
Abstract
The ability of cisplatin (cis-diamminedichloroplatinum II) toxicity to induce acute kidney injury (AKI) has attracted attention and concern for a long time, but the molecular mechanism of action for cisplatin is not clear. MicroRNA-483 is involved in several diseases, such as tumorigenesis and osteoarthritis, but its renal target and potential role in AKI are unknown. In this study, we explored the pathogenic role and underlying mechanism of miR-483-5p in cisplatin-induced AKI, using transgenic mice, clinical specimen, and in vitro cell line. We found that miR-483-5p was significantly upregulated by cisplatin in a cisplatin-induced mouse model, in serum samples of patients who received cisplatin therapy, and in NRK-52E cells. Overexpression of miR-483-5p in mouse kidneys by stereotactic renal injection of lentiviruses mediated miR-483-5p or generation of conditional miR-483-overexpressing transgenic mice accentuated cisplatin-induced AKI by increasing oxidative stress, promoting apoptosis, and inhibiting autophagy of tubular cells. Furthermore, our results revealed miR-483-5p directly targeted to GPX3, overexpression of which rescued cisplatin-induced AKI by inhibiting oxidative stress and apoptosis of tubular cells, but not by regulating autophagy. Collectively, miR-483-5p is upregulated by cisplatin and exacerbates cisplatin-induced AKI via negative regulation of GPX3 and contributing oxidative stress and tubular cell apoptosis. These findings reveal a pathogenic role for miR-483-5p in cisplatin-induced AKI and suggest a novel target for the diagnosis and treatment of AKI.
Similar content being viewed by others
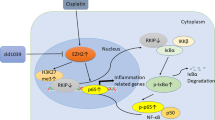


Introduction
Acute kidney injury (AKI) is a worldwide public health problem associated with increased morbidity and mortality1. AKI has been estimated to affect >13.3 million patients and results in ~1.7 million deaths worldwide each year. Other than dialysis, no therapeutic interventions reliably improve survival, limit injury, or speed recovery1,2. Ischemia and renal toxic reagents are the main inducers of AKI. Cisplatin, a platinum-based compound, is a commonly used and highly effective chemotherapeutic agent utilized for the treatment of a variety of solid tumors. Despite the effectiveness of cisplatin, nephrotoxicity and AKI are the most common side effect. Greater than 25% of patients treated with cisplatin develop renal failure, which necessitates discontinuation of treatment3,4. In the kidneys, cisplatin accumulates at high concentrations in renal cells (~5 times higher concentration than the blood), which accounts for ~90% of kidney volume. The tubules are responsible for reabsorption and secretion of various solutes, thus damage to this part of the nephron is a key determinant of AKI. Decades of research have resulted in significant insight into the pathogenesis of cisplatin-induced AKI, including tubular cell injury and death, vascular damage, oxidative stress, and an inflammatory response5,6; however, the molecular basis underlying cisplatin nephrotoxicity remains unclear. A better understanding of the molecular mechanisms underlying this process will facilitate identification of a diagnostic marker and therapeutic target, and is essential to improve the quality of life among cancer patients receiving cisplatin chemotherapy.
MicroRNAs (miRNAs) are small noncoding RNA molecules 22–25 nucleotides in length that negatively regulate translation of target mRNAs. MiRNAs normally bind to the 39-untranslated region (39UTR) of the target mRNAs, leading to inhibition of translation and/or degradation of mRNA7. Recent work has shown an important role played by miRNAs in various cellular processes, ranging from cell growth and proliferation to cell death8,9. Notably, dysregulation of miRNAs has emerged as a critical pathogenic factor in a variety of human diseases10. miR-483 is a multifunctional miRNA that regulates cell proliferation, differentiation, and migration in adipocytes, pancreatic beta cells, and various tumor cells11,12. An increased level of miR-483-5p was reported in the kidney of a unilateral ureteral obstruction (UUO) mouse model of renal fibrosis13 and the serum of patients with rejection after transplantation14; however, whether or not miR-483 plays a role in the regulation of AKI is unknown.
In the present study, we investigated the roles and mechanism underlying miR-483-5p in cisplatin-induced AKI using a miR-483-5p transgenic mice model. Our findings suggest that miR-483-5p is upregulated by cisplatin and contributes to oxidative stress and the apoptosis of renal tubular cells by inhibiting glutathione peroxidase 3 (GPX3) in cisplatin-induced AKI.
Material and methods
Patients and serum collection
From June to December of 2016, serum samples of five patients before and after 3 days of cisplatin chemotherapy were collected from the Department of Oncology of NanFang Hospital (Guangzhou, China). All samples were frozen at −80 °C until use. All research involving human participants was approved by the Ethics Committee of Nanfang Hospital Affiliated to Southern Medical University (Guangzhou, China), and informed consent was obtained from all patients.
Animals
The rtTA mouse line(Gt(ROSA)26Sortm1(rtTA*M2)Jae) was purchased from Jackson Laboratory (Jax no. 006965; Bar Harbor, ME, USA). Transgenic miR-483 mice were generated at Cyagen Biosciences (Suzhou, Jiangsu, China). The plasmid, pRP.ExSi-TRE-Pri-Mir483, which contains pri-miR-483, was purified and microinjected into C57BL/6J F2 mouse oocytes, then surgically transferred into pseudopregnant C57BL/6J dams. Genotyping was performed according to the Jackson Laboratory instructions. The pri-miR- 483 transgene and rtTA inducer allele were termed TG 483 mice. Mice negative for pri-miR-483 or rtTA, or both, were designated as experimental negative control groups. TG 483 and control mice were treated with 2 mg/mL of doxycycline (Sigma, St. Louis, MO, USA) in drinking water from 6 weeks of age.
AKI models
Male mice (8–10 weeks of age) were intraperitoneally injected with a single dose of cisplatin (15 mg/kg) and then sacrificed at 72 h. All animal experiments were approved by the Southern Medical University Committee on the Use and Care of Animals and were performed in accordance with the Committee guidelines and regulations.
Stereotactic injection of lentiviruses in mouse kidney
Lentivirus (GenePharma, Suzhou, Jiangsu, China) was injected into renal parenchyuma at four sites with 20 μl of lentivirus per kidney using a 33G needle and a micro-syringe (Hamilton, Reno, NEV, USA). Lentivirus-mediated miR-483-5p (5′-AAGACGGGAGAAGAGAAGG GAG-3′ [1 × 109 TU/ml]), NC (5′-TTCTCCGAACGTGT CACGTTTC-3′ [1 × 109 TU/ml]), or GPX3 (NM_001329860.1, 1 × 109 TU/ml). AKI was induced in all experimental mice on day 14 after the injection of lentivirus.
Renal function, histopathology, and TUNEL assay
The BUN level was measured using an automatic biochemistry analyzer (IDEXX Catalyst Dx; IDEXX Laboratories, Westbrook, ME, USA). The kidneys were fixed in 4% paraformaldehyde overnight, then processed using paraffin wax. The sections (2 μm) of each kidney were used for hematoxylin and eosin staining. The 4-μm sections of each kidney were used for the TUNEL assay (Promega, Madison, WI, USA) and immunohistochemistry (IHC) detection of GPX3 (1:50; Abcam, Cambridge, MA, USA). The criteria of tubular damage was scored according to the Paller method. For each kidney, 100 cortical tubules from at least 10 different areas were scored. Higher scores represented more severe damage, with points given for the presence and extent of tubular epithelial cell flattening (1 point), loss of the brush border (1 point), cell membrane bleb formation (1 or 2 points), interstitial edema (1 point), cytoplasmic vacuolization (1 point), cell necrosis (1 or 2 points), and tubular lumen obstruction (1 or 2 points).The images were obtained using a AXIO Scope A1 (Zeiss, Oberkochen, Germany). The sections were mounted with medium containing DAPI (Thermo Fisher Scientific, Waltham, MA, USA), images were obtained using a AXIO Scope A1, and images of cells were using a FluoView FV1000 confocal microscope (Olympus, Tokyo, Japan). For quantification, ten fields were randomly selected from each tissue section to count the TUNEL-positive cells.
Transmission electron microscopy
C57BL/6 mice were anesthetized and the kidneys were removed and immediately fixed in 2.5% glutaraldehyde. After standard electron microscopy sample preparation, the processed samples were photographed and analyzed using an H-7650 transmission electron microscope (Hitachi, Tokyo, Japan).
Western blot analysis
Kidney and cell lysates were analyzed by SDS-PAGE and transferred to a nitrocellulose (NC) membrane (GE Healthcare Life Sciences, Little Chalfont, Buckinghamshire, UK). The antibodies used were anti-p53 (1:1000; Abclonal Technology, Woburn, MA, USA), anti-caspase-3 (1:1000; Cell Signal Technology, Danvers, MA, USA), anti-LC3B (1:1000; Cell Signal Technology), anti-malondialdehyde (1:1000; Abcam), anti-GPX3 (1:500; Abclonal Technology), anti-GAPDH (1:3000; Beijing Ray Antibody Biotech, Beijing, China).
miR-483-5p in situ hybridization
The sequences of the probes (Exiqon, Vedbaek, Denmark) for mmu-miR-483-5p containing the locked nucleic acid and digoxigenin-modified bases were as follows: /5DigN/CTCCCTTCTTTCCTCCCGTCTT/3Dig_N/. The signals were detected using anti-digoxigenin-AP (Roche, Basel, Switzerland). NBT/BCIP was used as the chromogen and images were obtained using an AXIO Scope A1.
RNA isolation, cDNA synthesis, and real-time PCR
Total RNA was isolated from cells or tissues using RNAiso Plus (TaKaRa Bio, Tokyo, Japan), mouse serum using miRcute Serum/plasma miRNA isolation kit (Tiangen, Guangzhou, China) and human serum using TRIzol LS Reagent (Invitrogen, Carlsbad, CA, USA) . RNA was stored at 80 °C. To determine the miR- 483-5p level(mmu-miR-483-5p-FO, 5′-CCACCTAAGACGGGAGAAGA-3′;mmu-miR-483-5p-RE, 5′-TATGGTTGTTGTGCTCTCTGACTC-3′;hsa-miR-483-5p-FO, 5′-AGAGCACAAGACGGGAGGAA-3′;hsa-miR-483-5p-RE, 5′-TATGGTTGTTCACGACTCCTTCAC-3′), we used the Mmu-miR-483-5p hairpin-it real-time PCR kit and the U6 snRNA real-time PCR normalization kit (GenePharma, Suzhou, Jiangsu, China). To analyze KIM-1 (forward primer, 5′-ATGCCCATCTTCTGCTTGTCA-3′; reverse primer, 5′-CCTTGTAGTTGTGGGTCTTCT-3′) and GAPDH (forward primer, 5′-TGTGTCCGTCGTGGATCTGA-3′; reverse primer, 5′-TTGCTGTTGAAGTCGCAGGAG-3′) levels, we used Takara reverse transcription reagents and Real-time PCR Mix (TaKaRa Bio) on the StepOnePlus Real-time PCR Instrument (Invitrogen).The U6 and GAPDH genes were used as endogenous controls to normalize for differences in the amount of total miRNA and mRNA, and cel-miR-39-3p exogenous controls to normalize for differences in the mouse and human serum.
Luciferase assay
GPX3 mRNA 3′UTR containing the miR-483-5p binding sequences for the mouse GPX3 gene (GenBank: NM_001329860.1) were amplified by PCR from mouse cDNA. The fragment of GPX3 mRNA 3′UTR was amplified by forward primer (5′-CCGCTCGAGTCATTCTTGGGGCCAGCATC-3′) and reverse primer (5′-ATTTGCGGCCGCGGGGGTGTTGAGATACCAGTG-3′). The PCR product was then subcloned into the XhoI and NotI (Thermo Fisher Scientific) cloning sites of psiCHECK-2 empty vector (Promega). Binding region mutations were achieved using primers, as follows: GPX3-mu forward primer, 5′-CCCCTGAGGGGCAGAGTGGTGCCA-3′; and GPX3-mu reverse primer, 5′-AAGGGCTGGCACCACTCTGCCCCTCA-3′. The GPX3 mRNA 3’UTR expression plasmids were co-transfected with miR-483-5p mimics or negative control into NRK-52E cells, and luciferase assays were performed with the dual-luciferase reporter assay system (Promega) according to the manufacturer’s instructions. Luminescent signals were quantified by a luminometer (Glomax; Promega).
Cell line DNA transfection and miRNA interference
The rat renal epithelial cell line, NRK-52E, was cultured in maintenance medium consisting of DMEM (Corning, Cambridge, MA, USA) supplemented with 10% fetal bovine serum (Gibco, Grand Island, NY, USA). The cells were maintained under standard cell culture conditions (5% CO2 and 95% humidity). MiR-483-5p mimics (GenePharma) and plasmids were transfected using Lipofectamine3000 following the manufacturer’s instructions (Invitrogen).
Statistical analyses
Experiments were repeated at least three times. All statistical analyses were performed using SPSS software (version 23; IBM, Armonk, NY, USA). Statistical significance was defined as a p < 0.05 and tests were two-tailed. Comparisons between two groups were made using t test. Comparisons between multiple groups were made by one-way ANOVA, followed by Tukey test. Samples from the clinic were analyzed using the Wilcoxon test.
Results
miR-483-5p is upregulated in the mouse model of cisplatin-induced AKI
We first established a cisplatin-induced AKI mouse model and detected the level of miR-483-5p in the kidneys. C57BL/6 male mice were injected intraperitoneally with cisplatin (15 mg/kg) and euthanized 8, 12, 24, or 72 h later. Cisplatin-induced significant weight loss in mice in a time-dependent manner from 12 to 72 h after cisplatin administration (Fig. 1A). Blood urea nitrogen level increased significantly 72 h after cisplatin administration (Fig. 1B). A significant increase in expression of Kim-1 mRNA, a sensitive early indicator of AKI15, was observed in the kidneys from 24 to 72 h after cisplatin (15 mg/kg) administration in a time-dependent manner (Fig. 1C). By Q-PCR a marked increase in miR-483-5p expression in the kidneys (Fig. 1D) and serum (Fig. 1E) was also detected 72 h after cisplatin (15 mg/kg) administration. Seventy-two hours after cisplatin administration, we also observed morphologic and histologic changes in the kidney. There were no obvious differences between mice with cisplatin administration and controls in terms of kidney weight or global appearance(data not shown). Hematoxylin and eosin staining showed tubular epithelial cell flattening, brush border loss, and cell membrane bleb formation 72 h after cisplatin administration (Fig. 1F). By in situ hybridization miR-483-5p was observed upregulated within tubular epithelial cells 72 h after cisplatin administration (Fig. 1G).

C57BL/6 mice were intraperitoneally injected with cisplatin (15 mg/kg) and then sacrificed at 8, 12, 24, and 72 h. A The body weight decreased 8–72 h after cisplatin treatment (n = 4). B Blood urea nitrogen level decreased 24–72 h after cisplatin treatment (n = 4). C Real-time PCR analysis showed increased KIM-1 level in the renal cortex 24–72 h after cisplatin treatment. The relative expression of KIM-1 was normalized to GAPDH (n = 4). D Real-time PCR analysis showed increased miR-483-5p level in the renal cortex 24–72 h after cisplatin treatment. The relative expression of the mature miR-483-5p was normalized to U6 (n = 4). E Real-time PCR analysis showed increased miR-483-5p level in the serum 72 h after cisplatin treatment. F Representative H&E images. Scale bar = 20 μm. G In situ hybridization analysis showed upregulated expression of miR-483-5p in the mouse renal cortex 72 h after 15 mg/kg of cisplatin treatment. Scale bar = 20 μm. t test was used for two comparisons. One-way ANOVA was used for multiple comparisons. All data are presented as the means ± SD. ns no significant difference; *P < 0.05; **P < 0.01; ***P < 0.001.
miR-483-5p overexpression aggravates cisplatin-induced AKI in C57BL/6 mice
The gain of function of lentivirus-mediated gene transfer was used to overexpress miR-483-5p in the kidneys of C57BL/6 mice. miR-483-5p expression was increased markedly in the kidney tissue infused with lentivirus-mediated miR-483-5p(LV- miR-483-5p) compared with negative control (NC) (Fig. 2A), which did not influence body weight (Fig. 2B), the level of blood urea nitrogen (Fig. 2C) and the histologic appearance (Fig. 2D) without cisplatin. After cisplatin was administered, the mice infused with LV-miR-483-5p exhibited more severe kidney injury. Compared with the controls(LV-NC), the levels of urea nitrogen in the blood and KIM-1 in the kidney were increased more significantly (Fig. 2C, F), more renal tubule vacuolation degeneration and cell death were observed (Fig. 2D) and the tubular injury score according to Paller’s method was higher after cisplatin treatment (Fig. 2E). These results suggest that elevation of miR-483-5p in the kidney aggravates cisplatin-induced AKI.

C57BL/6 mice were transduced with lentivirus-mediated miR-483-5p (LV-miR-483-5p) or negative control (LV-NC) into the kidney, followed by treatment with cisplatin (15 mg/kg) and then sacrificed at 72 h. A Real-time PCR analysis of the miR-483-5p level in the renal cortex (n = 3–4). B The body weight (n = 3–4). C Blood urea nitrogen level (n = 3–4). D Representative H&E images. E Tubular injury score was assessed according to the Paller method (n = 3). Scale bar = 20 μm. F Real-time PCR analysis of KIM-1 level in the renal cortex (n = 3). One-way ANOVA was used for multiple comparisons. All data are presented as the means ± SD. ns no significant difference; *P < 0.05; **P < 0.01; ***P < 0.001.
miR-483-5p overexpression aggravates cisplatin-induced AKI in TG 483 mice
We generated miR-483 transgenic mice to identify the role of miR-483-5p in cisplatin-induced AKI (Fig. 3A). The expression of miR-483 is induced by a doxycycline (Dox) tet-on system in TG 483 mice (Fig. 3B). We administered 2 mg/mL of Dox in drinking water to mice 6 weeks of age. The levels of miR-483-5p in the kidney were upregulated after exposure to Dox for 2 weeks (Fig. 3C). 72 h after cisplatin treatment, TG 483 mice exhibited more severe renal damage with higher levels of urea nitrogen in the blood and KIM-1 in the kidney (Fig. 3E, F), and much more renal tubule vacuolation degeneration and cell death compared to the littermate controls (Fig. 3H, G). Cisplatin-induced significant weight loss both in TG 483 mice and the littermate controls mice (Fig. 3D).

miR-483 transgenic (TG 483) mice were generated, identified, and induced to express miR-483-5p by treatment with doxycycline (Dox) at 6 week for 2 weeks, followed by treatment with cisplatin (15 mg/kg) and then sacrificed at 72 h. A Identification of gene miR-483 and rtTA by PCR. B Schematic representation of the generation of miR-483 transgenic (TG 483) mice. C Real-time PCR analysis of miR-483-5p levels in the renal cortex of TG 483 and control mice (n = 3–4). D The body weight as indicated (n = 3–4). E Blood urea nitrogen level (n = 3–4). F Real-time PCR analysis of the KIM-1 level in the renal cortex in TG 483 mice compared to control mice (n = 4). H Representative H&E images. G Tubular injury score was assessed according to the Paller method. Scale bar = 20 μm. One-way ANOVA was used for multiple comparisons. All data are presented as the means ± SD. ns no significant difference; *P < 0.05; **P < 0.01; ***P < 0.001.
Overexpression of miR-483-5p promotes apoptosis and inhibits autophagy of renal tubular cells induced by cisplatin in vivo
After 72 h of cisplatin treatment, an increased number of apoptotic terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL)-positive tubular epithelial cells and an elevated activity of the cleaved active form of caspase-3 were detected by TUNEL kit analysis and Western blot, suggesting that cisplatin induces apoptosis of tubular epithelial cells (Fig. 4A, B, E). When miR-483-5p was overexpressed by LV-miR-483-5p infusion or in TG 483 mice, cisplatin-induced increased apoptosis of tubular epithelial cells compared with negative controls (Fig. 4A, C, D, F, G). We also found that cisplatin-induced autophagosomes in tubular epithelial cells under a transmission electron microscope, and enhanced expression of LC3B in the kidney by western bolt, suggesting that cisplatin induces autophagy of tubular epithelial cells (Fig. 4A, H). When miR-483-5p was overexpressed with LV-miR-483-5p infusion or in TG 483 mice, LC3B expression was significantly inhibited, suggesting that miR-483-5p alleviate autophagy of tubular epithelial cells in cisplatin-induced AKI mice. Together, our data suggest that miR-483-5p plays an important pathophysiologic role in the kidneys during cisplatin-induced AKI, which aggravates cisplatin-induced AKI by promoting apoptosis and inhibiting autophagy of tubular epithelial cells.

C57BL/6 mice with LV-miR-483-5p infused into the kidney or TG483 mice, were treated with cisplatin(15 mg/kg) C57BL/6 mice were intraperitoneally injected with cisplatin (15 mg/kg) and then sacrificed at 72 h. A Western blotting analysed the expression of p53, cleaved-caspase-3, and LC3B in the renal cortex. B C57BL/6 mice were treated with cisplatin (15 mg/kg), then sacrificed at 72 h, and apoptosis was assessed in the kidney by TUNEL staining. Arrows indicate apoptotic cells. Scale bar = 50 μm. C C57BL/6 mice infused with LV-miR-483-5p or LV-NC into the kidney were treated with cisplatin(15 mg/kg), then sacrificed at 72 h, and apoptosis was assessed in the kidney by TUNEL staining. Arrows indicate apoptotic cells. Scale bar = 50 μm. D TG 483 and control mice were treated with cisplatin (15 mg/kg), then sacrificed at 72 h, and apoptosis was assessed in the kidney by TUNEL staining. Arrows indicate apoptotic cells. Scale bar = 50 μm. E–G Quantification of cell apoptosis. The average number of apoptotic cells per section were calculated. (n = 3–5). H Transmission electron microscopic analyses revealed autophagosomes in C57BL/6 mice 72 h after cisplatin (15 mg/kg) treatment. Red arrow indicates autophagosomes. Scale bars = 500 nm. t test was used for two comparisons. All data are presented as the means ± SD. ns no significant difference; *P < 0.05; **P < 0.01; ***P < 0.001.
Overexpression of miR-483-5p promotes apoptosis and inhibits autophagy of NRK-52E cells induced by cisplatin
To characterize the effects of cisplatin on tubular epithelial cells, we carried out in vitro experimentation using the renal tubular epithelial cell line, NRK-52E. When treated with low-dose cisplatin (0–15 μM) for 12 h, miR-483-5p was not upregulated in the cell (Fig. 5B). As the dose of cisplatin was added (up to 30 μM) for 0–12 h, miR-483-5p was markedly increased in a time-dependent manner (Fig. 5A). With 30 μM cisplatin treatment for 12 h, NRK-52E cells were induced to undergo significant apoptosis, as shown by an increased number of apoptotic TUNEL-positive tubular epithelial cells (Fig. 5D, E) and elevated activity of the cleaved active form of caspase-3 (Fig. 5C). When miR-483-5p mimics were transfected into cells, apoptosis was aggravated with 30 μM cisplatin for 12 h compared with NC controls (Fig. 5C, E). Upregulation of LCIII by 30 μM cisplatin for 12 h was inhibited by miR-483-5p mimics transfection, showing that miR-483-5p decrease the autophagy of NRK-52E cells induced by cisplatin (Fig. 5C). These data suggest that miR-483-5p overexpression aggravated apoptosis, but attenuated autophagy of NRK-52E cells induced by cisplatin.

A Real-time PCR analysis of miR-483-5p levels in NRK-52E cells after 8 and 12 h of treatment with 30 µM cisplatin (n = 3–4). B Real-time PCR analysis of miR-483-5p levels in NRK-52E cells after 0–30 µM cisplatin treatment for 12 h (n = 3). C NRK-52E cells were transfected with miR-483-5p mimics, followed by 0, 7.5, 15, and 30 µM cisplatin treatment for 12 h. Western blot analysed the levels of GPX3, cleaved-caspase-3, and LC3B. E NRK-52E cells were transfected with miR-483-5p mimics, followed by treatment with 30 µM cisplatin for 12 h. Apoptosis was assessed by TUNEL staining. Red arrow indicates TUNEL-positive cells. Scale bar = 300 μm. D Quantification of cell apoptosis. The average number of apoptotic cells in each well of a 6-well plate were calculated (n = 3–5). One-way ANOVA was used for multiple comparisons. All data are presented as the means ± SD. ns no significant difference; *P < 0.05; **P < 0.01; ***P < 0.001.
miR-483-5p directly targets GPX3
To gain further insight into the mechanisms by which miR-483-5p plays important pathophysiologic roles in the kidneys during cisplatin-induced AKI, we used Web-based target prediction software programs (Target Scan, miRWalk, miRanda, and PITA) to predict the potential targets of miR-483-5p. GPX3 was selected because GPX3 belongs to the glutathione peroxidase family, which functions in the detoxification of hydrogen peroxide. Using the luciferase assay, we identified that nucleotides 570-576 of the 3′UTR of GPX3 from mice are complementary to seed sequences of miR-483-5p, which inhibited the luciferase activity of a reporter containing the wild-type (WT) GPX3 3′ UTR, but had no effect on the reporter with a mutated 3′UTR (Fig. 6A, B). Furthermore, expression of GPX3 protein in NRK-52E cells was downregulated by miR-483-5p mimics transfection (Figs. 6C, 5C). We next determined the role of miR-483-5p in regulating GPX3 in vivo. As expected, GPX3 was downregulated in the kidney of mice induced AKI by cisplatin, which was negatively correlated with miR-483-5p level in the kidney during cisplatin-induced AKI (Fig. 6D). Importantly, injection of LV3-miR-483-5p to the kidney contributed to reduced GPX3 level in the kidney. Moreover, cisplatin treatment further downregulated GPX3 level in the kidney (Fig. 6E). Furthermore, GPX3 was also significantly downregulated in TG 483 mice, and showed lower level during cisplatin-induced AKI (Fig. 6F). Taken together, these data demonstrate that GPX3 is a direct target of miR-483-5p during cisplatin-induced AKI.

A miR-483-5p aligned with the 3′UTR of GPX3 mRNA. B NRK-52E cells were co-transfected with a reporter carrying a mutant or wild-type GPX3 3′UTR and miR-483-5p mimics or its control, followed by luciferase assay (n = 3). C Western blot analysis of GPX3 expression in NRK-52E cells transfected with miR-483-5p mimics. D Immunohistochemical analysis of GPX3 expression in the kidneys in cisplatin-induced AKI in C57BL/6 mice (n = 3). Scale bar = 20 μm. E C57BL/6 mice transduced with LV-miR-483-5p or LV-NC were treated with cisplatin(15 mg/kg), then sacrificed at 72 h, followed by immunohistochemical analysis of GPX3 expression in the kidney (n = 3). Scale bar = 20 μm. F Immunohistochemical analysis of GPX3 expression in kidneys of TG 483 and control mice 72 h after treatment with cisplatin (15 mg/kg). Scale bar = 20 μm. One-way ANOVA was used for multiple comparisons. All data are presented as the means ± SD. ns no significant difference; *P < 0.05; **P < 0.01; ***P < 0.001.
GPX3 overexpression rescues cisplatin-induced AKI in TG 483 mice
The function of GPX3 in AKI was further examined in vivo. Lentivirus-mediated GPX3 (LV-GPX3) or negative control (LV-NC) was injected into the renal cortex of TG 483 mice. The expression of GPX3 in the kidney of TG 483 mice was markedly elevated based on Western blot analysis and IHC (Fig. 7A, B). As expected, GPX3 overexpression in the kidneys of TG 483 mice rescued cisplatin-induced AKI in TG 483 mice. As shown in Fig. 7C–G, the administration of cisplatin to TG 483 mice induced more severe renal dysfunction and kidney damage compared to controls, which was manifest as increased levels of BUN in the blood and KIM-1 in the kidney and aggravated renal tubule vacuolation degeneration and cell death in the kidney (Fig. 7D–G). It is interesting that aggravated renal dysfunction and kidney injury was completely rescued by overexpression of GPX3 in the kidney of TG 483 mice (Fig. 7D–G). Together, our data suggest that miR-483-5p plays an important role in cisplatin-induced AKI by targeting GPX3.

A Western blot analyses of GPX3 expression in the NRK-52E cell infected with lentivirus-mediated GPX3 (LV-GPX3) or negative control (LV-NC). B TG483 mice were transduced with LV-GPX3 or LV-NC into the kidney, followed by treatment with cisplatin (15 mg/kg) and then sacrificed at 72 h. Representative micrographs of immunostaining for GPX3 in the kidney are presented (n = 3). Scale bar = 20 μm. C–E TG483 mice were transduced with LV-GPX3 or LV-NC into the kidney, followed by treatment with cisplatin (15 mg/kg) and then sacrificed at 72 h. The body weight (C), blood nitrogen level (D), and Real-time PCR analysis of KIM-1 level in the renal cortex are present (n = 3). G Representative H&E images. F Tubular injury score was calculated according to the Paller method (n = 3). Scale bar = 20 μm. One-way ANOVA was used for multiple comparisons. All data are presented as the means ± SD. ns no significant difference; *P < 0.05; **P < 0.01; ***P < 0.001.
miR-483-5p promotes renal tubular cells apoptosis in cisplatin-induced AKI by targeting GPX3
To determine if apoptosis and autophagy involved in cisplatin-induced AKI are regulated by miR-483-5p targeting GPX3, we overexpressed GPX3 in renal tubular cells by LV-GPX3 lentivirus injection into the kidney of TG 483 mice, then cisplatin was or was not administered to mice. We found that cisplatin induced more renal tubular cells apoptosis in the kidney of TG 483 mice compared with the controls, because higher level of the cleaved active form of caspase-3 (Fig. 8A) and more apoptotic TUNEL-positive tubular epithelial cells were detected in the kidney of TG483 mice (Fig. 8C, E). However, overexpression of GPX3 in the kidney significantly inhibited apoptosis in the kidney of TG483 mice (Fig. 8A, C, E). These data suggest that miR-483-5p promotes renal tubular cells apoptosis in cisplatin-induced AKI by targeting GPX3. We further investigated if miR-483-5p promoted apoptosis in NRK-52E cells upon cisplatin treatment by targeting GPX3. As shown in Fig. 8B, D, F, NRK-52E cells were transfected with miR-483-5p mimics, followed by 30 μM cisplatin treatment for 12 h. Compared with controls, more NRK-52E cells underwent apoptosis with miR-483-5p mimics, which was rescued by GPX3 lentivirus infection either (Fig. 8B, D, F). We also determined the expression of p53, a molecule closely linked to apoptosis. We showed that in vivo and in vitro, p53 was induced by cisplatin and further upregulated when miR-483-5p was overexpressed. While overexpression of GPX3 completely rescued the upregulation (Fig. 8A, B). These data suggest that during cisplatin-induced AKI, miR-483-5p promoted renal tubular cells apoptosis and aggravated the kidney injury by targeting GPX3.

A TG483 mice were transduced with LV-GPX3 or LV-NC into the kidney, followed by treatment with cisplatin (15 mg/kg) and then sacrificed at 72 h. Levels of p53, cleaved-caspase-3, and LC3B in the renal cortices were determined by western blot. B NRK-52E cells were transfected with miR-483-5p mimics or LV-GPX3, followed by treatment with 30 µM cisplatin for 12 h. Levels of p53, cleaved-caspase-3, and LC3B in NRK-52E cells were determined by western blot. C, E TG483 and control mice were transduced with LV-GPX3 or LV-NC into the kidney, followed by treatment with cisplatin (15 mg/kg) and then sacrificed at 72 h. Cell apoptosis was detected using a TUNEL kit. Red arrow indicates TUNEL-positive cells (E). The average number of apoptotic cells per section was calculated (C) (n = 3).Scale bar = 50 μm. D, F NRK-52E cells were infected with LV-GPX3 or LV-NC, followed by treatment with 30 µM cisplatin for 12 h. Cell apoptosis was detected using a TUNEL kit. Red arrow indicates apoptotic cells (F). Quantification data are presented. The average number of apoptotic cells per section was calculated (D) (n = 3) Scale bar = 300 μm.One-way ANOVA was used for multiple comparisons. All data are presented as the means ± SD. ns no significant difference; *P < 0.05; **P < 0.01; ***P < 0.001.
In addition to apopotosis, autophagy was also induced in the renal tubular cells by cisplatin in vivo and in vitro, based on the upregulation of LC3B by cisplatin (Fig. 8A, B). miR-483-5p overexpression inhibited the autophagy of renal tubular cells and GPX3 overexpression could not rescue this process (Fig. 8A, B). These data suggest that during cisplatin-induced AKI, miR-483-5p inhibited renal tubular cells autophagy and aggravated the kidney injury without targeting GPX3.
miR-483-5p increases oxidative stress in cisplatin-induced AKI by targeting GPX3
Because oxidative stress is closely associated with cisplatin-induced renal damage16, we investigated the association between miR-483-5p and oxidative stress in cisplatin-treated TG 483 mice and NRK-52E cells. Without cisplatin, malondialdehyde in the renal cortex was detected at a very low level in TG 483 mice and controls using an anti-malondialdehyde protein carrier antibody. Following cisplatin administration, TG 483 mice had a much higher malondialdehyde level, which could be rescued by GPX3 lentivirus infection (Fig. 9A). In NRK-52E cells, both MDA and ROS levels were increased significantly with 30 µM cisplatin for 12 h, and miR-483-5p mimics further enhanced their levels. As expected, GPX3 lentivirus infection significantly decreased MDA and ROS levels (Fig. 9B, C). Our data suggest that miR-483-5p aggravates oxidative stress by targeting GPX3 in cisplatin-induced AKI.

A TG483 mice and the controls were transduced with LV-GPX3 or LV-NC into the kidney, followed by treatment with cisplatin (15 mg/kg) or not, and then sacrificed at 72 h. Levels of malondialdehyde protein carrier in the renal cortices were determined by western blot. B NRK-52E cells were transfected with negative control (NC+ LV-NC), miR-483-5p mimics, miR-483-5p mimics, and LV-GPX3, followed by 30 µM cisplatin treatment for 12 h or not.Intracellular MDA levels were determined with a Lipid Peroxidation MDA Assay Kit (Beyotime Biotechnology, Shanghai, China). n = 3. C NRK-52E cells were transfected with negative control (NC+ LV-NC), miR-483-5p mimics, miR-483-5p mimics and LV-GPX3, followed by 30 µM cisplatin treatment for 12 h or not. Intracellular ROS levels were determined with a ROS Assay Kit (NanJing JianCheng Bioengineering Institute, Nanjing, China). n = 3. One-way ANOVA was used for multiple comparisons. All data are presented as the means ± SD. ns no significant difference; *P < 0.05, **P < 0.01; ***P < 0.001.
miR-483-5p is elevated in the serum of cancer patients after chemotherapies with cisplatin
In our preliminary study, serum samples were collected from 5 cancer patients before and 48 h after chemotherapy with cisplatin. Although the blood urea nitrogen and creatinine levels did not differ significantly before and after chemotherapy with cisplatin, the level of KIM-1 in the serum increased significantly after chemotherapy (Fig. 10A–C). KIM-1 is a transmembrane glycoprotein of the renal proximal tubule cell that belongs to the immunoglobulin superfamily. It has been reported that KIM-1 is a sensitive early indicator of AKI, which increases rapidly and usually earlier than BUN and serum creatinine in response to damaged proximal tubule cells. Using semiquantitative real-time PCR, we further demonstrated that miR-483-5p in the serum of patients increased significantly 72 h after chemotherapy with cisplatin (Fig. 10D). These results suggest that miR-483-5p be an alternative indicator of AKI (Fig. 11).

A, B Blood urea nitrogen and creatinine levels of cancer patients before and after 72 h of chemotherapy with cisplatin. C Serum KIM-1 level was measured using an ELISA Kit (Elabscience, Wuhan, Hubei, China). D Serum miR-483-5p level was analysed by Real-time PCR. The relative expression of the mature miR-483-5p was normalized to cel-miR-39-3p. Samples from the clinic were analyzed using the Wilcoxon test. *P < 0.05 (n = 5).

Our study reveals foremost that miR-483-5p is upregulated by cisplatin treatment, elevated miR-483-5p exacerbates oxidative stress by targeting GPX3, then up-regulates the expression of p53, activates caspase-3, and as a result, aggravates apoptosis of renal tubular cells. In addition, after cisplatin treatment, protective autophagy in renal tubular cells is inhibited by upregulated miR-483-5p, without targeting GPX3. Finally, the promotion of apoptosis and inhibition of autophagy in renal tubular cells aggravate cisplatin-induced AKI. Together, we demonstrate that miR-483-5p is a key and upstream mediator regulating AKI induced by cisplatin and may serve as a new target for diagnosis and therapy of AKI.
Discussion
As the main morphologic and functional unit for the kidney, the functional state of tubular cells play critical roles in the pathophysiology of kidney, as well as the recovery from injury17. In the kidneys, cisplatin accumulates at high concentrations in renal tubular cells (~5 times higher than the blood), causing tubular cell injury and death, which is a key determinant of AKI4. Therefore, a more complete understanding of the mechanism by which cisplatin affects tubular epithelial cells has become an area of research interest for the development of novel diagnosis and therapies for AKI. In this study we first reported upregulation of miR-483-5p and the pathogenic role in mediating oxidative stress and subsequent cell death and autophagy in renal tubular epithelial cells of cisplatin-induced AKI in a mouse model, which verified the increased expression and potential relationship with the pathogenesis of human AKI induced by cisplatin. We further demonstrated that miR-483-5p promotes cisplatin-induced AKI by targeting GPX3 and overexpression of GPX3 by virus injection prevents cisplatin-induced AKI in mice.
MicroRNA-483-5p has been reported to be involved in several diseases, such as cancer and osteoarthritis12,18,19. An increased level of miR-483-5p has been reported in the kidneys of a UUO mouse model of renal fibrosis13 and the serum of patients with rejection after transplantation14; however, whether or not miR-483 plays a role in the regulation of AKI is unknown. In this study we took advantage of a cisplatin-induced AKI mouse model using C57BL/6J mice and first reported a prominent upregulation of miR-483-5p primarily in the renal tubular cells after acute injury. In serum samples from patients treated with cisplatin, we also showed that upregulation of miR-483-5p in human serum which is more sensitive to indicate kidney injury than the blood urea nitrogen and creatinine levels. These results suggest that miR-483-5p is related to AKI. Our data demonstrated that overexpression of miR-483-5p in the kidneys of mice following lentivirus injection or in a transgenic mouse model induced more severe kidney injury after cisplatin treatment. These data suggest that miR-483-5p promotes cisplatin-induced AKI. Our results demonstrated that GPX3 is a direct target of miR-483-5p in regulating the progression of cisplatin-induced AKI. Interestingly, GPX3 overexpression in the kidneys of TG 483 mice rescued cisplatin-induced AKI in TG 483 mice, thus anti-miR-483-5p treatment alleviates AKI induced by cisplatin. These data indicate that miR-483-5p in tubular cells may be a key mediator of cisplatin-induced AKI, and therefore might serve as a potential therapeutic target for treating AKI.
The next question that was addressed was how miR-483-5p accentuates AKI. In our three mouse models of cisplatin treatment, we noted tubular epithelial cell flattening, brush border loss, and cell membrane bleb formation (Fig. 1). Further study showed that apoptosis was significantly induced in tubular epithelial cells (Fig. 4). Overexpression of miR-483-5p increased apoptosis of tubular epithelial cells induced by cisplatin (Fig. 4A, C, D, F, G). The in vitro findings also confirmed the miR-483-5p effect. More importantly, our results demonstrated that GPX3 overexpression rescued apoptosis of renal tubular cells induced by cisplatin, which confirmed that GPX3 might serve as a potential therapeutic target for treating AKI. Not only apoptosis, but also autophagy was detected in the cisplatin-induced AKI models. Autophagy is a highly conserved lysosomal degradation pathway that eliminates protein aggregates and dysfunctional organelles20,21. During cellular stress, autophagy is activated and serves primarily as an adaptive mechanism for cell survival, but sometimes may serve as an injured effect. It has been reported that in nephrotoxic AKI, autophagy is induced to protect against tubular cell injury and death22,23,24,25. In our research, cisplatin-induced autophagy both in vivo and in vitro to protect against tubular cell injury and death. Cisplatin was shown to induce a autophagosomes in tubular epithelial cells of the NRK-52E cell line under a transmission electron microscope (Fig. 4E). Expression of LC3B in the kidney was also significantly upregulated in vivo (Fig. 4A). Overexpression of miR483-5p in the kidney was associated with significant inhibition of LC3B expression. Unlike apoptosis, GPX3 overexpression did not rescue autophagy of renal tubular cells induced by cisplatin in vivo and in vitro. Thus, we believed that miR-483-5p promoted AKI, not only by promoting renal tubular cell apoptosis, but also by down-regulating autophagy, the process of which does not target GPX3. We further found that p53 was significantly increased in the kidney following treatment with cisplatin in all three mouse models. Overexpression of miR-483-5p in the kidney by means of virus injection or the transgenic method significantly increased the p53 level in the kidney (Fig. 4A). We also found that p53 was induced by cisplatin and overexpression of miR-483-5p increased the p53 level significantly in vitro and in vivo (Fig. 8A, B). Of note, p53 is a transcription factor that has been shown to be closely related to apoptosis and also regulates autophagy26,27. Recent studies have concluded that the p53 pathway plays a critical protective role in the pathogenesis of cisplatin-induced AKI28,29,30. Our results indicate that miR-483-5p regulates tubular cell apoptosis and autophagy via the p53 pathway.
Reactive oxygen species (ROS) and subsequent oxidative stress are largely involved in the development of AKI and are thought to be the conduit by which injury is transmitted to tubular cells5,31,32,33. Our results demonstrated that miR-483-5p aggravates oxidative stress induced by cisplatin in vivo and in vitro; anti-miR-483-5p treatment through GPX3 overexpression rescues this response. GPX3 belongs to the glutathione peroxidase family, most of which are produced in the renal proximal tubular cells, then secreted into the serum so that GPX3 is also referred to as plasma glutathione peroxidase34,35. GPX3 mainly functions in the detoxification of hydrogen peroxide and plays a role in protecting the kidney from oxidative damage36,37. We hypothesized that cisplatin induced the generation of ROS, stimulated the expression of p53, then promoted apoptosis of tubular epithelial cells, which resulted in AKI, at least in part.
MiR-483-5p has been reported to be an oncogene of various cancers, such as lung cancer18, adrenocortical cancer38, and nasopharyngeal carcinoma39. High expression of miR-483-5p could enhance epithelial ovarian cancer resistance to platinum-based chemotherapy by decreasing TAOK1 expression40. Whether inhibition of miR-483-5p will promote cancer treatment with cisplatin deserves futher investigation.
In conclusion, we demonstrate foremost that miR-483-5p is upregulated by cisplatin in vivo and in vitro. Overexpression of miR-483-5p by lentivirus or transgenic technology accentuated cisplatin-induced AKI in mice. The mechanism involved may be that after cisplatin treatment, elevated miR-483-5p exacerbates oxidative stress by targeting GPX3, then up-regulates the expression of p53, activates caspase-3, and as a result, promotes apoptosis of renal tubular cells. In addition, after cisplatin treatment, protective autophagy in renal tubular cells is inhibited by upregulated miR-483-5p, without targeting GPX3. Finally, the promotion of apoptosis and inhibition of autophagy in renal tubular cells aggravate cisplatin-induced AKI. Together, we demonstrate that miR-483-5p is a key and upstream mediator regulating AKI induced by cisplatin and may serve as a new target for diagnosis and therapy of AKI.
Data availability
The datasets analyzed during the current study are available from the corresponding author on reasonable request after publication.
References
Rewa, O. & Bagshaw, S. M. Acute kidney injury-epidemiology, outcomes and economics. Nat. Rev. Nephrol. 10, 193–207 (2014).
Bellomo, R., Kellum, J. A. & Ronco, C. Acute kidney injury. Lancet 380, 756–766 (2012).
Manohar, S. & Leung, N. Cisplatin nephrotoxicity: a review of the literature. J. Nephrol. 31, 15–25 (2018).
Ozkok, A. & Edelstein, C. L. Pathophysiology of cisplatin-induced acute kidney injury. BioMed Res. Int. 2014, 967826 (2014).
Holditch, S. J., Brown, C. N., Lombardi, A. M., Nguyen, K. N. & Edelstein, C. L. Recent advances in models, mechanisms, biomarkers, and interventions in cisplatin-induced acute kidney injury. Int. J. Mol. Sci. 20, 3011 (2019).
Volarevic, V. et al. Molecular mechanisms of cisplatin-induced nephrotoxicity: a balance on the knife edge between renoprotection and tumor toxicity. J. Biomed. Sci. 26, 25 (2019).
Bartel, D. P. MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233 (2009).
Ebert, M. S. & Sharp, P. A. Roles for microRNAs in conferring robustness to biological processes. Cell 149, 515–524 (2012).
Tufekci, K. U., Meuwissen, R. L. & Genc, S. The role of microRNAs in biological processes. Methods Mol. Biol. 1107, 15–31 (2014).
Paul, P. et al. Interplay between miRNAs and human diseases. J. Cell. Physiol. 233, 2007–2018 (2018).
Fu, H. et al. Identification of human fetal liver miRNAs by a novel method. FEBS Lett. 579, 3849–3854 (2005).
Wang, H. et al. Intra-articular delivery of Antago-miR-483-5p inhibits osteoarthritis by modulating Matrilin 3 and tissue inhibitor of metalloproteinase 2. Mol. Ther. 25, 715–727 (2017).
Li, N. Q. et al. Expression of intronic miRNAs and their host gene Igf2 in a murine unilateral ureteral obstruction model. Braz. J. Med. Biol. Res. 48, 486–492 (2015).
Sui, W. et al. Serum microRNAs as new diagnostic biomarkers for pre- and post-kidney transplantation. Transplant. Proc. 46, 3358–3362 (2014).
Sabbisetti, V. S. et al. Blood kidney injury molecule-1 is a biomarker of acute and chronic kidney injury and predicts progression to ESRD in type I diabetes. J. Am. Soc. Nephrol. 25, 2177–2186 (2014).
Dugbartey, G. J., Peppone, L. J. & de Graaf, I. A. An integrative view of cisplatin-induced renal and cardiac toxicities: Molecular mechanisms, current treatment challenges and potential protective measures. Toxicology 371, 58–66 (2016).
Johansson, M. E. Tubular regeneration: when can the kidney regenerate from injury and what turns failure into success? Nephron Exp. Nephrol. 126, 76 (2014).
Song, Q. et al. miR-483-5p promotes invasion and metastasis of lung adenocarcinoma by targeting RhoGDI1 and ALCAM. Cancer Res. 74, 3031–3042 (2014).
Wang, H. et al. Chondrocyte mTORC1 activation stimulates miR-483-5p via HDAC4 in osteoarthritis progression. J. Cell. Physiol. 234, 2730–2740 (2019).
Ohsumi, Y. Historical landmarks of autophagy research. Cell Res. 24, 9–23 (2014).
Yang, Z. & Klionsky, D. J. Eaten alive: a history of macroautophagy. Nat. Cell Biol. 12, 814–822 (2010).
Livingston, M. J. & Dong, Z. Autophagy in acute kidney injury. Semin. Nephrol. 34, 17–26 (2014).
Kaushal, G. P. & Shah, S. V. Autophagy in acute kidney injury. Kidney Int. 89, 779–791 (2016).
Havasi, A. & Dong, Z. Autophagy and tubular cell death in the kidney. Semin. Nephrol. 36, 174–188 (2016).
Tang, C. et al. PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against renal ischemia-reperfusion injury. Autophagy 14, 880–897 (2018).
Nikoletopoulou, V., Markaki, M., Palikaras, K. & Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 1833, 3448–3459 (2013).
Hashimoto, N., Nagano, H. & Tanaka, T. The role of tumor suppressor p53 in metabolism and energy regulation, and its implication in cancer and lifestyle-related diseases. Endocr. J. 66, 485–496 (2019).
Dutta, R. K. et al. Beneficial effects of Myo-inositol oxygenase deficiency in cisplatin-induced AKI. J. Am. Soc. Nephrol. 28, 1421–1436 (2017).
Yang, A. et al. p53 induces miR-199a-3p to suppress mechanistic target of rapamycin activation in cisplatin-induced acute kidney injury. J. Cell. Biochem. 120, 17625–17634 (2019).
Ranganathan, P. et al. UNC5B receptor deletion exacerbates tissue injury in response to AKI. J. Am. Soc. Nephrol. 25, 239–249 (2014).
Lu, C. Y. & de Albuquerque Rocha, N. Oxidative stress and metabolism: the NF-Erythroid 2 p45-related factor 2: Kelch-like ECH-associated protein 1 system and regulatory T lymphocytes in ischemic AKI. J. Am. Soc. Nephrol. 26, 2893–2895 (2015).
Kaushal, G. P., Chandrashekar, K. & Juncos, L. A. Molecular interactions between reactive oxygen species and autophagy in kidney disease. Int. J. Mol. Sci. 20, 3791 (2019).
Chen, X., Wei, W., Li, Y., Huang, J. & Ci, X. Hesperetin relieves cisplatin-induced acute kidney injury by mitigating oxidative stress, inflammation and apoptosis. Chem. Biol. Interact. 308, 269–278 (2019).
Olson, G. E. et al. Extracellular glutathione peroxidase (Gpx3) binds specifically to basement membranes of mouse renal cortex tubule cells. Am. J. Physiol. Renal Physiol. 298, F1244–F1253 (2010).
Burk, R. F., Olson, G. E., Winfrey, V. P., Hill, K. E. & Yin, D. Glutathione peroxidase-3 produced by the kidney binds to a population of basement membranes in the gastrointestinal tract and in other tissues. Am. J. Physiol. Gastrointest. Liver Physiol. 301, G32–G38 (2011).
Xu, W. et al. Differential expression of genes associated with the progression of renal disease in the kidneys of liver-specific glucokinase gene knockout mice. Int. J. Mol. Sci. 14, 6467–6486 (2013).
Fan, Y. et al. Mechanism of ginsenoside Rg1 renal protection in a mouse model of d-galactose-induced subacute damage. Pharm. Biol. 54, 1815–1821 (2016).
Agosta, C. et al. MiR-483-5p and miR-139-5p promote aggressiveness by targeting N-myc downstream-regulated gene family members in adrenocortical cancer. Int. J. Cancer 143, 944–957 (2018).
Zheng, X.-H. et al. Plasma microRNA profiling in nasopharyngeal carcinoma patients reveals miR-548q and miR-483-5p as potential biomarkers. Chin J. Cancer 33, 330–338 (2014).
Rattanapan, Y. et al. High expression of miR−483-5p predicts chemotherapy resistance in epithelial ovarian cancer. Microrna 10, 51–57 (2021).
Acknowledgements
This work was supported by National Natural Sciences Foundation of China (31371186, 31000633, 81302230), Guangdong Natural Science Foundation (2019A1515011458, 2014A030313296), Science and Technology Program of Guangzhou, China (201607010081), the Guangdong Province Outstanding Young Teacher Training funds, the China Postdoctoral Science Foundation (2013M542159). We thank Prof. Xiaochun Bai for contributing suggestions.
Author information
Authors and Affiliations
Contributions
M.L. devised the conceptual ideas. Y.X., J.L., and M.L. contributed to study design. Y.X., X.Z., W.G., X.X., W.P., Y.L., and Y.Z. carried out the cell culture experiments. Y.X., J.L., and W.P. performed the animal experiments. L.L. collected the data from the cancer patients receiving chemotherapies with cisplatin. Y.X., J.L., and M.L. contributed to the discussion and interpretation of the results. M.L. took the lead in writing the paper. Y.X., C.Z., and J.L. contributed to paper editing. All authors approved the final paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All research involving human participants was approved by the Ethics Committee of Nanfang Hospital Affiliated to Southern Medical University (Guangzhou, China), and informed consent was obtained from all patients.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Xia, Y., Pan, W., Xiao, X. et al. MicroRNA-483-5p accentuates cisplatin-induced acute kidney injury by targeting GPX3. Lab Invest 102, 589–601 (2022). https://doi.org/10.1038/s41374-022-00737-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-022-00737-3
This article is cited by
-
A cisplatin conjugate with tumor cell specificity exhibits antitumor effects in renal cancer models
BMC Cancer (2023)
-
MiR-483-5p downregulation alleviates ox-LDL induced endothelial cell injury in atherosclerosis
BMC Cardiovascular Disorders (2023)
