
Abstract
Fibroma of tendon sheath (FTS) is an uncommon benign fibroblastic/myofibroblastic neoplasm that typically arises in the tenosynovial tissue of the distal extremities. Histologically, it is a well-circumscribed proliferation of spindle cells within collagenous stroma with peripheral slit-like vessels. Most examples are relatively hypocellular and more densely collagenous than nodular fasciitis; however, a cellular variant has been described, which has considerable morphologic overlap with nodular fasciitis and has been shown to harbor USP6 translocations in a subset of cases. The incidence of these rearrangements and the identity of the USP6 fusion partners have not been described in detail. In this study we evaluate 13 cases of cellular fibroma of tendon sheath by anchored multiplex PCR/next generation sequencing in order to detect potential gene fusions. Nucleic acids of adequate quality were obtained in 11 cases, demonstrating gene fusions in 7/11 (64%), all of which involve USP6 with a variety of partners, including PKM, RCC1, ASPN, COL1A1, COL3A1, and MYH9. Some unusual histomorphologic findings were present in a subset of cases including palisading growth pattern, epithelioid cells, and osteoclast-like multinucleated giant cells, particularly in the tumors with PKM and ASPN gene partners. Overall, the findings support a biologic relationship between cellular fibroma of tendon sheath and other lesions within the spectrum of USP6-rearranged neoplasms, particularly nodular fasciitis.
Similar content being viewed by others

Introduction
Geschickter and Copeland first coined the term fibroma of tendon sheath (FTS) in 1936. They characterized these lesions as encapsulated, easily excised masses, which rarely attained a size larger than that of a hen’s egg [1]. They demonstrated a low risk of recurrence, male predominance, and association with tenosynovial tissues, most commonly in the hands and wrists [2,3,4]. Histologically, FTS appears as a well-circumscribed, often lobulated, paucicellular neoplasm composed of cytologically bland fibroblasts/myofibroblasts within a variably collagenous matrix containing slit-like clefts and vascular spaces [2,3,4]. More than four decades after the initial description of these neoplasms, Chung and Enzinger noted a subset of examples showing morphologic overlap with nodular fasciitis including increased cellularity, vaguely fascicular architecture, and a prominent myxoid matrix with extravasation of red blood cells. The terminology “cellular fibroma of tendon sheath” (CFTS) was used to refer to such examples [3].
Fusions involving the USP6 gene with various partner genes have been described in a variety of neoplasms of bone and soft tissue, nearly all of which behave in an indolent manner, with rare exception. These include nodular fasciitis [5,6,7], cranial fasciitis [5], fibro-osseous pseudotumor of digits [6, 7], myositis ossificans [6, 8], and aneurysmal bone cyst [9, 10]. In addition, Carter et al. recently reported the presence of USP6 rearrangements in 6 of 9 cases of CFTS using fluorescent in-situ hybridization technique; however, fusion partners were not identified in that study [11].
In this study, we aim to better characterize the molecular features of CFTS and explore the relationship between these lesions and other neoplasms harboring USP6 gene rearrangements. Using an anchored multiplex PCR/next generation sequencing platform (Archer® FusionPlex), we describe the presence of multiple gene fusions involving USP6 in 7 of 13 (64%) cases of CFTS, as well as the morphologic features associated with these lesions.
Materials and methods
Case selection
With prior Institutional Review Board (IRB) approval, we searched our surgical pathology archives for lesions diagnosed as “FTS” or “CFTS” between 2000 and 2018. All available histologic slides were reviewed by three pathologists (JGM, BLH, RWR) in order to confirm appropriate tumor classification and further evaluate for any distinguishing histologic features. CFTS was defined as a tumor having identifiable histologic features of classic FTS, specifically areas of low cellularity with spindled to stellate cells, densely collagenous stroma, and slit-like vessels or clefts, but also having regions of increased cellularity and often a nodular fasciitis-like growth pattern, including a tissue culture-like growth pattern of fibroblasts/myofibroblasts, collagenous and myxoedematous stroma, and extravasation of red blood cells. Cases fulfilling histologic criteria for CFTS were selected for anchored PCR/Next generation sequencing studies, as described below. Demographic, clinical and pathologic characteristics, as well as pertinent clinical follow-up, were recorded.
Anchored PCR/NGS
Unstained slides or tissue curls were obtained from formalin-fixed paraffin-embedded tissue blocks from all cases, from which total nucleic acid was extracted using AllPrep DNA/RNA FFPE Kit according to the manufacturer’s recommended protocol (Qiagen, Valencia, CA). First- and second-strand complementary DNA (cDNA) synthesis was performed. A library of DNA fragments was constructed for targeted capture of cDNA from gene transcripts using Archer® FusionPlex® sarcoma kit according to the manufacturer’s recommended protocol (ArcherDx, Boulder, CO). The targeted 26-gene panel included ALK, EPC1, GLI1, MKL2, PLAG1, TAF15, USP6, CAMTA1, EWSR1, HMGA2, NCOA2, ROS1, TCF12, YWHAE, CCNB3, FOXO1, JAZF1, NTRK3, SS18, TFE3, CIC, FUS, MEAF6, PDGFB, STAT6, and TFG. A library of DNA fragments was constructed for targeted capture of cDNA from fusion genes using Archer® FusionPlex® Sarcoma kit (ArcherDX, Boulder, CO). The library was quantitated using quantitative PCR and normalized for next generation sequencing. Paired-end sequencing of the enriched library was performed using Mid Output v2 (Illumina) chemistry on a NextSeq sequencer according to the manufacturer’s recommended protocol (Illumina, Inc. San Diego, CA). FASTQ files with base call and quality information of minimum 1.5 million paired-end sequence reads were processed using ArcherDx Analysis software to annotate gene fusions and variants found within these genes (http://archerdx.com/). Human Genome build GRCh37 (hg19) was used. The pre-sequencing quality control (QC) threshold of 29 cycles of quantitative PCR assay was used. Four main QC parameters used after sequencing included on target deduplication ratio (>3:1), minimum average number unique start sites per control gsp2 (>10), minimal number of breakpoint-spanning reads supporting the fusion (>5), and minimum percentage of reads per GSP2 supporting the fusion (>10%).
Results
Patient characteristics
Our archive search identified a total of 40 lesions for which the diagnosis was either FTS or CFTS. After histologic review, 13 tumors fulfilled histologic criteria for classification as CFTS. The affected patients included 10 men and 3 women, with an average age of 40.6 years (range 28–63 years). Tumor locations included the fingers/hand (n = 10), wrist (n = 1), ankle (n = 1) and lower leg (n = 1), with a mean tumor size of 1.6 cm (range 1.0–2.3 cm). All tumors were surgically excised, with local recurrence in a single case 1 year after the initial procedure and subsequent repeat excision. The detailed clinicopathologic features for each tumor are provided in Table 1.
Anchored PCR/NGS
Of the 13 examples of CFTS, 11 yielded nucleic acids of adequate quality for testing. A fusion gene was detected in 7 (64%) of the 11 tumors tested. All fusion genes involved USP6 with various gene fusion partners including PKM (n = 1), RCC1 (n = 1) ASPN (n = 1), COL1A1 (n = 1), COL3A1 (n = 1), and MYH9 (n = 2). In all cases, the USP6 breakpoints were located within the 5′ UTR with preservation of the entire USP6 coding sequence and fusion to the promoter region of each corresponding fusion gene partner (see Fig. 1).

The coding region of USP6 is preserved in each case and fused to the promoter region of each respective partner.
Histologic features and correlation with fusion genes
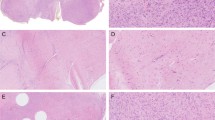
All 13 examples of CFTS fulfilled the histologic features described above in “Materials and methods” section. In addition, a subset of tumors displayed some unusual histologic findings. A palisading growth pattern was evident in the tumors harboring PKM, ASPN, and RCC1 gene fusion partners. This growth pattern was not a diffuse finding throughout any tumor but was most prominent in the tumor with PKM-USP6 and only focal and less well-developed in the tumor with RCC1-USP6. Scattered cells with increased eosinophilic cytoplasm and a somewhat epithelioid appearance were present in the tumor with PKM-USP6 (see Fig. 2). These epithelioid cells were also focally present in one tumor that tested negative for gene fusion. Osteoclast-like multinucleated giant cells were present in four tumors but were most prominent in the tumor with PKM-USP6. Enlarged fibroblasts with multiple nuclei resembling so called floret cells were present in the tumor harboring a COL3A1-USP6 fusion gene. Mitotic activity was variable, ranging from inconspicuous to up to seven figures per ten high-power fields. Tumors with PKM, MYH9, and ASPN fusions partners notably showed the most prominent mitotic activity. Even in those tumors with unusual histologic findings, characteristic features of FTS were also present in variable proportions, most typically identifiable near the periphery of the tumors, allowing for classification as CFTS. We found no significant difference in tumor size or recurrence rate between lesions with USP6 gene rearrangements and those without. Examples of the histologic findings are shown in Figs. 2 and 3.

a An example of an area of increased cellularity with palisading growth in CFTS with ASPN-USP6 fusion as well as a representative image b from the periphery of the same lesion showing features of classic fibroma of tendon sheath. c CFTS with PKM-USP6 fusion showing palisading growth as well as multinucleated giant cells and d scattered enlarged epithelioid cells with eosinophilic cytoplasm. e CFTS with RCC1-USP6 fusion showing vague palisading growth and f periphery of the tumor showing features of classic fibroma of tendon sheath including hypocellularity with abundant collagenized stroma and long slit-like vessels.

CFTS with MYH9-USP6 fusion showing histomorphologic features of a nodular fasciitis in a central cellular area as well as b peripheral features a classic fibroma of tendon sheath. c CFTS with COL3A1-USP6 fusion showing enlarged multinucleated fibroblasts resembling floret cells. d CFTS in which no fusion was detected (Case 8) showing a cellular region with scattered eosinophilic cells similar to those seen in the tumor with PKM-USP6 fusion. Other regions of this tumor showed nodular-fasciitis like growth as well as peripheral features of classic fibroma of tendon sheath.
Discussion
FTS is an uncommon fibroblastic/myofibroblastic neoplasm that most commonly arises in the distal upper extremities of young adults. These lesions are typically small in size and uncommonly recur after conservative excision. A cellular variant of this neoplasm has been described as having morphologic overlap with nodular fasciitis, while retaining areas with features of classic FTS [3]. There is no known correlation between morphologic variants and different clinical outcomes.
In our study, we utilized an anchored multiplex PCR/next generation sequencing platform (Archer® FusionPlex) to describe gene rearrangements involving USP6 in 64% of tested examples of CFTS. USP6 (Ubiquitin specific protease 6) is a primate-specific gene residing on chromosome 17p13. It serves as a de-ubiquitinating enzyme with a diverse set of functions including protein turnover, intracellular trafficking, inflammatory signaling, and cell transformation; however, its physiologic functions are not fully understood [12]. Given its expression in the fetal brain, USP6 has been postulated to participate in neural development, and translocations in this gene have been associated with developmental disorders [13, 14]. Although its role in tumorigenesis is not entirely clear, its overexpression has been linked to upregulation of multiple cellular functions, including activation of matrix metalloproteinases, leading to increased angiogenesis and changes in vascular permeability [12]. Increased expression of USP6 has also been shown to upregulate the activity of c-Jun and its downstream targets through deubiquitinization [15], as well as to increase the activity of NF-κβ [16].
In all cases of CFTS in which we detected a gene fusion, the first exon of the partner gene was fused to exons 1 or 2 of USP6, preserving the promoter of the former and coding region of the latter in a mechanism known as promoter swapping, which has been previously shown to lead to the constitutional overexpression of the USP6 protein and the development of neoplasia [17]. The fusion partners in our cases varied and included PKM, RCC1, ASPN, COL3A1, COL1A1, and MYH9. To our knowledge, fusions involving PKM, RCC1, and ASPN with USP6 have not been previously reported in the literature and represent novel findings. PKM (pyruvate kinase M1/2) encodes for the M isoform of pyruvate kinase, a glycolytic enzyme that appears to play a complex role in embryogenesis, regeneration and multiple types of cancer [18, 19]. The product of RCC1 (regulator of chromosome condensation 1) participates in multiple functions in chromatin assembly and mitotic spindle assembly [20, 21]. ASPN (asporin) encodes for a proteoglycan involved in chondrogenesis and collagen mineralization, which has been postulated to influence the development of osteoarthritis [22]. Fusions of USP6 with COL1A1 and MYH9, on the other hand, have been well described in a wide variety of lesions [6, 8, 9, 17, 23, 24], while those involving COL3A1 have been reported in a subset of cranial fasciitis [5].
We also describe unusual histomorphologic features among our series including palisading growth, enlarged eosinophilic cells, and multinucleated giant cells. These features appeared to be most notable in, but not entirely restricted to, tumors harboring USP6 fusions with PKM, ASPN, and RCC1 partners. Demonstrating any definitive correlation between fusion partners and morphologic features, however, would require further studies.
Prior to the discovery of gene fusions involving USP6, the biological nature of lesions such as nodular fasciitis and primary aneurysmal bone cyst was unclear. Given their localized nature, reported association with trauma in the former, and co-existence with other bone lesions in the latter, both entities were hypothesized to represent reactive processes. However, the description of recurrent gene rearrangements involving USP6, most often fused with CDH11 in aneurysmal bone cyst and most often fused with MYH9 in nodular fasciitis, allowed for their classification as neoplasms [9, 10]. Since then, several other fusion partners of USP6 have been described in both neoplasms [17, 23,24,25,26]. Over time, rearrangements involving USP6 and an increasing variety of fusion partners have also been described in a number of neoplasms including cranial fasciitis [5], fibro-osseous pseudotumor of digits [6, 7], myositis ossificans [6, 8] and, most recently, CFTS [11]. Of note, the vast majority of neoplasms with USP6 rearrangement behave in an indolent or benign nature; however, two examples of tumors harboring an amplified PPPRP3-USP6 fusion gene have been described as showing nodular fasciitis-like morphology and exhibiting malignant clinical behavior [27, 28]. A diagram summarizing a review of neoplasms reported as harboring USP6 rearrangements is provided in Fig. 4.

In our series, we describe six different fusion partners in a subset of CFTS, three of which are novel (highlighted in purple). Key: ABC aneurysmal bone cyst, CF cranial fasciitis, CFTS cellular fibroma of tendon sheath, FOPD fibro-osseous pseudotumor of digits, MO myositis ossificans, NF nodular fasciitis.
In contrast to CFTS, few cytogenetic alterations have been described in classic FTS, including t(2;11)(q31–32;q12) and t(9;11)(p24;q13–14), which were detected in two cases [29, 30]. The former rearrangement has also been described in collagenous fibroma (desmoplastic fibroblastoma), raising the possibility that these two lesions may be biologically related [31, 32].
Given their significant morphologic overlap as well as shared gene fusions, the distinction between some cases of CFTS and nodular fasciitis can be challenging and may also be subjective. Not only does CFTS often contain areas resembling nodular fasciitis, but long-standing nodular fasciitis can develop dense, hyalinized, fibrosis resembling that seen in FTS. Furthermore, intra-articular examples of nodular fasciitis harboring MYH9-USP6 fusions have been reported [33, 34]. While one could consider that intra-articular nodular fasciitis may be more appropriately classified, at least by some diagnostician’s standards, as CFTS based on location or involvement of tendon sheath/retinaculum, our findings also lend credence to the notion that many USP6 rearranged neoplasms exist on a morphologic spectrum and that definitive distinction in some cases may not be possible.
In conclusion, the detection of USP6 gene rearrangements in a subset of CFTS has provided insight into the pathogenesis of these neoplasms and supports the notion that these entities represent a site-specific manifestation of the broadening family of USP6 driven neoplasms. While there is histomorphologic overlap between CFTS and nodular fasciitis as well as other neoplasms with USP6 rearrangement, a subset of the tumors in our series showed distinct morphologic features and novel gene fusions, possibly representing a separate class of USP6 rearranged neoplasms.
References
Geschickter CF, Copeland M. Tumors of bone. 3rd ed. Philadelphia: JB Lippincott; 1949.
Pulitzer DR, Martin PC, Reed RJ. Fibroma of tendon sheath. A clinicopathologic study of 32 cases. Am J Surg Pathol. 1989;13:472–9.
Chung EB, Enzinger FM. Fibroma of tendon sheath. Cancer. 1979;44:1945–54.
Smith PS, Pieterse AS, McClure J. Fibroma of tendon sheath. J Clin Pathol. 1982;35:842–8.
Paulson VA, Stojanov IA, Wasman JK, Restrepo T, Cano S, Plunkitt J, et al. Recurrent and novel USP6 fusions in cranial fasciitis identified by targeted RNA sequencing. Mod Pathol. 2020;33:775–78.
Svajdler M, Michal M, Martinek P, Ptakova N, Kinkor Z, Szepe P, et al. Fibro-osseous pseudotumor of digits and myositis ossificans show consistent COL1A1-USP6 rearrangement: a clinicopathological and genetic study of 27 cases. Hum Pathol. 2019;88:39–47.
Flucke U, Shepard SJ, Bekers EM, Tirabosco R, van Diest PJ, Creytens D, et al. Fibro-osseous pseudotumor of digits—expanding the spectrum of clonal transient neoplasms harboring USP6 rearrangement. Ann Diagn Pathol. 2018;35:53–5.
Flucke U, Bekers EM, Creytens D, van Gorp JM. COL1A1 is a fusionpartner of USP6 in myositis ossificans—FISH analysis of six cases. Ann Diagn Pathol. 2018;36:61–2.
Erickson-Johnson MR, Chou MM, Evers BR, Roth CW, Seys AR, Jin L, et al. Nodular fasciitis: a novel model of transient neoplasia induced by MYH9-USP6 gene fusion. Lab Investig. 2011;91:1427–33.
Oliveira AM, Perez-Atayde AR, Inwards CY, Medeiros F, Derr V, Hsi BL, et al. USP6 and CDH11 oncogenes identify the neoplastic cell in primary aneurysmal bone cysts and are absent in so-called secondary aneurysmal bone cysts. Am J Pathol. 2004;165:1773–80.
Carter JM, Wang X, Dong J, Westendorf J, Chou MM, Oliveira AM. USP6 genetic rearrangements in cellular fibroma of tendon sheath. Mod Pathol. 2016;29:865–9.
Oliveira AM, Chou MM. The TRE17/USP6 oncogene: a riddle wrapped in a mystery inside an enigma. Front Biosci. 2012;4:321–34.
Ou Z, Jarmuz M, Sparagana SP, Michaud J, Decarie JC, Yatsenko SA, et al. Evidence for involvement of TRE-2 (USP6) oncogene, low-copy repeat and acrocentric heterochromatin in two families with chromosomal translocations. Hum Genet. 2006;120:227–37.
Tentler D, Johannesson T, Johansson M, Rastam M, Gillberg C, Orsmark C, et al. A candidate region for Asperger syndrome defined by two 17p breakpoints. Eur J Hum Genet. 2003;11:189–95.
Li L, Yang H, He Y, Li T, Feng J, Chen W, et al. Ubiquitin-Specific Protease USP6 Regulates the Stability of the c-Jun Protein. Mol Cell Biol. 2017;38:e00320–17.
Pringle LM, Young R, Quick L, Riquelme DN, Oliveira AM, May MJ, et al. Atypical mechanism of NF-kappaB activation by TRE17/ubiquitin-specific protease 6 (USP6) oncogene and its requirement in tumorigenesis. Oncogene. 2012;31:3525–35.
Patel NR, Chrisinger JSA, Demicco EG, Sarabia SF, Reuther J, Kumar E, et al. USP6 activation in nodular fasciitis by promoter-swapping gene fusions. Mod Pathol. 2017;30:1577–88.
Dayton TL, Jacks T, Vander Heiden MG. PKM2, cancer metabolism, and the road ahead. EMBO Rep. 2016;17:1721–30.
Desai S, Ding M, Wang B, Lu Z, Zhao Q, Shaw K, et al. Tissue-specific isoform switch and DNA hypomethylation of the pyruvate kinase PKM gene in human cancers. Oncotarget. 2014;5:8202–10.
Furuta M, Hori T, Fukagawa T. Chromatin binding of RCC1 during mitosis is important for its nuclear localization in interphase. Mol Biol Cell. 2016;27:371–81.
Moore W, Zhang C, Clarke PR. Targeting of RCC1 to chromosomes is required for proper mitotic spindle assembly in human cells. Curr Biol. 2002;12:1442–7.
Kizawa H, Kou I, Iida A, Sudo A, Miyamoto Y, Fukuda A, et al. An aspartic acid repeat polymorphism in asporin inhibits chondrogenesis and increases susceptibility to osteoarthritis. Nat Genet. 2005;37:138–44.
Lam SW, Cleton-Jansen AM, Cleven AHG, Ruano D, van Wezel T, Szuhai K, et al. Molecular analysis of gene fusions in bone and soft tissue tumors by anchored multiplex PCR-based targeted next-generation sequencing. J Mol Diagn. 2018;20:653–63.
Oliveira AM, Perez-Atayde AR, Dal Cin P, Gebhardt MC, Chen CJ, Neff JR, et al. Aneurysmal bone cyst variant translocations upregulate USP6 transcription by promoter swapping with the ZNF9, COL1A1, TRAP150, and OMD genes. Oncogene. 2005;24:3419–26.
Sekoranja D, Bostjancic E, Salapura V, Mavcic B, Pizem J. Primary aneurysmal bone cyst with a novel SPARC-USP6 translocation identified by next-generation sequencing. Cancer Genet. 2018;228-229:12–6.
Warren M, Xu D, Li X. Gene fusions PAFAH1B1-USP6 and RUNX2-USP6 in aneurysmal bone cysts identified by next generation sequencing. Cancer Genet. 2017;212-213:13–8.
Teramura Y, Yamazaki Y, Tanaka M, Sugiura Y, Takazawa Y, Takeuchi K, et al. Case of mesenchymal tumor with the PPP6R3-USP6 fusion, possible nodular fasciitis with malignant transformation. Pathol Int. 2019;69:706–9.
Guo R, Wang X, Chou MM, Asmann Y, Wenger DE, Al-Ibraheemi A, et al. PPP6R3-USP6 amplification: Novel oncogenic mechanism in malignant nodular fasciitis. Genes Chromosomes Cancer. 2016;55:640–9.
Dal Cin P, Sciot R, De Smet L, Van den Berghe H. Translocation 2;11 in a fibroma of tendon sheath. Histopathology. 1998;32:433–5.
Nishio J, Iwasaki H, Nagatomo M, Naito M. Fibroma of tendon sheath with 11q rearrangements. Anticancer Res. 2014;34:5159–62.
Sciot R, Samson I, van den Berghe H, Van Damme B, Dal Cin P. Collagenous fibroma (desmoplastic fibroblastoma): genetic link with fibroma of tendon sheath? Mod Pathol. 1999;12:565–8.
Bernal K, Nelson M, Neff JR, Nielsen SM, Bridge JA. Translocation (2;11)(q31;q12) is recurrent in collagenous fibroma (desmoplastic fibroblastoma). Cancer Genet Cytogenet. 2004;149:161–3.
Igrec J, Brčić I, Igrec R, Bergovec M, Kashofer K, Fuchsjäger M, et al. Intraarticular nodular fasciitis of the knee with MHY9-USP6 fusion: a case report. Int J Surg Pathol. 2020. [published online ahead of print]
Miyama A, Kuratsu S, Takenaka S, Yoshimura M, Yoneda G, Yamada Y, et al. Two case reports of intra-articular nodular fasciitis of the knee confirmed by MYH9-USP6 gene fusion expression. J Orthop Sci. 2019;S0949-2658:30379–8.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mantilla, J.G., Gross, J.M., Liu, Y.J. et al. Characterization of novel USP6 gene rearrangements in a subset of so-called cellular fibroma of tendon sheath. Mod Pathol 34, 13–19 (2021). https://doi.org/10.1038/s41379-020-0621-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-020-0621-1
This article is cited by
-
Multimodality imaging features of USP6-associated neoplasms
Skeletal Radiology (2023)
-
The pathogenesis of coronavirus-19 disease
Journal of Biomedical Science (2022)
-
Fibroma of tendon sheath is defined by a USP6 gene fusion—morphologic and molecular reappraisal of the entity
Modern Pathology (2021)
-
Nodular fasciitis of the breast: clinicopathologic and molecular characterization with identification of novel USP6 fusion partners
Modern Pathology (2021)
-
Intraarticular nodular fasciitis—detection of USP6 gene fusions in three cases by targeted RNA sequencing
Virchows Archiv (2021)
