
Abstract
Mesenchymal tumors driven by NTRK fusions are clinically and morphologically heterogeneous. With an increasing number of clinicopathological entities being associated with NTRK fusions, the diagnostic and predictive value of the identification of NTRK fusions is uncertain. Recently, mesenchymal tumors in the gastrointestinal tract with NTRK fusions were described as gastrointestinal stromal tumors (GIST), but the nosology of such neoplasms remains controversial. We report eight mesenchymal tumors involving the gastrointestinal tract with NTRK1 or NTRK3 rearrangements. The tumors occurred in six children and two adults, five males and three females (age range 2 months–55 years; median 3.5 years), and involved the small intestine (n = 4), stomach (n = 2), rectum (n = 1), and mesentery (n = 1). Clinical outcomes were variable, ranging from relatively indolent (n = 2) to aggressive diseases (n = 2). Morphologically, the tumors were heterogeneous and could be classified in the following three groups: (1) infantile fibrosarcoma involving the gastrointestinal tract (n = 4), enriched for NTRK3 fusions; (2) low-grade CD34-positive, S100 protein-positive spindle-cell tumors, associated with NTRK1 fusions (n = 2); and (3) unclassified high-grade spindle-cell sarcomas, with NTRK1 fusions (n = 2). By immunohistochemistry, the tumors demonstrated diffuse pan-TRK expression, of variable intensity, and lacked a specific line of differentiation. Four cases expressed CD34, which was coexpressed with S100 protein in three cases. Expression of SOX10, KIT, and DOG1 was consistently absent. Molecular genetic testing identified TPM3–NTRK1 (n = 3), TPR–NTRK1, LMNA–NTRK1, and ETV6–NTRK3 (n = 2), and SPECC1L–NTRK3 in-frame gene fusions. We conclude that the evaluation of mesenchymal spindle-cell neoplasms of the gastrointestinal tract without a definitive line of differentiation should include interrogation of NTRK alterations, particularly in pediatric patients. Mesenchymal tumors of the gastrointestinal tract with NTRK rearrangements are clinically and morphologically heterogeneous, and few, if any, seem related to GIST.
Similar content being viewed by others

Introduction
Oncogenic gene fusions involving the neurotrophic receptor tyrosine kinase genes NTRK1, NTRK2, and NTRK3 (“NTRK fusions”) are major oncogenic drivers in multiple tumor types. In some tumor types, such as infantile fibrosarcoma, NTRK fusions are present in most cases and are considered defining molecular features that determine tumor biology. In others, such as colorectal adenocarcinoma, NTRK fusions are present in a very small fraction of cases, without distinctive clinical or morphological features. In any case, the presence of a functional clonal NTRK fusion is associated with high response rates to TRK inhibitors, which may therefore be clinically relevant for some aggressive tumors.
From a diagnostic perspective, the relevance of NTRK fusions is variable, depending on the clinicopathological context. In mesenchymal neoplasia, NTRK fusions were first identified in infantile fibrosarcoma and cellular congenital mesoblastic nephroma, two clinical presentations of a rare pediatric malignant spindle-cell sarcoma with distinctive morphology, driven by clonal oncogenic tyrosine kinase fusions, most commonly ETV6–NTRK3 fusions. Evidence of NTRK3 or ETV6 rearrangement in a highly cellular spindle-cell sarcoma affecting an infant was considered specific for these entities, and has been widely used to support these histopathologic diagnoses [1, 2]. More recently, NTRK fusions have been described in small series of other rare mesenchymal tumors that may arise at various soft tissue and visceral locations in children and adults, such as lipofibromatosis-like neural tumors [3], inflammatory myofibroblastic tumors [4], uterine and cervical sarcomas with “fibrosarcoma”-like features [5,6,7], spindle-cell sarcomas with myopericytic growth pattern [8], and spindle-cell sarcomas resembling malignant peripheral nerve sheath tumors [9, 10]. While subsets of these tumors have been described as novel entities, others have been difficult to classify due to the lack of distinctive clinicopathological features. In addition, at least two spindle-cell sarcomas with NTRK fusions involving the gastrointestinal tract have been recently described as molecular variants of gastrointestinal stromal tumor (GIST), albeit with limited morphologic and immunophenotypic support for this diagnosis [11, 12]. Herein, we present the clinical, morphological, immunophenotypic, and molecular genetic features of eight gastrointestinal mesenchymal tumors with NTRK fusions and discuss the features that warrant their distinction from GIST.
Materials and methods
Eight cases were retrieved through a retrospective review of the authors’ institutional and consultation archives, and re-reviewed. Four cases had been classified as infantile fibrosarcoma involving the gastrointestinal tract at the time of diagnosis; four additional cases were identified as unclassified mesenchymal tumors involving the gastrointestinal tract, in which molecular studies identified rearrangements of NTRK1 and NTRK3. Some aspects of three cases have been previously published (cases 2, 3, and 7) [13, 14]. Additional information, such as clinical follow-up and response to therapy, was extracted from the medical record or provided by referring pathologists. Morphologic features were assessed on hematoxylin and eosin-stained slides. All testing was performed in CLIA approved laboratories and after pertinent approval from institutional review boards.
Immunohistochemistry (IHC)
All cases were assessed for protein expression with commercially available antibodies to the following antigens, using formalin-fixed, paraffin-embedded tissue sections and routine laboratory protocols: pan-TRK, CD117 (KIT), DOG1, S100 protein, and CD34. pan-TRK IHC was performed using the commercially available clone EPR17341 antibody (Abcam, Cambridge, MA, USA), which reacts with the conserved C-terminal region of the tropomyosin receptor kinase proteins A, B, and C. Positive staining was identified and included all patterns (membranous, cytoplasmic, perinuclear, or nuclear). Additional IHC performed at the time of diagnosis in subsets of cases included: SOX10 (n = 5), SMA (n = 4), STAT6 (n = 2), desmin (n = 2), pan-keratin (n = 1), HMB45 (n = 1), MiTF (n = 1), melan-A (n = 1), WT1 (n = 1), GFAP (n = 1), claudin-1 (n = 1), EMA (n = 1), GLUT1 (n = 1), SDHB (n = 1), and H3K27me3 (n = 1).
DNA and RNA sequencing
Sequencing was performed on clinically approved sequencing platforms at the institutions involved, all of which interrogate structural rearrangements in the three NTRK genes, NTRK1, NTRK2, and NTRK3. Four tumors (cases 1, 2, 3, and 7) were analyzed using hybrid capture DNA-based targeted panels (UCSF500 Cancer Gene Panel and OncoPanel) that include probes for exons and select introns of NTRK1, NTRK2, and NTRK3, as well as ETV6 exonic and intronic probes [15, 16]. Two cases (cases 5 and 8) were analyzed using an in-house developed assay that utilizes RNA targeted sequencing involving single primer extension target enrichment and unique molecular indices technologies to identify rearrangements. Reference transcript variants were NM_002529.3 (NTRK1), NM_006180.4 (NTRK2), and NM_001012338 (NTRK3). Case 6 was sequenced with the TruSight RNA Fusion panel (Illumina, San Diego, CA, USA), as previously described [17]. Case 4 was sequenced using GeneTrails Comprehensive Solid Tumor Panel™, a panel from Knight Diagnostics that involves both a DNA and an RNA component.
Fluorescence in situ hybridization (FISH)
Two cases (cases 1 and 2) were analyzed with FISH using commercial break-apart FISH probes for ETV6 in accordance with standard protocols in our laboratories.
Results
Table 1 summarizes the clinicopathological, immunohistochemical, and molecular genetic features of eight gastrointestinal mesenchymal tumors with NTRK fusions.
Clinicopathological features
The patients were six children and two adults (age range 2 months–55 years old, median: 3.5 years), five male and three females. Tumor size ranged from 3.0 to 12.5 cm (mean: 8.1 cm). Tumors involved the small bowel [4], stomach [2], rectum [1], and mesentery [1]. Clinical follow-up was available for four of the eight patients (patients 2, 5, 7, and 8). Outcomes were variable, ranging from indolent tumors (patients 5 and 8) to aggressive sarcomas (patients 2 and 7). Patient 2 died of complications arising from local extension of the tumor (size: 12.5 cm). Patient 5 underwent surgical resection of the mass with negative findings on a 4-month follow-up CT scan; the patient developed a plasma cell disorder a month after follow-up, but imaging did not suggest tumor recurrence. Of the two morphologically high-grade tumors, one patient has been sequentially enrolled in clinical trials of TRK inhibitors (patient 7) [14]; she is alive, with metastatic disease, 50 months after initial diagnosis. Patient 8, a 7-year old boy treated with larotrectinib, has no evidence of disease after 3.5 months of follow-up; repeat CT and PET scans did not identify new lesions.
Microscopic morphology
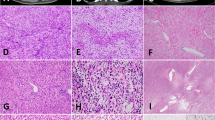
The microscopic appearances were heterogeneous, both between tumors and, in some cases, within them. Four tumors (cases 1–4) were composed of relatively monomorphic proliferations of small spindled cells, with inconspicuous cytoplasm and round-to-ovoid nuclei with regular contours and fine chromatin, arranged in dense sheets or in vague fascicles (Fig. 1). Despite similarities, each of these four cases showed variable features, such as focally prominent stromal blood vessels, patchy chronic inflammation, and focal necrosis (case 1); focal round-cell morphology (case 2); focal hyalinization (case 3, Fig. 1e); and ectatic “hemangiopericytoma-like” vessels (case 4). Infiltrative growth through preexisting muscle and glandular structures was frequently encountered. The mitotic index was variable: low in two cases but ranging up to 44 mitotic figures/10 high-power fields (HPFs) in case 2. The morphologic features of these four cases are those of infantile fibrosarcoma.

a Highly cellular proliferation of monomorphic spindle cells with eosinophilic cytoplasm and hyperchromatic ovoid nuclei (case 1). b Cellular proliferation of short spindled cells with abundant mitotic figures (44/10 HPF) (case 2). This tumor demonstrated locally aggressive growth, such that the patient died within 2 weeks of presentation. c Cellular fascicles of monomorphic spindle cells (case 3). d Cytologically bland spindle-cell proliferation in vaguely fascicular arrangement (case 4). e Spindle cells with inconspicuous cytoplasm with stromal and perivascular hyalinization (case 3). f Highly infiltrative proliferation of monomorphic spindle-cell dissecting through glands in the antral mucosa (case 3).
Case 5 consisted of a monomorphic proliferation of bland spindled cells haphazardly arranged (Fig. 2a). An unusual feature of this case was the presence of numerous intracytoplasmic vacuoles (“pseudolipoblasts”), mimicking the appearance of some GIST (Fig. 2b). Case 6 consisted of a dense and regionally heterogeneous proliferation of spindled cells, with limited nuclear atypia, arranged in a variety of architectural patterns, including fascicular, storiform, and “herringbone” patterns of variable cellularity (Fig. 2c, d). Infiltration through perigastric fat was prominent, mimicking the so-called “honeycomb” infiltration of subcutaneous fat in dermatofibrosarcoma protuberans.

a Monomorphic spindle-cell population with intracytoplasmic vacuoles (b), mimicking lipoblasts or GIST cells (case 5). c Storiform pattern of growth, focally reminiscent of dermatofibrosarcoma protuberans (case 6); in other areas of the same tumor, long fascicles of spindle cells resembling desmoid or low-grade MPNST (d).
Two cases (cases 7 and 8) exhibited high-grade morphological features (Fig. 3), including variability in cell size and nuclear atypia, either throughout the tumor (case 7) or more limited in extent with transition from low-grade spindle-cell areas to focal high-grade epithelioid and pleomorphic areas (case 8). Both neoplasms were intramural lesions and lacked any adipocytic component.

a Highly infiltrative malignant spindled cell tumor (case 7). Some areas showed myxoid stroma generating an epithelioid or round-cell morphology (b). Case 8 demonstrated transition from low-grade spindle-cell areas (c) to high-grade areas resembling pleomorphic sarcoma (d).
Immunohistochemistry
Pan-TRK IHC showed diffuse expression in all cases, with variable intensity (strong: 5, moderate: 3), both in cytoplasmic and nuclear patterns (Table 1; Figs. 4 and 5). Four cases exhibited CD34 expression, with coexpression of S100 protein in three cases (cases 5, 6, and 8) (Fig. 4); SOX10 expression was consistently absent. CD117 and DOG1 expression was undetectable in all cases (Fig. 5). Multifocal expression of SMA was present in cases 2 and 3. Pertinent markers were performed to rule out alternative potential diagnoses (Table 2): case 1 lacked expression of desmin, WT1, and GFAP; case 5 lacked expression of claudin-1, GLUT1, EMA, and STAT6; case 6 lacked expression of desmin, STAT6, ALK, or pan-keratin markers; and case 8—a high-grade tumor—was MiTF, HMB45, and melan-A negative and retained SDHB and H3K27me3 expression.

TRK expression was diffused in all cases with variable intensity: cytoplasmic staining of strong intensity shown in case 7 (a) and moderately strong cytoplasmic staining in case 1 (b). CD34 (c) and S100 (d) were coexpressed in three tumors (case 6). SOX10 immunohistochemistry was consistently negative.

a A proliferation of bland spindle cells with palely eosinophilic cytoplasm and occasional perinuclear vacuoles may raise a morphological diagnosis of GIST (case 5). b TRK immunohistochemistry demonstrated diffuse and strong expression in this tumor with a TPM3–NTRK1 fusion. None of the tumors in this series expressed KIT (c) or DOG1 (d).
Molecular genetic alterations in NTRK genes
Genomic rearrangements or gene fusions involving NTRK genes were identified in seven cases, involving NTRK1 in five tumors and NTRK3 in two tumors. The 3′ end of the NTRK gene sequence was juxtaposed to the 5′ sequence of a series of partner genes, all of which have been previously implicated in NTRK fusions in different tumor types [18, 19]. One tumor with characteristic clinicopathological features of infantile fibrosarcoma showed a rearrangement of ETV6 (case 1), consistent with an ETV6–NTRK3 fusion.
The most common fusion type was TPM3–NTRK1 (n = 3) followed by ETV6–NTRK3 (n = 2). Additional fusions, observed in a single tumor each, included TPR–NTRK1, LMNA–NTRK1, and SPECC1L–NTRK3. The latter SPECC1L–NTRK3 fusion was also amplified, as detected by an increased number of reads mapping to NTRK3 by DNA sequencing, corresponding to the presence of multiple copies. The breakpoints of all NTRK fusions occurred 5′ to the sequence encoding the kinase domain of the NTRK protein implicated, supporting the formation of a functional fusion transcript preserving the entire tyrosine kinase domain. Additional rearrangements potentially leading to functional fusion genes were not identified.
NTRK3 fusions were detected only in cases with morphological features of infantile fibrosarcoma. There was no additional detectable correlation between fusion variant and clinicopathological or immunohistochemical features.
Discussion
We describe a series of eight mesenchymal tumors with NTRK fusions involving the gastrointestinal tract, demonstrating substantial heterogeneity in clinical, morphological, and immunohistochemical features. Three groups of tumors with NTRK fusions were identified in this series, spanning a clinical spectrum from clinically benign to lethal, despite sharing a similar driver oncogenic gene fusion. These groups are (1) infantile fibrosarcoma involving the gastrointestinal tract, enriched for NTRK3 fusions; (2) morphologically low-grade CD34-positive, S100 protein-positive spindle-cell tumors, associated with NTRK1 fusions; and (3) unclassified high-grade spindle-cell sarcomas, which may have epithelioid and pleomorphic areas, with NTRK1 fusions.
Although the GI tract is an infrequent location for infantile fibrosarcoma, multiple cases have been described in the ileum, colon, and rectum [20,21,22]. Many intestinal infantile fibrosarcomas present with a characteristic clinical picture of congenital intestinal perforation and meconium peritonitis [23, 24]. As in patient 2 in this series, clinical complications of these tumors often occur due to local invasion and direct involvement of the gastrointestinal tract. Molecularly, NTRK3 fusions predominate in this group, in particular ETV6–NTRK3, but NTRK1 fusions similar to the one observed in case 3 have been documented [25].
Overlap also exists between other tumors described herein and their somatic soft tissue counterparts. Low-grade spindle-cell soft tissue tumors with coexpression of CD34 and S100 protein seem to be enriched for NTRK1 fusions, and may represent a clinicopathological entity, although there is substantial morphological heterogeneity within this group [10]. Case 5 could be interpreted as a deep-seated counterpart of the superficially located lipofibromatosis-like neural tumors [3]. Case 6 showed in part similar features, with a wider range of morphological patterns. The variably cellular fascicles of spindled cells showed striking regional architectural variation and focal mild nuclear atypia. Different areas resembled dermatofibrosarcoma protuberans, desmoid fibromatosis, or low-grade MPNST. All of these potential differential diagnoses could be readily excluded by integrating clinical, morphological, and immunohistochemical findings, but illustrate the remarkable intratumoral morphologic heterogeneity of some NTRK1-rearranged mesenchymal tumors.
Subsets of NTRK1 and NTRK3-rearranged soft tissue sarcomas show high‐grade morphological features, and are typically composed of cellular fascicles of mitotically active, hyperchromatic spindled cells, often with necrosis [8, 10]. Depending on their immunophenotype, some of these tumors have been described in the literature as “MPNST‐like”—due to expression of S100 protein, with or without CD34 coexpression [10]—or “fibrosarcoma‐like”—with or without CD34 expression and absence of S100 protein [6, 8, 10, 25]. Similar to low-grade NTRK1-rearranged tumors, there is substantial intratumoral heterogeneity in this group. Cases 7 and 8 of this series are morphologically high-grade sarcomas but did not show evidence of a specific line of differentiation or similarities to recognized sarcoma histotypes. Consequently, cases 7 and 8 could be considered “fibrosarcoma-like,” although in the absence of distinctive morphologic or immunophenotypic features, we favor the designation “unclassified high-grade spindle-cell sarcoma with NTRK1 rearrangement.” As dramatically illustrated by case 7, in this small subset of high-grade sarcomas, the identification of NTRK fusions may be of great clinical and therapeutic relevance.
Of special interest is the relationship of the tumors described herein with GIST. In addition to morphological differences, the absence of KIT and DOG1 immunohistochemical expression permit the distinction of NTRK-rearranged tumors from GIST, and highlights important phenotypic differences between these tumor types. At least two gastrointestinal tumors with ETV6–NTRK3 fusions have been described as GIST in the literature [11, 12], leading some authors to advocate for the interrogation of NTRK fusions in so-called “quadruple negative” GIST—GISTs lacking mutations in KIT, PDGFRA, BRAF/KRAS/NF1, and SDH-subunits. It is not possible to independently evaluate the diagnosis of “GIST” for one of these two cases, as pertinent morphological and immunohistochemical information was not provided [12]. The other published case, however, a high-grade epithelioid tumor of the rectum in a 44-year old man, demonstrated diffuse KIT (CD117) and DOG1 expression, consistent with the diagnosis of GIST [11]. Given the pleiotropism of ETV6–NTRK3 oncogenic fusion, with transforming capacity in neoplasia as heterogeneous as infantile fibrosarcoma, secretory breast carcinoma, mammary analog secretory carcinoma of salivary glands, or radiation-associated thyroid cancer [26,27,28], it is certainly conceivable that this fusion drives the formation of intestinal GIST in unusual cellular contexts. If so, this is an extraordinarily rare occurrence, because our review of targeted sequencing data from 738 GIST (including 312 from the authors’ institutions, and 426 from the publicly available AACR Project GENIE dataset) did not identify any cases with NTRK rearrangements. Rigorous clinicopathological characterization of purported GISTs from patients that have been enrolled in clinical trials evaluating TRK inhibitors would be of great interest to expand these observations.
All five 5′ partner genes involved in NTRK fusions in the cases presented here have been implicated previously in NTRK fusions in published cases. All of them contribute oligomerization domains to the fusion oncoprotein (coiled-coil domains from TPM3, SPECC1L, LMNA, TPR, and a SAM/PNT domain from ETV6), which likely lead to constitutive kinase activation and sustained oncogenic signaling. In this series, the pattern of pan-TRK immunohistochemical staining did not correlate with the predicted subcellular localization of the 5′ member of the fusion, suggesting that IHC may not be a reliable method to identify NTRK fusion variants. Similarly, NTRK1 and NTRK3 oncoproteins did not show differential subcellular distribution in these cases, as assessed by IHC. Despite the high sensitivity of pan-TRK IHC observed in this series, recent studies have documented reduced sensitivity—particularly in tumors with NTRK3 fusions [29]—as well as limited specificity due to TRK expression in other round and spindle-cell sarcomas, including tumors with BCOR, YWHAE, and ALK rearrangements [29,30,31]. These findings indicate that the diagnostic use of pan-TRK IHC requires careful contextual interpretation and, in many instances, confirmation with molecular methods.
In summary, the results of the present study show that gastrointestinal mesenchymal tumors with NTRK1 or NTRK3 rearrangements demonstrate substantial clinical and morphological heterogeneity, and are generally unrelated to GIST. In addition to infantile fibrosarcoma, NTRK fusions may drive both low-grade and high-grade spindle-cell mesenchymal tumors in the gastrointestinal tract, with a nondistinctive immunophenotype and no clear line of differentiation. Interrogation of NTRK alterations should be considered during evaluation of such tumors with unclear line of differentiation, particularly in pediatric patients. Identification of an NTRK oncogenic fusion in a high-grade mesenchymal tumor of the gastrointestinal tract may provide a clinically useful therapeutic target in selected cases.
References
Knezevich SR, McFadden DE, Tao W, Lim JF, Sorensen PH. A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nat Genet. 1998;18:184–7.
Chung EB, Enzinger FM. Infantile fibrosarcoma. Cancer. 1976;38:729–39.
Agaram NP, Zhang L, Sung YS, Chen CL, Chung CT, Antonescu CR, et al. Recurrent NTRK1 gene fusions define a novel subset of locally aggressive lipofibromatosis-like neural tumors. Am J Surg Pathol. 2016;40:1407–16.
Alassiri AH, Ali RH, Shen Y, Lum A, Strahlendorf C, Deyell R, et al. ETV6-NTRK3 is expressed in a subset of ALK-negative inflammatory myofibroblastic tumors. Am J Surg Pathol. 2016;40:1051–61.
Croce S, Hostein I, Longacre TA, Mills AM, Perot G, Devouassoux-Shisheboran M, et al. Uterine and vaginal sarcomas resembling fibrosarcoma: a clinicopathological and molecular analysis of 13 cases showing common NTRK-rearrangements and the description of a COL1A1-PDGFB fusion novel to uterine neoplasms. Mod Pathol. 2019;32:1008–22.
Chiang S, Cotzia P, Hyman DM, Drilon A, Tap WD, Zhang L, et al. NTRK fusions define a novel uterine sarcoma subtype with features of fibrosarcoma. Am J Surg Pathol. 2018;42:791–8.
Rabban JT, Devine P, Sangoi AR, Poder L, Alvarez E, Davis JL, et al. NTRK fusion cervical sarcoma: a report of 3 cases, emphasizing morphological and immunohistochemical distinction from other uterine sarcomas, including adenosarcoma. Histopathology. 2020;77:100–11.
Haller F, Knopf J, Ackermann A, Bieg M, Kleinheinz K, Schlesner M, et al. Paediatric and adult soft tissue sarcomas with NTRK1 gene fusions: a subset of spindle cell sarcomas unified by a prominent myopericytic/haemangiopericytic pattern. J Pathol. 2016;238:700–10.
Suurmeijer AJ, Dickson BC, Swanson D, Zhang L, Sung YS, Huang HY, et al. The histologic spectrum of soft tissue spindle cell tumors with NTRK3 gene rearrangements. Genes Chromosomes Cancer. 2019;58:739–46.
Suurmeijer AJH, Dickson BC, Swanson D, Zhang L, Sung YS, Cotzia P, et al. A novel group of spindle cell tumors defined by S100 and CD34 co-expression shows recurrent fusions involving RAF1, BRAF, and NTRK1/2 genes. Genes Chromosomes Cancer. 2018;57:611–21.
Brenca M, Rossi S, Polano M, Gasparotto D, Zanatta L, Racanelli D, et al. Transcriptome sequencing identifies ETV6-NTRK3 as a gene fusion involved in GIST. J Pathol. 2016;238:543–9.
Shi E, Chmielecki J, Tang CM, Wang K, Heinrich MC, Kang G, et al. FGFR1 and NTRK3 actionable alterations in “Wild-Type” gastrointestinal stromal tumors. J Transl Med. 2016;14:339.
Rudzinski ER, Lockwood CM, Stohr BA, Vargas SO, Sheridan R, Black JO, et al. Pan-Trk immunohistochemistry identifies NTRK rearrangements in pediatric mesenchymal tumors. Am J Surg Pathol. 2018;42:927–35.
Hemming ML, Nathenson MJ, Lin JR, Mei S, Du Z, Malik K, et al. Response and mechanisms of resistance to larotrectinib and selitrectinib in metastatic undifferentiated sarcoma harboring oncogenic fusion of NTRK1. JCO Precis Oncol. 2020;4:79–90.
Garcia EP, Minkovsky A, Jia Y, Ducar MD, Shivdasani P, Gong X, et al. Validation of OncoPanel: a targeted next-generation sequencing assay for the detection of somatic variants in cancer. Arch Pathol Lab Med. 2017;141:751–8.
Kline CN, Joseph NM, Grenert JP, van Ziffle J, Talevich E, Onodera C, et al. Targeted next-generation sequencing of pediatric neuro-oncology patients improves diagnosis, identifies pathogenic germline mutations, and directs targeted therapy. Neuro Oncol. 2017;19:699–709.
Dickson BC, Swanson D. Targeted RNA sequencing: a routine ancillary technique in the diagnosis of bone and soft tissue neoplasms. Genes Chromosomes Cancer. 2019;58:75–87.
Gatalica Z, Xiu J, Swensen J, Vranic S. Molecular characterization of cancers with NTRK gene fusions. Mod Pathol. 2019;32:147–53.
Cocco E, Scaltriti M, Drilon A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol. 2018;15:731–47.
Parmar V, Peters RT, Cheesman E, Edi-Osagie N, Craigie RJ. Congenital infantile fibrosarcoma of the colon: a case series and literature review. Pediatr Surg Int. 2014;30:1079–85.
van Niekerk ML, Nel WA, Slavik T. Infantile fibrosarcoma of the ileum presenting with congenital bowel obstruction. J Pediatr Surg. 2010;45:461–2.
Buccoliero AM, Castiglione F, Rossi Degl’Innocenti D, Maio V, Taddei A, Sardi I. et al. Congenital/Infantile fibrosarcoma of the colon: morphologic, immunohistochemical, molecular, and ultrastructural features of a relatively rare tumor in an extraordinary localization. J Pediatr Hematol Oncol. 2008;30:723–7.
Berrebi D, Fournet JC, Boman F, Fabre M, Philippe-Chomette P, Branchereau S, et al. Intestinal congenital/infantile fibrosarcoma: a new clinico-pathological entity? Pediatr Surg Int. 2015;31:375–9.
Rizkalla H, Wildgrove H, Quinn F, Capra M, O’Sullivan MJ. Congenital fibrosarcoma of the ileum: case report with molecular confirmation and literature review. Fetal Pediatr Pathol. 2011;30:156–60.
Davis JL, Lockwood CM, Stohr B, Boecking C, Al-Ibraheemi A, DuBois SG, et al. Expanding the spectrum of pediatric NTRK-rearranged mesenchymal tumors. Am J Surg Pathol. 2019;43:435–45.
Tognon C, Knezevich SR, Huntsman D, Roskelley CD, Melnyk N, Mathers JA, et al. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell. 2002;2:367–76.
Skalova A, Vanecek T, Sima R, Laco J, Weinreb I, Perez-Ordonez B, et al. Mammary analogue secretory carcinoma of salivary glands, containing the ETV6-NTRK3 fusion gene: a hitherto undescribed salivary gland tumor entity. Am J Surg Pathol. 2010;34:599–608.
Leeman-Neill RJ, Kelly LM, Liu P, Brenner AV, Little MP, Bogdanova TI, et al. ETV6-NTRK3 is a common chromosomal rearrangement in radiation-associated thyroid cancer. Cancer. 2014;120:799–807.
Solomon JP, Linkov I, Rosado A, Mullaney K, Rosen EY, Frosina D, et al. NTRK fusion detection across multiple assays and 33,997 cases: diagnostic implications and pitfalls. Mod Pathol. 2020;33:38–46.
Yamamoto H, Nozaki Y, Kohashi K, Kinoshita I, Oda Y. Diagnostic utility of pan‐Trk immunohistochemistry for inflammatory myofibroblastic tumours. Histopathology. 2020;76:774–8.
Kao Y, Sung Y, Argani P, Swanson D, Alaggio R, Tap W, et al. NTRK3 overexpression in undifferentiated sarcomas with YWHAE and BCOR genetic alterations. Mod Pathol. 2020;33:1341–9.
Acknowledgements
The authors are indebted to the following pathologists who kindly contributed case materials and clinical follow-up information when available: Roanh Le Dinh, Hanoi, Vietnam; Lucas F. Abrahao-Machado, Botucatu, Brazil; Xiaohua Qian, Boston, USA; Geling Li, Alabama, USA; and Jeffrey Schowinsky, Colorado, USA. This work was supported by the SARC LMSARC research fund (AM-E); the Brigham and Women’s Hospital Program in Precision Medicine (AM-E); and the GIST Cancer Research Fund (GCRF) (JAF).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Presented in part at the 109th United States and Canadian Pathology Meeting, March 2020, Los Angeles, CA, USA
Rights and permissions
About this article

Cite this article
Atiq, M.A., Davis, J.L., Hornick, J.L. et al. Mesenchymal tumors of the gastrointestinal tract with NTRK rearrangements: a clinicopathological, immunophenotypic, and molecular study of eight cases, emphasizing their distinction from gastrointestinal stromal tumor (GIST). Mod Pathol 34, 95–103 (2021). https://doi.org/10.1038/s41379-020-0623-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-020-0623-z
This article is cited by
-
NTRK2 expression in gastrointestinal stromal tumors with a special emphasis on the clinicopathological and prognostic impacts
Scientific Reports (2024)
-
Evaluation of NTRK expression and fusions in a large cohort of early-stage lung cancer
Clinical and Experimental Medicine (2024)
-
English version of Japanese Clinical Practice Guidelines 2022 for gastrointestinal stromal tumor (GIST) issued by the Japan Society of Clinical Oncology
International Journal of Clinical Oncology (2024)
-
Japanese Society of Medical Oncology/Japan Society of Clinical Oncology/Japanese Society of Pediatric Hematology/Oncology-led clinical recommendations on the diagnosis and use of tropomyosin receptor kinase inhibitors in adult and pediatric patients with neurotrophic receptor tyrosine kinase fusion-positive advanced solid tumors
International Journal of Clinical Oncology (2023)
-
New Tyrosine Kinase Inhibitors for the Treatment of Gastrointestinal Stromal Tumors
Current Oncology Reports (2022)
