
Abstract
LncRNAs and miRNAs are correlated with the pathogenesis of myocardial ischemia-reperfusion injury (MIRI). Whether lncRNA ROR or miR-185-5p plays a crucial role in MIRI is still unclear. In in-vitro, human cardiac myocytes (HCMs) were treated with hypoxia/reoxygenation (H/R). Wistar rats were used to set up an in-vitro I/R model by means of recanalization after ligation. Evaluation of the myocardial injury marker lactate dehydrogenase (LDH) in HCMs cells was performed. The expression of miR-185-5p and ROR, IL-1β, and IL-18 were detected by qRT-PCR. ELISA was also performed to evaluate the secretion of IL-1β and IL-18. Western blotting was carried out to determine CDK6, NLRP3, GSDMD-N, ASC, and cleaved-caspase1 protein expression. The relationship between miR-185-5p and CDK6 or ROR was confirmed by a dual-luciferase reporter assay. Our findings revealed that H/R treated HCMs showed a significantly decreased miR-185-5p expression and increased expression of CDK6 and ROR. ROR knockdown reduced H/R induced pyroptosis and inflammation, while knockdown of miR-185-5p accelerated the effect. Furthermore, miR-185-5p was negatively regulated and absorbed by ROR in HCMs. Overexpression of miR-185-5p reversed the H/R-induced cell pyroptosis and upregulation of LDH, IL-1β, and IL-18. In HCMs, miR-185-5p was also negatively regulated and related to CDK6 expression. Moreover, overexpression of CDK6 significantly inhibited the effects of miR-185-5p mimics on the inflammatory response and pyroptosis of HCMs. Knockdown of ROR alleviated H/R-induced myocardial injury by elevating miR-185-5p and inhibiting CDK6 expression. Taken together, our results show that the ROR/miR-185-5p/CDK6 axis modulates cell pyroptosis induced by H/R and the inflammatory response of HCMs.
Similar content being viewed by others


Introduction
Cardiovascular events induced by myocardial ischemia-reperfusion injury (MIRI) are one of the leading causes of death in patients with coronary heart disease (CHD) and acute myocardial infarction (AMI)1,2. The harm caused by MIRI to patients includes the development of CHD, myocardial infarction, and cardiac surgery3. However, the pathological mechanism of MIRI has not been elaborated yet. Pyroptosis is programmed cell death mediated by caspase1 and characterized by the release of cell contents and the formation of membrane pore and swelling4. Pyroptosis played a vital role in the progression of various diseases, such as autoimmune diseases, infectious diseases, and cardiovascular and cerebrovascular diseases5.
Long non-coding RNAs (lncRNAs) are non-protein coding transcripts of more than 200 nucleotides6. The role of lncRNA ROR has been extensively studied in various malignant diseases. Studies have shown that upregulation of ROR activated the TGF-β pathway and promotes cell growth and cancer invasion7. ROR affects docetaxel resistance of cancer cells through epithelial-mesenchymal transition (EMT), indicating that it may be a therapeutic target8. Recently, lncRNAs were correlated with ischemia-reperfusion (I/R) injury. ROR promotes the phosphorylation of ERK1/2 and p38, and aggravates the cardiomyocyte apoptosis induced by hypoxia/reoxygenation (H/R)9. It has been reported that the inflammatory response and HCMs apoptosis induced by H/R was regulated by ROR10. However, the role and underlying mechanism of ROR in MIRI needs to be further investigated.
MicroRNAs (miRNAs) of 20–23 nucleotides have no protein-coding ability. miRNAs inhibit the translation of target genes or directly degrades target genes by binding to the 3’-UTR of mRNA molecules11. Recent studies show that miRNAs play a vital role in the development of MIRI. Transverse aortic constriction reduced the expression of miR-185 in cardiac hypertrophy, which illustrated an anti-hypertrophic effect12. Additionally, miR-185 was down-expressed in myocardial tissues, which also reduced apoptosis by regulation of SOCS2 expression in myocardial ischemia (MI) mice13. Patients with dilated cardiomyopathy had higher plasma levels of miR-185 than the healthy14. In our pilot experiment, miR-185-5p and ROR were demonstrated to have target binding sites using bioinformatic approaches, but their regulatory mechanisms in MIRI were still unclear.
This study aimed to explore the underlying molecular mechanism and role of ROR, miR-185-5p, and cyclin-dependent kinase 6 (CDK6) in MIRI in order to provide a new therapeutic option for the treatment of MIRI.
Materials and methods
Cell culture and treatment
The human cardiac myocyte (HCM) cell lines isolated from the human heart, were obtained from ScienCell Research Laboratories (#6200, San Diego, CA, USA). The HCM cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Flow Laboratories Inc., Rockville, MD) containing 5% foetal bovine serum (FBS; Invitrogen, Cergy Pontoise, France) and 1% penicillin/streptomycin (Gibco, CA, USA). 293 T cells were obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA) and cultured in DMEM medium containing D-glucose (4.5 g/L) and 5% FBS. Before the experiment, the sample cells were quickly thawed in a water bath at 37 °C for 1–2 min. Then, 10 mL medium was added and the cells were centrifuged at 1000 rpm for 5 min. The cell pellet was re-suspended in the prepared medium. For normal conditions, all cells were cultured in an incubator with 5% CO2 at 37 °C.
To establish an in-vitro I/R injury system, cells were first exposed to hypoxia (InvivO2 hypoxic cabinet; 1% O2, 5% CO2, and 94% N2) for 1 h, and then normal oxygen for 2 h. At the same time, the cells in the control group (without FBS and glucose-deprived DMEM) were maintained under normoxic conditions (21% O2, 5% CO2, and 74% N2) for 3 h15.
Cell transfection
MiR-185-5p mimics/inhibitor and sh-ROR and corresponding controls (mimics NC, inhibitor NC, and sh-NC) were all synthesized by GenePharma Corporation (Shanghai, China). Then, we amplified and cloned the full-length sequence of ROR and CDK6 into the expression vector pcDNA3.1 (vector) to obtain overexpression-ROR (OE-ROR) plasmid or overexpression-CDK6 (OE-CDK6) plasmid, which were respectively transfected into HCMs using Lipofectamine 2000 (Invitrogen, CA) according to the manufacturer’s instruction.
Dual-luciferase assay
The relationship between miR-185-5p and ROR or CDK6 was detected by a double-luciferase assay. The wild type (WT) sequences or mutant (MUT) sequences of the binding sites with miR-185-5p in ROR or CDK6 3’-UTR were cloned downstream of the luciferase report gene of pmirGLO reporter vector (Promega, US). Mutants were generated through the KOD Plus Mutagenesis kit (TOYOBO, Osaka, Japan) according to the manufactures’ instructions. The 293 T cells were co-transfected miR-185-5p mimics/inhibitor or NC mimics. A total of 48 h after transfection, luciferase assays were carried out using a Bright-Glo™ Luciferase Assay System (Promega, US) and normalized to Renilla luciferase activity.
Establishment of an in-vivo Wistar rat myocardial I/R model
The animal study was approved by the ethical committee of Second Affiliated Hospital of Nanchang University. A total of 72 male Wister rats (250–300 g, Shanghai Laboratory Animal Center, Chinese Academy of Science, China) were used to establish an in-vivo model of myocardial I/R. A total of 72 rats were used in three replicates, and 24 rats were included in every replicate. All of 24 rats were randomly into the Sham group, I/R group, I/R + sh-NC group, and I/R + sh-ROR group (six rats each group). The lentivirus used in the study was commercially constructed by Bio-Link (Shanghai, China). Lentiviral vectors encoding ROR (sh-ROR) or NC as control (sh-NC) were constructed using routine procedures16. Lv-ROR and Lv-NC were injected into the tail vein, and then the myocardial I/R model was established. All tested rats were treated with 45 min of transitory ligation followed by 3 h of reperfusion. The rats in Sham group only received sham operation. After 72 h of the surgery, all rats were sacrificed, and myocardium tissues were obtained for further analysis.
Triphenyl tetrazolium chloride (TTC) staining
To determine the role of ROR in myocardial I/R rats, TTC staining was performed. The heart sections were placed in 1% TTC for 15 min at room temperature. The slices were inverted once every 5 min to avoid over-staining. And then rinsed three times with distilled water. The images were collected to measure infarct size and area using ImageJ software. The infarct area is white.
Hematoxylin and eosin (HE) staining and Masson staining
The histopathological examination of myocardial tissue was stained with hematoxylin-eosin (HE). The left ventricle of the heart sample was placed in a 10% formaldehyde solution, dehydrated with ethanol gradient, embedded in paraffin, and cut into 4 μm sections. After the sample was removed, it was stained with hematoxylin and eosin. Then, the slices were observed under an optical microscope (Leica Microsystems, Wetzlar, Germany).
For Masson staining, the rat heart tissue sections were firstly used to stain cell nuclei for 5 min by Wiegert’s iron hematoxylin solution (Sigma-Aldrich, St. Louis, MO, USA). After rinsing 3 times with distilled water, the sections were stained with 0.7% Masson-Ponceau-acid fuchsin staining solution (Sigma-Aldrich) for 10 min. After rinsing in 2% glacial acetic acid and differentiated in phosphomolybdic acid for 4 min, the sections were directly stained with 2% aniline blue dye solution (Sigma-Aldrich). After dehydration with ethanol series, remove with xylene, install with neutral resin, and capture the image of the stained section with an optical microscope17,18.
Detection of lactate dehydrogenase (LDH), IL-18, and IL-1β activity
After rat reperfusion and cell treatment, plasma samples and media were collected from coronary effluent and cell culture, respectively. Lactate dehydrogenase (LDH) (Nanjing Jiancheng Bioengineering Company) was used to detect serum lactate dehydrogenase (LDH) activity. Commercial enzyme-linked immunosorbent assay (ELISA) kits (Jining Shiye, Shanghai, China) were used to detect the expression of IL-18 and IL-1β in HCMs and myocardium tissues. Absorbance was measured at 450 nm.
Propidium iodide (PI) staining
To measure the actual percentage of cell death, we performed PI single staining to detect cell death. Briefly, PI staining solution at 50 µg/mL was prepared. Cells were collected and washed in PBS, fixed in pre-cooled 70% ethanol, and added ethanol dropwise to the cell pellet while vortexing. The cells were added 200 µL PI (50 µg/mL stock solution) and incubated at room temperature for 15 min. For dead cells, PI acted by intercalating with cellular DNA and emitting red fluorescence.
Quantitative real-time polymerase chain reaction (qRT-PCR)
The relative expression of ROR, IL-1β, IL-18, miR-185-5p, and CDK6 was analyzed by qRT-PCR. Total RNA was extracted according to Trizol (Takara) experimental method. RNA quality and quantity were assessed with agarose gel electrophoresis and A260/A280 ratio with a spectrophotometer (Nano Drop Technologies, Inc, Rockland, DE). The A260/A280 ratios for all RNA preparations were about 1.8∼2.0. Taqman MicroRNA Reverse Transcription Kit and Taqman® MicroRNA Assay Kit (Applied Biosystems, Foster City, CA, US) were applied for cDNA synthesis and qRT-PCR, respectively. The reaction conditions: 95 °C for 10 min, followed by 38 cycles of 95 °C for 10 s, and 58 °C for 60 s. U6 and GAPDH were used as endogenous controls, respectively.
Western blotting
6 well plates of HCMs were lysed using RIPA lysis buffer (Beyotime, China) containing protease inhibitor cocktail. Protein was quantified by a BCA assay kit (Bio-Rad, USA). Then, 30 μg protein was separated by SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis). The separated proteins were transferred to the PVDF membrane and then blocked with 5% skim milk. The membrane was then stored in the primary antibody against NLRP3, GSDMD-FL (full length), GSDMD-N (N-terminal), ASC, pro-caspase1, and cleaved-caspase1 and incubated overnight at 4 °C. All primary antibodies were obtained from Abcam (UK). The membrane was then incubated with horseradish peroxidase-labeled secondary antibody. The expression of each protein was analyzed ECL and detection system (Millipore). The expression of proteins was normalized by GAPDH.
Statistical analysis
All data in our study are expressed as the mean ± standard deviation (SD) and analyzed by Graphpad 6.0 (GraphPad Software, CA). Differences between the two groups were analyzed by student’s t-test, and differences among the multiple groups were analyzed by analysis of variance (ANOVA). Each experiment was carried out at least three times. The difference was considered statistically significant at P < 0.05.
Results
ROR regulates pyroptosis and inflammation induced by H/R
To investigate the effect of ROR in H/R treated HCMs, the ROR expression in H/R treated HCMs was analyzed by qRT-PCR. Fig. 1A indicates that α-sarcometric actin (cardiomyocyte marker) was strongly positive in HCM cells. ROR expression was more highly upregulated in H/R treated HCMs than in the control group (Fig. 1B). The effects of transfection with ROR in HCMs were confirmed by qRT-PCR analysis. ROR expression was decreased in HCMs after transfection with sh-ROR and increased in HCMs after transfection with OE-ROR (Fig. 1C). LDH levels were higher in the H/R group than the normoxia group, but the knockdown of ROR partially reversed this effect. In addition, overexpression of ROR further promoted the release of LDH in HCMs (Fig. 1D). To measure the actual percentage of cell death, we performed PI single staining. The results show that PI-positive cells were significantly increased in HCM cells treated with H/R, while knockdown of ROR inhibited the increase of PI-positive cells, whereas overexpression of ROR promoted the increase of PI-positive cells (Fig. 1E). Additionally qRT-PCR and ELISA experiments demonstrated that H/R exposure promoted IL-18 and IL-1β in HCMs treated with H/R (Fig. 1F-I). However, this effect was counteracted by ROR inhibition and further promoted by ROR overexpression (Fig. 1F-I). Additionally, overexpression of ROR promoted the expression of NLRP3, cleaved-caspase1, ASC, and GSDMD-N, indicating that overexpression of ROR promoted the occurrence of cell pyroptosis (Fig. 1J). Conversely, ROR knockdown inhibited the H/R-induced upregulated expression of GSDMD-N, NLRP3, ASC, and cleaved-caspase1, but had no significant effect on the protein levels of pro-caspase1 and GSDMD-FL (Fig. 1J). These results indicate that inhibition of ROR expression reduces pyroptosis and inflammation induced by H/R, while overexpression reverses the effect.

HCMs were transfected with Vector, OE-ROR, H/R + Vector, H/R + OE-ROR, H/R + sh-NC, H/R + sh-ROR. A Immunofluorescence assay to detect α-Sarcometric actin. B Relative expression level of ROR in HCMs after H/R or normoxia treatment by qRT-PCR. C Efficiency analysis of ROR knockdown and overexpression. D The release level of LDH. E The actual percentage of cell death detected by PI single staining. F, G The expression of IL-1β and IL-18 in HCMs. H, I The concentrations of IL-1β and IL-18 in HCMs were evaluated by ELISA. J The levels of protein NLRP3, GSDMD-N, GSDMD-FL, ASC, cleaved-caspase1, Pro-caspase1, and GAPDH in HCMs by western blotting. Each experiment was replicated three times. *P < 0.05, **P < 0.01, ***P < 0.001.
ROR inhibits miR-185-5p expression in HCMs
We further explored the target genes for ROR. ROR inhibition increased miR-185-5p expression in HCMs, while ROR overexpression showed the opposite result (Fig. 2A). miR-185-5p mimics/inhibitor experiments showed that miR-185-5p expression was promoted after transfection and inhibited after miR-185-5p inhibitor transfection (Fig. 2B). Complementary binding sites between miR-185-5p and ROR were observed by bioinformatics analysis (Fig. 2C). The interaction between miR-185-5p and ROR in HCMs was verified by a dual-luciferase reporter assay. MiR-185-5p mimics decreased the luciferase activity of ROR-WT cells, miR-185-5p inhibitor promoted the luciferase activity of ROR-WT cells, while no differences were observed in the ROR-MUT groups (Fig. 2D). These findings revealed that ROR in HCMs absorbed and negatively regulated the expression of miR-185-5p.

A qRT-PCR analyzed the expression of miR-185-5p in HCMs treated with sh-ROR or OE-ROR. B qRT-PCR analyzed the expression of miR-185-5p in HCMs treated with miR-185-5p mimics/inhibitor. C The binding site between ROR and miR-185-5p. D Interaction between ROR and miR-185-5p was verified by dual-luciferase reporter assay. Each experiment was replicated three times. *P < 0.05, **P < 0.01, ***P < 0.001.
MiR-185-5p inhibition counteracteds the effect of ROR knockdown in H/R-induced HCMs
To further study the functions of miR-185-5p, miR-185-5p expression was examined in H/R-exposure HCMs. qRT-PCR showed that H/R treatment inhibited miR-185-5p expression (Fig. 3A). MiR-185-5p inhibitor transfection further promoted LDH activity and reversed the inhibition of ROR knockdown on LDH activity (Fig. 3B). The actual percentage of cell death was detected by PI single staining (Fig. 3C). The qRT-PCR and ELISA assays revealed that H/R-induced IL-1β and IL-18 expression was significantly promoted by miR-185-5p inhibitor at mRNA level or protein level. Further, miR-185-5p inhibitor rescued the inhibition of IL-1β and IL-18 expression caused by ROR knockdown (Fig. 3D-G). Western blotting revealed that miR-185-5p inhibition further promoted the protein expression of H/R-induced NLRP3, GSDMD-N, ASC and cleaved-caspase1, but had no significant effect on the protein levels of pro-caspase1 and GSDMD-FL (Fig. 3H). Furthermore, knockdown of miR-185-5p reversed the inhibitory effect of ROR knockdown (Fig. 3H), indicating that ROR regulated H/R-induced pyroptosis and inflammation induced by targeting miR-185-5p.

HCM cells were transfected with sh-NC + inhibitor NC, sh-ROR + inhibitor NC, sh-NC + miR-185-5p inhibitor or sh-ROR + miR-185-5p inhibitor. A The expression level of miR-185-5p in HCMs was detected by qRT-PCR. B The release level of lactate dehydrogenase (LDH). C The actual percentage of cell death detected by PI single staining. D, E The expression of IL-1β and IL-18 in HCMs were evaluated by qRT-PCR. F, G The concentrations of IL-1β and IL-18 in HCMs were evaluated by ELISA. H The protein levels were detected by western blotting. Each experiment was replicated three times. *P < 0.05, **P < 0.01, ***P < 0.001.
MiR-185-5p directly targets CDK6 in HCMs
To confirm the binding relationship between miR-185-5p and CDK6 in HCMs, the CDK6 expression in HCMs after miR-185-5p overexpression or inhibition was analyzed by qRT-PCR and western blotting. As illustrated in Fig. 4A, B, miR-185-5p mimics significantly inhibited the mRNA and protein expression of CDK6, and miR-185-5p inhibitor significantly stimulated the mRNA and protein expression of CDK6. Bioinformatics analysis predicted the binding sites between miR-185-5p and CDK6 (Fig. 4C). Dual-luciferase reporter assay displayed that miR-185-5p mimics inhibited the activity and that miR-185-5p inhibitor promoted the activity in the CDK6-WT group, but there were no significant differences in the CDK6-MUT groups (Fig. 4D). All the results above revealed that miR-185-5p can negatively regulate and directly target CDK6.

A, B The expression of CDK6 was analyzed by qRT-PCR and Western blotting. C The binding site of miR-185-5p and CDK6. D Dual-luciferase reporter assay. Each experiment was replicated three times. *P < 0.05, **P < 0.01, ***P < 0.001.
MiR-185-5p inhibits H/R-induced pyroptosis and inflammation by inhibiting CDK6
Next, we investigated whether miR-185-5p regulated H/R-induced apoptosis and inflammation of HCMs through CDK6. First, we overexpressed CDK6 in HCMs, and the CDK6 mRNA and protein expression were significantly increased in HCMs transfected with CDK6 overexpressing vector (Fig. 5A, B). Overexpression of miR-185-5p inhibited the increased LDH activity and PI-positive cells induced by H/R. Overexpression of CDK6 promoted the increase of LDH activity and PI-positive cells, and reversed the effect of miR-185-5p overexpression (Fig. 5C, D). Overexpression of miR-185-5p or CDK6 inhibited or promoted the H/R-induced expression IL-1β and IL-18, respectively (Fig. 5E–I). Moreover, CDK6 overexpression deteriorated the effect of miR-185-5p overexpression on IL-1β and IL-18 induced by H/R (Fig. 5E–I). After miR-185-5p overexpression, the expression of NLRP3, GSDMD, ASC, and Caspase 1 was inhibited after H/R induction. CDK6 overexpression further promoted the expression of GSDMD-N, NLRP3, ASC, and cleaved-aspase1, and inhibited the effect of miR-185-5p overexpression (Fig. 5I). Our results illustrated that miR-185-5p inhibited pyroptosis and inflammation in H/R HCMs through the regulation of CDK6.

HCM cells were transfected with mimics NC + vector, mimics NC + OE-CDK6, H/R + mimics NC + Vector, H/R + miR-185-5p mimics + vector, H/R + mimics NC + OE-CDK6 or H/R + miR-185-5p mimics + OE-CDK6. A qRT-PCR analysis of CDK6 expression in HCMs after CDK6 overexpressed. B Western blotting analysis of CDK6 protein level in HCMs after CDK6 overexpressed. C The release level of lactate dehydrogenase (LDH) in HCMs. D The actual percentage of cell death detected by PI single staining. E, F The expression of IL-1β and IL-18 in HCMs was evaluated by qRT-PCR. G, H The concentrations of IL-1β and IL-18 were evaluated by ELISA. I Western blotting detected the related protein levels in HCMs. Each experiment was replicated three times. *P < 0.05, **P < 0.01, ***P < 0.001.
Knockdown of ROR alleviates I/R-induced myocardial injury by elevating miR-185-5p to inhibit CDK6 expression
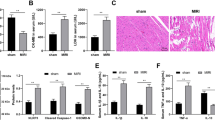
To determine the role of ROR in myocardial I/R rats, TTC staining was performed to measure infarct size and area. The infarct size was significantly increased in the I/R group (22.10 ± 4.66%) compared with the sham group (3.43 ± 1.70%) (Fig. 6A). ROR knockdown significantly decreased the infarction infarct area (5.90 ± 2.23% in I/R + sh-ROR group vs 21.20 ± 5.07% in I/R + sh-NC group) (Fig. 6A). Then, the changes of cardiomyocyte morphology were analyzed by H&E and Masson staining. Cells were arranged orderly and intact in the sham group, and no pathological changes such as necrosis or hyperplasia were observed (Fig. 6B). However, the myocardial fiber structures in the I/R group and I/R + sh-NC group were disordered with amounts of inflammatory cells infiltrated, and the myocardial cells swollen, necrotic and apoptotic (Fig. 6B). Knockdown of ROR reduced heart muscle damage (Fig. 6B). Compared with the sham group, the myocardial fibers in the I/R group and I/R + sh-NC group showed disordered arrangement and abundant collagen deposition (Fig. 6C). Knockdown of ROR reduces the reaction described above (Fig. 6C). Compared with the sham group, the expression of ROR and CDK6 in the I/R group and I/R + sh-NC group were significantly increased, and the expression of miR-185-5p was significantly decreased (Fig. 6D–F). Knockdown of ROR reversed this effect, and the expression levels of ROR and miR-185-5p in both the I/R group and I/R + sh-NC group were decreased and increased compared with I/R + sh-ROR group (Fig. 6D–F). All above indicated that knockdown of ROR alleviated I/R-induced myocardial injury by elevating miR-185-5p to inhibiting CDK6 expression.

A After reperfusion, hearts were harvested, sliced, and stained with TTC to examine the infarct size of the myocardium. The histopathological analysis of myocardial tissue was stained with (B) HE staining and (C) Masson staining. Scale bar, 100 μm. D–F The expression levels of ROR, miR-185-5p, and CDK6 expression in myocardial tissues were evaluated by qRT-PCR. Every group has 6 Wister rats. *P < 0.05, **P < 0.01, ***P < 0.001.
Inhibition of ROR in rats reduced I/R-induced pyroptosis and inflammation via the miR-185-5p /CDK6 axis
Further, we analyzed whether ROR modulated pyroptosis and inflammation by suppression of miR-185-5p/CDK6. In the I/R group and I/R + sh-NC group, the LDH activity increased, and ROR knockdown reversed the I/R-induced LDH activity (Fig. 7A). Additionally, the expression of IL-1β and IL-18 was increased in the I/R group and I/R + sh-NC group, and ROR knockdown inhibited the I/R-induced upregulation of IL-1β and IL-18 (Fig. 7B–E). Moreover, protein NLRP3, GSDMD-N, ASC cleaved-caspase1, and CDK6 expression were increased in the I/R group and I/R + sh-NC group, which were conversely decreased after ROR knockdown (Fig. 7F). These data reveal that inhibition of ROR inhibited I/R-induced pyroptosis and inflammation via the miR-185-5p/CDK6 axis.

A The release level of lactate dehydrogenase (LDH) analysis. B, C The expression of IL-1β and IL-18 in HCMs. D, E The concentrations of IL-1β and IL-18 in HCMs were evaluated by ELISA. F Western blotting analyzed the protein levels. Every group has 6 Wister rats. *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
To date, the underlying mechanism of MIRI is still unclear. The mortality due to cardiovascular disease in 2012 is approximately 17,500,000 people9. Myocardial I/R can cause cardiomyocyte damage and apoptosis19. Cardiovascular disease is the primary cause of hospitalization and mortality worldwide20. Recently, the correlation between the expression of lncRNA and MIRI revealed that lncRNA played a key role in MIRI21. In the study, our findings demonstrated that inhibition of ROR inhibited I/R-induced pyroptosis and inflammation via the miR-185-5p /CDK6 axis.
LncRNAs are important regulators in biological processes. Yu et al. found that knockdown of lncRNA AK139328 enhanced the expression of miR-204-3p and suppressed autophagy of cardiomyocytes, and thus attenuated MIRI in diabetic mice17. As previously described, lncRNA H19 relieved MIRI by targeting miR-22-3p22. The study also demonstrated that NEAT1 contributed to MIRI by activating the MAPK pathway23. Yin et al. found that knockdown of SNHG12 protected cerebral I/R injury by the regulation of miR-199a expression [10]. LncRNA ROR absorbed miR-138 to aggravate H/R-induced cardiomyocyte apoptosis via upregulation of Mst124. ROR was initially identified as a regulator of human-induced pluripotent stem cell reprogramming, which played a role in various tumors25. Recently, a study showed that MIRI can be aggravated by ROR9. Zhang et al. revealed that ROR was highly expressed in MI and hypoxia injury, and ROR relieved myocardial cerebral I/R injury via p38/MAPK signal pathway9. In our study, an in-vivo MIRI model was established and LncRNA ROR was highly expressed in the MIRI model rats. Inhibition of ROR expression reduced H/R-induced pyroptosis and inflammation, while overexpression of ROR was conversely the phenomenon. Inhibition of ROR in rats reduced I/R-induced pyroptosis and inflammation. We conclude that LncRNA ROR played a vital role in the progression of MIRI.
MiRNAs are involved in the pathogenesis mechanism of kinds of cardiovascular diseases26. Studies have found that 220 human miRNAs are expressed in heart tissues27. MiR-133 is specifically expressed in the heart and plays a vital regulatory role in heart development, myocardial apoptosis, and myocardial remodeling28. In a recent, the expression of miR-1 and myocardial cell apoptosis were significantly increased by negatively regulating its target genes HSP60 and HSP7029. Additionally, miR-27a-5p was related to liver ischemia-reperfusion injury and miR-27a-5p overexpression may alleviate cell apoptosis in H/R injury by targeting Bach1 in vitro30. H/R treatment regulated the expression of some miRNAs. MiR-148b-3p was involved in regulating H/R-induced injury of cardiomyocytes in vitro through modulating SIRT7/p53 signaling31. The expression of miR-148b-3p was induced by H/R treatment, and miR-148b-3p interference alleviated H/R injury31. As a tumor suppressive, miR-185 was a key mediator to hypoxia in the cellular response, which directly affected angiogenesis32. MiR-185 was poorly expressed in myocardial tissues, and alleviated cardiomyocytes apoptosis by regulating the expression of SOCS213. In the current study, the expression of miR-185-5p was decreased after H/R treatment. miR-185-5p was confirmed a target gene of ROR. Inhibition of miR-185-5p counteracted the effect of ROR knockdown in H/R-induced HCMs, Moreover, our results demonstrated that miR-185-5p inhibited H/R-induced pyroptosis and inflammation.
The cell cycle kinase CDK6 is a cyclin-dependent kinase id s transcriptional regulator with distinct properties from its closely homologous CDK4B33,34,35,36,37. CDK6 may regulate the transcription of many genes on basis of its kinase activity38. It has been reported that miR-1 improved myocardial hypertrophy by mediating down-regulation of CDK639. Studies also showed that trypanosine A regulated the HCM cell cycle by inducing miR-129-5p to inhibit the expression of CDK6 in HCM cardiomyocytes40. Similarly, our findings revealed that CDK6 overexpression reversed the down-regulation of LDH activity and inflammatory factors induced by miR-185-5p mimics in HCMs under H/R exposure. Overall, we demonstrated that CDK6 functions as a pro-inflammatory mediator in HCMs induced by H/R, and that miR-185-5p attenuated the inflammatory response and pyroptosis by suppressing the expression of CDK6.
In conclusion, the ROR/miR-185-5p/CDK6 axis was closely related to the regulation of inflammatory and pyroptosis induced by H/R in HCMs. Our study, therefore, provides potential therapeutic targets for MIRI.
Data availability
All data generated or analyzed during this study are included in this article. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Whelan, R. S., Kaplinskiy, V. & Kitsis, R. N. Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu. Rev. Physiol. 72, 19–44 (2010).
González-Montero, J., Brito, R., Gajardo, A. I. & Rodrigo, R. Myocardial reperfusion injury and oxidative stress: Therapeutic opportunities. World J. Cardiol. 10, 74–86 (2018).
Chen, D. L. et al. Long non-coding RNA UICLM promotes colorectal cancer liver metastasis by acting as a ceRNA for microRNA-215 to regulate ZEB2 expression. Theranostics. 7, 4836–4849 (2017).
Ball, D. P. et al. Caspase-1 interdomain linker cleavage is required for pyroptosis. Life Sci. Alliance 3, e202000664 (2020).
Jiang, C., Shi, R., Chen, B., Yan, X. & Tang, G. Casticin elicits inflammasome-induced pyroptosis through activating PKR/JNK/NF-κB signal in 5-8F cells. Biomed Pharmacother. 123, 109576 (2020).
Flynn, R. A. & Chang, H. Y. Long noncoding RNAs in cell-fate programming and reprogramming. Cell Stem Cell 14, 752–761 (2014).
Hou, L. et al. Long noncoding RNA ROR promotes breast cancer by regulating the TGF-β pathway. Cancer Cell Int. 18, 142 (2018).
Pan, Y. et al. Long noncoding RNA ROR regulates chemoresistance in docetaxel-resistant lung adenocarcinoma cells via epithelial mesenchymal transition pathway. Oncotarget. 8, 33144–33158 (2017).
Zhang, W., Li, Y. & Wang, P. Long non-coding RNA-ROR aggravates myocardial ischemia/reperfusion injury. Braz. J. Med. Biol. Res. 51, e6555 (2018).
Liang, Y. P. et al. The lncRNA ROR/miR-124-3p/TRAF6 axis regulated the ischaemia reperfusion injury-induced inflammatory response in human cardiac myocytes. J. Bioenergetics Biomembranes. 51, 381-392 (2019).
Anfossi, S., Fu, X., Nagvekar, R. & Calin, G. A. MicroRNAs, regulatory messengers inside and outside cancer cells. Adv. Exp. Med. Biol. 1056, 87–108 (2018).
Kim, J. O. et al. miR-185 plays an anti-hypertrophic role in the heart via multiple targets in the calcium-signaling pathways. PloS One. 10, e0122509 (2015).
Li, Y. et al. Bone marrow mesenchymal stem cells-derived exosomal microRNA-185 represses ventricular remolding of mice with myocardial infarction by inhibiting SOCS2. Int. Immunopharmacol. 80, 106156 (2020).
Yu, M. et al. Circulating miR-185 might be a novel biomarker for clinical outcome in patients with dilated cardiomyopathy. Sci. Rep. 6, 33580 (2016).
Ye, B. et al. Emodin alleviates myocardial ischemia/reperfusion injury by inhibiting gasdermin D-mediated pyroptosis in cardiomyocytes. Drug Des. Devel. Ther. 13, 975–990 (2019).
Mattila, M., Koskenvuo, J., Söderström, M., Eerola, K. & Savontaus, M. Intramyocardial injection of SERCA2a-expressing lentivirus improves myocardial function in doxorubicin-induced heart failure. J. Gene Med. 18, 124–133 (2016).
Yu S. Y., Dong B., Fang Z. F., Hu X. Q. & Tang L. Knockdown of lncRNA AK139328 alleviates myocardial ischaemia/reperfusion injury in diabetic mice via modulating miR-204-3p and inhibiting autophagy. J. Cell. Mol. Med. 22, 4886–4898 (2018).
Liu, M., Liu, J., Zhang, L., Geng, Q. & Ge, Y. Antidepressant-like effects of ginseng fruit saponin in myocardial infarction mice. Biomed. Pharmacother. 115, 108900 (2019).
Yellon, D. M. & Hausenloy, D. J. Myocardial reperfusion injury. N. Engl. J. Med. 357, 1121–1135 (2007).
Barnett, P. & van den Hoff, M. J. Cardiac regeneration: different cells same goal. Med. Biol. Eng. Comput. 49, 723–732 (2011).
Ong, S. B. et al. Non-coding RNAs as therapeutic targets for preventing myocardial ischemia-reperfusion injury. Expert Opin. Ther. Targets. 22, 247-261 (2018).
Zhang, B. F., Chen, J. & Jiang, H. LncRNA H19 ameliorates myocardial ischemia-reperfusion injury by targeting miR-22-3P. Int. J. Cardiol. 278, 224 (2019).
Du, X. J. et al. NEAT1 promotes myocardial ischemia-reperfusion injury via activating the MAPK signaling pathway. J. Cell Physiol. 234, 18773–18780 (2019).
Hu, Y. H. et al. Long non-coding RNA ROR sponges miR-138 to aggravate hypoxia/reoxygenation-induced cardiomyocyte apoptosis via upregulating Mst1. Exp. Mol. Pathol. 114, 104430 (2020).
Lu, R., Chen, J., Kong, L. & Zhu, H. Prognostic value of lncRNA ROR expression in various cancers: a meta-analysis. Biosci. Rep. 38, BSR20181095 (2018).
Zong, L. & Wang, W. CircANXA2 promotes myocardial apoptosis in myocardial ischemia-reperfusion injury via inhibiting miRNA-133 expression. Biomed. Res. Int. 2020, 8590861 (2020).
Wang, J. X. et al. MicroRNA-103/107 regulate programmed necrosis and myocardial ischemia/reperfusion injury through targeting FADD. Circ. Res. 117, 352–363 (2015).
Wang, S., Cheng, Z., Chen, X. & Xue, H. microRNA-135a protects against myocardial ischemia-reperfusion injury in rats by targeting protein tyrosine phosphatase 1B. J. Cell Biochem. 120, 10421–10433 (2019).
Xu, C. et al. The muscle-specific microRNAs miR-1 and miR-133 produce opposing effects on apoptosis by targeting HSP60, HSP70 and caspase-9 in cardiomyocytes. J. Cell Sci. 120, 3045–3052 (2007).
Xing, Y. & Li, J. MiR-27a-5p regulates apoptosis of liver ischemia-reperfusion injury in mice by targeting Bach1. J. Cell Biochem. 119, 10376–10383 (2018).
Sun, M. et al. MicroRNA-148b-3p is involved in regulating hypoxia/reoxygenation-induced injury of cardiomyocytes in vitro through modulating SIRT7/p53 signaling. Chem. Biol. Interact. 296, 211–219 (2018).
Qu, F. et al. MicroRNA-185 suppresses proliferation, invasion, migration, and tumorigenicity of human prostate cancer cells through targeting androgen receptor. Mol. Cell. Biochem. 377, 121–130 (2013).
Kollmann, K. et al. A kinase-independent function of CDK6 links the cell cycle to tumor angiogenesis. Cancer Cell 24, 167–181 (2013).
Scheicher, R. et al. CDK6 as a key regulator of hematopoietic and leukemic stem cell activation. Blood. 125, 90–101 (2015).
Uras, I. Z. et al. Palbociclib treatment of FLT3-ITD+ AML cells uncovers a kinase-dependent transcriptional regulation of FLT3 and PIM1 by CDK6. Blood. 127, 2890–2902 (2016).
Handschick, K. et al. Cyclin-dependent kinase 6 is a chromatin-bound cofactor for NF-κB-dependent gene expression. Mol. Cell. 53, 193–208 (2014).
Hydbring, P., Malumbres, M. & Sicinski, P. Non-canonical functions of cell cycle cyclins and cyclin-dependent kinases. Nat. Rev. Mol. Cell Biol. 17, 280–292 (2016).
Tigan, A. S., Bellutti, F., Kollmann, K., Tebb, G. & Sexl, V. CDK6-a review of the past and a glimpse into the future: from cell-cycle control to transcriptional regulation. Oncogene. 35, 3083–3091 (2016).
Yuan, W. et al. CDK6 mediates the effect of attenuation of miR-1 on provoking cardiomyocyte hypertrophy. Mol. Cell Biochem. 412, 289–296 (2016).
Majumdar, G. & Raghow, R. Trichostatin A induces a unique set of microRNAs including miR-129-5p that blocks cyclin-dependent kinase 6 expression and proliferation in H9c2 cardiac myocytes. Mol. Cell Biochem. 415, 39–49 (2016).
Acknowledgements
We would like to give our sincere gratitude to the reviewers for their constructive comments.
Funding
This work was supported by Natural Science Foundation of Jiangxi Province (20202BABL206070, 20202BBG73025), and National Natural Science Foundation of China (82060344).
Author information
Authors and Affiliations
Contributions
J.S., Y.M.Z., Q.L., and Y.L. designed the study; Y.H., C.L., and H.J. conducted the experiments; G.X. and R.X. participated in writing the manuscript; X.X. and S.Y. were responsible for the statistical analysis. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
Animal studies were approved by the ethical committee of Second Affiliated Hospital of Nanchang University.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sun, J., Zhu, YM., Liu, Q. et al. LncRNA ROR modulates myocardial ischemia-reperfusion injury mediated by the miR-185-5p/CDK6 axis. Lab Invest 102, 505–514 (2022). https://doi.org/10.1038/s41374-021-00722-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-021-00722-2
This article is cited by
-
LncRNA RNA ROR Aggravates Hypoxia/Reoxygenation-Induced Cardiomyocyte Ferroptosis by Targeting miR-769-5p/CBX7 Axis
Biochemical Genetics (2023)
-
Circulating small extracellular vesicle-encapsulated SEMA5A-IT1 attenuates myocardial ischemia–reperfusion injury after cardiac surgery with cardiopulmonary bypass
Cellular & Molecular Biology Letters (2022)
