
Abstract
Fibroma of tendon sheath (FTS) is an uncommon benign myofibroblastic neoplasm that arises in association with tenosynovial tissue. Fusions of the USP6 gene have been recently documented in a proportion of so-called “cellular FTS” but not in “classic FTS”. It remains unknown whether FTS can be defined by a USP6 fusion, regardless of cellularity, and what are USP6 fusion-negative “cellular FTS”. Furthermore, FTS with low cellularity seems to be frequently confused with desmoplastic fibroblastoma. We performed a comprehensive analysis, including targeted RNA sequencing, of 58 consecutive cases originally diagnosed as FTS (n = 49), desmoplastic fibroblastoma (n = 6), or nodular fasciitis (n = 3); the latter two at the predilection sites for FTS. After review of the original slides, 28 lesions were morphologically classified as FTS (13 “classic” and 15 “cellular”) and 23 as desmoplastic fibroblastoma. Among originally diagnosed FTS at the more cellular end of the spectrum, we identified seven lesions that shared many morphologic features of FTS but, in addition, showed several distinct morphologic features consistent with myofibroma, such as myoid appearance, branching thin-walled vessels, and perivascular growth. Targeted RNA sequencing showed a USP6 fusion in 17 of 18 analyzed FTS, regardless of cellularity, 0 of 5 desmoplastic fibroblastomas and 0 of 4 myofibromas. MYH9, COL1A1, and ASPN were identified as fusion partners in three cases each, and MIR22HG, CTNNB1, SPARC, CAP1, EMP1, LINC00152, NR1D1, and RAB1A in a single case each. FTS, regardless of cellularity, can be defined by a USP6 fusion with a variety of fusion partners. More cellular lesions exhibiting some morphologic features of FTS but lacking a USP6 fusion tend to be myofibromas.
Similar content being viewed by others

Introduction
Fibroma of tendon sheath (FTS) is an uncommon benign (myo)fibroblastic neoplasm that arises in association with tenosynovial tissue, predominantly in the fingers, hands, and wrists [1]. The term was coined by Geschickter and Copeland in 1936, and the largest series was reported by Chung and Enzinger in 1979, followed by a number of series in the eighties [1,2,3,4,5,6,7,8,9]. It is a relatively well-circumscribed, often lobulated or multinodular lesion, composed of cytologically bland spindle or stellate myofibroblasts within a variably collagenous matrix. Slit-like vessels, particularly at the periphery of the lesion are an almost constant finding. Cellularity varies among lesions and within the same lesion, from paucicellular sclerotic to relatively cellular [1, 10]. FTS, in particular at the more cellular end of the spectrum, shows a morphologic overlap with nodular fasciitis and such lesions have also been referred to as “cellular FTS” [3, 10, 11]. It has been suggested that some more cellular examples of FTS actually represent nodular fasciitis and some cases of nodular fasciitis reported on acral sites may represent or are indistinguishable from cellular FTS [2, 12, 13].
Nodular fasciitis is a benign, usually self-limiting myofibroblastic tumor with a wide anatomic distribution and a predilection for subcutaneous tissue [14]. It is characterized by fusions involving the USP6 gene [11, 13, 15,16,17]. Taking into account morphologic similarities between nodular fasciitis and FTS at the more cellular end of the spectrum, Carter et al. [3] identified a USP6 gene rearrangement by fluorescence in situ hybridization (FISH) in six of nine cases of “cellular FTS”, but they did not detect a USP6 rearrangement in any of 10 paucicellular (classic) FTS. “Cellular FTS” were defined as containing at least 15% moderately to highly cellular, fascicle forming areas. In 2020, Mantilla et al. analyzed 13 “cellular FTS” by anchored multiplex PCR (AMP) sequencing and identified a USP6 fusion in 7 of 11 cases with a successful analysis, including MYH9 (in two cases), COL3A1, COL1A1, ASPN, RCC1, and PKM as fusion partners [10].
Hypocellular and sclerotic FTS shares certain morphologic features with desmoplastic fibroblastoma [18, 19]. Desmoplastic fibroblastoma is a rare benign myofibroblastic tumor with a wide anatomic distribution and a predilection for subcutaneous tissue and skeletal muscles of the proximal extremities and trunk [18, 19]. It is a relatively hypocellular and hypovascular lesion, composed of bland stellate and spindle myofibroblasts dispersed in a collagenous or fibroedematous matrix [18, 19]. A significant proportion of desmoplastic fibroblastomas has been described on acral sites (14 of 63 and 11 of 25 cases in two series), where they are frequently associated with tendon sheaths [18, 19]. There is often some increased cellularity in desmoplastic fibroblastoma and dilated vascular spaces may sometimes be present [18]. Because of morphologic similarities between FTS and desmoplastic fibroblastomas, some desmoplastic fibroblastomas on acral sites are likely misdiagnosed as FTS [18, 19]. Desmoplastic fibroblastoma is characterized by a 11q12 rearrangement, which leads to upregulation of the FOSL1 gene [20]. In a recent study, diffuse immunohistochemical nuclear staining for FOSL1 was noted in all cases of desmoplastic fibroblastomas, in contrast to FTS [19]. A 11q12 rearrangement has also been identified in some cases of FTS; however, this finding may reflect a morphologic overlap between hypocellular FTS and desmoplastic fibroblastoma [21, 22].
Because only a limited number of “cellular FTS” has been analyzed for the presence of a USP6 fusion and the morphologic definition of the “cellular” variant of FTS (in relation to “classic FTS” and nodular fasciitis) is arbitrary, several questions remain: (1) Do “classic” and “cellular” variants of FTS represent the same or two different entities?; (2) Could FTS be defined by a USP6 fusion regardless of cellularity?; (3) Is the spectrum of USP6 fusion partners in FTS similar to or different from the spectrum in nodular fasciitis?; (4) How can a clear distinction between FTS and desmoplastic fibroblastoma be made?; (5) Do “cellular FTS” that are negative for a USP6 fusion represent another entity?. To address these questions, we analyzed a series of 58 unselected consecutive lesions diagnosed as either FTS, desmoplastic fibroblastoma, or nodular fasciitis.
Materials and methods
Case identification and selection
All consecutive cases of lesions that were originally diagnosed as either FTS, desmoplastic fibroblastoma, or nodular fasciitis (the latter two on acral sites that represent predilection sites for FTS) at the Institute of Pathology, Faculty of Medicine, University of Ljubljana, from 2000 to 2020, were identified through a computer-based search. After review of original slides and exclusion of lesions that could be classified as other entities (four sclerosing perineuriomas, four palmar fibromatoses, five lesions with a non-specific fibrous morphology), 58 excised lesions were identified and included in the study. Our goal with this approach was to identify and include the entire morphologic spectrum of FTS and its potential mimics. The study was approved by the Institutional Review Board (ID 3/20).
Morphologic assessment
All lesions were independently reviewed by two of the authors (B.L., J.P.) and classified morphologically prior to molecular and immunohistochemical analyses, as either FTS, desmoplastic fibroblastoma, or nodular fasciitis, according to the latest WHO classification [14]. Features in favor of FTS were: variation in cellularity, prominent vascularization with slit-like vascular spaces at the periphery, and multilobulated growth. Features in favor of desmoplastic fibroblastoma were relatively uniform cellularity, the predominance of stellate-shaped cells, and the absence of prominent slit-like vascular spaces at the periphery [18, 19]. Because many FTS displayed at least focal features of nodular fasciitis, in particular, at the more cellular end of the spectrum, and the extent of these features was distributed in a fairly continuous spectrum, we could not make a clear distinction between FTS and nodular fasciitis. We, therefore, classified all lesions within the spectrum of FTS and nodular fasciitis as FTS. As we hypothesized that some more cellular lesions diagnosed as FTS (and lacking a USP6 fusion) may represent some other entity, we focused on identifying unusual morphologic features in FTS [3, 10]. We noted that some lesions, originally diagnosed as FTS at the more cellular end of the spectrum, although displaying many features of FTS (including variation in cellularity, multilobulated growth, slit-like vascular spaces at the periphery), showed at least focally two or more of the following features: myoid appearance, myoid nodules, pseudochondroid morphology, branching thin-walled vessels, perivascular growth or cellular areas at the periphery with primitive ovoid to epithelioid cells. The morphology of these lesions was suggestive of myofibroma [23, 24]. There was disagreement in morphologic diagnosis between the two pathologists in five lesions—four lesions with a differential diagnosis between FTS and desmoplastic fibroblastoma and one lesion with a differential diagnosis between FTS and myofibroma. A consensus diagnosis was made after reviewing the cases together.
In all lesions, regardless of morphologic diagnosis, the following pathologic features were assessed: maximum macroscopic diameter, circumscription, lobulation, presence of slit-like vessels at the periphery, branching thin-walled (hemangiopericytoma-like) vessels, osteoclast-like giant cells, keloid-like fibers, number of mitoses per 10 high power fields and cellularity and its variation. Cellularity was graded as hypocellular sclerotic, low or moderate-to-high, in relation to the extent of corresponding areas represented in a given tumor.
Immunohistochemistry
Immunohistochemical staining was performed in automatic immunostainers: Benchmark Ultra (FOSL1 and desmin) or Benchmark XT (beta-catenin, CD34, H-caldesmon, cytokeratin AE1AE3, smooth muscle actin, S100 protein), (Ventana Medical Systems Inc., Tucson, AZ). The primary antibodies used are shown in Supplemental Table 1.
Individual immunohistochemical stains were performed either in all cases or in unselected consecutive most recent cases. Nuclear staining for FOSL1 was evaluated and scored as 4+ (≥75% nuclei positive), 3+ (≥50 to <75% nuclei positive), 2+ (≥10 to <50% nuclei positive), 1+ (≥1 to <10% nuclei positive) or 0 (0%).
Molecular analysis
Total RNA and DNA were obtained from formalin-fixed and paraffin-embedded tissue (FFPE), enriched in most cellular areas using a 0.6-mm needle (punch) (Manual Tissue Arrayer MTA, Beecher, Estigen, Estonia). DNA and RNA were isolated using the Promega automated system and the Maxwell RSC DNA Plus FFPE kit or Maxwell RSC RNA FFPE kit (Promega, Madison, WI), according to the manufacturer’s protocol, but with some modifications—overnight proteinase K digestion and omitting the DNase treatment step in RNA isolation. The quality and concentration of isolated DNA/RNA were determined spectrophotometrically on NanoDrop-One and fluorometrically on Qubit 3.0 (both from Thermo Fisher Scientific, Waltham, MA).
Anchored multiplex RNA sequencing
Targeted RNA sequencing was performed using the AMP approach on the Ion Torrent S5 NGS system (Thermo Fischer Scientific) following a protocol described previously [25, 26]. Libraries were prepared using up to 250 ng of total RNA and an Archer FusionPlex Sarcoma kit (ArcherDx, Boulder, CO), according to the manufacturer’s recommended protocol for IonTorrent. To assess the amplifiable cDNA in the analyzed samples, we performed the PreSeq RNA QC Assay using iTaq Universal SYBR Green Supermix (Biorad, Hercules, CA) and used the cycle threshold (Ct) cutoff value of ≤28.5 cycles for continuing with the library preparation. Libraries were quantified using an Ion Library TaqMan quantitation kit (Thermo Fisher Scientific). We used the automated IonChef system (Thermo Fisher Scientific) for clonal amplification of the libraries (enrichment PCR) and chip loading. Sequencing analysis and further raw data processing of RNA libraries were performed on the Ion S5 system (Thermo Fisher Scientific). Output UBAM files were uploaded onto and processed using Archer Analysis Software version 6.0.3 to annotate gene fusions and variants found within target genes (the list of genes analyzed with the FusionPlex Sarcoma kit is available at https://archerdx.com). Human Genome build GRCh37 (hg19) was used as reference genome.
Quantitative PCR for FOSL1 mRNA expression
For FOSL1 mRNA expression analysis, cDNA was generated with SuperScript VILO cDNA Synthesis Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Endogenous controls were 18S, B2M, and GAPDH. Calculations of expression differences were done using the comparative Ct method (ie, dCT method), whereby each sample was normalized to the geometric mean expression of endogenous controls [27]. A more negative dCt value in our calculations means a lower expression level of FOSL1 in tumor samples. See Supplemental File for a more-detailed description and assay information.
Detection of PDGFRB mutation status
For determining the mutation status of PDGFRB in cases, diagnosed as myofibromas [28, 29], we carried out PCR amplification and bidirectional Sanger sequencing of PDGFRB exons 11, 12, 14, 18, and 21 (NM_002609.4, GRCh38). See Supplemental File for primer sequences and a more-detailed method description.
Statistical analysis
Statistical analysis was performed using IBM SPSS Statistics 24 software (IBM Corporation, New York, USA). Differences in expression levels of the FOSL1 gene among tumor groups were tested with one-way analysis of variance and the post hoc Fisher’s least significant difference test.
Results
Pathologic features
Of the 58 lesions, 28 were morphologically classified as FTS, 23 as desmoplastic fibroblastomas, and 7 as myofibromas (Table 1). Originally, 25 of 28 FTS were diagnosed as FTS and 3 as nodular fasciitis. Seventeen of 23 desmoplastic fibroblastoma were originally diagnosed as FTS and 6 (all after 2012) as desmoplastic fibroblastomas. All seven myofibromas were originally diagnosed as FTS, including three cases in which FTS was favored but myofibroma was suggested as a differential diagnosis.
Cellularity varied in FTS, from hypocellular sclerotic areas to areas of moderate-to-high cellularity. Areas of low cellularity predominated in most of the lesions (Tables 1 and 2, Figs. 1 and 2). Areas of moderate-to-high cellularity were identified in 19 lesions and represented more than 15% of the tumor in 15 lesions. Areas consistent with features of nodular fasciitis were present in many FTS, in particular at the more cellular end of the spectrum. However, slit-like vessels at the periphery of the lesions were at least focally identified in all 28 cases and 21 of 28 lesions were multilobulated, features characteristic of FTS and not typically found in nodular fasciitis [14]. Of the 28 FTS, 13, therefore, corresponded to “classic” and 15 to “cellular” FTS [3]. A few osteoclast-like giant cells were identified in two FTS.

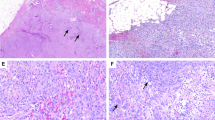
Case #6. A multilobulated cellular lesion with slit-like vascular spaces and focal keloid-like fibers (A, B). Case #5. A vaguely multilobulated, hypocellular lesion with central cystic degeneration (C, lower right). The lesion is reminiscent of a desmoplastic fibroblastoma, except for more prominent vessels (D). Case #3. A relatively cellular lesion with some variation in cellularity, focal slit-like vascular spaces, and an adjacent tenosynovial sheath (E, lower right). The lesion is relatively cellular and vascularized, with nodular fasciitis-like features (F).

Representative images of case #2 (A), case # 13 (B), case #15 (C, D) and case #8 (E, F). Note the variation in cellularity and relatively prominent vessels in all cases. Note keloid-like fibers—focal in A and extensive in F. A lesion diagnosed as fibroma of tendon sheath (G, H) but lacking a USP6 fusion may be a scar rather than a fibroma of tendon sheath. Note poor demarcation from the surrounding fibrous tissue (H).
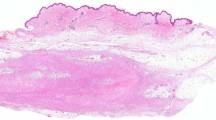
Hypocellular sclerotic areas predominated in desmoplastic fibroblastomas and a small area of moderate cellularity was identified in only one lesion (Table 1, Fig. 3). Although there was some variation in cellularity within individual desmoplastic fibroblastoma, it was much less pronounced than in FTS [18, 19]. There were areas in desmoplastic fibroblastomas indistinguishable from hypocellular sclerotic areas or areas with low cellularity in FTS. However, desmoplastic fibroblastomas tended to be better circumscribed, less cellular, and less lobulated than FTS. Although universally present in FTS, slit-like vessels were identified in only four (17%) desmoplastic fibroblastomas and were much less conspicuous than in FTS.

A relatively hypocellular and hypovascular lesion (A), with a small area of moderate cellularity (B). Different areas of another lesion (C–E) with diffuse nuclear positivity for FOSL1 in tumor cells (F). Note relatively finely fibrillar fibroedematous to focally myxoid (D) matrix and a relatively uniform population of stellate cells in both lesions.
Pathologic features of seven lesions classified as myofibromas are presented in Tables 1 and 3 and Fig. 4. There were two distinct morphologic patterns—myoid spindle cells with a focal pseudochondroid appearance (present in six lesions) and relatively cellular areas of primitive ovoid to epithelioid cells and perivascular growth (present in six lesions). A biphasic morphology with both patterns coexisted in five lesions. Relatively large numbers of osteoclast-like giant cells were identified in five (71%) cases and were usually most prominent in cellular areas of ovoid cells.

Case #3. A multilobulated lesion with variation in cellularity (A, B). Note the hypocellular area of myoid cells (right) and the cellular area of ovoid cells (left) in B. A cellular area at the periphery, with tumor protruding into slit-like vessels (C). A cellular area of ovoid to epithelioid tumor cells and osteoclast-like giant cells (D). Case #6. Myoid morphology with prominent branching vessels (E) and a more cellular area at the periphery composed of ovoid tumor cells and osteoclast-like giant cells, protruding into a slit-like vessel (F). Note the transition of a myoid pseudochondroid area (upper right) into a more cellular area (lower left). Case #4. A myoid lesion with variation in cellularity and prominent branching vessels (G). A cellular nodule at the periphery with perivascular growth and osteclast-like giant cells (H).
Immunohistochemical features
Immunohistochemical findings are summarized in Table 1. Smooth muscle actin was the only myogenic marker consistently expressed to a variable extent in all tested FTS, desmoplastic fibroblastomas, and myofibromas. FOSL1 expression could be successfully analyzed in 56 lesions. Twenty-six of 27 analyzed FTS were completely negative for FOSL1. In one FTS, there was focally mild positivity in <10% of spindle cells (1+). There was mild focal nuclear positivity in endothelial cells in 4 of 27 cases. FOSL1 was positive in 17 of 22 desmoplastic fibroblastomas (77%), with a 4+ positivity in 15 and 3+ positivity in two cases. Five cases were completely negative. In one case with 4+ positivity, there were areas of FOSL1 expression in all tumor cells and areas with a complete absence of FOSL1 expression. There was focal mild positivity in less than 10% of tumor cells (1+) in three of seven myofibromas.
Targeted RNA sequencing
We performed targeted RNA sequencing in 27 cases, including 18 FTS, 5 desmoplastic fibroblastomas, and 4 myofibromas. The analysis was successful in all cases. It revealed a USP6 gene fusion in 17 of 18 lesions classified as FTS, with 11 different fusion partners (Table 2). The only negative case was located on the dorsum of the hand in a 75-year-old female. It was poorly demarcated, exhibited a relatively cellular fascicular proliferation of monotonous spindle cells with prominent keloid-like fibers (Fig. 2G, H) and was the only FTS case focally positive for FOSL1. We reviewed it knowing the molecular results. Although it displayed features of an FTS/nodular fasciitis, it could potentially represent a scar rather than an FTS.
No USP6 gene fusion was detected in desmoplastic fibroblastomas and myofibromas. All 17 fusion-positive cases of FTS showed a fusion of the 5′-UTR (promoter) region of a partner gene to the first exon of the USP6 gene. In 15 cases, the chromosomal location (hg19) of the breakpoint in the USP6 gene was identified as chr17:5033231 (NM_004505.2). In 11 fusion-positive cases of FTS, we detected a second fusion transcript, including involvement of exon2 of the USP6 gene in nine cases (Table 2, Fig. 5). In two cases, we detected a fusion transcript containing intron1 of the SPARC and CAP1 gene partners. The different fusion transcripts identified are probably related to alternative splicing of mRNA at post-trascriptional level, although there is a possibility of the occurrence of two different chromosomal rearrangements.

In each case, the fusion transcript retains the entire coding region of the USP6 gene, which is fused to the promoter region (exon1) of the respective gene partner.
FOSL1 gene expression
We analyzed the expression of the FOSL1 gene in 32 cases, including 18 FTS, 10 desmoplastic fibroblastomas and four myofibromas (Fig. 6). Expression levels of FOSL1 were significantly higher in the desmoplastic fibroblastomas (mean dCt = −3.51) than in FTS and myofibromas (p = 0.004). Further subgroup comparison showed significantly higher expression levels of FOSL1 in desmoplastic fibroblastomas than in FTS alone (mean value dCt = −6.65, p = 0.004). Comparing desmoplastic fibroblastomas to myofibromas showed a less significant difference (mean dCt = −6.17, p = 0.049). FTS and myofibromas showed similar expression of FOSL1 (p = 0.7).

Data under the bars represent immunohistochemical scores of FOSL1 expression (NS, immunohistochemical analysis not successful).
PDGFRB mutation status
We analyzed the presence of PDGFRB mutation in seven myofibromas, including all four of the USP6 fusion-negative cases (Tables 1 and 3). Mutations in the PDGFRB gene were detected in three of seven myofibromas. All three cases harbored multiple variants (see Table 3), located predominantly in the juxta-membrane (exon12) and the kinase domains (exons 14 and 18), which are the classic hotspots for oncogenic mutations in the PDGFRB gene.
Discussion
To investigate how FTS and its potential mimics could be better defined, we analyzed 58 lesions located on acral sites that were originally diagnosed as FTS, desmoplastic fibroblastoma, or nodular fasciitis. We showed that a large proportion of the lesions originally diagnosed as FTS (17 of 52) at the hypocellular end of the spectrum actually represent desmoplastic fibroblastomas and they can be distinguished from FTS morphologically and genetically. Among originally diagnosed FTS at the more cellular end of the spectrum, we identified seven lesions that shared many morphologic features of FTS but, in addition, showed several distinct morphologic features consistent with myofibroma.
Of the 28 FTS, 13 could be classified as “classic” and 15 as “cellular”, but we find this distinction arbitrary and potentially confusing [3, 10, 13]. Seventeen of 18 consecutive lesions diagnosed as FTS and analyzed by targeted RNA sequencing harbored a USP6 fusion regardless of their cellularity (see Table 2). The only USP6 fusion-negative case may represent a scar rather than an FTS. In a previous study, a USP6 fusion was identified in six of nine “cellular FTS” but none of “classic FTS” [3]. The absence of USP6 fusions in “classic FTS” could be explained by the lower sensitivity of FISH analysis to detect a USP6 fusion compared with targeted RNA sequencing in lesions with low cellularity [17]. Alternatively, at least some lesions diagnosed as “classic FTS” in this study may actually be desmoplastic fibroblastomas [3]. In another recent study, the presence of a USP6 fusion was analyzed in 13 “cellular FTS” by targeted RNA sequencing utilizing the same test as in our study. A USP6 fusion was detected in 7 of 11 successfully analyzed cases [10]. Our results show that FTS represents a distinct entity, defined by a USP6 fusion and exhibiting a continuous spectrum of cellularity, from predominantly hypocellular sclerotic lesions to lesions with predominantly moderate-to-high cellularity. It is likely that in the previous studies [3, 10], “cellular FTS” lacking a USP6 fusion represented some other entities, including myofibroma. An illustration of a USP6-negative case with ovoid cells and accompanying osteoclast-like giant cells probably corresponds to the cellular areas of our lesions classified as myofibromas [10].
Among 17 FTS with a proven USP6 fusion, we identified MYH9, COL1A1, and ASPN as fusion partners in three cases each, and MIR22HG, CTNNB1, SPARC, CAP1, EMP1, LINC00152, NR1D1, and RAB1A in a single case each. The last five genes are novel fusion partners of the USP6 gene, not previously documented in any USP6-associated human neoplasm [11]. In our and a previous series combined, an MYH9-USP6 fusion was detected in 5 of 24 (21%) FTS, in contrast to 70% in nodular fasciitis [10, 13, 15, 16]. In addition to the five novel fusions discovered in our study, ASPN-USP6, RCC1-USP6, and PKM1-USP6 fusions have so far been identified only in FTS [10]. It is very likely that the list of potential fusion partners in FTS will grow. All the five novel USP6 fusion gene partners, CAP1, EMP1, LINC00152, NR1D1, and RAB1A, seem to be involved in important biological processes [30,31,32,33,34,35,36,37,38,39,40,41,42,43]. Only NR1D1 has been previously reported as a fusion gene partner in a single case of pediatric soft tissue tumor with an NR1D1-MAML1 fusion [30]. Structurally, all detected fusions consisted of the entire coding sequence of the USP6 gene and the promoter of a partner gene (Fig. 5). As demonstrated in different tumor types, a promoter-swapping mechanism leads to increased transcription of the USP6 gene, inducing cell transformation [13, 25, 26].
Our study confirms that FTS represents an entity within the family of USP6-associated neoplasms, which includes FTS, aneurysmal bone cyst, myositis ossificans, fibro-osseous pseudotumor of the digits, and nodular fasciitis [11]. Many FTS display at least focal morphologic features of nodular fasciitis. Indeed, morphologically and genetically FTS is closely related to nodular fasciitis and it may therefore be interpreted as a variant of nodular fasciitis [3, 10, 13]. However, there are several clinicopathologic and molecular aspects that justify keeping FTS as a separate entity. FTS arises on acral sites (almost exclusively on the hand), frequently in association with tendon sheaths, tends to be multilobulated, and is characterized by a universal presence of slit-like vessels. FTS are generally twice as common in males than in females, whereas nodular fasciitis generally shows an equal sex distribution [1, 14, 19]. Finally, USP6 fusion partners and their frequency in FTS seem to differ from fusion partners in nodular fasciitis. Although MYH9 is a USP6 fusion partner in 70% of nodular fasciitis, MYH9-USP6 fusion constitutes only ~20% of fusions in FTS [10].
Most (17 of 23) desmoplastic fibroblastomas in our series were originally diagnosed as FTS. Only in recent years have we recognized desmoplastic fibroblastoma more frequently as a tumor that should be differentiated from FTS on acral sites. Similar to our data, there is strong evidence, based on descriptions, location, illustrations, and genetic data, that several cases reported as “classic FTS” are actually desmoplastic fibroblastomas [1, 3, 19, 21, 22]. FTS are only slightly more common than desmoplastic fibroblastomas on acral sites [19]. When pathologists are aware that a large proportion of desmoplastic fibroblastomas arise on acral sites, the two lesions can be fairly reliably distinguished on morphologic grounds [18, 19]. Desmoplastic fibroblastomas tend to be more sclerotic and less lobulated, exhibit less variation in cellularity, tumor cells tend to be stellate-shaped and slit-like vessels at the periphery are usually absent or inconspicuous [19]. As shown in our series, a “fibrous” lesion on the foot, ankle or toes is likely to be a desmoplastic fibroblastoma rather than FTS. Clinically, desmoplastic fibroblastomas tend to be larger and occur in older patients than FTS [19].
The morphologic distinction between desmoplastic fibroblastomas and FTS can be substantiated by immunohistochemical and molecular testing. FOSL1 immunohistochemistry can be used to differentiate desmoplastic fibroblastoma from FTS [19, 20]. Although in a previous study, 25 of 25 desmoplastic fibroblastomas exhibited diffuse positivity [19], a similar extent of positivity was observed in 15 of 22 desmoplastic fibroblastomas in our series. In an additional two desmoplastic fibroblastomas, >50% of tumor cells expressed FOSL1, whereas five cases were completely negative, despite showing the typical morphology of desmoplastic fibroblastoma. Although our study shows that mRNA expression levels of FOSL1 are generally significantly higher in desmoplastic fibroblastomas than in FTS, there is some overlap and relatively poor correlation with FOSL1 expression at the protein level as evidenced by immunohistochemical results. Immunohistochemistry seems to be superior (more reliable and easier to perform) to mRNA expression analysis in differentiating between desmoplastic fibroblastoma and FTS. In contrast to a consistent presence of USP6 fusion in FTS, an absence of USP6 fusion in all five desmoplastic fibroblastomas tested by targeted RNA sequencing in our series provides further evidence that desmoplastic fibroblastoma is a distinct entity and not an end-stage sclerotic phase of FTS [19].
Seven lesions in our series originally diagnosed as FTS displayed several morphologic features in favor of myofibroma rather than FTS, such as myoid morphology, pseudochondroid appearance, perivascular growth, and branching vessels [23, 24, 28, 44]. Because of poor circumscription, cellular areas, and poorly developed or absent myoid nodules, six of the seven myofibromas could be designated as myofibromas with atypical features [23, 24, 28]. Most of the myofibromas in our series displayed peculiar morphologic features, not typically documented in myofibromas, in particular, prominent perivascular growth of primitive ovoid cells in six cases and prominent osteoclast-like giant cells in five cases [23]. Of note, 6 of the 24 myofibromas with atypical features reported by Linos et al [23] were located on distal extremities and a large number of osteoclast-like cells were identified in one lesion. PDGFRB gene mutations involving exons 12 or 14 were identified in 70% of myofibromas [28]. We identified PDGFRB mutations in three of seven lesions diagnosed as myofibromas, further supporting the diagnosis of myofibroma [28]. There were no evident morphologic differences between PDGFRB mutation-positive and negative cases (see Table 3). None of the four analyzed myofibromas harbored a USP6 fusion.
In conclusion, FTS is characterized by variations in cellularity and can be defined by a USP6 fusion with different fusion partners (13 identified so far, including eight USP6 fusion partners identified only in FTS). Although FTS belongs to the family of USP6-associated neoplasms and exhibits some morphologic overlap with nodular fasciitis, distinct clinicopathologic features, and differences in the USP6 fusion partners warrant the classification of FTS as a separate entity. Hypocellular lesions that are often diagnosed as FTS but lack a USP6 fusion tend to be desmoplastic fibroblastomas. Desmoplastic fibroblastomas can be differentiated from FTS by morphologic features and strong diffuse expression of FOSL1 in most cases. More cellular lesions exhibiting some morphologic features of FTS but lacking a USP6 fusion tend to be myofibromas. When unusual morphologic features are present, such as myoid morphology, branching vessels, infiltrative growth, pseudochondroid appearance, perivascular growth, cellular areas of primitive ovoid cells or prominent osteoclast-like giant cells, molecular or FISH tests to detect a USP6 fusion may be appropriate to confirm a diagnosis of FTS. The absence of a USP6 fusion in such lesions probably excludes a diagnosis of FTS.
Data availability
All data generated or analyzed during this study are included in this published article.
References
Chung EB, Enzinger FM. Fibroma of tendon sheath. Cancer. 1979;44:1945–54.
Pulitzer DR, Martin PC, Reed RJ. Fibroma of tendon sheath. A clinicopathologic study of 32 cases. Am J Surg Pathol. 1989;13:472–9.
Carter JM, Wang X, Dong J, Westendorf J, Chou MM, Oliveira AM. USP6 genetic rearrangements in cellular fibroma of tendon sheath. Mod Pathol. 2016;29:865–9.
Lundgren LG, Kindblom LG. Fibroma of tendon sheath. A light and electron-microscopic study of 6 cases. Acta Pathol Microbiol Immunol Scand A. 1984;92:401–9.
Smith PS, Pieterse AS, McClure J. Fibroma of tendon sheath. J Clin Pathol. 1982;35:842–8.
Hashimoto H, Tsuneyoshi M, Daimaru Y, Ushijima M, Enjoji M. Fibroma of tendon sheath: a tumor of myofibroblasts. A clinicopathologic study of 18 cases. Acta Pathol Jpn. 1985;35:1099–107.
Jablokow VR, Kathuria S. Fibroma of tendon sheath. J Surg Oncol. 1982;19:90–92.
Azzopardi JG, Tanda F, Salm R. Tenosynovial fibroma. Diagn Histopathol. 1983;6:69–76.
Al-Qattan MM. Fibroma of tendon sheath of the hand: a series of 20 patients with 23 tumours. J Hand Surg Eur Vol. 2014;39:300–5.
Mantilla JG, Gross JM, Liu YJ, Hoch BL, Ricciotti RW. Characterization of novel USP6 gene rearrangements in a subset of so-called cellular fibroma of tendon sheath. Mod Pathol. 2020;34:13–19.
Hiemcke-Jiwa LS, van Gorp JM, Fisher C, Creytens D, van Diest PJ, Flucke U. USP6-associated neoplasms: a rapidly expanding family of lesions. Int J Surg Pathol. 2020;28:816–25.
Hornick JL, Fletcher CD. Intraarticular nodular fasciitis-a rare lesion: clinicopathologic analysis of a series. Am J Surg Pathol. 2006;30:237–41.
Patel NR, Chrisinger JSA, Demicco EG, Sarabia SF, Reuther J, Kumar E, et al. USP6 activation in nodular fasciitis by promoter-swapping gene fusions. Mod Pathol. 2017;30:1577–88.
WHO Classification of Tumours editorial board. Soft Tissue and Bone Tumours. 5 edn (International Agency for Research on Cancer, 2020).
Amary MF, Ye H, Berisha F, Tirabosco R, Presneau N, Flanagan AM. Detection of USP6 gene rearrangement in nodular fasciitis: an important diagnostic tool. Virchows Arch. 2013;463:97–98.
Erickson-Johnson MR, Chou MM, Evers BR, Roth CW, Seys AR, Jin L, et al. Nodular fasciitis: a novel model of transient neoplasia induced by MYH9-USP6 gene fusion. Lab Invest. 2011;91:1427–33.
Lam SW, Cleton-Jansen AM, Cleven AHG, Ruano D, van Wezel T, Szuhai K, et al. Molecular analysis of gene fusions in bone and soft tissue tumors by anchored multiplex PCR-based targeted next-generation sequencing. J Mol Diagn. 2018;20:653–63.
Miettinen M, Fetsch JF. Collagenous fibroma (desmoplastic fibroblastoma): a clinicopathologic analysis of 63 cases of a distinctive soft tissue lesion with stellate-shaped fibroblasts. Hum Pathol. 1998;29:676–82.
Kato I, Yoshida A, Ikegami M, Okuma T, Tonooka A, Horiguchi S, et al. FOSL1 immunohistochemistry clarifies the distinction between desmoplastic fibroblastoma and fibroma of tendon sheath. Histopathology. 2016;69:1012–20.
Macchia G, Trombetta D, Moller E, Mertens F, Storlazzi CT, Debiec-Rychter M, et al. FOSL1 as a candidate target gene for 11q12 rearrangements in desmoplastic fibroblastoma. Lab Invest. 2012;92:735–43.
Nishio J, Iwasaki H, Nagatomo M, Naito M. Fibroma of tendon sheath with 11q rearrangements. Anticancer Res. 2014;34:5159–62.
Dal Cin P, Sciot R, De Smet L., Van den Berghe. Translocation 2;11 in a fibroma of tendon sheath. Histopathology. 1998;32:433–5.
Linos K, Carter JM, Gardner JM, Folpe AL, Weiss SW, Edgar MA. Myofibromas with atypical features: expanding the morphologic spectrum of a benign entity. Am J Surg Pathol. 2014;38:1649–54.
Oudijk L, den Bakker MA, Hop WC, Cohen M, Charles AK, Alaggio R, et al. Solitary, multifocal and generalized myofibromas: clinicopathological and immunohistochemical features of 114 cases. Histopathology. 2012;60:E1–11.
Sekoranja D, Zupan A, Mavcic B, Martincic D, Salapura V, Snoj Z, et al. Novel ASAP1-USP6, FAT1-USP6, SAR1A-USP6, and TNC-USP6 fusions in primary aneurysmal bone cyst. Genes Chromosomes Cancer. 2020;59:357–65.
Sekoranja D, Bostjancic E, Salapura V, Mavcic B, Pizem J. Primary aneurysmal bone cyst with a novel SPARC-USP6 translocation identified by next-generation sequencing. Cancer Genet. 2018;228–9:12–16.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8.
Agaimy A, Bieg M, Michal M, Geddert H, Markl B, Seitz J, et al. Recurrent somatic PDGFRB mutations in sporadic infantile/solitary adult myofibromas but not in angioleiomyomas and myopericytomas. Am J Surg Pathol. 2017;41:195–203.
Arts FA, Sciot R, Brichard B, Renard M, Rocca Serra A, Dachy G., et al. PDGFRB gain-of-function mutations in sporadic infantile myofibromatosis. Hum Mol Genet. 2017;26:1801–10.
Komatsu M, Yamamoto N, Kawamoto T, Kawakami Y, Hara H, Uemura S, et al. Soft tissue tumor with novel NR1D1-MAML1 fusion in a pediatric case. Virchows Arch. 2020;477:891–5.
Hasan R, Zhou GL. The cytoskeletal protein cyclase-associated protein 1 (CAP1) in breast cancer: context-dependent roles in both the invasiveness and proliferation of cancer cells and underlying cell signals. Int J Mol Sci. 2019;20:2653.
Zhang H, Ramsey A, Xiao Y, Karki U, Xie JY, Xu J, et al. Dynamic phosphorylation and dephosphorylation of cyclase-associated protein 1 by antagonistic signaling through cyclin-dependent kinase 5 and cAMP are critical for the protein functions in actin filament disassembly and cell adhesion. Mol Cell Biol. 2020;40:e00282–00219.
Liu Y, Ding Y, Nie Y, Yang M. EMP1 promotes the proliferation and invasion of ovarian cancer cells through activating the MAPK pathway. Onco Targets Ther. 2020;13:2047–55.
Wang J, Li X, Wu H, Wang H, Yao L, Deng Z, et al. EMP1 regulates cell proliferation, migration, and stemness in gliomas through PI3K-AKT signaling and CD44. J Cell Biochem. 2019;120:17142–50.
Aries IM, Jerchel IS, van den Dungen RE, van den Berk LC, Boer JM, Horstmann MA, et al. EMP1, a novel poor prognostic factor in pediatric leukemia regulates prednisolone resistance, cell proliferation, migration and adhesion. Leukemia. 2014;28:1828–37.
Everett LJ, Lazar MA. Nuclear receptor Rev-erbalpha: up, down, and all around. Trends Endocrinol Metab. 2014;25:586–92.
Bugge A, Feng D, Everett LJ, Briggs ER, Mullican SE, Wang F, et al. Rev-erbalpha and Rev-erbbeta coordinately protect the circadian clock and normal metabolic function. Genes Dev. 2012;26:657–67.
Galamb O, Kalmar A, Sebestyen A, Danko T, Kriston C, Furi I, et al. Promoter hypomethylation and increased expression of the long non-coding RNA LINC00152 support colorectal carcinogenesis. Pathol Oncol Res. 2020;26:2209–23.
Yang Y, Sun X, Chi C, Liu Y, Lin C, Xie D, et al. Upregulation of long noncoding RNA LINC00152 promotes proliferation and metastasis of esophageal squamous cell carcinoma. Cancer Manag Res. 2019;11:4643–54.
Huang Y, Luo H, Li F, Yang Y, Ou G, Ye X, et al. LINC00152 down-regulated miR-193a-3p to enhance MCL1 expression and promote gastric cancer cells proliferation. Biosci Rep. 2018;38:BSR20171607.
Zhang L, Wang Y, Su H. Long non-coding RNA LINC00152/miR-613/CD164 axis regulates cell proliferation, apoptosis, migration and invasion in glioma via PI3K/AKT pathway. Neoplasma. 2020;67:762–72.
Westrate LM, Hoyer MJ, Nash MJ, Voeltz GK. Vesicular and uncoated Rab1-dependent cargo carriers facilitate ER to Golgi transport. J Cell Sci. 2020;133:jcs239814.
Li Z, Li Y, Jia Y, Ding B, Yu J. Rab1A knockdown represses proliferation and promotes apoptosis in gastric cancer cells by inhibition of mTOR/p70S6K pathway. Arch Biochem Biophys. 2020;685:108352.
John I, Fritchie KJ. What is new in pericytomatous, myoid, and myofibroblastic tumors? Virchows Arch. 2020;476:57–64.
Funding
The authors acknowledge financial support from the Slovenian Research Agency (research core funding No. P3-0054).
Author information
Authors and Affiliations
Contributions
J.P., A.M., A.Z., and K.D. performed study concept and development of methodology, statistical analysis, and writing of the paper. All authors provided acquisition, analysis, and interpretation of data, reviewed and approved the final paper.
Corresponding author
Ethics declarations
Ethics approval consent to participate
The study was approved by the Institutional Review Board (ID 3/20). The study was performed in accordance with the Declaration of Helsinki.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Pižem, J., Matjašič, A., Zupan, A. et al. Fibroma of tendon sheath is defined by a USP6 gene fusion—morphologic and molecular reappraisal of the entity. Mod Pathol 34, 1876–1888 (2021). https://doi.org/10.1038/s41379-021-00836-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-021-00836-4
This article is cited by
-
Pathology of intra-articular tumours and tumour-like lesions: pearls, pitfalls and rarities from a general surgical pathology practice
Skeletal Radiology (2024)
-
Multimodality imaging features of USP6-associated neoplasms
Skeletal Radiology (2023)
