
Abstract
NUTM1 gene rearrangements were originally identified in NUT carcinoma. Recently, NUTM1 has been discovered to rearrange with a variety of gene partners in malignancies of diverse location and type. Only one NUTM1-rearranged tumor occurring in the colon has been reported. Herein we report five such tumors. The five tumors occurred in four females and one male, ranging from 38 to 67 years of age (median 51 years). The masses occurred in the colon (cecum, descending, sigmoid) and ileocecal valve region, measuring 2.5–20 cm in size (median 7 cm). Four patients had metastases at presentation (liver, n = 4; lymph nodes, n = 3). Histologically, the lesions arose in the submucosa, infiltrating into the mucosa and muscularis propria, and grew in fibrosarcoma-like fascicles and sheets of epithelioid or rhabdoid cells, with foci of hyalinized to vaguely osteoid-like matrix. The tumors were composed of relatively monomorphic, spindled to epithelioid cells with focal rhabdoid morphology, hyperchromatic nuclei, and small nucleoli. Mitotic activity was usually low (range 1–14/10 HPF; median 5/10 HPF); necrosis was present in two cases. Variable keratin expression and uniform nuclear NUT expression was present; KIT/DOG1 were negative and SMARCB1/SMARCA4 were retained. Next-generation sequencing identified MXD4-NUTM1 rearrangement in all cases (breakpoints: MXD4 exon 5, NUTM1 exons 2 or 3). Follow-up showed one of the four patients who presented with metastases to be dead of disease at 30 months; the other three patients were alive with metastatic disease. The final patient is disease-free, 5 months after diagnosis. NUTM1-rearranged colorectal sarcomas have characteristic morphologic, immunohistochemical, and molecular genetic features, suggesting that they represent a distinct entity within the family of NUTM1-rearranged neoplasia. A NUTM1-rearranged tumor should be considered for any difficult-to-classify submucosal spindle cell neoplasm of the gastrointestinal tract, in particular keratin-positive tumors showing an unusual combination of fibrosarcomatous, epithelioid to rhabdoid and hyalinized morphologies. Recognition of MXD4-NUTM1 rearranged sarcomas may be therapeutically important, even though best treatment is currently elusive/unknown.
Similar content being viewed by others

Introduction
Involvement of the NUTM1 gene in human neoplasia was first reported by Kubonishi and co-workers in 1991 in an aggressive thymic carcinoma harboring a t(15;19) translocation [1]. In 2003, French and colleagues identified the BRD4-NUTM1 fusion gene resulting from this translocation [2], and in 2004 this same group of investigators codified NUTM1-rearranged carcinoma as a distinct entity, typically occurring in midline location in children and young adults, and having a very poor prognosis [3]. NUT carcinoma is characterized histologically by primitive epithelioid cells showing foci of “abrupt” keratinization and is considered a variant of squamous cell carcinoma [4].
Subsequently, it has become apparent that rearrangements of the NUTM1 gene are not limited to NUT carcinoma and may also be seen in poroma/porocarcinoma, B-lymphoblastic leukemia/lymphoma, central nervous system embryonal tumor, myeloid neoplasm with eosinophilia and rearrangement of PDGFRA, and a variety of apparently undifferentiated sarcomas [5,6,7,8,9]. In contrast to NUT carcinomas, which typically show rearrangements of NUTM1 with BRD4 or BRD3 [4], these less common types of NUTM1-rearranged neoplasia have more diverse fusion partners, including the MGA, MXD1, MXI1, CIC, and MXD4 genes [9,10,11,12,13,14,15,16,17,18]. Fusion of NUTM1 to any of these partners results in overexpression of NUTM1 protein, detectable by immunohistochemistry [19].
NUTM1-rearranged sarcomas have been reported in a variety of different somatic soft tissue locations, with isolated cases also reported in visceral locations, including the stomach, kidney, brain, ovary, and colon [9, 11, 20, 21]. Prompted by a recent case of NUTM1-rearranged sarcoma occurring in the colon, and mimicking other colorectal spindle cell tumors, we reviewed our collective experience with five well-characterized examples of NUTM1-rearranged colorectal sarcoma.
Methods
Case procurement
Following Institutional Review Board approval, we searched our institutional and consultation archives for NUTM1-rearranged neoplasms arising in the gastrointestinal tract, identifying five cases. All available routinely stained and immunohistochemical slides were re-reviewed. Clinical information including follow-up was obtained from contributing pathologists and clinicians. Some details of one of these cases (Case 3) have previously been reported [9]; additional morphologic, immunohistochemical, and clinical information was obtained for this case.
Immunohistochemistry
Selected immunohistochemical studies were performed at Mayo Clinic utilizing formalin-fixed, paraffin-embedded tissue sectioned at 4 μm. After deparaffinization, the sections were processed on the Ventana BenchMarkXT (Ventana, Roche Diagnostics, Indianapolis, Indiana, USA) using antibodies against the following antigens: pankeratins (clone OSCAR, dilution 1:100, Biolegend, San Diego, California, USA), pankeratins (AE1/AE3 cocktail, dilution 1:100, Dako, Santa Clara, California, USA), high-molecular-weight keratins (clone 34betaE12, 1:100, Dako, Santa Clara, California, USA), NUT (clone C52B1, 1:45, Cell Signaling Technology, Danvers, Massachusetts, USA), S100 protein (polyclonal, 1:200, Leica/Novocastra, Buffalo Grove, Illinois, USA), KIT (clone YR145, 1:100, Cell Marque, Rocklin, California, USA), DOG1 (clone K9, 1:100, Leica/Novocastra, Buffalo Grove, Illinois, USA), SMARCB1/INI1 (clone 25/BAF47, dilution 1:800, BD transduction laboratories, San Jose, California, USA), and SMARCA4/BRG1 (clone EPR3912, 1:100, Abcam, Cambridge, Massachusetts, USA). All sections were then counterstained with hematoxylin.
Genetic analyses
RNA extracted from formalin-fixed, paraffin-embedded tissue was the source for gene fusion analysis for all cases. The three cases from Mayo Clinic were analyzed for gene fusions using a Mayo Clinic developed gene translocation assay available through Mayo Clinic Labs (Rochester, Minnesota, USA, mayocliniclabs.com). This assay utilizes PCR-based next-generation sequencing to detect translocations of 138 gene targets including NUTM1. One case was analyzed at Caris Life Sciences (Irving, Texas, USA) using similar technology and previously described methods [9]. The last case was studied at University Hospital, Basel, Switzerland using the Custom ArcherTM Fusion Plex Panel (Boulder, Colorado, USA), targeting translocations involving 51 gene targets.
Results
Clinical features
Table 1 summarizes the clinicopathologic features of the reported cases. The tumors occurred in four women and one man, ranging from 38–67 years of age (median 44 years). The tumors occurred in the ileocecal valve region, cecum, descending colon and sigmoid colon, and measured 2.5–20 cm (median 3.5 cm).
Clinical follow-up information was obtained for all patients, with a median follow-up duration of 12.5 months (range 5–30 months). Metastatic disease at presentation occurred in four patients, with three having lymph node and liver involvement, and one having only lymph node involvement (Fig. 1). The latter patient developed liver metastases shortly after surgical resection of the primary tumor. Of these three patients, one died from disease 30 months after presentation; the others are alive with disease 10 and 15 months after diagnosis, respectively. Case 2 was alive without metastatic disease 5 months after surgery. Case 5 is too recent for follow-up, but had liver, lymph nodes and extensive mesenteric, omental, and intraabdominal visceral involvement at presentation.

A Coronal image of an abdominal CT scan with intravenous and oral contrast, showing a colo-colic intussusception with the tumor as the lead point (arrow) and adjacent lymph node metastases (asterixis). B Axial image of the abdominal follow up CT scan, demonstrating multiple hypodense liver lesions, suspicious for metastases (arrows). C Axial MRI images of the liver with a hepatocyte specific contrast agent verifying multiple metastases (arrow on largest lesion) with high signal on T2 weighted images (a), lack of contrast enhancement during the hepatobiliary phase (b) and restricted diffusion ((c), high b value image; (d) apparent diffusion coefficient).
Pathologic findings
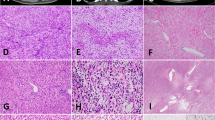
Grossly, the masses were described as generally circumscribed, with a white-tan to gray, whorled appearance on sectioning. Histologically, the five lesions were centered in the submucosa but showed diffuse infiltration of the lamina propria and muscularis propria. Their morphologic features were quite similar, and displayed three distinctive patterns, including (1) intersecting fascicles of relatively monomorphic spindled cells (fibrosarcomatous pattern) (Fig. 2), (2) a sheet-like proliferation of primitive epithelioid to rhabdoid cells (epithelioid/rhabdoid pattern) (Fig. 3), and (3) nests and cords of tumor cells within abundant hyalinized collagen (hyalinized/nested pattern), reminiscent of areas that might be seen in sclerosing epithelioid fibrosarcoma or a malignant myoepithelial lesion (Fig. 4). As noted in Table 1, the relative percentage of these three patterns varied from case to case, with predominance of the fibrosarcomatous pattern in two cases, relatively equal percentages of the fibrosarcomatous and epithelioid/rhabdoid patterns in two cases, and of the rhabdoid pattern in one. The hyalinized/nested pattern appeared to represent an intermediate stage in cases showing modulation from fibrosarcomatous to epithelioid/rhabdoid features.

A NUTM1-rearranged sarcoma of the colon, presenting as a submucosal mass, and B displaying chiefly the spindle cell, “fibrosarcomatous” pattern of growth. C Higher power view of relatively monotonous, hyperchromatic spindled cells with a modest amount of eosinophilic cytoplasm. D Area of transition from fibrosarcomatous to “hyalinized/nested” pattern.

A Colonic NUTM1-rearrranged sarcoma, displaying predominantly the hyalinized/nested pattern. B “Amianthoid fiber-like” collagen was occasionally present. C In hyalinized areas, the cells could sometimes assume a more epithelioid and rhabdoid appearance, shown at higher power in D.

A In some tumors, such as this one, the “epithelioid/rhabdoid” pattern predominated. B Higher power view of ovoid tumor cells with eccentrically placed nuclei and eosinophilic cytoplasm. C Rhabdoid foci often displayed diminished cellular cohesion, somewhat mimicking a hematolymphoid neoplasm. D More spindled foci of tumor could also show rhabdoid cytology. E Necrosis was an uncommon finding.
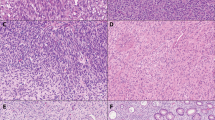
The fibrosarcomatous areas displayed a “herringbone” pattern and were composed of intersecting fascicles of uniform spindled cells with a modest amount of eosinophilic cytoplasm and irregular nuclear contours, with small nucleoli. Mild to at most nuclear pleomorphism was present. Similarly, the epithelioid areas in these tumors displayed minimal pleomorphism, although nuclear irregularity tended to be more pronounced and nucleoli more prominent. Subsets of tumor cells had a rhabdoid appearance, with eosinophilic cytoplasmic inclusions that displaced the nucleus. In three cases, nests of epithelioid to rhabdoid tumor cells were separated into small nests and strands of cells by hyalinized collagen, sometimes with an “amianthoid fiber-like” appearance. The tumor vasculature was well-developed, ranging from thick-walled, hyalinized vessels, to arcades of smaller vessels cuffed by tumor cells. Mitotic activity was generally low, ranging from 1 to 14 mitoses per 10 high power fields (median: 5 mitoses per 10 high power fields). Geographic necrosis was present in two cases and absent in the others. Metastatic lesions showed similar morphology (Fig. 5).

Metastases at presentation were common, to sites such as the liver (A), lymph nodes (B), and serosal surfaces, here involving the appendix (C).
Immunohistochemical features
The immunohistochemical data are summarized in Table 2. As expected, all tumors expressed NUT protein in >75% of cells in a “speckled” nuclear pattern, although the intensity of staining varied within and between tumors (Fig. 6). Keratin expression was seen in four of five cases, with two cases showing relatively diffuse staining (>50% of cells) and two containing only rare keratin-positive cells. Importantly, the neoplastic cells were entirely negative for markers of gastrointestinal stromal tumor (KIT and DOG1) and showed retained expression of SMARCB1 and SMARCA4, despite having rhabdoid morphology and expressing keratins. A wide variety of other tested markers was negative or non-contributory.

A All tumors expressed NUT protein in a nuclear pattern. B Keratin expression, here with the AE1/AE3 antibody cocktail, was present in all tumors but variable in extent. C Despite showing rhabdoid morphology and keratin expression, all tumors displayed retained expression of SMARCB1 (shown) and SMARCA4 (not shown). D A variety of other markers, including KIT and DOG1, was negative. Here a negative desmin immunostain highlights diffuse infiltration of the muscularis propria by tumor cells.
Genetic findings
Next-generation sequencing demonstrated MDX4-NUTM1 gene fusions in all cases, with MDX4 exon 5-NUTM1 exon 3 in two cases, and MDX exon 5-NUTM1 exon 2 in the others (Fig. 7). Genomic breakpoints for the three Mayo Clinic cases were MXD4 exon 5 Chr4:g.2252811 (three cases), NUTM1 exon 2 Chr15:g.3464017 (two cases), and NUTM1 exon 3 Chr15:g.34640170 (one case).

Spanning reads identified by the targeted RNA-seq sarcoma fusion panel support the presence of an MXD4-NUTM1 fusion. The column in the middle of the reads and the labeling at the bottom of the figure show where exon 5 of the MXD4 gene (transcript NM_00645) is joined to exon 3 of the NUTM1 gene (transcript NM_001284292). There were 124 supporting reads for the fusion, which is predicted to be in-frame.
Discussion
Although rearrangements of the NUTM1 gene were originally associated with carcinomas, it is now clear that gene fusions involving this locus also characterize a variety of essentially undifferentiated spindle cell, round cell, and epithelioid malignancies, best regarded as sarcomas. Inclusive of the five tumors that comprise the present report, we are aware of 28 reported NUTM1-rearranged sarcomas. The clinicopathologic features of our 5 cases and of 24 previously reported cases are detailed in Tables 1 and 3, respectively. One patient (Table 1, Case 3; Table 3, Case 13) is included in both tables.
The NUT midline carcinoma family member 1 (NUTM1) gene, located on the long arm of chromosome 15, is normally expressed in the testis and participates in spermatogenesis by altering histone acetylation [9, 22]. In NUT carcinoma, NUTM1 is most often rearranged with one of two Bromodomain and Extra-Terminal family genes, BRD4 (66% of cases) or BRD3 (25% of cases), and less commonly with other genes. Fusion of NUTM1 with BRD3/BRD4 leads to abnormal proliferation and arrest of cellular differentiation, mediated by binding of the BRD3/4-NUTM1 fusion protein product to acetylated lysine moieties on histones, followed by recruitment of p300, a histone acetyltransferase [23]. Aberrant p300-mediated histone acetylation results in overexpression of a variety of oncogenes, including MYC and TP63 [4, 24].
Although BRD3/4-containing fusions may be seen in NUTM1-rearranged sarcomas, such molecular events are present in only a small minority of cases (5 of 28, 18%), with MAX family genes (e.g., MXD4, MGA, MXD1) comprising the largest group of NUTM1 partners (12 of 28, 43%) and CIC-containing tumors forming the next largest subset (8 of 28, 29%). This is in obvious contrast to NUTM1 carcinoma, in which BRD3/4-rearranged tumors are far and away most common [4]. The MAX dimerization protein 4 (MXD4) gene is a member of the MAX-interacting transcription factor network, also involved in the regulation of MYC. MXD4/MAX heterodimers bind to E-box DNA sequences in target promoters, leading to repression of MYC-induced transcription [25]. As illustrated in the present study, the MXD4-NUTM1 fusion also results in nuclear localization of NUT protein, presumably with downstream signaling effects similar to those of BRD3/4-NUTM1 fusions [9].
Although NUTM1-rearranged sarcomas may occur in patients of any age (median 38 years of age; range 3–71 years) and are equally common in males and females (15 males; 12 females), tumors harboring MAX family rearrangements appear to have a predilection for visceral locations, including the gastrointestinal tract and lung. Oddly, MXD4-NUTM1 fusions are to date exclusively seen in colorectal tumors.
Morphologically, NUTM1-rearranged sarcomas share certain features regardless of fusion type, consisting of monomorphic, relatively bland round, epithelioid to rhabdoid and spindled cells, frequently in association with hyalinized or amianthoid fiber-like collagen. Certain trends are suggested here as well, with fibrosarcomatous and hyalinized/nested morphology seemingly most often present in MAX family-rearranged tumors, and CIC-rearranged tumors typically consisting of round to rhabdoid cells. Unlike BRD3/4-rearranged NUTM1 carcinomas, keratin expression is rare in NUTM1-rearranged sarcomas, but when present seems chiefly a feature of MAX family-rearranged lesions, especially those harboring MXD4-NUTM1. Obviously, keratin expression may be seen in sarcomas (e.g., epithelioid sarcoma, synovial sarcoma) and does not suggest that MXD4-NUTM1 tumors are better regarded as carcinomas. NUT protein expression is a feature of all NUTM1-rearranged sarcomas, although in our experience the intensity of staining is less than that seen in NUT carcinomas. There does not seem to be an association between fusion type and outcome and the overall prognosis is quite poor for patients with NUTM1-rearranged sarcoma, with metastatic disease reported in 14 of 22 (64%) patients and only 7 of 24 (29%) patients alive without disease at last follow-up.
The differential diagnosis for NUTM1-rearranged sarcoma in the colorectal region is broad and includes a variety of common and unusual tumors that may show spindle cell, hyalinized or epithelioid morphology, and keratin expression. Sarcomatoid carcinomas typically display greater pleomorphism than do NUTM1-rearranged sarcomas and will often be associated with an adenoma and foci of conventional adenocarcinoma. Although sarcomatoid mesotheliomas may be relatively monomorphic and hyalinized, they typically show more diffuse keratin expression, express markers of mesothelial differentiation (e.g., WT1, calretinin) and lack NUT protein expression. Aberrant keratin expression is quite rare in gastrointestinal stromal tumors [26], which generally do not contain abundant collagen and almost always express KIT and DOG1. Monophasic synovial sarcoma is exceptionally rare in the colon [27], has somewhat different cytomorphology with wiry collagen and alternating zones of hyper and hypocellularity, and displays only scattered keratin-positive cells. In difficult cases, molecular genetic demonstration of SS18/SS18L1-SSX1/2/4 fusion and absent MXD4-NUTM1 is confirmatory of synovial sarcoma. Although both low-grade fibromyxoid sarcoma and sclerosing epithelioid fibrosarcoma may be considered for NUTM1-rearranged tumors dominated by the fibrosarcomatous and hyalinized/nested patterns, expression of MUC4 and demonstration of rearrangements of FUS/EWSR1 and CREB3L2/CREB3L1 should allow these distinctions without great difficulty [28]. Predominantly epithelioid/rhabdoid NUTM1-rearranged sarcomas lack SMARCB1 or SMARCA4 loss, greatly assisting in their distinction from exceptionally rare SMARCB1/SMARCA4-deficient colonic carcinomas and epithelioid sarcomas [29]. Expression of ALK protein in a perinuclear pattern characterizes epithelioid inflammatory myofibroblastic sarcoma [30]. In general, expression of NUT protein is restricted to NUTM1-rearranged neoplasia, and this immunohistochemical study may be very helpful in the differential diagnosis of difficult-to-classify colorectal tumors.
In summary, we have described the clinicopathologic, immunohistochemical, and molecular genetic features of 5 NUTM1-rearranged colorectal sarcomas. The distinctive pathologic features of these tumors and the consistent presence of MXD4-NUTM1 fusions suggest that these unusual lesions represent a distinct entity, under the overall umbrella of NUTM1-rearranged neoplasia. Distinction of NUTM1-rearranged colorectal sarcoma from potential morphologic mimics is important, as there is considerable clinical interest in the development of therapeutic agents for the treatment of patients with often-lethal NUTM1-rearranged neoplasia, with for example the p300 inhibitor A-485 showing some efficacy in cell lines [31].
Data availability
The data for this study are available upon request of the corresponding author.
References
Kubonishi I, Takehara N, Iwata J, Sonobe H, Ohtsuki Y, Abe T, et al. Novel t(15;19)(q15;p13) chromosome abnormality in a thymic carcinoma. Cancer Res. 1991;51:3327–8.
French CA, Miyoshi I, Kubonishi I, Grier HE, Perez-Atayde AR, Fletcher JA. BRD4-NUT fusion oncogene: a novel mechanism in aggressive carcinoma. Cancer Res. 2003;63:304–7.
French CA, Kutok JL, Faquin WC, Toretsky JA, Antonescu CR, Griffin CA, et al. Midline carcinoma of children and young adults with NUT rearrangement. J Clin Oncol. 2004;22:4135–9.
French CA. NUT carcinoma: clinicopathologic features, pathogenesis, and treatment. Pathol Int. 2018;68:583–95.
Sekine S, Kiyono T, Ryo E, Ogawa R, Wakai S, Ichikawa H, et al. Recurrent YAP1-MAML2 and YAP1-NUTM1 fusions in poroma and porocarcinoma. J Clin Invest. 2019;129:3827–32.
Parra O, Kerr DA, Bridge JA, Loehrer AP, Linos K. A case of YAP1 and NUTM1 rearranged porocarcinoma with corresponding immunohistochemical expression: review of recent advances in poroma and porocarcinoma pathogenesis with potential diagnostic utility. J Cutan Pathol. 2020. https://doi.org/10.1111/cup.13832.
Cheng Z, Luo Y, Zhang Y, Wang Y, Chen Y, Xu Y, et al. A novel NAP1L4/NUTM1 fusion arising from translocation t(11;15)(p15;q12) in a myeloid neoplasm with eosinophilia and rearrangement of PDGFRA highlights an unusual clinical feature and therapeutic reaction. Ann Hematol. 2020;99:1561–4.
Pincez T, Landry JR, Roussy M, Jouan L, Bilodeau M, Laramée L, et al. Cryptic recurrent ACIN1-NUTM1 fusions in non-KMT2A-rearranged infant acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2020;59:125–30.
Stevens TM, Morlote D, Xiu J, Swensen J, Brandwein-Weber M, Miettinen MM, et al. NUTM1-rearranged neoplasia: a multi-institution experience yields novel fusion partners and expands the histologic spectrum. Mod Pathol. 2019;32:764–73.
McEvoy CR, Fox SB, Prall OWJ. Emerging entities in NUTM1-rearranged neoplasms. Genes Chromosomes Cancer. 2020;59:375–85.
Diolaiti D, Dela Cruz FS, Gundem G, Bouvier N, Boulad M, Zhang Y, et al. A recurrent novel MGA-NUTM1 fusion identifies a new subtype of high-grade spindle cell sarcoma. Cold Spring Harb Mol Case Stud. 2018;4.
Mangray S, Kelly DR, LeGuellec S, Fridman E, Aggarwal S, Shago M, et al. Clinicopathologic features of a series of primary renal CIC-rearranged sarcomas with comprehensive molecular analysis. Am J Surg Pathol. 2018;42:1360–9.
Schaefer IM, Dal Cin P, Landry LM, Fletcher CDM, Hanna GJ, French CA. CIC-NUTM1 fusion: a case which expands the spectrum of NUT-rearranged epithelioid malignancies. Genes Chromosomes Cancer. 2018;57:446–51.
Le Loarer F, Pissaloux D, Watson S, Godfraind C, Galmiche-Rolland L, Silva K, et al. Clinicopathologic features of CIC-NUTM1 sarcomas, a new molecular variant of the family of CIC-fused sarcomas. Am J Surg Pathol. 2019;43:268–76.
Mantilla JG, Ricciotti RW, Chen E, Hoch BL, Liu YJ. Detecting disease-defining gene fusions in unclassified round cell sarcomas using anchored multiplex PCR/targeted RNA next-generation sequencing-molecular and clinicopathological characterization of 16 cases. Genes Chromosomes Cancer. 2019;58:713–22.
Chien YW, Hsieh TH, Chu PY, Hsieh SM, Liu ML, Lee JC, et al. Primary malignant epithelioid and rhabdoid tumor of bone harboring ZNF532-NUTM1 fusion: the expanding NUT cancer family. Genes Chromosomes Cancer. 2019;58:809–14.
Goto T, Arai Y, Shibata T, Oyama T, Yoshida A. Sarcoma with MGA-NUTM1 fusion in the lung: an emerging entity. Virchows Arch. 2020;476:317–22.
Underwood CIM, Cardona DM, Bentley RC, Shen G, Feng X, Jour G, et al. Epithelioid hyalinizing sarcoma with MGA-NUTM1 fusion. Am J Clin Pathol. 2020;154:859–66.
Haack H, Johnson LA, Fry CJ, Crosby K, Polakiewicz RD, Stelow EB, et al. Diagnosis of NUT midline carcinoma using a NUT-specific monoclonal antibody. Am J Surg Pathol. 2009;33:984–91.
Dickson BC, Sung YS, Rosenblum MK, Reuter VE, Harb M, Wunder JS, et al. NUTM1 gene fusions characterize a subset of undifferentiated soft tissue and visceral tumors. Am J Surg Pathol. 2018;42:636–45.
Tamura R, Nakaoka H, Yoshihara K, Mori Y, Yachida N, Nishikawa N, et al. Novel MXD4-NUTM1 fusion transcript identified in primary ovarian undifferentiated small round cell sarcoma. Genes Chromosomes Cancer. 2018;57:557–63.
Shiota H, Barral S, Buchou T, Tan M, Couté Y, Charbonnier G, et al. Nut directs p300-dependent, genome-wide H4 hyperacetylation in male germ cells. Cell Rep. 2018;24:3477–3487.e6.
Salati M, Baldessari C, Bonetti LR, Messina C, Merz V, Cerbelli B, et al. NUT midline carcinoma: current concepts and future perspectives of a novel tumour entity. Crit Rev Oncol Hematol. 2019;144:102826.
McEvoy CR, Holliday H, Thio N, Mitchell C, Choong DY, Yellapu B, et al. A MXI1-NUTM1 fusion protein with MYC-like activity suggests a novel oncogenic mechanism in a subset of NUTM1-rearranged tumors. Lab Invest. 2020. https://doi.org/10.1038/s41374-020-00484-3.
Cascón A, Robledo M. MAX and MYC: a heritable breakup. Cancer Res. 2012;72:3119–24.
Lopes LF, Bacchi CE. Cytokeratin expression in gastrointestinal stromal tumor: a clinicopathologic and immunohistochemical study of 687 cases. Appl Immunohistochem Mol Morphol. 2012;20:8–12.
Parfitt JR, Xu J, Kontozoglou T, Oluwafemi AR, Driman DK. Primary monophasic synovial sarcoma of the colon. Histopathology. 2007;50:521–3.
Folpe AL. “Hey! Whatever happened to hemangiopericytoma and fibrosarcoma?” An update on selected conceptual advances in soft tissue pathology which have occurred over the past 50 years. Hum Pathol. 2020;95:113–36.
Agaimy A, Daum O, Markl B, Lichtmannegger I, Michal M, Hartmann A. SWI/SNF complex-deficient undifferentiated/rhabdoid carcinomas of the gastrointestinal tract: a series of 13 cases highlighting mutually exclusive loss of SMARCA4 and SMARCA2 and frequent co-inactivation of SMARCB1 and SMARCA2. Am J Surg Pathol. 2016;40:544–53.
Marino-Enriquez A, Wang WL, Roy A, Lopez-Terrada D, Lazar AJ, Fletcher CD, et al. Epithelioid inflammatory myofibroblastic sarcoma: an aggressive intra-abdominal variant of inflammatory myofibroblastic tumor with nuclear membrane or perinuclear ALK. Am J Surg Pathol. 2011;35:135–44.
Zhang X, Zegar T, Lucas A, Morrison-Smith C, Knox T, French CA, et al. Therapeutic targeting of p300/CBP HAT domain for the treatment of NUT midline carcinoma. Oncogene. 2020;39:4770–9.
Author information
Authors and Affiliations
Contributions
All authors contributed to data collection and the writing of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethics approval and consent to participate
Approval for this study was granted by the Institutional Review Boards of the participating institutions. Waiver of consent was granted.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Van Treeck, B.J., Thangaiah, J.J., Torres-Mora, J. et al. NUTM1-rearranged colorectal sarcoma: a clinicopathologically and genetically distinctive malignant neoplasm with a poor prognosis. Mod Pathol 34, 1547–1557 (2021). https://doi.org/10.1038/s41379-021-00792-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-021-00792-z
