
Abstract
Undifferentiated pleomorphic sarcoma (UPS) and malignant peripheral nerve sheath tumor (MPNST) are aggressive soft tissue sarcomas that do not respond well to current treatment modalities. The limited availability of UPS and MPNST cell lines makes it challenging to identify potential therapeutic targets in a laboratory setting. Understanding the urgent need for improved treatments for these tumors and the limited cellular models available, we generated additional cell lines to study these rare cancers. Patient-derived tumors were used to establish 4 new UPS models, including one radiation-associated UPS—UPS271.1, UPS511, UPS0103, and RIS620, one unclassified spindle cell sarcoma—USC060.1, and 3 new models of MPNST—MPNST007, MPNST3813E, and MPNST4970. This study examined the utility of the new cell lines as sarcoma models by assessing their tumorigenic potential and mutation status for known sarcoma-related genes. All the cell lines formed colonies and migrated in vitro. The in vivo tumorigenic potential of the cell lines and corresponding xenografts was determined by subcutaneous injection or xenograft re-passaging into immunocompromised mice. USC060.1 and UPS511 cells formed tumors in mice upon subcutaneous injection. UPS0103 and RIS620 tumor implants formed tumors in vivo, as did MPNST007 and MPNST3813E tumor implants. Targeted sequencing analysis of a panel of genes frequently mutated in sarcomas identified TP53, RB1, and ATRX mutations in a subset of the cell lines. These new cellular models provide the scientific community with powerful tools for detailed studies of tumorigenesis and for investigating novel therapies for UPS and MPNST.
Similar content being viewed by others


Introduction
Soft tissue sarcomas are rare malignancies originating from the mesoderm. There exist more than 50 histological subtypes based on their tissues of origin. Undifferentiated pleomorphic sarcoma (UPS) comprises a subset of 5 to 15% of soft tissue sarcomas for which clinicians cannot determine the tissue of origin from analysis of the tumor’s molecular and histologic characteristics1,2,3. A subset of UPS, unclassified spindle cell sarcomas (USC), are rare high-grade malignancies with an unknown cell of origin4,5. The standard of treatment for UPS is surgical resection. For patients in whom surgery is not feasible, chemotherapy and radiation therapy may be used, but they have low-to-moderate response rates6,7. UPS is characterized by a high level of genomic instability, as indicated by its complex karyotype8,9,10, yet specific recurring genetic alterations are uncommon11. UPS has a 5-year overall survival rate of 60% and a 10-year overall survival rate of 48%12. Malignant peripheral nerve sheath tumors (MPNSTs), which comprise 2% of all soft tissue sarcomas1, are another rare and genomically unstable sarcoma13,14. Half of MPNSTs occur in patients with the autosomal dominant cancer predisposition syndrome neurofibromatosis type 1 (NF1; OMIM # 162200). Approximately 40% of MPNST cases are sporadic, and the remaining 10% are associated with previous radiation therapy15,16,17. Chemotherapy and radiation therapy are ineffective for treating MPNSTs2,18,19; therefore, surgical resection remains the standard treatment20,21,22,23. MPNSTs have a high rate of local recurrence24,25. The 5-year survival rate is 35 to 50%, and the 10-year survival rate is 7.5%, stressing the need for improved treatment options17,26. Limited treatment options are available for patients with UPS, USC and MPNST, largely because the mechanisms involved in the progression of these heterogeneous sarcomas require further investigation17,27,28,29.
UPS and MPNST both have complex karyotypes, and no specific genomic alterations are unique to the tumor type9,10,30,31. UPS has complex cytogenetic rearrangements in 30 to 35% of the genome32, but to date, no specific alterations have been useful for classifying UPS. Loss of chromosome 13q, which leads to inactivation of the retinoblastoma (RB) pathway, is frequently observed in UPS33,34,35, as are losses of genes in the TP53/ARF/MDM2 pathway36,37. A study of soft tissue sarcoma by The Cancer Genome Atlas Research Network10 reported several somatic copy number alterations, specifically deep deletions in RB1 in 16%, TP53 in 16%, and CDKN2A in 20% of UPS. The mutation burden was low in soft tissue sarcomas, an average of 1.06 per Mb in 206 tumors. The significantly mutated genes in UPS included RB1 and potential driver truncating mutations of NF110. MPNST’s complex genomic profile includes copy number aberrations, like microdeletions of NF1 in 5 to 20% of cases38,39, as well as CDKN2A inactivation40,41, and TP53 mutations. Recently, mutations in the genes of the polycomb repressive complex 2 (PRC2) components SUZ12 and EED and the loss of histone 3 lysine 27 trimethylation (H3K27me3) were proposed as markers for NF1-related and sporadic MPNSTs28,42.
Cell line models that can form tumors in mice are valuable tools to advance the understanding of soft tissue sarcomas with no known genetic drivers such as UPS and MPNST. UPS cell lines have been developed by our laboratory (UPS186 and RIS81929) and others43,44, some with the ability to form tumors when injected subcutaneously into mice45,46. Well-studied NF1-associated MPNST cell lines are also available within the scientific community, including SNF02.2 and SNF94.3, both derived from a metastatic site in the lung, SNF96.2, isolated from a recurrent MPNST; and S462, ST88-14, T265, and MPNST64247,48,49,50. Longo et al. recently developed and characterized a sporadic MPNST cell line, 2XSB51. The NF1-associated MPNST cell lines listed above can form tumors in mice, allowing for in vivo study of these difficult-to-treat sarcomas47,48,52,53,54. Multiple new NF1-MPNST cell lines and PDX models, with corresponding clinical and genomic annotations, are now available for preclinical studies55. However, further development of UPS and MPNST resources is warranted because cell lines from genomically complex sarcomas are underrepresented in cancer cell line databases such as the Cancer Cell Line Encyclopedia and Catalog Of Somatic Mutations In Cancer56,57. While some sarcoma cell lines are listed in large-scale systematic databases like The Cancer Dependency Map (depmap)58, they have not been studied as extensively as those from other lineages. Broad systematic approaches that include gene-dependency screens and small molecule sensitivity data (among other—omics data analyses)58,59, when applied to poorly understood sarcomas like those presented here could identify unique targets and avenues for drug discovery.
This study aimed to generate and characterize new cellular models of UPS and MPNST that will deepen our understanding of the aberrant cellular processes of these sarcomas known to lack clear genetic or molecular markers. The UPS and MPNST cell lines generated in this study will be available to the scientific community to facilitate the study of these debilitating tumors.
Materials and methods
UPS and MPNST primary culture and cell lines
We obtained approval from the Institutional Review Board of The University of Texas MD Anderson Cancer Center and patients’ informed written consent before establishing the cell lines described in this study. The cell lines were isolated from surgical specimens from patients at MD Anderson Cancer Center. A sarcoma pathologist confirmed the final histology of the tumors at MD Anderson Cancer Center. A portion of the original specimen was fixed in formalin and paraffin-embedded, followed by immunohistochemical (IHC) staining for hematoxylin and eosin to visualize the samples. Isolation of tumor cells for RIS620, UPS271.1, and UPS511 was described previously by May et al.29 USC060.1 and UPS0103 cells were isolated as described by Peng et al.60. Briefly, fresh sterile samples from resected tumors were minced in Dulbecco’s modified Eagle medium (DMEM; Corning, cat # 10013CV and incubated with collagenase type I (3%; Sigma-Aldrich, cat # C-0130), DNase I (0.02%; Sigma-Aldrich, cat # D-4527), and hyaluronidase (1.5 mg/ml; Sigma-Aldrich, cat # H-3506) at 37 °C for 2 to 4 h. After digestion, the sample was strained through a mesh screen (BD Biosciences, cat # 352360), and undigested tissue was discarded. The sample was centrifuged, washed, resuspended in phosphate-buffered saline (PBS; Corning, Corning, NY, cat # 21040CV), and gently transferred to tubes containing 10 ml of a 100% polysucrose/sodium diatrizoate solution (Histopaque-1077; Sigma-Aldrich, cat # H8889) overlaid with 15 ml of 75% Histopaque-1077. After centrifugation at 4 °C for 30 min at 1200 × g, the tumor cells in the top interface above the 75% Histopaque were collected and plated on cell culture plates. After establishing cells that grew in a monolayer, the morphology of the cell lines was evaluated by microscopy.
UPS cells were cultured in DMEM supplemented with 20% FBS, 1% penicillin/streptomycin (100 U/ml), and 1% L-alanyl-L-glutamine (glutagro; Corning, cat # 25-015-CI). The USC060.1 and UPS271.1 nomenclature denotes that these cell lines were established from a patient-derived xenograft tumor that arose from a subcutaneous injection of either USC060.1 cells or UPS271.1 cells into a mouse. MPNST3813E cells and MPNST007 cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin (100 U/ml). MPNST4970 was cultured in DMEM/F12 (Corning, cat # 10090CV) supplemented with 20% FBS, 1% penicillin/streptomycin (100 U/ml), 1% glutagro, and 1% nonessential amino acids (Corning, cat # 25-025-CI), and Schwann cell growth supplement (ScienCell Research Laboratories, cat # 1752). Isolation of MPNST642 cells was described previously47, and this cell line was characterized further in this study by sequencing analysis. Cells were cultured in a humidified incubator at 37 °C with 5% CO2. Cells were tested for mycoplasma every 6 months.
Doubling time was measured by determining cell confluence using an Incucyte system (Essen Bioscience, Ann Arbor, MI). UPS or MPNST cells were plated in 24-well plates or 96-well plates at 10 to 30% confluence and imaged every 4 h until they reached confluency. The doubling time was calculated using a nonlinear regression fit for exponential growth in Prism 7 software (GraphPad Software, La Jolla, CA).
Short tandem repeat (STR) DNA profiling was performed using a Powerplex 16 HS kit (Promega, cat # DC2101). Genomic DNA was extracted from the tumor specimens and the cultured cell lines using a QIAmp mini DNA isolation kit (Qiagen, cat # 51304). Sixteen loci were screened for regions of microsatellite instability with defined trinucleotide or tetranucleotide repeats within the chromosomes. The STR results for each cell line were authenticated against the corresponding primary tumor’s STR profile and internal database of over 4000 public profiles and profiles unique to cell lines developed by MDACC investigators. To comply with the Health Insurance Portability and Accountability Act of 1996 and MD Anderson institutional policy, only a selection of human loci is shown in Table 2. The alleles from 8 human loci are reported in this study: AMEL, CSF1PO, D13S317, D16S539, D5S818, D7S820, TH01, and TPOX.
Normal Schwann cells (NSC) served as control cells. NSC were cultured in Schwann Cell Medium (ScienCell Research Laboratories, cat # 1700 and 1701). Human bone-marrow-derived mesenchymal stem cells (MSC) were cultured in mesenchymal stem cell medium (PromoCell cat # C12974 and C28009) and used as a reference for MDM2 amplification. DDLPS-224A cells were cultured in DMEM supplemented with 20% FBS and 1% penicillin/streptomycin (100 U/ml) and used as a positive control for MDM2 amplification61.
Clonogenicity and migration assays
Clonogenicity was assayed as described by May et al.29. Briefly, cells were seeded onto 6-well plates at low densities of 5000 cells for UPS cell lines and 500 to 2000 cells for MPNST cells (depending on the cell line) and were allowed to form colonies for 14 to 30 days. Images were obtained using a digital scanner (Konica Minolta, Tokyo, Japan) or a BZ-X800 microscope (Keyence Corporation, Itasca, Illinois). Migration assays were performed using a modified Boyden chamber assay with Transwell inserts containing polyethylene terephthalate membranes with 8-µm pores (Corning, cat # 354578). For clonogenicity and migration assays, cells were fixed with 5% glutaraldehyde and stained with 0.1% crystal violet in 20% methanol. For migration assays, Images at ×20 magnification were taken with a Nikon microscope (Tokyo, Japan) or a BZ-X800 microscope (Keyence Corporation, Itasca, Illinois).
Protein expression assays
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis and Western blotting were performed as described by May et al.29. Briefly, proteins were extracted from cultured cells in radioimmunoprecipitation buffer (RIPA) containing protease inhibitors (Sigma-Aldrich, cat # 11697498001). Thirty micrograms of protein were loaded on to 4 to 15% precast polyacrylamide gels (Bio-Rad, cat# 4561085) gels for PRC2 components and p53. Acid extraction following the standard protocol (Abcam) was performed on cultured cells to isolate histones, and 3 μg of protein was separated on 4 to 20% precast polyacrylamide gels (Bio-Rad, cat # 4561095) for detection of histone H3 and H3K27me3. Proteins were transferred onto low-fluorescence polyvinylidene fluoride membranes using a semidry transfer method according to the manufacturer’s instructions (Bio-Rad, cat # 1704274). Membranes were blocked in 5% nonfat dry milk before probing with the specific antibodies listed in Supplementary Table 1. Horseradish peroxidase-conjugated secondary antibodies were detected by enhanced chemiluminescence (GE Healthcare, cat # RPN2236) and imaged using a Chemidoc imager (Bio-Rad).
In vivo xenograft studies
The MD Anderson Institutional Animal Care and Use Committee approved all animal procedures. We proceeded with patient-derived xenograft (PDX) implant generation when we received <1 g of residual tumor; if the residual tumor weighed >2 g, we generated a PDX implant and established a cell line. For PDX implantation experiments, 6-week-old male or female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (Jackson Laboratory, Bar Harbor, ME) were implanted with pieces of MPNST007, MPNST3813E, RIS620, or UPS0103 tumor (~3–5 mm in diameter) in the flank. For injections of USC060.1 cells, 3 × 106 cells in 1:1 PBS:Matrigel (Corning, cat #354248) (0.1 ml/mouse) Trypan blue viable cells were injected subcutaneously into the flank of 6-week-old male or female NSG mice. For the UPS511 cell line, 5 × 106 Trypan blue viable cells in 1:1 PBS:Matrigel (Corning, cat #354248) (0.1 ml/mouse) were injected subcutaneously into the flank of 6-week-old male or female NSG mice. Mice were monitored for tumor size and well-being and were euthanized when tumors reached an approximate volume of 1000–2000 mm3. Tumor growth was measured once per week using a caliper. The mice were not assessed for metastases. Figures show data from at least five representative animals. Resected tumors were weighed, fixed in formalin and paraffin-embedded for IHC studies. IHC studies including hematoxylin and eosin were performed to compare the xenograft specimens to the original patient tumors. The percentage uptake was calculated by dividing the number of mice that grew tumors by the total number of mice implanted or injected with cells. The latency period was estimated from the tumor growth data.
Sequencing
Targeted sequencing was performed for a panel of 4 genes frequently mutated in UPS and MPNST according to TCGA: TP53, ATRX, RB1 and CDKN2A, using genomic DNA isolated from cultured cells with a Qiagen QIAmp mini DNA isolation kit10,62. DNA was fragmented and bait-captured according to the manufacturer’s guidelines63. Libraries were sequenced on a HiSeq2000 or HiSeq4000 sequencer (Illumina, San Diego, CA) with 76-bp paired-end reads for MPNST007, MPNST3813E, UPS0103, and RIS620; and 76-bp single reads for USC060.1, UPS271.1, and UPS511. The raw data were converted to fastq format and aligned to the reference genome (hg19) with the Burroughs–Wheeler Aligner64. Median target coverage was 256 × to 763× for MPNST007, MPNST3813E, MPNST4970; and UPS0103 cells and 207× to 230× for USC060.1, UPS271.1, and UPS511 cells. Mutations were called against a common normal lung sample using the MuTect and Pindel tools65,66. Sanger sequencing of TP53 was performed with genomic DNA from the original MPNST specimen using either an ABI 3730XL or 3730 DNA sequencer (Thermo Fisher Scientific, Waltham, MA) with a Big Dye Terminator cycle sequencing chemistry kit (Thermo Fisher Scientific, cat # 4337455) according to the manufacturer’s guidelines.
Copy number alteration analysis
MDM2 copy number analysis was performed using the MDM2 TaqMan Copy Number Assay Hs02970282_cn (Thermo Fisher Scientific cat # 4400291) and the reference assay for TERT (Thermo Fisher Scientific cat # 4403316) according to manufacturer’s guidelines. Results were analyzed using CopyCaller Software v2.1 (Thermo Fisher Scientific, Waltham, MA).
Results
Establishment and morphology of UPS and MPNST cell lines
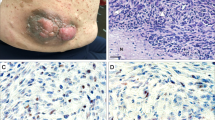
All cell lines were established from cells isolated from residual resected tumors. The UPS cell lines were generated from recurrent or metastatic tumors, and the MPNST tumors (all three NF1-associated) were generated from primary or recurrent tumors (Table 1). A sarcoma pathologist at MD Anderson Cancer Center confirmed the final histology of the tumors (AJL). The STR profiles of the established cell lines matched those of the original patient tumors, indicating that each of the cell lines was derived from the tumor of origin, and there was no cross-contamination with other cell lines. The hematoxylin and eosin staining of the original specimens showed a mixture of tumor cells and stromal cells in UPS and MPNST tumors (Fig. 1a, lower panels). The cells were all mononuclear. USC060.1 and UPS511 cell lines had an elongated spindle-like shape, while UPS271.1 and UPS0103 cell lines were polygonal and RIS620 cells exhibited both polygonal and spindle shapes (Fig. 1a, upper panels). MPNST007 cell lines had an elongated spindle shape, whereas MPNST3813E cells showed more polygonal morphology in comparison, as did MPNST4970 cells (Fig. 1a, upper panels). Table 2 shows STR profiles from the resected tumor and corresponding cell line.

a Bright-field images of UPS and MPNST cell lines (upper panels) and images of hematoxylin and eosin (H&E)-stained sections of the original patient tumors (lower panels), ×20 magnification. Scale bar represents 100 µm. b MDM2 copy number analysis in UPS cell lines. MSC mesenchymal stem cells, were used as a reference. DDLPS-224A cells were used as a positive control for MDM2 amplification. TERT was used as the reference gene for the MDM2 copy number assay. N = 2, graph shows calculated copy number; error bars indicate copy number range. c Immunoblots showing expression of EZH2, SUZ12, EED, RbAP46/48, and H3K27me3 in normal Schwann cells (NSC), MPNST007, MPNST3813E, MPNST642, and MPNST4970 cells. EZH2, SUZ12, EED, and RbAP46/48 levels were normalized to actin (middle panels). H3K27me3 expression was normalized to H3 levels (lower two panels).
As of November 2021, USC060.1 has been carried to passage 50, UPS271.1 to passage 36, UPS511 to passage 35, and UPS0103 to passage 17. MPNST007 and MPNST3813E have been taken over passage 100, MPNST4970 has been carried to passage 41. Our laboratory will continue to passage the lower passage UPS and MPNST cell lines to determine whether they can be carried to over 90 passages.
Misclassification of the UPS tumors as dedifferentiated liposarcoma (DDLPS) was ruled out by MDM2 copy number analysis using genomic DNA isolated from the established cell lines. Bone marrow-derived mesenchymal stem cells (MSC) served as the reference for MDM2 copy number, as they represented a potential cell of origin for UPS. DDLPS-224A, a DDLPS cell line with known amplification of MDM2, served as a positive control61. No amplification of MDM2 was detected in the UPS cell lines, providing evidence that the specimens from which these cell lines were derived were indeed UPS and not misclassified DDLPS.
Protein expression
Western blotting was used to assess the expression of histone marks and proteins known to be associated with MPNST: H3K27me3, and the PRC2 members SUZ12, EED, RbAp46/48, and EZH2. NSCs, considered the cell of origin for MPNST, served as control cells. The MPNST007 and MPNST4970 cell lines expressed higher levels of EZH2 than did the control NSCs. The MPNST cells were also assayed for the absence of H3K27me3, which is considered a potential diagnostic marker of NF1-associated MPNSTs28. The H3K27me3 mark was present in MPNST007 cells and MPNST4970 but not in MPNST642 or MPNST3813E cells (Fig. 1c, lower panels). These findings suggested that MPNST007 and MPNST4970 are PRC2-competent, and MPNST642 and MPNST3813E are PRC2-incompetent.
Cell proliferation rate
The proliferation rate for the UPS and MPNST cell lines was determined by measuring relative confluence (Fig. 2a, b). For the UPS cell lines, the doubling time ranged from approximately 33.3 to 279.8 h. USC060.1 cells had the shortest doubling time (33.3–53.5 h), and the doubling time of UPS271.1 cells was similar to that of USC060.1 cells (48.8–56.6 h). UPS511 cells and RIS620 cells had intermediate doubling times (90.6–92.0 h and 151.5–174.8 h, respectively). The UPS0103 cell line had the slowest doubling time of the UPS cells in this study, at 279.8 h. Of the MPNST cell lines, MPNST3813E cells doubled in 36.3 h (range 34.6 to 39.7 h). MPNST007 cells doubled in 49.4 to 58.9 h, and MPNST4970 doubled in ~84.6 h. These results showed that the cell lines established in the current study have a wide range of doubling times, reflecting these tumors’ heterogeneous nature.

Doubling time for UPS (a) and MPNST (b) cell lines. Cells were plated and allowed to grow until they reached a growth plateau. Relative confluence was measured using an Incucyte device (Essen Bioscience). c Clonogenicity assay for UPS and MPNST cells. For UPS cells, 5000 cells were seeded and allowed to grow for the indicated number of days. For MPNST cells, 500 to 2000 cells were seeded and allowed to grow for 16 days. Migration of UPS cells and MPNST cells, ×20 magnification. d Targeted sequencing results for selected sarcoma genes. Refer to the results section for details on the mutations identified.
In vitro tumorigenic potential of the new UPS and MPNST cell lines
The UPS and MPNST cell lines were assessed for clonogenic and migratory potential. For clonogenic assays, cells were seeded at a low density and allowed to form colonies for up to 16 days. All the UPS cell lines formed colonies. USC060.1, UPS271.1, and RIS620 formed small colonies, UPS511 formed large distinct colonies, and UPS0103 formed diffuse colonies (Fig. 2c, upper panels). MPNST007, and MPNST3813E formed distinct colonies, while MPNST4970 cells formed more diffuse colonies. Further analysis of tumorigenic potential was performed by measuring the migration of the established cell lines in a modified Boyden chamber assay. All the UPS and MPNST cell lines exhibited the ability to migrate toward a chemoattractant (Fig. 2c, lower panels). These results showed that the newly established cell lines could form colonies and migrate.
DNA mutation analysis of selected cancer genes
The mutation status of genes frequently mutated in UPS according to the TCGA, including ATRX, CDKN2A, RB1, and TP53, was assessed using high-coverage targeted sequencing with a minimum median target coverage of ~200×10,62,67,68. Figure 2d summarizes the mutations found in the cell lines. UPS271.1, UPS511, UPS0103 cell lines had no mutations in the cancer genes listed above. The USC060.1 cell line harbored a mutation in RB1: c.C1574G; p.A525G. This mutation has been reported in hemangioblastomas and acute myeloid leukemia69,70. The RIS620 cell line carried a TP53 mutation at c.G818A; p.R273H, that was documented in lung cancer, esophageal cancer, pancreatic carcinoma, hematological malignancies, ovarian and endometrial cancers, and gastric cancer33,71,72,73,74,75,76. Mutation status for EED, EZH2, NF1, and SUZ12 in the MPNST cell lines was reported by Kochat et al.68.
In vivo tumorigenicity studies
The in vivo tumor-forming capacity of the established cell lines was determined by either subcutaneous injection or tumor implantation. For USC060.1, and UPS511 cell lines, 3–5 million cells were injected subcutaneously into the flank of NSG mice, and tumor growth was measured weekly (Fig. 3a, b). For the implantation experiments, UPS0103, RIS620, MPNST007, and MPNST3813E, tumor pieces of ~3–5-mm3 were embedded into the flank of NSG mice, which were then monitored for tumor growth (Fig. 3c–f). Mice were sacrificed when the tumors reached a volume of 1000 to 2000 mm3. Hematoxylin and eosin staining of the xenograft tumors was performed on slides cut from formalin-fixed, paraffin-embedded blocks (Fig. 3a–f, right panels). Cells from the xenografts had similar morphology as cells from the parental tumors (H&E insets). The percentage of uptake of tumor cells and latency period are shown in Table 3. Overall, the cell line injection and implant xenograft models had uptake ranging from 70 to 100%. The latency period was the longest for UPS511 (50 days) cells and the shortest for RIS620 cells (23 days) (Table 3). The UPS271.1 and MPNST4970 cell lines show a heterologous growth pattern for the generation of xenografts that is currently under investigation.

a–d Tumor growth after UPS cell line injection or implantation in five representative animals (left panels). Images of hematoxylin and eosin (H&E)-stained sections of xenograft tumors at ×20 magnification (right panels). Scale bar indicates 100 µm. Inset shows original patient tumor image at ×20 magnification. e, f Tumor growth after MPNST cell line implantation in five representative animals (left panels). Images of H&E-stained sections of xenograft tumors at ×20 magnification (right panels). Scale bar indicates 100 µm. Inset shows original patient tumor image at ×20 magnification.
Discussion
Primary tumor cell lines established from patient tumors can recapitulate the original tumor for further study in a preclinical setting. The goal of the present study was to generate and characterize cellular and xenograft models for the study of 2 genomically complex sarcomas, UPS and MPNST. Including models like these in the latest systematic approaches58 or other large-scale methods will reveal previously unknown targetable vulnerabilities in sarcomas. We successfully established 4 UPS cell lines and one USC cell line. UPS was previously classified as malignant fibrous histiocytoma (MFH); the disadvantage of using any cell lines generated before the renaming of MFH as UPS is that they could be other sarcoma subtypes1,77,78. UPS cell lines with pathological confirmation are needed to overcome this complication. The UPS cell lines described in this study were derived from patient tumors classified as UPS or USC by a sarcoma pathologist at MD Anderson Cancer Center.
The establishment of new cancer cell line models presents some difficulties; most primary cultures fail to grow due to senescence after several months in culture. A recent study suggested the success rate is 15% in establishing sarcoma cell lines from resected tumors46. The soft tissue sarcoma cell lines described here have been in culture for ~1–2 years at this writing, showing no indication of senescence. Cell line contamination can be an issue for slow-growing cell lines (doubling times over 24 h) like those presented here. We confirmed no contamination in the cell lines based on the matching STR profiles for each cell line and the corresponding parental tumor. For the cell lines cultured from the initial patient-derived xenograft (USC060.1 and UPS271.1), targeted sequencing of the tumor samples showed over 95% of reads mapped to the human reference genome, ruling out the possibility of potential mouse cell contamination. Moreover, six of the eight established cell lines successfully formed xenograft tumors when administered to immunocompromised mice.
Targeted sequencing was performed on selected known cancer-related genes to identify genetic alterations in the established UPS and MPNST cell lines and provide an initial genomic characterization of the cell lines. Our finding that the genomes of the UPS and MPNST cell lines were not highly mutated agreed with the recent TCGA study of sarcomas10. In that study, the authors reported copy number losses in ATRX, CDKN2A, NF1, RB1, and TP53. PRC2 mutations, specifically inactivation of EED and SUZ12, have been documented in up to 90% of MPNSTs31,68,79,80. Of the 3 NF1-associated MPNST cell lines characterized in this study, MPNST642 carried a frameshift mutation (p.D675fs) in SUZ1268. Previous studies showed EZH2 is significantly upregulated in MPNST specimens compared to normal nerve cells, but not mutated68,81,82. The absence of EZH2 inactivating mutations in the NF1-MPNST cell lines described here (MPNST007 has a heterozygous gain of EZH2) supports the notion that functional EZH2 is required for uncontrolled tumor growth in MPNSTs68.
One drawback of the currently available large-scale analyses for these sarcoma subtypes is the relatively small number of samples in each study, which is expected due to their rarity (UPS, n = 44; MPNST, n = 5 in TCGA10). Creating more patient-derived UPS and MPNST resources gives researchers and clinicians much-needed tools to overcome the limited case numbers and available tumor materials. Patient-derived models that represent molecular subtypes of MPNST have multiple research and clinical applications in drug sensitivity studies, development of drug-resistant models, genetic/epigenetic manipulation, and large-scale—omics-based approaches. The UPS and MPNST PDX models established here can be used to interrogate tumor microenvironment, drug response, and mechanisms of drug resistance in vivo. The MPNST cell lines represent two subtypes of NF1-MPNST: those with intact PRC2 (MPNST007 and MPNST4970) and those lacking PRC2 activity (MPNST3813E). While an estimated 70–90% of MPNSTs feature loss of H3K27me3, the remaining 10–30% of MPNSTs retain functional PRC231,79. These subtypes can be leveraged in preclinical studies to elucidate the contribution of inactive PRC2 to MPNST progression, which remains unclear. From a personalized medicine standpoint, drug sensitivity data sets that include multiple MPNST subtypes (sporadic, NF1-associated, PRC2-wild type, PRC2 mutant, and potentially others) could help predict which treatments will work best for individuals based on the genetic makeup of their tumors. Similarly, insights into UPS’s genetic or epigenetic drivers and drug sensitivity could come from applying large-scale comprehensive -omics approaches to an extended pool of UPS models that represent the heterogeneous nature of this tumor type.
In summary, we successfully established several new cellular models for the study of UPS and MPNST. Furthermore, the initial characterization of the cell lines showed that they could be used as in vitro and in vivo models for further study of sarcoma etiology. Analysis of mutations in genes implicated in UPS and MPNST progression showed that the cell lines recapitulate known genetic alterations in UPS and MPNST. We hope that establishing these cellular models will allow researchers to take the next steps in investigating these debilitating soft tissue sarcomas in greater detail.
Data availability
All data collected and analyzed in this study are available.
References
Fletcher, C. D. M., World Health Organization and International Agency for Research on Cancer. WHO Classification of Tumours of Soft Tissue and Bone. 4th edn, (IARC Press, 2013).
Kim, A. et al. Malignant peripheral nerve sheath tumors state of the science: leveraging clinical and biological insights into effective therapies. Sarcoma, https://doi.org/10.1155/2017/7429697 (2017).
Fletcher, C. D. M. The evolving classification of soft tissue tumours: an update based on the new WHO classification. Histopathology 48, 3–12 (2006).
Mocellin, S. in Soft Tissue Tumors: A Practical and Comprehensive Guide to Sarcomas and Benign Neoplasms 779–783 (Springer International Publishing, 2021).
Guillou, L. & Aurias, A. Soft tissue sarcomas with complex genomic profiles. Virchows Arch. 456, 201–217 (2010).
Maki, R. G. et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J. Clin. Oncol. 25, 2755–2763 (2007).
Nascimento, A. F. & Raut, C. P. Diagnosis and management of pleomorphic sarcomas (so-called “MFH”) in adults. J. Surg. Oncol. 97, 330–339 (2008).
Lahat, G., Lazar, A. & Lev, D. Sarcoma epidemiology and etiology: potential environmental and genetic factors. Surg. Clin. North Am. 88, 451–481 (2008).
Steele, C. D. et al. Undifferentiated sarcomas develop through distinct evolutionary pathways. Cancer Cell 35, 441–456 (2019).
TCGA. Comprehensive and integrated genomic characterization of adult soft tissue sarcomas. Cell 171, 950–965 (2017).
Wardelmann, E. et al. Soft tissue sarcoma: from molecular diagnosis to selection of treatment. Pathological diagnosis of soft tissue sarcoma amid molecular biology and targeted therapies. Ann. Oncol. 21(Suppl 7), vii265–vii269 (2010).
Vodanovich, D. A., Spelman, T., May, D., Slavin, J. & Choong, P. F. M. Predicting the prognosis of undifferentiated pleomorphic soft tissue sarcoma: a 20-year experience of 266 cases. ANZ J. Surg. 89, 1045–1050 (2019).
Bridge, R. S. Jr et al. Recurrent chromosomal imbalances and structurally abnormal breakpoints within complex karyotypes of malignant peripheral nerve sheath tumour and malignant triton tumour: a cytogenetic and molecular cytogenetic study. J. Clin. Pathol. 57, 1172–1178 (2004).
Fletcher, C. D. et al. Correlation between clinicopathological features and karyotype in spindle cell sarcomas. A report of 130 cases from the CHAMP study group. Am. J. Pathol. 154, 1841–1847 (1999).
Friedman, J. M. Epidemiology of neurofibromatosis type 1. Am. J. Med. Genet. 89, 1–6, https://doi.org/10.1002/(SICI)1096-8628(19990326)89:13.0.CO;2-8 (1999).
Gupta, G. & Maniker, A. Malignant peripheral nerve sheath tumors. Neurosurg. Focus 22, E12 (2007).
Watson, K. L. et al. Patterns of recurrence and survival in sporadic, neurofibromatosis Type 1-associated, and radiation-associated malignant peripheral nerve sheath tumors. J. Neurosurg. 126, 319–329, https://doi.org/10.3171/2015.12.Jns152443 (2017).
Widemann, B. C. & Italiano, A. Biology and management of undifferentiated pleomorphic sarcoma, myxofibrosarcoma, and malignant peripheral nerve sheath tumors: state of the art and perspectives. J. Clin. Oncol. 36, 160–167 (2018).
Bradford, D. & Kim, A. Current treatment options for malignant peripheral nerve sheath tumors. Curr. Treat. Options Oncol. 16, 328 (2015).
Blakely, M. L. et al. The impact of margin of resection on outcome in pediatric nonrhabdomyosarcoma soft tissue sarcoma. J. Pediatr. Surg. 34, 672–675 (1999).
Carli, M. et al. Pediatric malignant peripheral nerve sheath tumor: the Italian and German soft tissue sarcoma cooperative group. J. Clin. Oncol. 23, 8422–8430 (2005).
Ducatman, B. S., Scheithauer, B. W., Piepgras, D. G., Reiman, H. M. & Ilstrup, D. M. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer 57, 2006–2021 (1986).
Farid, M. et al. Malignant peripheral nerve sheath tumors. Oncologist 19, 193–201 (2014).
Wong, W. W., Hirose, T., Scheithauer, B. W., Schild, S. E. & Gunderson, L. L. Malignant peripheral nerve sheath tumor: analysis of treatment outcome. Int. J. Radiat. Oncol. Biol. Phys. 42, 351–360 (1998).
Zou, C. et al. Clinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcome. Ann. Surg. 249, 1014–1022 (2009).
Anghileri, M. et al. Malignant peripheral nerve sheath tumors: prognostic factors and survival in a series of patients treated at a single institution. Cancer 107, 1065–1074 (2006).
Roland, C. L. et al. Analysis of clinical and molecular factors impacting oncologic outcomes in undifferentiated pleomorphic sarcoma. Ann. Surg. Oncol. 23, 2220–2228 (2016).
Sahm, F., Reuss, D. E. & Giannini, C. WHO 2016 classification: changes and advancements in the diagnosis of miscellaneous primary CNS tumours. Neuropathol. Appl. Neurobiol. https://doi.org/10.1111/nan.12397 (2017).
May, C. D. et al. Co-targeting PI3K, mTOR, and IGF1R with small molecule inhibitors for treating undifferentiated pleomorphic sarcoma. Cancer Biol. Ther. 18, 816–826 (2017).
Brohl, A. S., Kahen, E., Yoder, S. J., Teer, J. K. & Reed, D. R. The genomic landscape of malignant peripheral nerve sheath tumors: diverse drivers of Ras pathway activation. Sci. Rep. 7, 14992 (2017).
Zhang, M. et al. Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat. Genet. 46, 1170–1172 (2014).
Mertens, F. et al. Cytogenetic analysis of 46 pleomorphic soft tissue sarcomas and correlation with morphologic and clinical features: a report of the CHAMP Study Group. Chromosomes and MorPhology. Genes Chromosomes Cancer 22, 16–25 (1998).
Chibon, F. et al. The RB1 gene is the target of chromosome 13 deletions in malignant fibrous histiocytoma. Cancer Res. 60, 6339–6345 (2000).
Derre, J. et al. Leiomyosarcomas and most malignant fibrous histiocytomas share very similar comparative genomic hybridization imbalances: an analysis of a series of 27 leiomyosarcomas. Lab. Investig. 81, 211–215 (2001).
Mairal, A. et al. Loss of chromosome 13 is the most frequent genomic imbalance in malignant fibrous histiocytomas. A comparative genomic hybridization analysis of a series of 30 cases. Cancer Genet. Cytogenet. 111, 134–138 (1999).
Larramendy, M. L., Gentile, M., Soloneski, S., Knuutila, S. & Bohling, T. Does comparative genomic hybridization reveal distinct differences in DNA copy number sequence patterns between leiomyosarcoma and malignant fibrous histiocytoma? Cancer Genet. Cytogenet. 187, 1–11 (2008).
Perot, G. et al. Constant p53 pathway inactivation in a large series of soft tissue sarcomas with complex genetics. Am. J. Pathol. 177, 2080–2090 (2010).
Jenne, D. E. et al. Molecular characterization and gene content of breakpoint boundaries in patients with neurofibromatosis type 1 with 17q11.2 microdeletions. Am. J. Hum. Genet. 69, 516–527 (2001).
Riva, P. et al. NF1 microdeletion syndrome: refined FISH characterization of sporadic and familial deletions with locus-specific probes. Am. J. Hum. Genet. 66, 100–109 (2000).
Kourea, H. P., Orlow, I., Scheithauer, B. W., Cordon-Cardo, C. & Woodruff, J. M. Deletions of the INK4A gene occur in malignant peripheral nerve sheath tumors but not in neurofibromas. Am. J. Pathol. 155, 1855–1860 (1999).
Nielsen, G. P. et al. Malignant transformation of neurofibromas in neurofibromatosis 1 is associated with CDKN2A/p16 inactivation. Am. J. Pathol. 155, 1879–1884 (1999).
Cleven, A. H. et al. Loss of H3K27 tri-methylation is a diagnostic marker for malignant peripheral nerve sheath tumors and an indicator for an inferior survival. Mod. Pathol. 29, 582–590 (2016).
Genberg, M., Mark, J., Hakelius, L., Ericsson, J. & Nister, M. Origin and relationship between different cell types in malignant fibrous histiocytoma. Am. J. Pathol. 135, 1185–1196 (1989).
Oyama, R. et al. Establishment and proteomic characterization of a novel cell line, NCC-UPS2-C1, derived from a patient with undifferentiated pleomorphic sarcoma. In Vitro Cell. Dev. Biol. Anim. https://doi.org/10.1007/s11626-018-0229-7 (2018).
Becker, M. et al. Xenograft models for undifferentiated pleomorphic sarcoma not otherwise specified are essential for preclinical testing of therapeutic agents. Oncol. Lett. 12, 1257–1264 (2016).
Salawu, A. et al. Establishment and molecular characterisation of seven novel soft-tissue sarcoma cell lines. Br. J. Cancer 115, 1058–1068 (2016).
Lopez, G. et al. Autophagic survival in resistance to histone deacetylase inhibitors: novel strategies to treat malignant peripheral nerve sheath tumors. Cancer Res. 71, 185–196 (2011).
Spyra, M. et al. Cancer stem cell-like cells derived from malignant peripheral nerve sheath tumors. PLoS ONE 6, e21099 (2011).
Yan, N. et al. Farnesyltransferase inhibitors block the neurofibromatosis type I (NF1) malignant phenotype. Cancer Res. 55, 3569–3575 (1995).
Badache, A. & De Vries, G. H. Neurofibrosarcoma-derived Schwann cells overexpress platelet-derived growth factor (PDGF) receptors and are induced to proliferate by PDGF BB. J. Cell. Physiol. 177, 334–342, https://doi.org/10.1002/(SICI)1097-4652(199811)177:23.0.CO;2-9 (1998).
Longo, J. F. et al. Establishment and genomic characterization of a sporadic malignant peripheral nerve sheath tumor cell line. Sci. Rep. 11, 5690 (2021).
Ikuta, K. et al. Antitumor effects of 4-methylumbelliferone, a hyaluronan synthesis inhibitor, on malignant peripheral nerve sheath tumor. Int. J. Cancer 140, 469–479 (2017).
Perrin, G. Q. et al. Plexiform-like neurofibromas develop in the mouse by intraneural xenograft of an NF1 tumor-derived Schwann cell line. J. Neurosci. Res. 85, 1347–1357 (2007).
Turk, A. N., Byer, S. J., Zinn, K. R. & Carroll, S. L. Orthotopic xenografting of human luciferase-tagged malignant peripheral nerve sheath tumor cells for in vivo testing of candidate therapeutic agents. J. Vis. Exp. https://doi.org/10.3791/2558 (2011).
Pollard, K. et al. A clinically and genomically annotated nerve sheath tumor biospecimen repository. Sci. Data 7, 184–184, https://doi.org/10.1038/s41597-020-0508-5 (2020).
Barretina, J. et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 (2012).
Forbes, S. A. et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 45, D777–D783 (2017).
DepMap, Broad: DepMap 22Q1 Public. figshare. Dataset. https://doi.org/10.6084/m9.figshare.19139906.v1 (2022).
Tlemsani, C. et al. Abstract 212: Sarcoma-CellMiner: an extensive resource for patient-derived sarcoma cell line epigenetics, genomics and pharmacology. Cancer Res. 81, 212–212 (2021).
Peng, T. et al. An experimental model for the study of well differentiated and dedifferentiated liposarcoma; deregulation of targetable tyrosine kinase receptors. Lab. Investig. 91, 392–403 (2011).
Keung, E. Z. et al. Increased H3K9me3 drives dedifferentiated phenotype via KLF6 repression in liposarcoma. J. Clin. Investig. 125, 2965–2978 (2015).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 (2013).
Zhang, J. et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 346, 256–259 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219 (2013).
Ye, K., Schulz, M. H., Long, Q., Apweiler, R. & Ning, Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25, 2865–2871 (2009).
Cerami, E. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012).
Kochat, V. et al. Enhancer reprogramming in PRC2-deficient malignant peripheral nerve sheath tumors induces a targetable de-differentiated state. Acta Neuropathol. 142, 565–590 (2021).
Hirsch, P. et al. Clonal history of a cord blood donor cell leukemia with prenatal somatic JAK2 V617F mutation. Leukemia 30, 1756–1759 (2016).
Shankar, G. M. et al. Sporadic hemangioblastomas are characterized by cryptic VHL inactivation. Acta Neuropathol. Commun. 2, 167 (2014).
Casson, A. G. et al. p53 gene mutations in Barrett’s epithelium and esophageal cancer. Cancer Res. 51, 4495–4499 (1991).
Fenaux, P. et al. Mutations of the p53 gene in B-cell lymphoblastic acute leukemia: a report on 60 cases. Leukemia 6, 42–46 (1992).
Naito, M. et al. Detection of p53 gene mutations in human ovarian and endometrial cancers by polymerase chain reaction-single strand conformation polymorphism analysis. Jpn. J. Cancer Res. 83, 1030–1036 (1992).
Renault, B. et al. Base transitions are the most frequent genetic changes at P53 in gastric cancer. Cancer Res. 53, 2614–2617 (1993).
Ruggeri, B. et al. Human pancreatic carcinomas and cell lines reveal frequent and multiple alterations in the p53 and Rb-1 tumor-suppressor genes. Oncogene 7, 1503–1511 (1992).
Sakashita, A. et al. Mutations of the p53 gene in adult T-cell leukemia. Blood 79, 477–480 (1992).
Matushansky, I. et al. MFH classification: differentiating undifferentiated pleomorphic sarcoma in the 21st Century. Expert Rev. Anticancer Ther. 9, 1135–1144 (2009).
Fletcher, C. D. M., Gustafson, P., Rydholm, A., Willén, H. & Åkerman, M. Clinicopathologic re-evaluation of 100 malignant fibrous histiocytomas: prognostic relevance of subclassification. J. Clin. Oncol. 19, 3045–3050 (2001).
Lee, W. et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet. 46, 1227–1232 (2014).
De Raedt, T. et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 514, 247–251 (2014).
Zhang, P. et al. EZH2–miR-30d–KPNB1 pathway regulates malignant peripheral nerve sheath tumour cell survival and tumourigenesis. J. Pathol. 232, 308–318 (2014).
Xu, K. et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 338, 1465–1469 (2012).
Acknowledgements
The authors acknowledge the Transgenic Mouse Facility and the Surgical Oncology Histology Facility at MD Anderson Cancer Center. The authors also thank Amy Ninetto and the Department of Scientific Publications at MD Anderson Cancer Center for their assistance in editing this paper.
Funding
This study was supported in part by The Jay Vernon Jackson Sarcoma Research Fund, The Sally Kingsbury Sarcoma Research Foundation, Johnnie Morgan Lowe Endowment for Sarcoma Research, The Poulos Family, and the Ferrin Randall Zeitlin Foundation for Sarcoma Research. J.P.L. was supported by the National Institutes of Health (T32 CA 009599). C.B.D. was sponsored by the National Institutes of Health’s National Cancer Institute Partnership for Excellence in Cancer Research (U54CA096300/U54CA096297) to perform this research. A.D.B. was funded by The Jay Vernon Jackson Sarcoma Research Fund. S.M.L. was funded by The Sally Kingsbury Sarcoma Research Foundation. Support for the clinical sample collection was provided by the Ferrin R Zeitlin Foundation, Artz Cure Sarcoma Foundation, and MoJo’s Miracle. STR DNA fingerprinting was performed by the Cytogenetics and Cell Authentication Core, the Advanced Technology Genomics Core (ATGC) performed Sanger sequencing, and the Research Histology Core Laboratory performed immunohistochemistry (CCSG P30 CA016672).
Author information
Authors and Affiliations
Contributions
A.D.B.: conceptualization, data interpretation, experiments; S.M.L.: cell line generation, experiments; A.K.S.: experiments; J.P.L.: experiments, chart review; M.G.Y.: cell line generation, chart review, patient consenting, experiments; G.S.B.M.: cell line generation, chart review, patient consenting; C.B.D.B.: experiments; S.D.: experiments; T.N.: cell line generation, experiments; X.Y.M.: experiments; S.B.: experiments; B.A.M.: cell line generation, experiments; S.E.L.C.: cell line generation, experiments; J.A.L.: conceptualization; X.M.: bioinformatics analysis; X.S.: bioinformatics analysis; A.J.L.: pathological analysis; I.E.M.: chart review, patient care; J.M.S.: chart review, patient care; K.R.: conceptualization; D.L.: conceptualization; K.E.T.: conceptualization, data interpretation, funding acquisition, project administration.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
We obtained approval from the Institutional Review Board of The University of Texas MD Anderson Cancer Center and patients’ informed written consent before establishing the cell lines described in this study.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Bhalla, A.D., Landers, S.M., Singh, A.K. et al. Experimental models of undifferentiated pleomorphic sarcoma and malignant peripheral nerve sheath tumor. Lab Invest 102, 658–666 (2022). https://doi.org/10.1038/s41374-022-00734-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-022-00734-6
