mutational burden (TMB) (see later). Mutations of the RB1 gene as well as CDK4 and CDK6 amplifications (but not low-level copy number gains) were found in scattered cases of the ATRX-intact group (Fig. 1a).
Among less frequently altered genes previously described to be affected in diffuse gliomas, GABRA6, LRP1B, LZTR1, MYCN and NOTCH1 harbored P or LP mutations in scattered cases. Intriguingly, mutations of the DNMT3A gene were found in 4 cases, all of which were ATRX deficient (p = 0.0391, padj = 0.0652, Fisher’s exact test) (Fig. 1a, b).
High TMB was detected exclusively in cases #17 and #25, both of which were ATRX-deficient. Microsatellite instability (MSI) analysis revealed MSI-high (MSI-H) status only in case #25 (Fig. 2). In case #17, CGP was performed on a recurrent GB. Both the primary and recurrent tumors showed oligodendroglioma-like morphology (Fig. 3a,b) as well as ATRX loss (Fig. 3c,d). In the recurrent tumor sample, a P mutation of the MLH1 gene was found, accompanied by loss of both MLH1 and PMS2 immunoreactivities in the tumor cells (Fig. 3f,h). However, both MLH1 and PMS2 expressions were retained in the primary sample of the patient taken prior to radio-chemotherapy (Fig. 3e,g). A variant of uncertain significance (VUS) in the MSH6 gene was also detected, but did not result in MSH6 loss. In addition to mismatch repair deficiency (MMR-d), this tumor harbored 2 LP mutations and 1 VUS in the POLE gene. Case #25 (Fig. 4a,b) harbored a VUS in the MLH1 gene, but both MLH1 and PMS2 expressions were retained. In contrast, MSH2 and MSH6 expressions were lost in the tumor cells, without detectable genetic lesion in the encoding genes (Fig. 4c,d). VUSs in MSH2, MSH6 and MLH1 were also found in cases #15, #19 and #10, but without loss of the encoded proteins, high TMB or MSI-H (Fig. 2).

Summary of the main clinical characteristics and alterations affecting genes related to DNA repair, tumor mutational burden (TMB) and microsatellite instability (MSI) as well as results of mismatch repair protein immunohistochemical analyses in ATRX-deficient and ATRX-intact glioma cases (divided cells denote multiple variants in the same gene).

Oligodendroglioma-like morphology of case #17 shown by the H&E-stained sections from the (a) primary and (b) recurrent tumor resected following radio-chemotherapy (scale bar: 50 μm). (c and d) ATRX expression was lost in both samples. (e) MLH1 and (g) PMS2 were retained in the primary specimen, while both (f) MLH1 and (h) PMS2 were lost in the recurrent tumor. (i) NTRK2 fluorescent in situ hybridization showed break apart signals (white arrowheads) in the tumor cells validating the presence of NTRK2::LRRFIP2 and LRRFIP2::NTRK2 fusions revealed by next-generation sequencing (j).

(a) H&E-stained sections from case #25 showed scattered multinucleated giant cells (black asterisk) (scale bar: 50 μm). Immunohistochemistry revealed loss of (b) ATRX, (c) MSH2 and (d) MSH6.
A conspicuously high number of HR repair-related genes harbored alterations (usually SNVs) in our cohort (Fig. 2). Among these, PTEN and DNMT3A were already described. Regarding others, P or LP alterations occurred in five ATRX-deficient tumors (ARID1A in case #23, ARID2 in case #1, ATM in case #25, CHEK2 in case #21 and FANCA in case #17), but only in one ATRX-intact case (FANCI in case #10).
For the detailed lists of variants fulfilling the inclusion criteria in each case, see Supplementary Table S2. Tabular summary of the variants, variant types as well as the frequency of affected cases are demonstrated on Supplementary Fig. S3.
Discussion
In the study presented here, we analyzed the genetic patterns of ATRX-deficient IDH-wildtype and H3-wildtype adult high-grade gliomas in comparison with ATRX-intact GBs using CGP. To the best of our knowledge, this is the first complex genomic analysis specifically concentrating on this rare subgroup. In addition to confirming previous findings showing the mutual exclusivity of ATRX and pTERT mutations as well as the association of NF1 mutations and ATRX loss, our study revealed novel, previously unreported genomic patterns with potential clinical significance. PTEN mutations were absent, while DNMT3A variants were enriched in ATRX-deficient tumors highlighting previously unknown genetic characteristics of this subgroup. In addition, MMR-d occurred in two peculiar ATRX-deficient tumors and the pattern of altered HR repair-related genes were substantially different in the examined cohorts.
ATRX deficiency and pTERT hotspot mutations were previously shown to be mutually exclusive3, which was clearly confirmed in our study. The main biological consequence of ATRX loss is the induction of ALT, which is the most important mechanism of telomere length maintenance in IDH-mutant and H3-altered diffuse astrocytic gliomas12,13. However, ATRX deficiency is not sufficient to induce ALT by itself, and other factors are also necessary. An example of these is the inhibition of the histone demethylase KDM4B in H3 G34R- and IDH-mutant astrocytic gliomas, as it was recently described14. In IDH- and H3-wildtype gliomas, the mechanisms of ALT are less well characterized. From this respect, considerable enrichment of DNMT3A mutations in the ATRX-deficient cases is particularly interesting. Each detected mutation has been described previously in various tumors (c.2259G>A in myelodysplastic syndrome15, c.2119G>A in GB6, c.1502A>G and c.1661G>A in acute myeloid leukemia16,17). DNMT3A mutations are well-known prognostic factors in acute myeloid leukemia18, but only sporadic occurrence has been reported in gliomas6,19,20,21. Considering previous observations showing ALT-related phenotypes in DNMT-deficient cells22, the cooperation of ATRX and DNMT3A mutations in telomere length maintenance can be suspected.
Among changes affecting RTK genes, EGFR amplification, a molecular diagnostic criterion of GB, was apparently, but not significantly less frequent in the ATRX-deficient group. This observation is not surprising, considering the association of EGFR amplification and pTERT mutations in GB3. Complex genetic events affecting the 4q12 region, containing the PDGFRA and KIT genes, were also detected in some cases. The peculiar complexity of genetic lesions in this region was previously observed by our group23 and others as well24, but these are not clearly associated with ATRX loss. Presence of fusions affecting RTK genes were relatively frequent in our cohort. The SEPTIN14::EGFR, PTPRZ1::MET25, KDR::PDGFRA23 and FGFR3::TACC311 fusions are known from the literature, but the PDGFRA::ADGRL3, WASL::MET and DNAJB6::MET rearrangements involve previously unreported partner genes.
Among the members of PI3K-AKT pathway, PTEN is well known to harbor loss-of-function mutations in around 30–40% of GBs26. PTEN mutations occurred frequently in our ATRX-intact, but were absent in the ATRX-deficient tumors. According to the currently available data, PTEN mutations do not affect the prognosis of GB27, but may harbor potential predictive value. Clinical studies with immune checkpoint inhibitors produced generally disappointing results in primary and recurrent GB, but a minority of patients shows clinical response28. It was recently recognized that PTEN mutations are more frequent in recurrent GBs being resistant to PD-1 inhibitors29.
The association of NF1 mutation and ATRX loss as well as ALT was previously described in high-grade diffuse gliomas and is accompanied by poor prognosis30,31. In our cohort, NF1 mutations were more frequent in ATRX-deficient tumors, although the difference was not statistically significant. Other alterations related to the RAS–MAPK pathway including KIAA1549::BRAF fusion and BRAF p.V600E mutation occurred in two additional cases in the ATRX-deficient group. These data suggest that there is a trend towards RAS–MAPK pathway alterations in ATRX-deficient high-grade gliomas. According to recent observations, variants of RAS–MAPK pathway components, in particular, BRAF mutations, seem to be the most promising actionable targets in brain tumors32. Furthermore, abnormalities in this pathway are significantly enriched in GBs responding to immunotherapy29 and NF1 mutation has been shown to induce an immunosuppressive, macrophage-microglia rich microenvironment in gliomas33. Inhibition of the RAS–MAPK pathway could serve as a valuable therapeutic tool by inducing sensitivity to immune checkpoint blockade in various tumors including breast cancer34. Our results regarding the frequency of PTEN and RAS–MAPK pathway alterations suggest that this approach may have a rationale in ATRX-deficient IDH-wildtype adult gliomas and ATRX loss could be a potential marker to predict responder cases to immunotherapeutic interventions.
MMR-d and subsequent genetic hypermutability were recently shown to be associated with at least partial giant cell morphology in GBs and are regarded to be important predictors of sensitivity to immune checkpoint inhibitors20,35. NF1 mutations as well as ATRX and DNMT3A mutations were also shown to be significantly enriched in this GB variant suggesting a biological overlap with our group of interest. Tumors containing giant cells were remarkably, but not significantly enriched in our ATRX-deficient group, although only scattered giant cells were present in most of these cases. 6 of the 8 cases harboring NF1 and/or DNMT3A mutations as well as 1 of 2 cases showing MMR-d (case #25) contained giant cells supporting the abovementioned previous observations, although the correlations were less evident in our cohort.
One of the ATRX-deficient samples with MMR-d (case #17) was a recurrent tumor showing oligodendroglioma-like morphology and harboring fusions involving the NTRK2 and LRRFIP2 genes. This rearrangement increases the peculiarity of this case, considering the rarity of fusions involving the NTRK2 gene in adult high-grade gliomas36 and the previously unreported partner gene. An LRRFIP2::ALK fusion was recently described in a case of epithelioid fibrous histiocytoma37, but the exact mechanism by which LRRFIP2 activates its partner genes remains to be elucidated. Among the detected NTRK2-fusions, the most likely to carry biological relevance is LRRFIP2::NTRK2 involving the 3’ end of NTRK2 with the protein kinase domain. It is worth noting, that the co-occurance of NTRK2-rearrangement and ATRX loss raises the possibility that this tumor represents the novel GTAKA category8. Loss-of-function mutation of MLH1, accompanied MLH1 and PMS2 loss as well as high TMB are likely to be related to therapy-induced hypermutation in this case, since both MLH1 and PMS2 expressions were intact in the primary tumor sample38. This tumor was classified as MSI-L, confirming previous observations regarding the low sensitivity of microsatellite instability as a marker of MMR-d in gliomas38. POLE mutations were also detected, neither of which affected the exonuclease domain39, but 2 of them (c.4773G>A; p.W1591* and c.2908G>A; p.G970S) were regarded to be LP. POLE variants were documented to be associated with constitutional MMR-d-related gliomas40 and the consecutive defect of DNA-polymerase proofreading might also contribute to the high TMB in this case.
The other ATRX-deficient, primary GB showing de novo high TMB and concomitant MSI-H harbored an MLH1 mutation, which was classified as a VUS by our algorithm and was not accompanied by PMS2 loss. However, loss of MSH2 and MSH6 proteins was identified, without genetic alterations in their genes. Together with some similar previous observations20,38, this interesting finding raises the possibility of the existence of an epigenetic mechanism of MSH2 and MSH6 loss in gliomas, such as promoter hypermethylation41. Besides the already known constitutional and therapy-induced MMR gene defects, this is a possible new pathway of MMR gene inactivation in this group of tumors. Although results with immunotherapies in hypermutated gliomas were disappointing, potential utility of this approach still merits further investigation38.
The abovementioned connections between HR and ATRX deficiency are also intriguing from a therapeutic point of view. Recent preclinical data revealed an increased DNA-damage response and HR repair deficiency (HRD) in ATRX-deficient tumors, resulting in higher sensitivity to poly (ADP-ribose) polymerase (PARP)42,43 as well as ataxia telangiectasia and Rad3-related (ATR) inhibitors43,44. PARP inhibitors are emerging new therapeutic options in the treatment of primary and recurrent GB45, but further clinical evidence is necessary regarding their utility. The effects of these compounds are generally based on synthetic lethality in tumors showing HRD. The latter is rarely related to BRCA1/2 mutations in high-grade gliomas, in contrast to breast or ovarian carcinomas. Instead, other genetic lesions producing a similar phenotype (“BRCAness”) include IDH1/2 and PTEN mutations as well as EGFR and MYC overexpression46. According to our results, these changes are absent or rare in ATRX-deficient gliomas, but several mutations in other HR-related genes47,48 were found. Although the relevance of these non-BRCA gene mutations in the effects PARP inhibitors is not completely clarified, their synchronous presence with ATRX loss raises the possibility that ATRX-deficient high-grade gliomas are promising targets of PARP inhibitors.
According to recent clinical surveys, results of CGP have a low impact on the management of adult patients with high-grade gliomas49. Responses to molecularly targeted therapies are mostly related to well characterized driver mutations32. Detailed characterization of high-grade glioma subgroups is needed in order to facilitate future successful clinical trials and precisely predict responders. ATRX-deficient IDH-wildtype and H3-wildtype adult high-grade gliomas occur in small numbers even in large publicly available databases. Although somewhat limited by the low sample number, our study provides invaluable novel data regarding the unique genomic patterns of this subgroup being comparable to the histomorphological data as well as potential clinical significance. Our results suggest that these tumors may represent intriguing targets of immune checkpoint- and RAS–MAPK pathway inhibitor combinations as well as DNA repair inhibitors. Therefore, our findings could promote further studies towards more effective and personalized future therapies.
Methods
Tissue collection and clinical data
Formalin-fixed, paraffin-embedded (FFPE) tissue samples of the 25 included cases were obtained from the pathology departments of the following institutions: Semmelweis University (Department of Pathology and Experimental Cancer Research), County Hospital Győr, Petz Aladár Hospital and University of Szeged, from a 4-year-long period. At least two independent neuropathologists reviewed the hematoxylin and eosin (H&E) stained sections as well as available IHC and molecular data and confirmed the diagnosis according to the current WHO criteria1. All further molecular and IHC analyses were performed in the Department of Pathology and Experimental Cancer Research of the Semmelweis University. The IDH-wildtype and H3-wildtype status was confirmed by CGP in all cases (see below). Clinical data were obtained from the archives of the National Institute of Mental Health, Neurology and Neurosurgery and National eHealth Infrastructure.
Immunohistochemistry (IHC)
IHC studies were performed according to the standard laboratory protocols on 3 μm thick sections, using a Leica Bond-Max automated immunostaining system (Leica Biosystems, Danvers, MA). For the reactions presented here, the following primary antibodies were used: ATRX (Sigma-Aldrich; polyclonal, HPA001906, 1:500), MLH1 (Cell Marque, G168-7289, 1:100), PMS2 (Leica Biosystems; NCL-L-PMS2, 1:100), MSH2 (Cell Marque, G219-1129, 1:100), MSH6 (Leica Biosystems; NCL-L-MSH6, 1:100).
Comprehensive genomic profiling (CGP)
CGP was performed from FFPE samples using the Illumina TruSight Oncology 500 High-Throughput assay, then sequenced on an Illumina NextSeq 2000 NGS platform and finally analyzed with TruSight Oncology 500 Local App v2.1 as previously described23. Predetermined definition of TMB-high status was at least 10 mutations per megabase50. MSI status was determined by interrogation of 130 homopolymer MSI marker sites. MSI-H status was defined as ≥ 20%, MSI-low (MSI-L) as 20–1% and MS-stable (MSS) as < 1%, based on previous studies51.
Each alteration was classified according to the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG-AMP) five-tier system52. Only P, LP changes as well as VUSs were included into this study. Synonymous SNVs as well as non-synonymous SNVs with variant allele frequency (VAF) below 2% and / or total depth lower than 100× were excluded from the analysis32. Regarding CNVs, low-level copy number gain was defined as copy numbers between 3 and 5, while amplifications as 5 or higher53. Fusions were designated according to the of HUGO Gene Nomenclature Committee (HGNC) recommendations54, thus the 5′ fusion partners were listed first (before the double colon). Potential germline variants were excluded based on a gnomAD frequency of 1%.
pTERT mutation analysis
Difficulties regarding the detection of pTERT mutations are well known from the literature due to the GC-rich nature of this region55. Therefore, all cases were tested for pTERT hotspot variants (c.-124C>T and c.-146C>T) using Sanger sequencing on an ABI3500 platform (Thermo Fisher Scientific, Waltham, MA, USA) with custom-designed primers, namely forward: 5′-CACCCGTCCTGCCCCTTCACCTT-3′ and reverse: 5′-GGCTTCCCACGTGCGCAGCAGGA-3′.
Statistical analysis
Kaplan–Meier analysis, Mann–Whitney test and Fisher’s exact tests were performed using GraphPad Prism software version 8.0 (San Diego, CA, USA). CGP revealed mostly scattered or balanced alterations in the analyzed groups, therefore, the statistical analysis was restricted to the associations of pTERT, PTEN, DNMT3A and NF1 mutations as well as EGFR amplification with ATRX mutations. This was performed using Fisher’s exact tests followed by adjustment of p-values for multiple comparisons (padj) using the Benjamini–Hochberg method. In all procedures, p or padj < 0.05 was regarded to be level of statistical significance. The detailed oncoplot presented on Supplementary Fig. S3 was created using Maftools v2.14 R-package56.
Ethical approval and consent to participate
This retrospective study was conducted in accordance with the Declaration of Helsinki and informed consent was obtained from the participants. All procedures were approved by the National Scientific and Ethical Committee (TUKEB) of the Medical Research Council of Hungary (ETT) (statement: IV/51-1/2022/EKU).
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ALT:
-
Alternative lengthening of telomeres
- CGP:
-
Comprehensive genomic profiling
- CNV:
-
Copy number variation
- FFPE:
-
Formalin-fixed, paraffin-embedded
- FISH:
-
Fluorescent in situ hybridization
- GB:
-
Glioblastoma
- GTAKA:
-
Glioneuronal tumor with ATRX alteration, kinase fusion and anaplastic features
- H&E:
-
Hematoxylin and eosin
- HGAP:
-
High-grade astrocytoma with piloid features
- HR:
-
Homologous recombination
- HRD:
-
Homologous recombination repair deficiency
- IHC:
-
Immunohistochemistry
- InDel:
-
Insertion and deletion
- LP:
-
Likely pathogenic
- MMR-d:
-
Mismatch repair deficiency
- MSI:
-
Microsatellite instability
- NGS:
-
Next-generation sequencing
- OS:
-
Overall survival
- P:
-
Pathogenic
- RTK:
-
Receptor tyrosine kinase
- SNV:
-
Single nucleotide variant
- TMB:
-
Tumor mutational burden
- VAF:
-
Variant allele frequency
- VUS:
-
Variant of uncertain significance
- WHO:
-
World Health Organization
References
Louis, D. N. et al. The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro-oncology 23, 1231–1251. https://doi.org/10.1093/neuonc/noab106 (2021).
Khaddour, K., Johanns, T. M. & Ansstas, G. The landscape of novel therapeutics and challenges in glioblastoma multiforme: Contemporary state and future directions. Pharmaceuticals (Basel) https://doi.org/10.3390/ph13110389 (2020).
Killela, P. J. et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. U. S. A. 110, 6021–6026. https://doi.org/10.1073/pnas.1303607110 (2013).
Clynes, D., Higgs, D. R. & Gibbons, R. J. The chromatin remodeller ATRX: A repeat offender in human disease. Trends Biochem. Sci. 38, 461–466. https://doi.org/10.1016/j.tibs.2013.06.011 (2013).
Cesare, A. J. & Reddel, R. R. Alternative lengthening of telomeres: Models, mechanisms and implications. Nat. Rev. Genet. 11, 319–330. https://doi.org/10.1038/nrg2763 (2010).
Diplas, B. H. et al. The genomic landscape of TERT promoter wildtype-IDH wildtype glioblastoma. Nat. Commun. 9, 2087. https://doi.org/10.1038/s41467-018-04448-6 (2018).
Reinhardt, A. et al. Anaplastic astrocytoma with piloid features, a novel molecular class of IDH wildtype glioma with recurrent MAPK pathway, CDKN2A/B and ATRX alterations. Acta Neuropathol. 136, 273–291. https://doi.org/10.1007/s00401-018-1837-8 (2018).
Bogumil, H. et al. Glioneuronal tumor with ATRX alteration, kinase fusion and anaplastic features (GTAKA): A molecularly distinct brain tumor type with recurrent NTRK gene fusions. Acta Neuropathol. 145, 667–680. https://doi.org/10.1007/s00401-023-02558-0 (2023).
Leske, H. et al. MGMT methylation pattern of long-term and short-term survivors of glioblastoma reveals CpGs of the enhancer region to be of high prognostic value. Acta Neuropathol. Commun. 11, 139. https://doi.org/10.1186/s40478-023-01622-w (2023).
Krzywinski, M. et al. Circos: An information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. https://doi.org/10.1101/gr.092759.109 (2009).
Bielle, F. et al. Diffuse gliomas with FGFR3-TACC3 fusion have characteristic histopathological and molecular features. Brain Pathol. 28, 674–683. https://doi.org/10.1111/bpa.12563 (2018).
Ferreira, M. S. V. et al. Alternative lengthening of telomeres is the major telomere maintenance mechanism in astrocytoma with isocitrate dehydrogenase 1 mutation. J. Neuro-oncol. 147, 1–14. https://doi.org/10.1007/s11060-020-03394-y (2020).
Minasi, S. et al. Alternative lengthening of telomeres in molecular subgroups of paediatric high-grade glioma. Childs Nerv. Syst. 37, 809–818. https://doi.org/10.1007/s00381-020-04933-8 (2021).
Udugama, M. et al. Mutations inhibiting KDM4B drive ALT activation in ATRX-mutated glioblastomas. Nat. Commun. 12, 2584. https://doi.org/10.1038/s41467-021-22543-z (2021).
Papaemmanuil, E. et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 122, 3616–3627. https://doi.org/10.1182/blood-2013-08-518886 (2013).
Markova, J. et al. Prognostic impact of DNMT3A mutations in patients with intermediate cytogenetic risk profile acute myeloid leukemia. Eur. J. Haematol. 88, 128–135. https://doi.org/10.1111/j.1600-0609.2011.01716.x (2012).
Gaidzik, V. I. et al. Clinical impact of DNMT3A mutations in younger adult patients with acute myeloid leukemia: Results of the AML study group (AMLSG). Blood 121, 4769–4777. https://doi.org/10.1182/blood-2012-10-461624 (2013).
Ribeiro, A. F. et al. Mutant DNMT3A: A marker of poor prognosis in acute myeloid leukemia. Blood 119, 5824–5831. https://doi.org/10.1182/blood-2011-07-367961 (2012).
Fomchenko, E. I. et al. DNMT3A co-mutation in an IDH1-mutant glioblastoma. Cold Spring Harb. Mol. Case Stud. https://doi.org/10.1101/mcs.a004119 (2019).
Barresi, V. et al. IDH-wild type glioblastomas featuring at least 30% giant cells are characterized by frequent RB1 and NF1 alterations and hypermutation. Acta Neuropathol. Commun. 9, 200. https://doi.org/10.1186/s40478-021-01304-5 (2021).
Rong, T. et al. A rare manifestation of a presumed non-osteophilic brain neoplasm: Extensive axial skeletal metastases from glioblastoma with primitive neuronal components. Front. Oncol. 11, 760697. https://doi.org/10.3389/fonc.2021.760697 (2021).
Gonzalo, S. et al. DNA methyltransferases control telomere length and telomere recombination in mammalian cells. Nat. Cell Biol. 8, 416–424. https://doi.org/10.1038/ncb1386 (2006).
Bedics, G. et al. Germline MUTYH mutations and high-grade gliomas: Novel evidence for a potential association. Genes Chromosomes Cancer 61, 622–628. https://doi.org/10.1002/gcc.23054 (2022).
Holtkamp, N. et al. Characterization of the amplicon on chromosomal segment 4q12 in glioblastoma multiforme. Neuro-oncology 9, 291–297. https://doi.org/10.1215/15228517-2007-009 (2007).
Ferguson, S. D. et al. Targetable gene fusions associate with the IDH wild-type astrocytic lineage in adult gliomas. J. Neuropathol. Exp. Neurol. 77, 437–442. https://doi.org/10.1093/jnen/nly022 (2018).
Brennan, C. W. et al. The somatic genomic landscape of glioblastoma. Cell 155, 462–477. https://doi.org/10.1016/j.cell.2013.09.034 (2013).
Ohgaki, H. et al. Genetic pathways to glioblastoma: A population-based study. Cancer Res. 64, 6892–6899. https://doi.org/10.1158/0008-5472.CAN-04-1337 (2004).
Sener, U., Ruff, M. W. & Campian, J. L. Immunotherapy in glioblastoma: Current approaches and future perspectives. Int. J. Mol. Sci. https://doi.org/10.3390/ijms23137046 (2022).
Zhao, J. et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 25, 462–469. https://doi.org/10.1038/s41591-019-0349-y (2019).
Rodriguez, F. J. et al. Telomere alterations in neurofibromatosis type 1-associated solid tumors. Acta Neuropathol. Commun. 7, 139. https://doi.org/10.1186/s40478-019-0792-5 (2019).
Rodriguez, F. J., Vizcaino, M. A., Blakeley, J. & Heaphy, C. M. Frequent alternative lengthening of telomeres and ATRX loss in adult NF1-associated diffuse and high-grade astrocytomas. Acta Neuropathol. 132, 761–763. https://doi.org/10.1007/s00401-016-1619-0 (2016).
Luger, A. L. et al. Molecular matched targeted therapies for primary brain tumors-a single center retrospective analysis. J. Neuro-oncol. 159, 243–259. https://doi.org/10.1007/s11060-022-04049-w (2022).
Wang, Q. et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 32, 42-56 e46. https://doi.org/10.1016/j.ccell.2017.06.003 (2017).
Bedognetti, D., Roelands, J., Decock, J., Wang, E. & Hendrickx, W. The MAPK hypothesis: Immune-regulatory effects of MAPK-pathway genetic dysregulations and implications for breast cancer immunotherapy. Emerg. Top. Life Sci. 1, 429–445. https://doi.org/10.1042/ETLS20170142 (2017).
Barresi, V. et al. Ultra-mutation in IDH wild-type glioblastomas of patients younger than 55 years is associated with defective mismatch repair, microsatellite instability, and giant cell enrichment. Cancers https://doi.org/10.3390/cancers11091279 (2019).
Torre, M. et al. Molecular and clinicopathologic features of gliomas harboring NTRK fusions. Acta Neuropathol. Commun. 8, 107. https://doi.org/10.1186/s40478-020-00980-z (2020).
Mansour, B. et al. Epithelioid fibrous histiocytoma: Three diagnostically challenging cases with novel ALK gene fusions, unusual storiform growth pattern, and a prominent spindled morphology. Virchows Arch. 481, 751–757. https://doi.org/10.1007/s00428-022-03418-0 (2022).
Touat, M. et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 580, 517–523. https://doi.org/10.1038/s41586-020-2209-9 (2020).
Leon-Castillo, A. et al. Interpretation of somatic POLE mutations in endometrial carcinoma. J. Pathol. 250, 323–335. https://doi.org/10.1002/path.5372 (2020).
Shlien, A. et al. Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nat. Genet. 47, 257–262. https://doi.org/10.1038/ng.3202 (2015).
Nagasaka, T. et al. Somatic hypermethylation of MSH2 is a frequent event in Lynch Syndrome colorectal cancers. Cancer Res. 70, 3098–3108. https://doi.org/10.1158/0008-5472.CAN-09-3290 (2010).
George, S. L. et al. Therapeutic vulnerabilities in the DNA damage response for the treatment of ATRX mutant neuroblastoma. EBioMedicine 59, 102971. https://doi.org/10.1016/j.ebiom.2020.102971 (2020).
Garbarino, J., Eckroate, J., Sundaram, R. K., Jensen, R. B. & Bindra, R. S. Loss of ATRX confers DNA repair defects and PARP inhibitor sensitivity. Transl. Oncol. 14, 101147. https://doi.org/10.1016/j.tranon.2021.101147 (2021).
Yuan, M. et al. Therapeutic vulnerability to ATR inhibition in concurrent NF1 and ATRX-deficient/ALT-positive high-grade solid tumors. Cancers https://doi.org/10.3390/cancers14123015 (2022).
Sim, H. W., Galanis, E. & Khasraw, M. PARP inhibitors in glioma: A review of therapeutic opportunities. Cancers https://doi.org/10.3390/cancers14041003 (2022).
Xavier, M. A., Rezende, F., Titze-de-Almeida, R. & Cornelissen, B. BRCAness as a biomarker of susceptibility to PARP inhibitors in glioblastoma multiforme. Biomolecules https://doi.org/10.3390/biom11081188 (2021).
Lord, C. J. & Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 16, 110–120. https://doi.org/10.1038/nrc.2015.21 (2016).
O’Sullivan Coyne, G. et al. PARP inhibitor applicability: Detailed assays for homologous recombination repair pathway components. Onco Targets Ther. 15, 165–180. https://doi.org/10.2147/OTT.S278092 (2022).
Zeitlberger, A. M. et al. Next generation sequencing in adult patients with glioblastoma in Switzerland: A multi-centre decision analysis. J. Neuro-oncol. 158, 359–367. https://doi.org/10.1007/s11060-022-04022-7 (2022).
Marabelle, A. et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: Prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 21, 1353–1365. https://doi.org/10.1016/S1470-2045(20)30445-9 (2020).
Kang, S. Y. et al. Comparative analysis of microsatellite instability by next-generation sequencing, MSI PCR and MMR immunohistochemistry in 1942 solid cancers. Pathol Res Pract 233, 153874. https://doi.org/10.1016/j.prp.2022.153874 (2022).
Li, M. M. et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn 19, 4–23. https://doi.org/10.1016/j.jmoldx.2016.10.002 (2017).
Pritchard, C. C. et al. Validation and implementation of targeted capture and sequencing for the detection of actionable mutation, copy number variation, and gene rearrangement in clinical cancer specimens. J. Mol. Diagn. 16, 56–67. https://doi.org/10.1016/j.jmoldx.2013.08.004 (2014).
Bruford, E. A. et al. HUGO Gene Nomenclature Committee (HGNC) recommendations for the designation of gene fusions. Leukemia 35, 3040–3043. https://doi.org/10.1038/s41375-021-01436-6 (2021).
Lee, H. et al. Detection of TERT promoter mutations using targeted next-generation sequencing: Overcoming GC bias through trial and error. Cancer Res. Treat. 54, 75–83. https://doi.org/10.4143/crt.2021.107 (2022).
Mayakonda, A., Lin, D. C., Assenov, Y., Plass, C. & Koeffler, H. P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 28, 1747–1756. https://doi.org/10.1101/gr.239244.118 (2018).
Acknowledgements
The authors are grateful for the professional technical assistance to Krisztina Császár, Lajos László Hegyi, Dóra Kapczár, Mónika Paulusz, Anna Tamási and Erik Zajta. We are also grateful for the colleagues at the Department of Pathology of University of Szeged as well as County Hospital Győr, Petz Aladár Hospital for their help in tissue sample collection.
Funding
Open access funding provided by Semmelweis University. This work was funded by the EU's Horizon 2020 Research and Innovation Program under grant agreement No. 739593, by the Ministry of Innovation and Technology of Hungary from the National Research, Development and Innovation Fund, financed under the TKP2021-EGA-24 and TKP2021-NVA-11 funding schemes as well as the ÚNKP-22-3-II New National Excellence Program of the Ministry for Culture and Innovation from the National Research, Development and Innovation Fund.
Author information
Authors and Affiliations
Contributions
G.B., B.B., T.N. and G.P. performed the NGS assay and other molecular studies. P.Sz. collected the clinical data and contributed to the statistical analyses. B.S. designed the study and wrote the manuscript. N.K., H.R., L.R. and Cs.B. contributed to the design of the study and critically reviewed the manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bedics, G., Szőke, P., Bátai, B. et al. Novel, clinically relevant genomic patterns identified by comprehensive genomic profiling in ATRX-deficient IDH-wildtype adult high-grade gliomas. Sci Rep 13, 18436 (2023). https://doi.org/10.1038/s41598-023-45786-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-45786-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
