
Abstract
Recessive dystrophic epidermolysis bullosa (RDEB) is an intractable genetic disease of the skin caused by mutations in the COL7A1 gene. The majority of patients with RDEB harbor compound heterozygous mutations—two distinct mutations on each chromosome—without any apparent hotspots in the COL7A1 mutation pattern. This situation has made it challenging to establish a reliable RDEB mouse model with mutations that accurately mimic the genomic background of patients. Here, we established an RDEB mouse model harboring patient-type mutations in a compound heterozygous manner, using the CRISPR-based genome-editing technology i-GONAD. We selected two mutations, c.5818delC and E2857X, that have frequently been identified in cohorts of Japanese patients with RDEB. These mutations were introduced into the mouse genome at locations corresponding to those identified in patients. Mice homozygous for the 5818delC mutation developed severe RDEB-like phenotypes and died immediately after birth, whereas E2857X homozygous mice did not have a shortened lifespan compared to wild-type mice. Adult E2857X homozygous mice showed hair abnormalities, syndactyly, and nail dystrophy; these findings indicate that E2857X is indeed pathogenic in mice. Mice with the c.5818delC/E2857X compound heterozygous mutation presented an intermediate phenotype between the c.5818delC and E2857X homozygous mice. Single-cell RNA sequencing further clarified that the intrafollicular keratinocytes in c.5818delC/E2857X compound heterozygous mice exhibited abnormalities in cell cycle regulation. The proposed strategy to produce compound heterozygous mice, in addition to the established mouse line, will facilitate research on RDEB pathogenesis to develop a cure for this devastating disease.
Similar content being viewed by others

Introduction
Recessive dystrophic epidermolysis bullosa (RDEB) is an intractable genetic disorder caused by mutations in the COL7A1 gene1,2. COL7A1 encodes type VII collagen, which anchors the epidermis to the dermis in the basal membrane zone of the skin3,4. Consequently, genetic mutations that result in the loss of functional type VII collagen weaken the adhesion of the epidermis to the dermis, causing the epidermis to detach upon even minor physical force. Patients with RDEB suffer from repetitive blistering and healing, which lead to skin scarring5 that has a significant impact on the quality of life. In the most severe forms, scarring leads to the development of highly malignant squamous cell carcinoma, which may reduce lifespan6,7. There is currently no cure for RDEB; thus, the development of new, effective treatments is an urgent issue8.
Establishing an ideal model system that accurately recapitulates human clinical features is essential for pathophysiological studies and drug development9. The Col7a1-knockout mouse, which lacks exons 46–69 of wild-type Col7a1, was introduced as the first RDEB mouse model10. Col7a1-knockout mice develop severe blistering of the skin and die 1–2 weeks after birth. The Col7a1 hypomorphic mouse was also established as an RDEB model by inserting an artificial gene-targeting cassette in intron 3 of Col7a1, which suppresses the expression of type VII collagen to 10% of the normal level11. Col7a1 hypomorphic mice also present RDEB-like manifestations, including blistering, nail dystrophy, and mitten deformities. Although these models accurately recapitulate some of the clinical features observed in human RDEB, genomic alterations evoking disease phenotypes are not identical to those of human RDEB patients. Thus, new RDEB models that harbor patient-derived mutations are required.
Mimicking the human COL7A1 RDEB mutations in mice is technically challenging owing to the unique nature of the mutation pattern associated with this disease. Most RDEB patients have compound heterozygous COL7A1 mutations, with distinct mutations at each COL7A1 locus3,4. Thus, to closely mimic the human causal mutation pattern, the mouse model also needs to carry two independent mutations, for which the establishment of two separate mouse lines harboring one of these mutations is required. In addition, the mutations causing RDEB are broadly distributed across the COL7A1 gene, which is the second-largest gene in the human genome with 118 exons3. More than 100 pathogenic mutations of RDEB have been registered in ClinVar to date, and no mutation hotspots have been reported12. Moreover, each RDEB mutation causes variable phenotypes and outcomes, indicating that modeling the disease in a mouse requires the establishment of multiple genetically modified lines and thus is a time-, cost-, and labor-intensive process. Owing to these limitations, no compound heterozygous mouse model for RDEB has been reported.
The development of mouse genome manipulation technologies using the CRISPR system has significantly expanded access to new model systems13,14. Recently, the Oviductal Nucleic Acids Delivery (i-GONAD) method based on CRISPR technology was developed to generate genetically modified mice with less technical difficulty15. In this study, using the i-GONAD method, we attempted to generate a mouse model carrying human RDEB compound heterozygous mutations. As a proof-of-concept, we selected two COL7A1 mutations that are relatively common (approximately 5–20% cases depending on the study) in Japanese patients: c.5818delC and E2857X16,17. In humans, both mutations introduce a premature termination codon in COL7A1, resulting in truncation of type VII collagen. c.5818delC has been proposed to cause more severe phenotypes than E2857X because the c.5818delC mutation site is located within the triple helical domain-coding region of COL7A1, a critical domain for trimerization, whereas the E2857X mutation site is located at the 3’ end of COL7A118,19,20. Patients carrying both the mutations simultaneously (compound heterozygous) have a moderate to severe phenotype16. Furthermore, we performed single-cell transcriptome analysis to elucidate the impact of each mutation on skin integrity.
Material and methods
Mice
C57BL/6J mice (8–30-week-old males and 8–12-week-old females) were obtained from CLEA Japan (Tokyo, Japan). All mice were housed under a 12-h light-dark cycle and provided with solid food and filtered water. All animals were handled in accordance with the guidelines of the Animal Committee of Osaka University Graduate School of Medicine, which approved the experimental protocol.
i-GONAD
Genome editing with i-GONAD was conducted according to a published protocol15,21. In brief, males and females in estrus were mated at 16:00–17:00, and the plug was visually confirmed the next morning at 9:00–10:00. Day 0 of pregnancy was set at 0:00, and surgical procedures were performed on day 0.7 (16:00). The dorsal skin of the anesthetized female was incised to expose the oviduct. Approximately 1 µL of electroporation solution was injected into the oviduct lumen using a micropipette (made from a glass tube; GDC-1, Narishige, Tokyo, Japan) using a puller (PC-100, Narishige) and a mouthpiece. Immediately after solution injection, the oviduct was covered with a Kimwipe soaked in phosphate-buffered saline and sandwiched between tweezer-type electrodes (CUY652P2.5×4, NEPA GENE, Chiba, Japan). Electroporation was performed using a NEPA21 system (NEPA GENE) with the following parameters: 50 V poring pulse with 5 ms pulse, 50 ms pulse interval, 3 pulses, and 10% decay (±pulse orientation); and 10 V transfer pulse with 50 ms pulse, 50 ms pulse interval, 3 pulses, and 40% decay (±pulse orientation). After electroporation, the fallopian tube was returned to its original position, and the incision was sutured. Sanger sequencing was performed on the resulting pups to ensure that the mice carried the designed mutations. The mice selected for this study were backcrossed for two generations before they were used in the experiments22,23.
Immunostaining
The excised skin fragments were soaked in 4% paraformaldehyde overnight and then in 30% sucrose solution in phosphate-buffered saline overnight. The tissues were then embedded in Tissue-Tek OCT Compound (Sakura Finetek, Tokyo, Japan), frozen on dry ice, and stored at –80 °C until use. Sections of 6 µm thickness were incubated with 1% bovine serum albumin in Tris-buffered saline with Tween for 1 h at room temperature and then incubated with rabbit polyclonal anti-mouse COL7 antibody (1:100; made in the laboratory) at 4 °C overnight. Alexa Fluor 488-conjugated donkey anti-rabbit IgG (1:500; Abcam, Cambridge, UK) and DAPI solution (1:600; Dojindo, Kumamoto, Japan) were added as secondary antibodies, and the reaction was performed for 1 h. The sections were sealed using ProLong Gold Antifade Mountant (Thermo Fisher Scientific, Waltham, MA, USA). All images were acquired using an all-in-one BZ-X800 fluorescence microscope (Keyence, Osaka, Japan).
Single-cell RNA sequencing
The hair on the backs of the mice (2-week-old) was shaved and removed using a hair removal cream under anesthesia. The mice were euthanized, and a 1 × 3 cm section of the back skin was collected. The skin samples were shredded with scissors in 10% fetal bovine serum/RPMI1640 medium (Nacalai Tesque, Kyoto, Japan) containing 300 µg/ml Liberase Research Grade (Roche, Basel, Switzerland) and 5 U/ml recombinant DNase I (Takara Bio, Shiga, Japan). The shredded skin samples were dissociated by incubation at 37 °C for 40 min, with agitation every 10 min in 5 ml of the same medium as described above. After reaction neutralization, a single-cell suspension was obtained by passing through a 70-µm cell strainer and then through a 40-µm cell strainer. The suspension was washed twice and resuspended in 2% minimal essential medium-alpha containing fetal bovine serum and nucleosides but not phenol red (Thermo Fisher Scientific, Waltham, MA, USA).
A single-cell RNA sequence library was constructed as previously described24. The libraries were sequenced using the NextSeq 2000 platform. The read length was set to 20 (read 1) + 8 (i7) + 8 (i5) + 51 (read 2) bases.
Data analysis
Single-cell RNA-sequencing data were aligned to the mouse reference genome (Genome Reference Consortium Mouse Build 38, mm10), and gene counting was performed using STAR (v2.7.6a)25. The output table from STAR was further analyzed using Seurat v326. Low-quality cells were manually filtered out based on the number of transcripts, genes, and the percentage of mitochondrial genes per cell. After filtering, the data were processed using a Seurat common workflow (“CreateSeuratObject,” “NormalizeData,” “FindVariableFeatures,” “ScaleData,” “RunPCA,” “FindNeighbors,” “FindClusters,” “RunUMAP”). The results were visualized in uniform manifold approximation and projection (UMAP) format using the “DimPlot” function. To identify the cell types of each cluster, cluster biomarkers were calculated using the “FindMarkers” function. Gene expression levels of marker genes were visualized using the “dotplot” method of Scanpy (v1.7.1)27. For detailed analyses of keratinocytes, cells of clusters 0 and 3 were extracted and re-processed following the Seurat pipeline. To investigate the impact of a Col7a1 mutation on the subtypes of keratinocytes, differentially expressed genes were determined for each sub-cluster using the “FindMarkers” function of the “MAST” method28. Gene Ontology enrichment analysis was conducted using the R package Enrichr29 to interpret the functional roles of the differentially expressed genes.
Results
Establishing the mutant mouse lines 5818delC and E2857X using i-GONAD
To generate a compound heterozygous model for RDEB, we selected two pathogenic mutations, 5818delC and E2857X, which are relatively common in the Japanese RDEB cohort (Fig. 1a)16. We searched the corresponding genomic locations for each mutation in the mouse genome and designed guide RNAs and single-stranded oligo DNA nucleotides to introduce the c.5818delC- or E2857X-type mutation into the mouse genome (Fig. 1b–e). c.5818delC was introduced into mice by deleting the cytosine at c.5000 (P1934) of mouse Col7a1. Induction of the E2857X mutation was accomplished by changing V2849 to a termination codon (c.8545G>T, c.8546T>A in mouse Col7a1). Although there are slight differences in the genomic coordinates for c.5818 and E2857 between humans and mice, we retained the human mutation names “5818delC” and “E2857X” for simplicity in the presentation and interpretation of this proof-of-concept study. To establish mouse lines with each mutation, we first performed genome editing with C57/BL6 mice using i-GONAD. Sanger sequencing was performed to validate the DNA sequences around the modified regions. We successfully obtained six (33.3% of total obtained pups) and seven (50% of total obtained pups) correctly modified mice for 5818delC and E2857X, respectively (Table 1). We also observed some mice with unwanted insertions or deletions identified through Sanger sequencing; these mice were excluded from further experiments. These data indicate that the i-GONAD method could be used to introduce RDEB patient-derived mutations into mice.

a Human type VII collagen structure and locations of the 5818delC and E2857X mutations. b, c Comparisons of the human and mouse amino acid sequences of type VII collagen surrounding the two mutations. The asterisk indicates a non-conserved amino acid. d, e Design of the single-stranded oligo DNA nucleotides (ssODNs) and guide RNAs (gRNAs) for gene editing; the 5818delC or E2857X mutations are highlighted in red. A 5818delC-type mutation was introduced into mice by deleting C at the 5000th base of the coding sequence (5000delC), which is located within P1934. E2857X was introduced into mice by converting V2849 to a termination codon, specifically through the following changes: c.8545G>T, c.8546T>A. Small letters indicate the modifications that prevent re-cutting by Cas9.
Validation of the pathogenicity of 5818delC and E2857X in mice
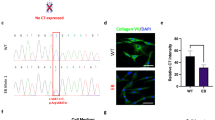
Next, we sought to confirm whether each mutation was pathogenic in mice. To this end, we chose mice with the 5818delC or E2857X mutation introduced in a heterozygous manner and backcrossed these mice at least twice with wild-type C57/BL6 mice to reduce the off-target effects of i-GONAD, followed by mating the backcrossed generation. We obtained heterozygous and homozygous mice with the expected Mendelian ratio for both mutations (Table 2). Interestingly, 5818delC homozygous mice died immediately after birth, whereas E2857X homozygous mice showed no survival defect; these findings correlated well with the clinical severity in RDEB patients (Fig. 2a, b). We also examined the formation of blistering, a characteristic complication in patients with RDEB. Extensive blister formation was consistently observed in 5818delC homozygous mice but not in E2857X mice (Fig. 2c, d). Next, we confirmed the expression levels of type VII collagen in the skin. As expected from their poor survival and extensive formation of blisters, 5818delC homozygous mice showed no obvious accumulation of type VII collagen at the dermal–epidermal junction, whereas E2857X homozygous mice showed a characteristic type VII collagen staining pattern at the dermal–epidermal junction (Fig. 2c, d). Although the phenotypes in E2857X homozygous mice were milder than those of 5818delC mice at a younger age, hair abnormalities, syndactyly, and nail dystrophy were observed in aged (45-week-old) E2857X homozygous mice (Fig. 2e, f). These data indicate that the 5818delC and E2857X mutations are pathogenic in mice.

a, b Survival analysis comparing wild-type, heterozygous, and homozygous mice. c, d Representative results showing the formation of blisters on the front paws (top). Immunostaining for type VII collagen (green) on the skin; nuclei were stained with DAPI (blue) (bottom). e Hair abnormality in aged (45-week-old) E2857X homozygous mice. f Nail dystrophy in aged (45-week-old) E2857X homozygous mice.
Generation of the RDEB compound heterozygous mouse model
After confirming the pathogenicity of 5818delC and E2857X in mice, we crossed E2857X homozygous mice and 5818delC heterozygous mice to generate 5818delC/E2857X compound heterozygous mice. The survival phenotype of 5818delC/E2857X compound heterozygous mice was milder than that of 5818delC homozygous mice but was more severe than that of E2857X homozygous mice (Fig. 3a). Extensive blistering was also observed in compound heterozygous mice, suggesting a severe form of the disease (Fig. 3b). Immunostaining of type VII collagen in the compound heterozygous mice showed weak but characteristic staining at the dermal–epidermal junction; this staining pattern could be attributed to the presence of the E2857X locus (Fig. 3b). Thus, we successfully generated viable RDEB compound heterozygous mice.

a Survival analysis. b Representative results showing the formation of blisters on the front paws (top). Immunostaining for type VII collagen (green) on the skin. The nucleus was stained with DAPI (blue) (bottom).
Single-cell transcriptome analysis of the RDEB compound heterozygous mouse model
Unlike other RDEB model mice, the compound heterozygous mouse model established in this study was based on genomic mutations identified in patients with RDEB. Therefore, we further characterized how these mutations hamper skin integrity in mice using single-cell RNA sequencing of wild-type and 5818delC/E2857X compound heterozygous mice. We used young (2-week-old) mice because the 5818delC/E2857X compound heterozygous mice had shorter expected survival than wild-type mice. After standard quality control and filtering steps, we recovered 3862 and 3957 cells from wild-type and 5818delC/E2857X compound heterozygous mice, respectively. The UMAP profiles of all cells detected in this study are shown in Fig. 4a. We identified ten clusters, and the cell types were annotated using known marker genes (Fig. 4b) described previously30,31. Clusters 0 and 3 were keratinocytes that expressed keratin-coding genes. Cluster 1 was annotated as fibroblast-like cells that expressed collagen genes. We also identified immune cells, vascular cells, muscle cells, and Schwann cells.

a UMAP representation of single-cell RNA-sequencing results for the skin from wild-type and compound heterozygous mice. b Heatmap showing the expression of marker genes in each cluster. The size of the dot indicates the fraction of cells expressing a given gene. The color indicates the magnitude of expression. c Cell proportion analysis of panel a between wild-type and compound heterozygous mice. Dots indicate the proportion for each replicate. d UMAP representation focusing on the keratinocyte fraction. e Heatmap showing the expression of marker genes in each cluster. f Differentially expressed genes in each cluster (wild-type vs. compound heterozygous mice). g Expression of genes demarcating the germinative layer cells. h Gene Ontology analysis for the differentially expressed genes from cluster 0. i UMAP representation focusing on the fibroblast-like cell fraction. j Cell proportion analysis of panel i between wild-type and the compound heterozygous mice. k Marker gene analysis of panel i.
By comparing the cell proportion, we found that the relative abundance of keratinocytes was reduced in the 5818delC/ E2857X compound heterozygous mice (Fig. 4c). In addition, as type VII collagen (COL7A1) is known to be functional in keratinocytes, we performed further analyses focusing on clusters 0 and 3. The cells in clusters 0 and 3 were extracted and re-clustered (Fig. 4d). The expression of marker genes for each sub-cluster is shown in Fig. 4e. Sub-clusters 0, 1, 3, 5, and 7 represent the inner layer, whereas sub-clusters 2, 6, and 8 represent the outer layer of the hair follicle. Interfollicular keratinocytes were detected as sub-cluster 4. To identify how the loss of functional type VII collagen affected skin integrity, we analyzed genes differentially expressed between wild-type and compound heterozygous mice in each sub-cluster (Fig. 4f). The most significant gene expression changes were observed in sub-cluster 0, which likely represents the germinative layer cells given the expression of Dcn and Mt2 (Fig. 4g)31. Gene Ontology analysis revealed that the differentially expressed genes in sub-cluster 0 were enriched in the biological process “cell cycle” (Fig. 4h), suggesting defects in proper hair follicle development in the compound heterozygous mice. In addition, the proportion of fibroblast-like cells increased in compound heterozygous mice (Fig. 4c). Further sub-cluster analysis of the fibroblast-like cells identified six sub-clusters (Fig. 4i). The proportion of cells in sub-cluster 2 showed the most prominent increase in compound heterozygous mice relative to wild-type mice (Fig. 4j). Marker gene analysis of these fibroblast-like cell sub-clusters revealed that Postn, which is upregulated in the skin of RDEB patients32, was strongly expressed in sub-cluster 2 (Fig. 4k), suggesting the possibility that compound heterozygous mice recapitulated the phenotype in humans.
Discussion
RDEB is a devastating genetic disease of the skin, and the development of a cure is eagerly anticipated. COL7A1-knockout mice and hypomorphic mice are commonly used models for RDEB research1,2. Although these models accurately recapitulate RDEB-related phenotypes, the genomic background of these mice does not match that of RDEB patients. In this study, using a rapid CRISPR/Cas9-based genome-editing technique, we established a compound heterozygous mouse model with the human COL7A1 mutations 5818delC and E2857X, and confirmed that the model manifested RDEB-related phenotypes. Therefore, this mouse model is useful for studying the pathogenesis of RDEB and the detailed function of COL7A1.
Establishing multiple genetically modified mouse lines is costly and time consuming33. Because there is no hotspot for RDEB-associated mutations and most patients have compound heterozygous mutations, it has been challenging to select the appropriate mutations to generate an RDEB mouse model. In this study, we utilized the recently developed i-GONAD, a CRISPR/Cas9-based genome-editing technique, to generate genetically modified mice15. As i-GONAD performs genome editing while the zygotes are still in the oviduct, this method eliminates multiple labor-intensive steps, including the isolation and ex vivo handling of embryos, microinjection of genome-editing tools, and preparation of pseudo-pregnant female mice for implanting genome-edited embryos. We have successfully demonstrated that i-GONAD can be used to effectively establish genetically modified mice with editing at multiple genome sites, which will be useful for the study of genetic diseases.
We have successfully demonstrated that E2857X is a pathogenic mutation in RDEB. Although E2857X is a pathogenic mutation in RDEB, it was unclear whether type VII collagen with E2857X alone could induce RDEB-related phenotypes. Because E2857X is located at the end of COL7A1, the impact of the deletion could be minimal. Saito et al. argued that, although type VII collagen with E2857X could be expressed at the skin, full-length type VII collagen is critical for the formation of fully functional anchoring fibrils20. The fact that mutations located closer to the C-terminus end than E2857X, such as Y2858H or P2906fs, are also registered as pathogenic mutations further supports the importance of having full-length type VII collagen to maintain skin integrity4. However, the detailed mechanisms of how E2857X affects proper fibril anchoring need to be addressed. For such studies, E2857X homozygous mice might serve as a useful tool.
In summary, we have provided a valuable workflow for establishing a mouse model of disease using i-GONAD. Given that precision medicine is becoming an increasingly recognized and important part of medical practice, it is critical to approach drug development by considering genetic differences among patients. The workflow presented herein will therefore serve as a fundamental tool for research on genetic diseases, particularly for diseases caused by compound heterozygous mutations.
Data availability
The sequencing data used in this study have been deposited and are available in GEO (GSE181357).
References
Bardhan, A. et al. Epidermolysis bullosa. Nat. Rev. Dis. Primers 6, 78 (2020).
Has, C. et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br. J. Dermatol. 183, 614–627 (2020).
Christiano, A. M. et al. Structural organization of the human type VII collagen gene (COL7A1), composed of more exons than any previously characterized gene. Genomics 21, 169–179 (1994).
Varki, R., Sadowski, S., Uitto, J. & Pfendner, E. Epidermolysis bullosa. II. Type VII collagen mutations and phenotype-genotype correlations in the dystrophic subtypes. J. Med. Genet. 44, 181–192 (2007).
Nystrom, A. & Bruckner-Tuderman, L. Injury- and inflammation-driven skin fibrosis: the paradigm of epidermolysis bullosa. Matrix Biol. 68-69, 547–560 (2018).
Fine, J. D., Johnson, L. B., Weiner, M., Li, K. P. & Suchindran, C. Epidermolysis bullosa and the risk of life-threatening cancers: the National EB Registry experience, 1986-2006. J. Am. Acad. Dermatol. 60, 203–211 (2009).
Guerra, L., Odorisio, T., Zambruno, G. & Castiglia, D. Stromal microenvironment in type VII collagen-deficient skin: the ground for squamous cell carcinoma development. Matrix Biol. 63, 1–10 (2017).
Has, C., South, A. & Uitto, J. Molecular therapeutics in development for epidermolysis bullosa: update 2020. Mol. Diagn. Ther. 24, 299–309 (2020).
Vandamme, T. F. Use of rodents as models of human diseases. J. Pharm. Bioallied. Sci. 6, 2–9 (2014).
Heinonen, S. et al. Targeted inactivation of the type VII collagen gene (Col7a1) in mice results in severe blistering phenotype: a model for recessive dystrophic epidermolysis bullosa. J. Cell Sci. 112(Pt 21), 3641–3648 (1999).
Fritsch, A. et al. A hypomorphic mouse model of dystrophic epidermolysis bullosa reveals mechanisms of disease and response to fibroblast therapy. J. Clin. Invest. 118, 1669–1679 (2008).
Landrum, M. J. et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067 (2018).
Shalem, O., Sanjana, N. E. & Zhang, F. High-throughput functional genomics using CRISPR-Cas9. Nat. Rev. Genet 16, 299–311 (2015).
Huijbers, I. J. Generating genetically modified mice: a decision guide. Methods Mol. Biol. 1642, 1–19 (2017).
Ohtsuka, M. et al. i-GONAD: a robust method for in situ germline genome engineering using CRISPR nucleases. Genome Biol. 19, 25 (2018).
Tamai, K. et al. Recurrent COL7A1 mutations in Japanese patients with dystrophic epidermolysis bullosa: positional effects of premature termination codon mutations on clinical severity. Japanese Collaborative Study Group on Epidermolysis Bullosa. J. Invest. Dermatol. 112, 991–993 (1999).
Sawamura, D. et al. Genetic studies of 20 Japanese families of dystrophic epidermolysis bullosa. J. Hum. Genet 50, 543–546 (2005).
Dang, N. & Murrell, D. F. Mutation analysis and characterization of COL7A1 mutations in dystrophic epidermolysis bullosa. Exp. Dermatol. 17, 553–568 (2008).
Koshida, S. et al. Hallopeau-Siemens dystrophic epidermolysis bullosa due to homozygous 5818delC mutation in the COL7A gene. Pediatr. Int. 55, 234–237 (2013).
Saito, M., Masunaga, T., Teraki, Y., Takamori, K. & Ishiko, A. Genotype-phenotype correlations in six Japanese patients with recessive dystrophic epidermolysis bullosa with the recurrent p.Glu2857X mutation. J. Dermatol. Sci. 52, 13–20 (2008).
Gurumurthy, C. B. et al. Creation of CRISPR-based germline-genome-engineered mice without ex vivo handling of zygotes by i-GONAD. Nat. Protoc. 14, 2452–2482 (2019).
Aoto, K. et al. ATP6V0A1 encoding the a1-subunit of the V0 domain of vacuolar H+-ATPases is essential for brain development in humans and mice. Nat. Commun. 12, 2107 (2021).
Wasylishen, A. R. et al. Daxx maintains endogenous retroviral silencing and restricts cellular plasticity in vivo. Sci. Adv. 6, eaba8415 (2020).
Ho, Y. T. et al. Longitudinal single-cell transcriptomics reveals a role for Serpina3n-mediated resolution of inflammation in a mouse colitis model. Cell Mol. Gastroenterol. Hepatol. 12, 547–566 (2021).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Finak, G. et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 16, 278 (2015).
Kuleshov, M. V. et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016).
Joost, S. et al. Single-cell transcriptomics reveals that differentiation and spatial signatures shape epidermal and hair follicle heterogeneity. Cell Syst. 3, 221–237 e229 (2016).
Joost, S. et al. The molecular anatomy of mouse skin during hair growth and rest. Cell Stem Cell 26, 441–457.e447 (2020).
Chacon-Solano, E. et al. Fibroblast activation and abnormal extracellular matrix remodelling as common hallmarks in three cancer-prone genodermatoses. Br. J. Dermatol. 181, 512–522 (2019).
Capecchi, M. R. Gene targeting in mice: functional analysis of the mammalian genome for the twenty-first century. Nat. Rev. Genet 6, 507–512 (2005).
Acknowledgements
We would like to thank Editage [http://www.editage.com] for editing and reviewing this manuscript for English language.
Funding
This study was supported by JSPS KAKENHI Grant Numbers JP21K08324 (T.S.) and JP19H03682 (K.T.) and a research fund from StemRIM Inc.
Author information
Authors and Affiliations
Contributions
T.S. and K.T. conceived the idea; S.T. and T.K. carried out the experiments. S.T., T.S., and K.T wrote, reviewed, and revised the manuscript; S.T, T.S, K.I, Y.Y, S.Y, S.M, and K.T. analyzed and interpreted the data and performed the statistical analysis. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
K.T. is a scientific founder of and received research funding from StemRIM. K.T. and T.S. are StemRIM stockholders. S.T., K.I., T.K., Y.Y., and S.Y. are employees of StemRIM.
Ethics approval and consent to participate
All animals were handled in accordance with the guidelines of the Animal Committee of Osaka University Graduate School of Medicine that approved the experimental protocol.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Takaki, S., Shimbo, T., Ikegami, K. et al. Generation of a recessive dystrophic epidermolysis bullosa mouse model with patient-derived compound heterozygous mutations. Lab Invest 102, 574–580 (2022). https://doi.org/10.1038/s41374-022-00735-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-022-00735-5
This article is cited by
-
A Review of CRISPR-Based Advances in Dermatological Diseases
Molecular Diagnosis & Therapy (2023)
