
Abstract
Recent evidence has shown that lipopolysaccharide (LPS)-induced aerobic glycolysis of lung fibroblasts is closely associated with the pathogenesis of septic pulmonary fibrosis. Nevertheless, the underlying mechanism remains poorly defined. In this study, we demonstrate that LPS promotes c-Jun N-terminal kinase (JNK) signaling pathway activation and endogenous tumor necrosis factor-α (TNF-α) secretion in pulmonary macrophages. This, in turn, could significantly promote aerobic glycolysis and increase lactate production in lung fibroblasts through 6-phosphofructo-2-kinase/fructose-2, 6-biphosphatase 3 (PFKFB3) activation. Culturing human lung fibroblast MRC-5 cell line with TNF-α or endogenous TNF-α (cell supernatants of macrophages after LPS stimulation) both enhanced the aerobic glycolysis and increased lactate production. These effects could be prevented by treating macrophages with JNK pathway inhibitor, by administering TNF-α receptor 1 (TNFR1) siRNA, PFKFB3 inhibitor, or by silencing PFKFB3 with fibroblasts-specific shRNA. In addition, the inhibition of TNF-α secretion and PFKFB3 expression prevented LPS-induced pulmonary fibrosis in vivo. In conclusion, this study revealed that LPS-induced macrophage secretion of TNF-α could initiate fibroblast aerobic glycolysis and lactate production, implying that inflammation-metabolism interactions between lung macrophages and fibroblasts might play an essential role in LPS-induced pulmonary fibrosis.
Similar content being viewed by others


Introduction
Pulmonary fibrosis is characterized by abnormal activation and proliferation of lung fibroblasts and excessive collagen deposition, which eventually leads to refractory respiratory failure and poor prognosis1,2. Sepsis is a life-threatening organ dysfunction caused by a dysregulated host response to infection3. As an essential component found in the cell wall of Gram-negative bacilli, LPS is a crucial factor for sepsis-associated acute respiratory distress syndrome and septic pulmonary fibrosis4,5,6,7. Recent studies revealed that aerobic glycolysis in lung tissues plays a key role during pulmonary fibrosis8,9,10 and septic pulmonary fibrosis11. Aerobic glycolysis, also known as the “Warburg effect”, first describes the phenomenon that tumor cells prefer glycolysis rather than oxidative phosphorylation for energy production to promote cell proliferation under aerobic conditions12,13. Likewise, lung fibroblasts also went through enhanced aerobic glycolysis and lactate production in LPS-induced pulmonary fibrosis14. In our previous study, we demonstrated that LPS could induce lung fibroblasts aerobic glycolysis, cause the production of lactate and promote the development of pulmonary fibrosis14. Macrophages represent the most abundant innate immune cells in the lung and have been shown to play an important role in the pathogenesis of pulmonary fibrosis15,16. Further investigations are required to understand the role of macrophages in the process of lung fibroblasts aerobic glycolysis and, more specifically, if the interactions between immune cells and structural cells contribute to the development of septic pulmonary fibrosis. In this study, we are applying cellular and mice models to mimic the process of LPS-induced pulmonary fibrosis and to explore the role of macrophages in lung fibroblasts aerobic glycolysis. This study revealed that LPS-induced macrophage secretion of TNF-α could initiate fibroblast aerobic glycolysis and lactate production, implying that inflammation-metabolism interactions between lung macrophages and fibroblasts might play an essential role in LPS-induced pulmonary fibrosis.
Materials and methods
Ethics statement and animals
Male C57BL/6 mice (eight-week-old; 20–25 g) were obtained from Shanghai SLAC Laboratory Animal, China. Animals were housed in a specific-pathogen-free environment with controlled temperature (22–24 °C), a 12 h/12 h light/dark cycle and free access to food and water. All experiments were approved by the Animal Care and Use Committee of Ren Ji hospital, Shanghai Jiao Tong University School of Medicine (Approval No: RJ2021-0126).
Cell lines and culture
The human lung fibroblast MRC-5 cell line was obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and cultured in Minimum Essential Medium (MEM, Hyclone, USA) containing 10% fetal bovine serum (FBS, Gibco, USA), 100 IU/ml penicillin and 100 IU/ml streptomycin. The cells were incubated at 37 °C with fully humidified atmosphere containing 5% CO2. The culture medium was changed every 2 days. When the cells reached 70–80% confluence, the adherent cells were washed with phosphate-buffered saline (PBS), detached by exposure to 0.25% trypsin for 2 min, and passaged at a dilution of 1:3. The murine macrophage cell line RAW264.7 was also obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Hyclone, USA) supplemented with 10% FBS, 100 IU/ml penicillin and 100 IU/ml streptomycin in a 5% CO2 humidified incubator at 37 °C.
Reagents and antibodies
LPS (Escherichia coli O127:B8) was purchased from Sigma (USA). The JNK signaling pathway inhibitor SP600125 (HY12041) was purchased from MedChemExpress(China), and the TNF-α inhibitor Lenalidomide (S1029) and the PFKFB3 inhibitor 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (3PO, S7639) were purchased from Selleck (USA). The primary antibodies used in this study were rabbit anti-PFKFB3 (ab181861, Abcam, USA), rabbit anti-GAPDH (D16H11, CST, USA), rabbit anti-β-tubulin (A12289, Abclonal, China), rabbit anti-p-JNK (#4668, CST, USA), rabbit anti-JNK (#9252, CST, USA), rabbit anti-TNFR1 (ab19139, Abcam, USA), and mouse anti-α-SMA (ab32575, Abcam, USA). Goat anti-mouse (A0216, Beyotime, China) and goat anti-rabbit (A0208, Beyotime, China) secondary antibodies were used as well.
Experimental grouping and design
The macrophages in the logarithmic phase of growth were seeded into 24-well culture plates at a density of 1 × 105 cells/well and treated with LPS (1 μg/mL). SP600125 was used to suppress the activation of the JNK pathway. Cells and supernatants were collected at 30 min or 8 h after the treatment.
MRC-5 cells were seeded into 24-well culture plates at a density of 2 × 104 cells/well. After the cells adhered and reached 70% confluency, the medium was changed to serum-free MEM to starve the cells overnight. Then the cells were challenged with recombinant human TNF-α and endogenous TNF-α (cell supernatants of macrophages after LPS stimulation). TNFR1-siRNA, 3PO, and PFKFB3-shRNA lentivirus were used to inhibit TNFR1 and PFKFB3 at the protein and gene levels. SP600125 was used to suppress the activation of the JNK pathway in macrophages. Cell and supernatants were collected at 3, 6, and 12 h after treatment.
Animal experiments were carried on male C57BL/6 mice. 5 mg/kg LPS was intraperitoneally injected for 5 days to establish a pulmonary fibrosis model. TNF-α inhibitor Lenalidomide and PFKFB3 inhibitor 3PO were injected intraperitoneally to inhibit the secretion of TNF-α and the expression of PFKFB3. Mice were euthanized with an overdose of pentobarbital for the collection of lungs, blood, and bronchoalveolar lavage fluid (BALF) at day 7 after LPS injection.
Knock down of TNFR and PFKFB3
The following two types of TNFRs have been reported: TNFR1 and TNFR2. Since TNFR1 is the dominant receptor on MRC-5 cells, we constructed TNFR1siRNAs to downregulate the expression of TNFR1 and block TNF-α downstream signaling. MRC-5 cells were seeded into 24-well plates at a density of 2 × 104 cells per well and cultured for 24 h. The cells were subsequently transfected with TNFR1 siRNA or negative control siRNA (NC-siRNA) (GenePharma, China) using Lipofectamine™ RNAiMAX Transfection Reagent and transfection medium (OptiMEM I Medium, Gibco, USA) according to the manufacturer’s instructions. A total of three primer sets were used as follows:
siRNA-1, F-5′-CGGCAUUAUUGGAGUGAAATT-3′ and R-5′-UUUCACUCCAAUAAUGCCGTT-3′;
siRNA-2, F-5′-CCUGCCAUGCAGGUUUCUUTT-3′ and R-5′-AAGAAACCUGCAUGGCAGGTT-3′;
siRNA-3, F-5′- CUCCUCUUCAUUGGUUUAATT-3′ and R-5′-UUAAACCAAUGAAGAGGAGTT-3′
The protein levels of TNFR1 were measured by western blot analysis in order to assess the efficiency of the lentiviral transfection.
Lentivirus vectors (Fubio Biological Technology, China) were transfected into MRC-5 cells to downregulate the expression of PFKFB3. At 48 h post transfection, green fluorescent protein was observed under a fluorescence microscope to confirm transfection efficiency. Then, the cells were selected by 2 μg/mL puromycin to establish a stable cell line. The level of PFKFB3 protein was measured by western blot. The primers used were: PFKFB3, F-5′-AGCTGACTC GCTACCTCAAC-3′, and R-5′-GTTGAGGTAGCGAGTC AGCT-3′.
Western blot analysis
The protein levels of TNFR1, JNK, p-JNK, PFKFB3, and α-SMA were detected by western blot. The cells were lysed on ice for 15 min in RIPA lysis buffer (Beyotime, China) containing 1% phenyl methyl sulfonyl fluoride (PMSF), protease inhibitor cocktail, and phosphatase inhibitor cocktail (KangChen, China). After centrifugation, the supernatants were collected and the protein quantification was determined using the BCA assay kit (Thermo scientific, USA). The proteins were separated by 12% SDS–polyacrylamide gel electrophoresis and transferred onto a polyvinylidene difluoride membrane (Millipore, Germany). Then, the membranes were incubated with appropriate primary and secondary antibodies, respectively. Subsequently, the immunoblots were detected by Image LabTM software (Bio-Rad, USA) using Enhanced ECL Chemiluminescent Substrate Kit (Vazyme, China).
Quantitative real-time PCR (qRT-PCR)
After 6 h of TNF-α treatment, the expression level of PFKFB3 mRNA in MRC-5 cells was evaluated using qRT-PCR. Total RNA was isolated from MRC-5 cells using an RNA Purification Kit (EZ Bioscience, USA) according to the manufacturer’s instruction. Complementary DNA synthesis was performed using Prime Script RT Master Mix (Takara, China), and real-time PCR was carried out on a Light Cycler 480 real-time PCR system (Roche, USA) using iTaq universal SYBR Green Supermix (Bio-Rad, Hercules, CA, USA). The primers were: GAPDH, F: TGGTGAAGGTCGGTGTGAAC and R: GCTCCTGGAAGATGGTGATGG; PFKFB3, F: ATTGCGGTTTTCGATGCCAC, R: GCCACAACTGTAGGGTCGT. The 2−ΔΔCt method was employed to calculate relative expression levels.
Real-time cell metabolism assay
XF-96 Extracellular Flux Analyzer (Seahorse Bioscience, Agilent Technologies, USA) was used to analyze real-time extracellular acidification rate (ECAR) according to the manufacturer’s instructions. For ECAR, 10 mM glucose, 1 μM oxidative phosphorylation inhibitor oligomycin, and 100 mM glycolytic inhibitor 2-DG were sequentially injected into each well at indicated time points. Data were analyzed by Seahorse XF-96 Wave software.
Measurement of TNF-α, PICP, and lactate levels
The cell supernatants were collected and centrifuged at 2000g for 5 min, and then stored at −80 °C. After irrigating the lung with 500 μL cold PBS, BALF samples were collected using a tracheal cannula and centrifuged at 2500 g for 10 min. The supernatants were stored at −80 °C.
The levels of TNF-α in the macrophage supernatants were measured with an ELISA kit (Multi sciences, China) according to the manufacturer’s instructions. The levels of lactate in MRC-5 cells supernatants and BALF were measured with a lactate assay kit (Nanjing Jiancheng Bioengineering, China) following the manufacturer’s instructions. The levels of PICP in the BALF were measured with an ELISA kit (Multi sciences, China) following the manufacturer’s instructions.
Lung histopathology
Lung tissue samples were fixed in 4% paraformaldehyde overnight, dehydrated, and embedded in paraffin. Five micrometer-thick sections were stained with hematoxylin and eosin (H&E) to assess lung morphological changes and Masson’s trichrome staining was performed to evaluate collagen deposition.
Statistical analysis
All experiments were performed at least three times and the data were analyzed using Graph Pad Prism7 software (USA) and presented as mean ± standard deviation (SD). One-way analysis of variance was used to determine the means of three or more treatment groups. Student’s t-test (two tailed) was used to compare the differences between two groups. A p value less than 0.05 (p < 0.05) was considered statistically significant.
Results
LPS induces endogenous TNF-α secretion in macrophages (RAW264.7) via the activation of JNK pathway
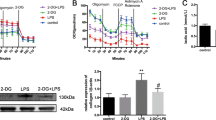
Western blot indicated that LPS stimulation induced JNK pathway activation in macrophages (RAW264.7) (Fig. 1A, B). The p-JNK/JNK ratio increased significantly following LPS stimulation compared with the control group. ELISA in Fig. 1C displays that LPS stimulation on macrophages (RAW264.7) lead to increased levels of TNF-α in the cell supernatants compared with the control group, while this effect could be reversed by JNK pathway inhibitor SP600125. We verified that through JNK pathway activation, LPS could promote TNF-α secretion in macrophages (RAW264.7).

The activation of JNK pathway was measured by western blot analysis; TNF-α concentration in cell culture supernatants was quantified by ELISA. A, B Protein levels of JNK, p-JNK following LPS treatment (1 μg/mL) of macrophages (RAW264.7) with or without a 30 min SP600125 pretreatment; **p < 0.01 vs. control group, ##p < 0.01 vs. LPS group. C Supernatant levels of TNF-α following LPS treatment of macrophages (RAW264.7) with or without a 30 min SP600125 pretreatment; **p < 0.01 vs. control group, ##p < 0.01 vs. LPS group. Mean values from triplicate experiments are both shown with SD error bars. The blots are representative of three independent experiments.
TNF-α promotes aerobic glycolysis and lactate production in lung fibroblasts
To investigate the mechanism underlying LPS-induced pulmonary fibrosis, we constructed a cell model of septic pulmonary fibrosis by culturing MRC-5 fibroblasts with supernatants of LPS-stimulated macrophages (RAW264.7) and the expression of α-SMA proved the success of the cell model (data not shown). We assessed the lactate levels of MRC-5 cells in cell culture supernatants, stimulated with different concentrations of exogenous TNF-α (1.25, 2.5, and 5 ng/ml). As shown in Fig. 2A, the lactate levels were higher in the TNF-α stimulation groups than the non-stimulation group. In addition, as TNF-α concentration was increased, lactate levels in the supernatants raised in a dose-dependent manner, although the differences were not significant. We constructed three siRNAs of TNFR1 and western blot analysis showed that siRNA-1 was the most efficient (Fig. 2B, C). As indicated by the lower levels of ECAR and less production of lactate (Fig. 2D, E), the downregulation of TNFR1 expression inhibited the aerobic glycolysis in the lung fibroblasts. To further examine the interactions between macrophages and fibroblasts in LPS-induced pulmonary fibrosis, MRC-5 fibroblasts were cultured with the supernatants of LPS-stimulated macrophages (RAW264.7). As shown in Fig. 2F, H, LPS-stimulated supernatants increased lactate production and promoted aerobic glycolysis in lung fibroblasts. This phenomenon could be alleviated by JNK pathway inhibitor SP600125 on macrophages (RAW264.7) or by the downregulation of TNFR1 expression on lung fibroblasts. These data are suggesting that, in lung fibroblasts, TNF-α secreted by macrophages (RAW264.7) promoted aerobic glycolysis and lactate production.

Lactate levels in cell culture supernatants of MRC-5 cells after different concentrations of exogenous TNF-α stimulation (1.25, 2.5 and 5 ng/ml) were quantified by Lactate Assay Kit (A); *p < 0.05, **p < 0.01 vs. non-stimulation group. The protein expression of TNFR1 was detected by Western blot to evaluate the efficiency of TNFR1-siRNA transfection (B, C); **p < 0.01 vs. control group. TNFR1-siRNA inhibited ECAR level and lactate production of MRC-5 cells increased by TNF-α (D, E); **p < 0.01 vs. control group, ##p < 0.01 vs. TNF-α group. MRC-5 fibroblasts were cultured with the supernatants of LPS-stimulated macrophages (RAW264.7). LPS-stimulated supernatants promoted ECAR level and increased lactate production, which could be alleviated by SP600125 on macrophages (RAW264.7) (F) and TNFR1-siRNA on MRC-5 cells (G, H); **p < 0.01 vs. control group, #p < 0.05 and ##p < 0.01 vs. LPS-Mφ group. The mean values from triplicate experiments are both shown with SD error bars. The blots are representative of three independent experiments. LPS- Mφ, supernatants of LPS-stimulated RAW264.7 macrophages; LPS + SP600125− Mφ, supernatants of LPS-stimulated RAW264.7 macrophages pretreating with SP600125.
TNF-α increases PFKFB3 expression in lung fibroblasts
PFKFB3 is a key enzyme in the aerobic glycolysis process, and we found that TNF-α stimulation could promote PFKFB3 expression in lung fibroblasts. As shown in Fig. 3A, B, lung fibroblasts were cultured in the presence of TNF-α for various periods of time. The expression of PFKFB3 protein was first increased at 6 h after treatment, and then decreased at 12 h. Downregulation of lung fibroblasts TNFR1 expression reversed the increased PFKFB3 mRNA and protein level after 6 h of TNF-α stimulation (Fig. 3C–E).

The PFKFB3 expression of MRC-5 cells was detected by western blot after incubated with TNF-α for various periods of time (A, B). TNFR1-siRNA inhibited PFKFB3 mRNA (C) and protein (D, E) levels of MRC-5 cells increased by TNF-α; *p < 0.05, **p < 0.01 vs. control group, #p < 0.05 and ##p < 0.01 vs. TNF-α group. Values are mean ± SD from triplicate experiments. The blots are representative of three independent experiments.
Inhibition of PFKFB3 precludes TNF-α-induced aerobic glycolysis and lactate production in lung fibroblasts
To investigate the role of PFKFB3, we constructed PFKFB3-shRNA lentivirus (PFKFB3 shRNA) to knock down PFKFB3 expression in lung fibroblasts. Figure 4A, B confirmed the transfection efficiency in MRC-5 cell lines. PFKFB3 shRNA transfection inhibited TNF-α-induced aerobic glycolysis in the lung fibroblasts, as indicated by the lower levels of ECAR and less production of lactate (see Fig. 4C, D).

The transfection efficiency of PFKFB3 shRNA in MRC-5 cell lines was confirmed by immunofluorescence (A) and western blot (B). TNF-α-increased ECAR level and lactate production of MRC-5 cells was inhibited by PFKFB3 shRNA (C, D) and 3PO (E, F); **p < 0.01 vs. control group, ##p < 0.01 vs. TNF-α group. Values are mean ± SD from triplicate experiments.
We then used 3PO, a selective inhibitor of PFKFB3. Likewise, pretreatment with 3PO significantly inhibited the aerobic glycolysis in the lung fibroblasts as indicated by lower levels of ECAR and less production of lactate (Fig. 4E, F). These findings are suggesting that PFKFB3 is essential in TNF-α-induced lung fibroblasts aerobic glycolysis.
Inhibition of TNF-α secretion and PFKFB3 expression precludes LPS-induced pulmonary fibrosis in vivo
To investigate whether inhibition of TNF-α secretion and PFKFB3 expression in vivo affects LPS-induced aerobic glycolysis and pulmonary fibrosis, mice were intraperitoneally pretreated with Lenalidomide (50 mg/kg) for 3 days or 3PO (70 mg/kg) for 5 days, followed by LPS (5 mg/kg) or the same dose of saline injection for 5 consecutive days. HE and Masson staining were performed at 7 days after LPS treatment. As shown in Fig. 5A, we observed typical pulmonary fibrosis in LPS group. HE staining showed that the lung tissue in LPS group manifested more obvious alveolar congestion and leukocytes infiltration compared with that of the control group, while Masson staining showed thickened alveoli septum and collagen deposition in LPS-induced lung tissue. These could be attenuated by pretreating with Lenalidomide and 3PO. Western blot analysis of α-SMA in lung tissue and BALF levels of PICP and lactate also confirmed that inhibition of TNF-α secretion and PFKFB3 prevents LPS-induced pulmonary fibrosis in vivo (Fig. 5B–I).

Eight-week-old male C57BL/6 mice were pretreated with Lenalidomide (50 mg/kg) for 3 days or 3PO (70 mg/kg) for 5 days followed by LPS (5 mg/kg) or the same dose of saline injection for 5 consecutive days (n = 6 for each group). Pulmonary fibrosis was measured by H&E and Masson staining (magnification, ×200) (A). Western blot was performed to detect the expression of α-SMA, PFKFB3 in lung tissues (B–G). BALF levels of PICP and lactate were determined by ELISA (H, I). Values are mean ± SD. *p < 0.05, **p < 0.01 vs. control group, #p < 0.05 and ##p < 0.01 vs. LPS group.
Discussion
In recent years, metabolic reprogramming of lung tissue in the pathogenesis of lung diseases has received widespread attention17. Evidences are suggesting that aerobic glycolysis in lung tissues is closely related to pulmonary fibrosis18. In our previous study we have demonstrated that LPS directly induced lung fibroblasts aerobic glycolysis, contributed to lactate production and promoted the development of pulmonary fibrosis14. Nevertheless, whether macrophages are involved in the process of lung fibroblasts aerobic glycolysis in septic pulmonary fibrosis remains poorly understood. The current study revealed that TNF-α secreted by LPS-stimulated macrophages induces aerobic glycolysis in lung fibroblasts and increases lactate production. Moreover, the inhibition of TNF-α downstream pathway and PFKFB3 expression prevents lung fibroblasts aerobic glycolysis and pulmonary fibrosis in vitro and in vivo (see Fig. 6).

LPS promotes JNK signaling pathway activation and endogenous TNF-α secretion in pulmonary macrophages, which could facilitate lung fibroblasts aerobic glycolysis and lactate production through PFKFB3 activation. This study suggests that inflammation-metabolism interactions between lung macrophages and fibroblasts might play an essential role in LPS-induced pulmonary fibrosis.
Macrophages are critical for phagocytosis of cell debris and pathogens and they activate lymphocytes or other immune cells to respond to pathogens. They also play an essential role in the development, metabolism, and maintenance of homeostasis. Pulmonary macrophages are displaying different functional phenotypes upon different stimuli and signals, and they are usually classically (M1) or alternatively (M2) activated macrophages. These two macrophage phenotypes differ in their biological behavior with regard to cell function, such as secretion of specific cytokines and other associated processes. It is reported that both M1 and M2 macrophages are playing significant roles in the pathogenesis of pulmonary fibrosis19. Following the exposure to LPS stimulation, the macrophages can be activated into their M1 phenotype and produce pro-inflammatory cytokines, such as TNF-α and interleukin-1β. In this study, we confirmed that LPS could induce macrophages to secrete TNF-α by activation of the JNK pathway. To identify the role of TNF-α in lung fibroblasts aerobic glycolysis, we challenged MRC-5 fibroblasts with exogenous TNF-α and supernatants of LPS-stimulated macrophages (endogenous TNF-α). Inhibition of TNF-α secretion by JNK pathway inhibitor SP600125 on macrophages and downregulation of TNFR1 expression by TNFR1 siRNA on MRC-5 fibroblasts both suppressed lung fibroblasts aerobic glycolysis and lactate production. These data are indicating that TNF-α secreted by macrophages are crucial in lung fibroblasts aerobic glycolysis.
PFKFB3 is a critical glycolytic enzyme that regulates glycolysis20. It is reported that the PFKFB3 expression in lung tissue is related to pulmonary fibrosis21. We further demonstrated that TNF-α upregulates the expression of PFKFB3 in lung fibroblasts while the inhibition of PFKFB3 by 3PO or PFKFB3 shRNA pretreatment precludes the TNF-α-induced aerobic glycolysis and lactate production. In addition, the above-mentioned hypothesis has been validated in the mouse model of LPS-induced pulmonary fibrosis. Inhibition of TNF-α secretion or PFKFB3 expression by pretreatment with Lenalidomide or 3PO intraperitoneally precludes LPS-induced pulmonary fibrosis in vivo.
The current study confirms that the inflammatory cytokine TNF-α, which is released by pulmonary macrophages following LPS stimulation, could promote lung fibroblasts aerobic glycolysis and lactate production. The interplay between lung macrophages and fibroblasts contributes to the development of septic pulmonary fibrosis. This evidence provides a new perspective on the mechanism of septic pulmonary fibrosis and could be a potential therapeutic target in the future.
It is also worth mentioning that high-mobility group box 1 (HMGB1) was also found to mediate M1 polarization of macrophages22. HMGB1 is a nuclear protein that acts as a crucial factor in pulmonary fibrosis23. Our previous study showed that LPS stimulation could induce lung fibroblast secretion of HMGB1 through the NF-κB pathway7. This may amplify the production of TNF-α in macrophages and boost the downstream effects on lung fibroblasts. Further studies should deepen the role of HMGB1 in the regulation of macrophage–fibroblast interactions following LPS stimulation.
Data availability
All datasets generated or analyzed during the current study are not publicly available as some of them are involved in our ongoing studies.
References
Meduri, G. U. & Eltorky, M. A. Understanding ARDS-associated fibroproliferation. Intensive Care Med. 41, 517–520 (2015).
Marshall, R., Bellingan, G. & Laurent, G. The acute respiratory distress syndrome: fibrosis in the fast lane. Thorax 53, 815–817 (1998).
Singer, M. et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 315, 801–810 (2016).
Zhou, W. Q., Wang, P., Shao, Q. P. & Wang, J. Lipopolysaccharide promotes pulmonary fibrosis in acute respiratory distress syndrome (ARDS) via lincRNA-p21 induced inhibition of Thy-1 expression. Mol. Cell. Biochem. 419, 19–28 (2016).
He, Z., Zhu, Y. & Jiang, H. Inhibiting toll-like receptor 4 signaling ameliorates pulmonary fibrosis during acute lung injury induced by lipopolysaccharide: an experimental study. Respir. Res. 10, 126 (2009).
Du, S. et al. Houttuynia cordata inhibits lipopolysaccharide-induced rapid pulmonary fibrosis by up-regulating IFN-gamma and inhibiting the TGF-beta1/Smad pathway. Int. Immunopharmacol. 13, 331–340 (2012).
Li, W. et al. High-mobility group box 1 accelerates lipopolysaccharide-induced lung fibroblast proliferation in vitro: involvement of the NF-kappaB signaling pathway. Lab. Investig. 95, 635–647 (2015).
Kang, Y. P. et al. Metabolic profiling regarding pathogenesis of idiopathic pulmonary fibrosis. J. Proteome Res. 15, 1717–1724 (2016).
Kottmann, R. M. et al. Lactic acid is elevated in idiopathic pulmonary fibrosis and induces myofibroblast differentiation via pH-dependent activation of transforming growth factor-beta. Am. J. Respir Crit Care Med. 186, 740–751 (2012).
Kottmann, R. M. et al. Pharmacologic inhibition of lactate production prevents myofibroblast differentiation. Am. J. Physiol. Lung Cell Mol. Physiol. 309, L1305–L1312 (2015).
Li, H. M. et al. Blockage of glycolysis by targeting PFKFB3 suppresses tumor growth and metastasis in head and neck squamous cell carcinoma. J. Exp. Clin. Cancer Res. 36, 7 (2017).
Maher, T. M. Aerobic glycolysis and the warburg effect. an unexplored realm in the search for fibrosis therapies? Am. J. Respir. Crit. Care Med. 192, 1407–1409 (2015).
Vander Heiden, M. G., Cantley, L. C. & Thompson, C. B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 (2009).
Hu, X. et al. PI3K-Akt-mTOR/PFKFB3 pathway mediated lung fibroblast aerobic glycolysis and collagen synthesis in lipopolysaccharide-induced pulmonary fibrosis. Lab. Investig. 100, 801–811 (2020).
Misharin, A. V. et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med. 214, 2387–2404 (2017).
McCubbrey, A. L. et al. Deletion of c-FLIP from CD11b(hi) Macrophages Prevents Development of Bleomycin-induced Lung Fibrosis. Am. J. Respir. Cell. Mol. Biol. 58, 66–78 (2018).
Zhao, H., Dennery, P. A. & Yao, H. Metabolic reprogramming in the pathogenesis of chronic lung diseases, including BPD, COPD, and pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 314, L544–l554 (2018).
Xu, J. et al. HMGB1 promotes HLF-1 proliferation and ECM production through activating HIF1-alpha-regulated aerobic glycolysis. Pulm. Pharmacol. Ther. 45, 136–141 (2017).
Zhang, L. et al. Macrophages: friend or foe in idiopathic pulmonary fibrosis? Respir. Res. 19, 170 (2018).
Minchenko, A. et al. Hypoxia-inducible factor-1-mediated expression of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3) gene. Its possible role in the Warburg effect. J. Biol. Chem. 277, 6183–6187 (2002).
Xu, J. et al. HMGB1 promotes HLF-1 proliferation and ECM production through activating HIF1-α-regulated aerobic glycolysis. Pulm. Pharmacol. Ther. 45, 136–141 (2017).
Tian, S. et al. HMGB1 exacerbates renal tubulointerstitial fibrosis through facilitating M1 macrophage phenotype at the early stage of obstructive injury. Am. J. Physiol. Renal. Physiol. 308, F69–F75 (2015).
Hamada, N. et al. The role of high mobility group box1 in pulmonary fibrosis. Am. J. Respir. Cell. Mol. Biol. 39, 440–447 (2008).
Author contributions
Q.X. and S.M. performed most of the experiments and were major contributors to this manuscript. Z.H. and S.X. designed the study and provided guidance for the implementation of the study. F.N. provided assistance in writing the manuscript. J.F. and Z.Z. took charge of data analysis. J.Z. performed part of the animal experiments. X.Q. helped with the cell metabolism assay. Y.G. participated in the design of the study and provided guidance for project implementation. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (NSFC, No.81970059 and 81770060) and the Natural Science Foundation of Shanghai 2020 “Science and Technology Innovation Action Plan” (20ZR1433000). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
All animals used were approved by the Animal Care and Use Committee of Ren Ji Hospital, Shanghai Jiao Tong University School of Medicine (RJ2021-0126).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Xu, Q., Mei, S., Nie, F. et al. The role of macrophage–fibroblast interaction in lipopolysaccharide-induced pulmonary fibrosis: an acceleration in lung fibroblast aerobic glycolysis. Lab Invest 102, 432–439 (2022). https://doi.org/10.1038/s41374-021-00701-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-021-00701-7
This article is cited by
-
PFKFB3 in neovascular eye disease: unraveling mechanisms and exploring therapeutic strategies
Cell & Bioscience (2024)
-
LPS-induced monocarboxylate transporter-1 inhibition facilitates lactate accumulation triggering epithelial-mesenchymal transformation and pulmonary fibrosis
Cellular and Molecular Life Sciences (2024)
-
Lipopolysaccharide alters VEGF-A secretion of mesenchymal stem cells via the integrin β3-PI3K-AKT pathway
Molecular & Cellular Toxicology (2024)
-
ASK1-ER stress pathway-mediated fibrotic-EV release contributes to the interaction of alveolar epithelial cells and lung fibroblasts to promote mechanical ventilation-induced pulmonary fibrosis
Experimental & Molecular Medicine (2022)
-
Non-coding RNA in idiopathic interstitial pneumonia and Covid-19 pulmonary fibrosis
Molecular Biology Reports (2022)
