
Abstract
Pigment epithelium-derived factor (PEDF) plays a role in protecting retinal pigment epithelial (RPE) cells from oxidative stress (OS), a causative factor of RPE cell death. Genetically modified mesenchymal stem cells (MSCs) can be used to treat critical and incurable retinal diseases. Here, we overexpressed PEDF in placenta-derived MSCs (PD-MSCsPEDF, PEDF+) using a nonviral gene delivery system and evaluated the characteristics of PD-MSCsPEDF and their potential regenerative effects on RPE cells damaged by H2O2-induced OS. PD-MSCsPEDF maintained their stemness, cell surface marker, and differentiation potential characteristics. Compared to naive cells, PD-MSCsPEDF promoted mitochondrial respiration by enhancing biogenesis regulators (e.g., NRF1, PPARGC1A, and TFAM) as well as antioxidant enzymes (e.g., HMOXs, SODs, and GPX1). Compared to OS-damaged RPE cells cocultured with naive cells, OS-damaged RPE cells cocultured with PD-MSCsPEDF showed PEDF upregulation and VEGF downregulation. The expression levels of antioxidant genes and RPE-specific genes, such as RPE65, RGR, and RRH, were significantly increased in RPE cells cocultured with PD-MSCsPEDF. Furthermore, OS-damaged RPE cells cocultured with PD-MSCsPEDF had dramatically enhanced mitochondrial functions, and antiapoptotic effects improved due to cell survival signaling pathways. In the H2O2-induced retinal degeneration rat model, compared to administration of the naive counterpart, intravitreal administration of PD-MSCsPEDF alleviated proinflammatory cytokines and restored retinal structure and function by increasing PEDF expression and decreasing VEGF expression. Intravitreal administration of PD-MSCsPEDF also protected retinal degeneration against OS by increasing antioxidant gene expression and regulating the mitochondrial ROS levels and biogenesis. Taken together, PEDF overexpression in PD-MSCs improved the mitochondrial activities and induced OS-damaged RPE cell regeneration by regulating the oxidative status and mitochondrial biogenesis in vitro and in vivo. These data suggest that genetic modification of PEDF in PD-MSCs might be a new cell therapy for the treatment of retinal degenerative diseases.
Similar content being viewed by others

Introduction
Retinal degenerative diseases are a group of heterogeneous conditions that include age-related macular degeneration (AMD), retinitis pigmentosa, and diabetic retinopathy [1]. Numerous factors, such as oxidative stress (OS), genetic diseases, or aging, have been suggested to contribute to the progressive degeneration of retinal neurons and retinal pigment epithelium (RPE) [2,3,4]. The RPE plays essential roles in photoreceptor function, especially photoreceptor renewal, including an OS response for maintaining retinal homeostasis [5]. RPE cells have evolved a protective mechanism against light-induced damage by absorbing light energy. However, increased OS and reactive oxygen species (ROS) production during aerobic metabolism in the RPE lead to photoreceptor death and irreversible vision loss [6]. Recently, stem cell therapy has been applied as a valuable cell therapy in various acute and chronic neurodegenerative conditions, including ocular degenerative disorders [7]. Embryonic stem cell- and induced pluripotent stem cell (iPSC)-derived RPE cells are safely and effectively used for cell transplantation to restore retinal function [8, 9]. However, the clinical use of these cells has several problems, such as low efficiency of differentiation into RPE cells, graft rejection and risk of tumorigenicity [10]. Alternatively, retinal progenitor cells are another source of cells used for stem cell therapy. These cells can migrate into retinal layers and develop various retinal cell types, but these cells also have their disadvantages, including the requirement of a sufficient number of donor cells for transplantation and limited functional integration of donor cells [11]. Therefore, mesenchymal stem cell (MSC)-based therapy, which can greatly influence cell restoration via paracrine activity, can offer a promising strategy to promote RPE regeneration and overcome the risk of stem cell transplantation. In particular, it was shown that human placenta-derived MSCs (PD-MSCs) obtained from fetal tissue are an alternative source of MSCs and have several advantages, including multipotent differentiation, strong immunosuppressive properties, easy access to abundant cells in vitro, and a lack of ethical concerns [12, 13]. Nevertheless, the mechanism of efficacy of MSC-based cell therapy is uncertain, and the next generation of MSCs with enhanced functions is needed to enhance the efficacy of cell therapy and to clarify the mechanism of treatment.
Recent studies have focused on strategies for function-enhanced MSCs to overcome the limited effectiveness of MSCs in offsetting the deleterious effects of OS induced by tissue injury. With the discovery of genes important for the repair of damaged tissues, genetic engineering of MSCs to express specific cellular factors is a promising approach to further enhance the therapeutic effects of MSCs [14]. Pigment epithelium-derived factor (PEDF; serpin family F member 1) is a 50 kDa secreted protein known as serpin F1 and was first identified in cultured fetal RPE cells [15]. PEDF is capable of various biological functions, including neurotropic and antiangiogenetic activities, as well as protective functions against OS in the RPE [16, 17]. Furthermore, this factor stimulates the growth and survival of photoreceptor cells of the retina and neurons of the central and peripheral nervous systems [18].
OS leads to apoptosis when the antioxidant capacity is insufficient. The RPE contains antiapoptotic and neuroprotective factors to support retina survival and maintenance. However, accumulated oxidative damage in RPE cells induces increased ROS production, resulting in mitochondrial dysfunction in the cells, which eventually contributes to the development of retinal diseases such as AMD [6]. Mitochondrial biogenesis and aerobic respiration are upregulated in MSCs during differentiation and OS [19, 20]. Mitochondrial biogenesis is controlled by peroxisome proliferative-activated receptor gamma coactivator 1 alpha (PPARGC1A) and is further activated by nuclear respiratory factor 1 (NRF1), transcription factor A, and mitochondrial (TFAM), which facilitate mitochondrial DNA (mtDNA) replication. This result is followed by increases in the ATP levels and upregulation of oxidative metabolism [21]. However, little is known about the impact of overexpressing PEDF in PD-MSCs (PD-MSCsPEDF, PEDF+) on mitochondrial function. Moreover, it is not yet known whether PD-MSCsPEDF provide a regenerative ability to RPE cells to counter OS in vitro and in vivo. Clarifying how PD-MSCsPEDF suppress the deleterious effects of OS on the RPE may help improve stem cell therapy for treating retinal degeneration.
In this study, we engineered PD-MSCs overexpressing PEDF (PD-MSCsPEDF) using a nonviral gene delivery system and evaluated the characteristics of PD-MSCsPEDF based on the functional enhancement of mitochondrial biogenesis, which in turn augments the regenerative potential of a damaged RPE compromised by H2O2-induced OS.
Materials and methods
Cell culture and gene transfection
Placentas were collected for research purposes by the Institutional Review Board (IRB) of CHA Gangnam Medical Center, Seoul, Republic of Korea (IRB 07–18). PD-MSCs were isolated as described previously [22] and cultured in α-modified minimal essential medium (HyClone, Logan, UT, USA) supplemented with 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, USA), 1% penicillin/streptomycin (P/S; Invitrogen), 1 μg/mL heparin (Sigma-Aldrich, St. Louis, MO, USA), and 25 ng/mL human fibroblast growth factor-4 (PeproTech, Rocky Hill, NJ, USA). The PEDF plasmid was inserted into the pcDNA3.1 vector containing the cytomegalovirus (CMV) promoter, digested with the Hind3/Xba1 restriction enzymes and confirmed by DNA sequencing (BIONEER, Daejeon, Republic of Korea). To induce overexpression of the PEDF gene, naive PD-MSCs (passage = 7) were transfected with the PEDF plasmid vector using a nonviral AMAXA Nucleofector system (Lonza, Basel) according to the manufacturer’s instructions. After transfection for 24 h, cells were selected by 200 μg/mL hygromycin. ARPE-19 cells (human retinal pigment epithelial cell line, CRL-2302™; ATCC, Manassas, VA, USA) were cultured in Dulbecco’s modified Eagle medium (DMEM; Invitrogen) supplemented with 10% FBS (Invitrogen) and 1% P/S (Invitrogen). Cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2.
Animal models and transplantation of PD-MSCs
Seven-week-old male Sprague-Dawley rats (Orient Bio, Inc., Seongnam, Republic of Korea) were housed under specific pathogen-free conditions. A hydrogen peroxide (H2O2) solution (Sigma-Aldrich) was diluted with balanced salt solution (BSS) to the final concentration (10 μg/μL); rats were intravitreally injected with the solution and the sham control group (Sham; n = 6) was injected with an equal volume of BSS. Retinal degeneration was induced by a H2O2 injection as previously described [23]. At 1 week post injection, naive PD-MSCs (Tx Naive; n = 6) and PD-MSCsPEDF (Tx PEDF+; n = 6) (2 × 105 cells, 9–10 passages) cells were stained with a PKH67 Fluorescent Cell Linker Kit (Sigma-Aldrich) and transplanted into the rats through an intravitreal injection, excluding the H2O2-injured groups (H2O2 1 w; n = 6 and H2O2 2 w; n = 6). After 1 week, the rats were sacrificed, and the retinas from the eyeballs and blood were harvested. In all animal experimental processes, the protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of CHA University, Seongnam, Korea (IACUC-200033).
Histopathological and immunohistochemistry analyses
To analyze the induction of retinal degeneration with a H2O2 injection and to analyze VEGF expression, eyeball specimens for each group (n = 6) were collected and fixed in 10% neutral buffered formalin for histological examination. All samples were embedded in paraffin, and 5-μm-thick sections were obtained for hematoxylin and eosin staining. To observe the degree of degeneration to evaluate VEGF expression, each section of the eyeball was stained with an anti-VEGF antibody (1:200; Novus Biologicals, Centennial, CO, USA) using immunohistochemistry (Supplementary Fig. 3). Reactions with a horseradish peroxidase-conjugated streptavidin-biotin complex (DAKO, Santa Clara, CA, USA) and 3,3-diaminobenzidine (EnVision Systems, Santa Clara, CA, USA) were performed to generate a chromatic signal. Mayer’s hematoxylin (DAKO) was used as a counterstain. Morphometric images of whole sections in the eyeball were viewed using a digital slide scanner (3DHISTECH, Ltd, Budapest, Hungary).
Multilineage differentiation
To confirm the mesodermal differentiation potential, PD-MSCsPEDF (passage = 5) were seeded in each plate (5 × 103 cells/cm2) with each differentiation induction medium for ~21 days using the StemPro Adipogenesis and Osteogenesis Differentiation Kit (Invitrogen) according to the manufacturer’s instructions. After 21 days, the cells were fixed in 4% paraformaldehyde and stained using oil red O (Sigma-Aldrich) for lipid droplet identification, and von Kossa (Sigma-Aldrich) staining with 5% silver nitrate was used to visualize calcium deposits. Each experiment was performed in triplicate.
FACS analysis
For immunophenotyping of cell surface antigens, PD-MSCsPEDF (passage = 3) were stained with fluorescein isothiocyanate (FITC)- and phycoerythrin (PE)-conjugated antibodies. Antibodies against CD34-PE, CD90-PE, HLA-ABC-FITC, HLA-DR-FITC (BD Bioscience, San Jose, CA, USA), CD13-PE (BioLegend, San Diego, CA, USA), CD105-FITC (R&D Systems, Minneapolis, MN, USA), and HLA-G-FITC (Abcam, Cambridge, UK) were analyzed with a FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ). A total of 10,000 events were acquired. All reactions were analyzed at least in triplicate.
Doubling time
To analyze the doubling time, PD-MSCsPEDF (passage = 1) were seeded in plates at a density of 5 × 103 cells/cm2, and the doubling time (TD) of the harvested cells was calculated using the following online algorithm (http://www.doubling-time.com): TD = t × log2/(logN1 − logN0), where N0 is the number of cells inoculated, N1 is the number of cells harvested, and t is the culture time in hours. The experiment was performed at least in triplicate.
Karyotyping
Karyotypes were analyzed in PD-MSCsPEDF and naive PD-MSCs using the G-banding technique. The karyotypes were interpreted according to the International System for Human Cytogenomic Nomenclature (ISCN 2016) by analyzing at least ten metaphase cells counting each vessel under a light microscope (Axioskop 2 plus, Zeiss, Germany) [24].
Reverse transcription-polymerase chain reaction (RT-PCR) and quantitative real-time PCR (qRT-PCR)
Total RNA was isolated using TRIzol LS reagent (Invitrogen) according to the manufacturer’s protocol. cDNA was synthesized by reverse transcriptase (RT) from total RNA (500 ng) using SuperScript III RT (Invitrogen). PCR amplification was mediated by h-Taq DNA polymerase (SolGent, Daejeon, Republic of Korea) according to the manufacturer’s instructions. The primer sequences are provided in Table 1.
qRT-PCR was performed with primers (Table 2) and SYBR Green PCR Master Mix (Roche, Basel) in the CFX Connect™ Real-Time System (Bio-Rad Laboratories, Hercules, CA, USA). The expression of each gene was quantified using the 2−∆∆CT method. All reactions were performed in triplicate.
Western blotting
The cell pellet was lysed in lysis buffer (Sigma-Aldrich). Each protein was separated by 6–15% SDS-PAGE and transferred to polyvinylidene difluoride membranes. Each membrane was blocked using tris-buffered saline with tween-20 containing 5% bovine serum albumin, and then incubated overnight 4 °C with primary antibodies: anti-HO1 (1:1000, Novus Biologicals), anti-SOD1 (1:1000, Cell Signaling Technology, Danvers, MA, USA), anti-Drp1 (1:1000, Cell Signaling Technology), anti-Opa1 (1:500, Abcam), anti-Catalase (1:1000; Abcam), anti-COX IV (1:3000, Cell Signaling Technology), anti-Caspase-3 (1:1000, Cell Signaling Technology), anti-Bcl2 (1:1000, Novus Biologicals), anti-Bax (1:500, Santa Cruz Biotechnology, Dallas, TX, USA), anti-PEDF-R (1:500, R&D Systems), anti-total AKT (1:1000, Cell Signaling Technology), anti-phospho AKT (1:1000, Cell Signaling Technology), anti-total JNK (1:1000, Cell Signaling Technology), anti-phospho JNK (1:1000, Cell Signaling Technology), anti-total ERK (1:1000, Cell Signaling Technology), anti-phospho ERK (1:1000, Cell Signaling Technology), and anti-GAPDH (1:3000, AbFrontier, Seoul, Republic of Korea). Membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (Bio-Rad Laboratories). The bands were detected using an enhanced chemiluminescence reagent (Bio-Rad Laboratories). The experiment was conducted at least in triplicate.
TEM analysis
Samples for electron microscopy were prepared based on the NCMIR protocol [25]. Briefly, cultured cells for individual groups were fixed with 2% PFA and 2.5% glutaraldehyde in 0.15 M cacodylate buffer (pH 7.4) and placed in cold cacodylate buffer containing 2% OsO4/1.5% potassium ferrocyanide for 1 h. Coverslips were incubated in 1% thiocarbohydrazide (Ted Pella) in ddH2O for 20 min, and then reacted in 2% aqueous OsO4 for 30 min. Thereafter, samples were en bloc stained with 1% uranyl acetate overnight and Walton’s lead aspartate solution for 30 min to enhance the membrane contrast. Cells were dehydrated through an ethanol series, and then embedded in Epon 812 resin (EMS). The coverslip was removed from the resin by dipping the block into liquid nitrogen. Ultrathin (70-nm thickness) sections were collected on 200-mesh grids. For each group, cells were imaged in a Tecnai F20 TEM (FEI) (×3600 magnification, 120 kV). To estimate the mitochondrial volume fraction, the point counting method was used with a point grid (+) superimposed on the cell, as described previously [24]. The mitochondrial area fraction was calculated based on the ratio of the number of coincident grid points to the total number of points in the reference area. In addition, the number of mitochondria per unit area was also measured. All morphological analyses were performed by researchers who were blind to the experimental conditions.
Enzyme-linked immunosorbent assay (ELISA)
The double antibody sandwiched method of VEGF and PEDF (Abcam) was adopted. Cell culture supernatant from each group was used according to the instruction procedures of each kit. Rat IFN-γ, IL-6, and IL-10 (Abcam) ELISA was also conducted from rat individual serum (n = 6). The OD at 450 nm was measured by a microplate reader. All reactions were performed in triplicate.
Measurement of cellular and mitochondrial ROS levels
To analyze the intracellular ROS level of ARPE-19 cells exposed to H2O2, cells were stained with 20 μM 2′,7′-dichlorofluorescein diacetate (DCF-DA) (Abcam) for 30 min at 37 °C. The fluorescence intensity of DCF-DA was measured at an excitation wavelength of 485 nm and an emission wavelength of 535 nm with a microplate reader (Tecan, Männedorf, Switzerland), and cells were observed by confocal microscopy (LSM 700). To confirm the mitochondrial ROS level, H2O2-injured rat retina and ARPE-19 cells were stained with 5 μM MitoSOX (Invitrogen) for 10 min at 37 °C and washed three times with Hank’s BSS. The fluorescence intensity of MitoSOX was measured at an excitation wavelength of 510 nm and an emission wavelength of 530 nm with a microplate reader (Tecan) and observed by confocal microscopy. The experiment was performed at least in triplicate.
Measurement of mitochondrial function
For mitochondrial respiration function, real-time measurements of the oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) of live PD-MSCsPEDF were analyzed using an XF24 Extracellular Flux Analyzer (Seahorse Bioscience, Santa Clara, CA, USA). Naive PD-MSCs and PD-MSCsPEDF (7000 cells/well) were seeded on an XF24 microplate (Seahorse Bioscience) and equilibrated in XF buffer (Seahorse Bioscience) in a non-CO2 incubator for 60 min for simultaneous analysis of the OCR and ECAR by repeating cycles of mixing (3 min), incubation (2 min), and measurement (3 min). For basal respiration measurements, naive PD-MSCs and PD-MSCsPEDF were sequentially treated with 0.5 µM oligomycin, 0.5 µM FCCP, and a 1 µM rotenone and antimycin A mixture, and the changes in respiration were recorded according to the manufacturer’s recommendation. All reactions were performed in triplicate. To measure the mitochondrial membrane potential in ARPE-19 cells, tetraethylbenzimidazolylcarbocyanine iodide (JC-1) cationic dye, which accumulates in energized mitochondria, was detected. The monomer form (Ex/Em: 514/529 nm) and J-aggregate form (Ex/Em: 585/590 nm) were represented by a fluorescence microplate reader (Tecan). From the cell lysate (20 μg) from ARPE-19 cells, ATP production was confirmed by an ATP (colorimetric) assay kit (Abcam). According to the manufacturer’s instructions, ATP concentrations were measured by a microplate reader (BioTek, Winooski, VT, USA) at 570 nm. Each experiment was conducted at least in triplicate.
mtDNA copy number assay
Genomic DNA (gDNA) was extracted from naive PD-MSCs and PD-MSCsPEDF to analyze the mtDNA copy number. qRT-PCR amplification was conducted with specific primers (Table 2) containing 250 ng gDNA, primers of nuclear DNA with FAM- and mtDNA with JOE-labeled quencher dye, 1X TaqMan Universal Master Mix (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s instructions. Data were analyzed in triplicate.
Lactate dehydrogenase (LDH) assay
To confirm the cytotoxicity of ARPE-19 cells, the LDH levels from the cell culture supernatant were measured using an LDH (colorimetric) assay kit (Abcam) according to the manufacturer’s recommendation. The absorbance at 450 nm was assessed with a microplate reader (BioTek). All reactions were performed in triplicate.
Cell viability assay
To analyze the optimization of H2O2 treatment in ARPE-19 cells (5 × 103 cells/cm2), cells were incubated in serum-free DMEM containing 0.5 mg/mL 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT; Sigma-Aldrich) at 37 °C for 4 h depending on the H2O2 concentration (100, 200, 300, 400, 500, 700, 900, and 1000 µM). The supernatant was removed, and the crystal violet was dissolved in DMSO. To confirm the proliferative ability of ARPE-19 cells, Cell Counting Kit-8 (CCK-8; Dojindo Molecular Technologies, Inc., Rockville, MD, US) solution was incubated with ARPE-19 cells at 37 °C for 2 h. The absorbance at 490 nm for MTT and 450 nm for CCK-8 was measured with a microplate reader (Tecan). All reactions were performed in triplicate.
Statistical analysis
The experimental statistics are expressed as the means ± standard deviations. Statistical analysis was performed using Student’s t test and one-way ANOVA followed by Dunnett’s post-hoc test, and the difference was considered statistically significant when the p value was less than 0.05. Each experiment was performed either in duplicate or triplicate.
Results
Characteristics of PEDF-overexpressing PD-MSCs (PD-MSCsPEDF, PEDF+)
PD-MSCsPEDF were successfully generated using a nonviral electroporation AMAXA system. A plasmid pcDNA3.1(+) containing the PEDF gene under the CMV promoter control as well as the GFP vector and antibiotic hygromycin was constructed (Fig. 1a) and transfected into naive PD-MSCs. To determine whether modified PD-MSCs are capable of secreting PEDF, ELISA was performed. Overexpression of PEDF in PD-MSCs showed an ~30-fold increase in PEDF secretion compared to overexpression in naive PD-MSCs and WI-38 (human normal lung fibroblast cell line) (Fig. 1b). To confirm the maintenance of cell characteristics after transfection, stemness markers (e.g., POU class homeobox 1; POU5F1, NANOG, SOX2), TERT, and immunomodulation HLA-G were analyzed in PD-MSCsPEDF at passages 1 and 6 by RT-PCR. As expected, the expression levels of POU5F1, NANOG, and SOX2 were higher in PD-MSCsPEDF than in naive PD-MSCs at both passages, but there was no difference in the expression levels of TERT and HLA-G between the two groups (Fig. 1c). Compared with the proliferative capacity of naive PD-MSCs, the proliferative capacity of PD-MSCsPEDF was increased by a shortened doubling time in cell cultures ranging from 24 to 72 h at passage 1 (Fig. 1d). To identify the immunophenotypes, the cell surface markers of PD-MSCsPEDF were analyzed by flow cytometry. PD-MSCsPEDF were positive for the expression of the MSC markers CD13, CD90, CD105, and MHC class I (HLA-ABC and HLA-G) but negative for the hematopoietic lineage markers CD34 and MHC class II (HLA-DR) (Fig. 1e), which was consistent with our previous report [13].

a Plasmid vector map of PEDF and control empty vector containing GFP. b Secreted PEDF levels in the cell culture supernatant of naive PD-MSCs and PD-MSCsPEDF using an ELISA for human PEDF. The human lung fibroblast WI-38 cell line was used as a negative control. *p < 0.05 vs. WI-38, #p < 0.05 vs. naive. c mRNA expression of stemness markers in naive PD-MSCs and PD-MSCsPEDF depending on passages. d Doubling time in naive PD-MSCs and PD-MSCsPEDF (passage = 1) at 12, 24, 48, and 72 h. *p < 0.05 vs. naive. e MSC surface markers (e.g., hematopoietic, nonhematopoietic and HLA family) in naive PD-MSCs and PD-MSCsPEDF using FACS analysis. f Karyotyping in naive PD-MSCs and PD-MSCsPEDF. g Representative image of von Kossa staining of PD-MSCsPEDF after osteogenic differentiation. Scale bar = 50 μm. h mRNA expression of osteogenic markers (e.g., BGLAP, COL1A1, and RUNX2) in undifferentiated (undiff.) and differentiated (diff.) PD-MSCsPEDF by qRT-PCR. *p < 0.05 vs. undifferentiated. i Oil red O staining in PD-MSCsPEDF after adipogenic differentiation. Scale bar = 50 μm. j mRNA expression of adipogenic markers (e.g., CFD, ADIPOQ, and PPARG) in undiff. and diff. PD-MSCsPEDF by qRT-PCR. *p < 0.05 vs. undifferentiated. Each experiment was conducted at least in triplicate. Data from each group are shown as the mean ± SD and were determined by Student’s t test.
Karyotype analysis demonstrated that PD-MSCsPEDF were diploid and genetically stable, as shown in the naive cell karyotype (Fig. 1f). To examine the ability of PD-MSCsPEDF to differentiate into osteogenic and adipogenic lineages, PD-MSCsPEDF were cultured in the appropriate induction media for 21 days. As shown in Fig. 1g, i, calcium depositions stained by von Kossa and lipid droplets stained by oil red O were observed in PD-MSCsPEDF after differentiation occurred. Osteogenic-specific markers (e.g., bone gamma-carboxyglutamate protein (BGLAP), collagen type 1 alpha 1 (COL1A1), and runt-related transcription factor 2 (RUNX2)) (Fig. 1h) and adipogenic-specific markers (e.g., complement factor D (CFD), adiponectin (ADIPOQ), and peroxisome proliferator-activated receptor gamma (PPARG)) (Fig. 1j) were expressed at high levels in differentiated PD-MSCsPEDF by qRT-PCR. These results suggest that PD-MSCsPEDF were successfully generated by a nonviral system and maintained characteristics similar to those of naive PD-MSCs.
PEDF overexpression enhances mitochondrial biogenesis in PD-MSCs
PEDF is capable of various biological activities, including protective functions against OS and proliferation of RPE cells and MSCs [26]. As shown in Fig. 1d, PEDF improved the proliferative capacity of PD-MSCs, which correlated with mitochondrial respiration. To investigate whether PD-MSCsPEDF affect mitochondrial function in PD-MSCs, we examined the mRNA expression levels of regulators and enzymes involved in mitochondrial metabolism. The expression levels of master regulators of mitochondrial biogenesis, NRF1, PPARGC1A, and TFAM, in PD-MSCsPEDF were significantly increased, and those of the antioxidant enzymes superoxide dismutases (SOD1 and SOD2) and glutathione peroxidase 1 (GPX1) in PD-MSCsPEDF were also increased compared to the expression levels in naive PD-MSCs, even though the heme oxygenase (HMOX1 and HMOX2) expression levels were unchanged (Fig. 2a).

a qRT-PCR analysis of gene expression of antioxidant enzymes (e.g., HMOX1, HMOX2, SOD1, SOD2, and GPX1) and regulators of mitochondrial biogenesis (e.g., NRF1, PPARGC1A, and TFAM) in naive PD-MSCs and PD-MSCsPEDF. *p < 0.05 vs. naive. b Western blot analysis of antioxidant enzymes (HMOX1, SOD1, catalase) and mitochondrial dynamics markers (DRP1 and OPA1) in cell lysates isolated from mitochondrial and cytosolic fractions. Internal controls were used by GAPDH for the cytosolic fraction and COX IV for the mitochondrial fraction. c, d OCR and ECAR of naive PD-MSCs and PD-MSCsPEDF after sequential treatment with 1 μM oligomycin, 0.5 μM carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) and 0.5 μM rotenone/antimycin A by a Seahorse XF24 analyzer. e, f Quantification of ATP production and maximal respiration of naive PD-MSCs and PD-MSCsPEDF. *p < 0.05 vs. naive. g mtDNA copy number assay in naive PD-MSCs and PD-MSCsPEDF by TaqMan qRT-PCR. The experiments were analyzed at least in triplicate. Data from each group are shown as the mean ± SD and were determined by Student’s t test.
In addition, mitochondrial and cytoplasmic proteins were independently isolated, and western blot analysis determined their protein expression levels. The antioxidant enzyme levels of HMOX1 and SOD1 increased in both the mitochondrial and cytosolic fractions and the level of catalase increased in the cytosolic fraction of PD-MSCsPEDF compared to those of naive cells. Compared to its naive counterpart, PD-MSCsPEDF had an accumulation of dynamin-1-like protein (DRP1) in the mitochondria, while OPA1 was decreased, which might cause mitochondrial fission (Fig. 2b, Supplementary Fig. 1). To further evaluate the mitochondrial bioenergetics of PD-MSCsPEDF, we tested the OCR and ECAR values in living cells using the Seahorse XF Mito Stress assay. OCR measurements after sequential injections of mitochondrial stressors (e.g., oligomycin, carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP) and rotenone/antimycin) were used to calculate basal respiration, ATP production and maximal respiration. Cellular mitochondrial respiration of PD-MSCsPEDF had more activity than that of naive PD-MSCs (Fig. 2c, d). Although oligomycin treatment induced increases in ATP production that were only slightly higher in PD-MSCsPEDF than in its naive counterpart, the maximal respiration in PD-MSCsPEDF was significantly increased compared with that in its naive counterpart (Fig. 2e, f). In addition, the mtDNA copy number was remarkably higher in PD-MSCsPEDF than in the naive counterpart (Fig. 2g). These data suggest that PEDF improves mitochondrial metabolism and cellular antioxidant activity in PD-MSCsPEDF.
PD-MSCsPEDF promote antioxidant activities and RPE-specific gene expression in oxidative stress-induced RPE cells by H2O2
OS is the most common cause of RPE injury. To evaluate the effects of PD-MSCsPEDF on OS-induced RPE cells, we used an H2O2-treated human RPE cell line (ARPE-19) as an OS-induced model in vitro. H2O2 treatment inhibited the cell viability of RPE cells in a concentration- and time-dependent manner according to an MTT assay (Supplementary Fig. 2). The optimal conditions for the oxidative damage model were determined at 200 μM H2O2 for 2 h. Under this condition, the RPE cell density decreased by 80%. Next, we conducted a coculture system of H2O2-treated RPE cells with naive PD-MSCs or PD-MSCsPEDF (Fig. 3a). We examined the expression of RPE-specific protein markers in H2O2-treated RPE cells cocultured with PD-MSCsPEDF. These markers include visual cycle proteins, retinal pigment epithelium-specific 65 kDa protein (RPE65), retinaldehyde binding protein 1 (RLBP1), retinal G protein coupled receptor (RGR), retinal pigment epithelium-derived rhodopsin homolog (RRH), and bestrophin 1 (BEST1). The decreased mRNA expression levels of RPE-specific genes in H2O2-treated RPE cells was significantly elevated under coculture conditions with PD-MSCsPEDF except for the integral membrane protein BEST1.

a Schematic of naive PD-MSCs and PD-MSCPEDF coculture with RPE cells (ARPE-19 cell line) exposed to H2O2 (200 μM) for 2 h. b RT-PCR analysis of RPE-specific markers (e.g., RPE65, BEST1, RLBP1, RGR, and RRH) in H2O2-treated RPE cells cocultured with naive PD-MSCs and PD-MSCsPEDF. β-actin was used as an internal control. c mRNA expression of ROS markers in H2O2-treated RPE cells cocultured with naive PD-MSCs and PD-MSCsPEDF by qRT-PCR. d, e PEDF and VEGF levels in cell culture supernatants of H2O2-treated RPE cells cocultured with naive PD-MSCs and PD-MSCsPEDF. #p < 0.05 vs. H2O2 treatment, *p < 0.05 vs. naive PD-MSCs or PD-MSCPEDF coculture groups, **p < 0.05 vs. naive. The experiments were conducted at least in triplicate. Data from each group are indicated as the mean ± SD, determined by Student’s t test.
In particular, RPE65 and RGR, known as the most important factors in retinal metabolism, in PD-MSCsPEDF were dramatically recovered and elevated compared with those in the untreated control group and H2O2-treated RPE cells (Fig. 3b). Antioxidant enzymes are known to protect against oxidative damage by counteracting high levels of ROS in cells [27]. To determine antioxidant activities in OS-induced RPE cells, mRNA expression levels of selected antioxidant enzymes, including HMOXs, SODs, and GPX1, were examined by qRT-PCR. We observed that the downregulated expression of these genes in RPE cells by H2O2 treatment was recovered and elevated under coculture conditions with naive PD-MSCs and PD-MSCsPEDF. In particular, the expression levels of HMOX2 and SOD2 in the PD-MSCPEDF coculture were higher than those in the naive coculture condition (Fig. 3c). Some research groups have reported that OS in RPE cells could induce the upregulation of VEGF and downregulate PEDF expression [28].
As shown in Fig. 3d, e, H2O2-treated RPE cells showed decreased PEDF and increased VEGF expression, which is consistent with the aforementioned study of oxidative status. In contrast, compared to naive cocultured cells, H2O2-treated RPE cells cocultured with PD-MSCsPEDF resulted in dramatic changes in both PEDF and VEGF expression, indicating upregulation of PEDF and downregulation of VEGF. These data suggest that PD-MSCPEDF coculture protects RPE cells from H2O2-induced oxidative damage through upregulated antioxidant expression, promotes RPE cell function through upregulated expression of RPE-specific markers, and inhibits the elevated VEGF expression level induced by H2O2 in RPE cells.
PD-MSCsPEDF decrease the cellular ROS levels in oxidative stress-induced RPE cells by H2O2
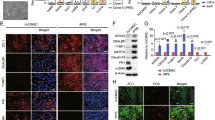
During OS-induced states, ROS production can increase dramatically. To further investigate the effect of PD-MSCsPEDF on the activities of cellular ROS and major antioxidant levels in H2O2-treated RPE cells, we analyzed the protein expression levels of intracellular antioxidants, including SOD1, HMOX1, and catalase, and the cellular ROS levels using western blotting and DCF-DA assays, respectively. As shown in Fig. 4a–d, the protein expression of SOD1, cytosolic Cu/Zn SOD, in H2O2-treated RPE cells was increased compared with the protein expression in cells cocultured with PD-MSCsPEDF or naive PD-MSCs, whereas decreased HMOX1 expression in H2O2-treated RPE cells was dramatically elevated in both coculture conditions with naive PD-MSCs and PD-MSCsPEDF. Catalase is a crucial antioxidant enzyme that catalyzes the conversion of hydrogen peroxide to water and molecular oxygen and is mostly located in peroxisomes.

Western blotting (a) and quantification analysis of the cellular antioxidant enzymes SOD1, HMOX1, and catalase (b–d) in H2O2-treated RPE cells cocultured with naive PD-MSCs and PD-MSCsPEDF. *p < 0.05 vs. naive PD-MSC or PD-MSCPEDF coculture groups, **p < 0.05 vs. naive. e, f Representative images and quantification by fluorescence microplate reader of cytosolic ROS levels in H2O2-treated RPE cells cocultured with naive PD-MSCs and PD-MSCsPEDF using 2′,7′-dichlorofluorescein diacetate (DCF-DA) assay. Scale bar = 100 μm. *p < 0.05 vs. naive PD-MSC or PD-MSCPEDF coculture groups. The experiments were conducted at least in triplicate. Data from each group are indicated as the mean ± SD and were determined by Student’s t test.
Under H2O2-induced OS in RPE cells, the expression level of catalase in coculture conditions with PD-MSCsPEDF was markedly increased compared with that in conditions with naive cells (Fig. 4d). In addition, the increased cytosolic ROS level induced by H2O2 treatment in RPE cells in coculture conditions with PD-MSCsPEDF was dramatically decreased compared to that in untreated control and cocultured cells with naive PD-MSCs (Fig. 4e, f). These data indicate that the PD-MSCPEDF coculture ameliorates the cellular antioxidant activities against OS.
PD-MSCsPEDF decrease the mitochondrial ROS levels and promote mitochondrial biogenesis in oxidative stress-induced RPE cells by H2O2
The elevated OS in the H2O2-treated RPE model had detrimental effects on cellular antioxidant capacity and increased ROS production. Mitochondria, an important cellular target of OS, are a major source of ROS in RPE cells. Increased ROS production under OS triggers dynamic changes in mitochondria, resulting in mitochondrial dysfunction. To analyze how PD-MSCsPEDF affect the mitochondrial function of OS-induced RPE cells, the mitochondrial superoxide ROS levels under live conditions were detected in RPE cells using MitoSOX staining. As shown in Fig. 5a, the mitochondrial ROS activity in PD-MSCsPEDF was significantly increased compared to that in control cells. In contrast, coculture of naive PD-MSCs or PD-MSCsPEDF with H2O2-treated RPE cells decreased the mitochondrial ROS levels. Moreover, the ROS levels in mitochondria were more alleviated in the PD-MSCPEDF coculture than in the naive PD-MSC coculture (Fig. 5b).

a, b Representative images and quantitative analysis of mitochondrial ROS levels in H2O2-treated RPE cells cocultured with naive PD-MSCs and PD-MSCsPEDF using the MitoSOX assay. Scale bar = 100 μm. c qRT-PCR analysis of mitochondrial biogenesis markers in H2O2-treated RPE cells cocultured with naive PD-MSCs and PD-MSCsPEDF. d ATP production assay. Representative TEM images of mitochondrial morphology (e), mitochondrial ratio (f), and the number of mitochondria per unit of cytoplasmic area (g). h Mitochondrial membrane potential by JC-1 fluorescence assay in H2O2-treated RPE cells cocultured with naive PD-MSCs and PD-MSCsPEDF and (i) cristae density by TEM analysis. Scale bar = 2 μm. *p < 0.05 vs. naive PD-MSC or PD-MSCPEDF coculture groups, **p < 0.05 vs. naive. Independent experiments were analyzed at least in triplicate. Data from each group are expressed as the mean ± SD and were determined by Student’s t test and ANOVA.
We next analyzed the mRNA expression of the mitochondrial biogenesis-related genes NRF1, PPARGC1A, and TFAM. In H2O2-treated RPE cells, the expression of these genes was significantly downregulated (Fig. 5c). However, PD-MSCPEDF coculture promoted the gene expression of PPARGC1A and TFAM but not of NRF1. As expected, PD-MSCPEDF coculture led to a significant increase in the ATP content in H2O2-treated RPE cells (Fig. 5d). To analyze whether H2O2 treatment affects the morphology and distribution of mitochondria, we estimated the cytoplasmic volume fraction occupied by mitochondria using TEM. Compared to controls, H2O2-injured RPE cells decreased the volume fraction of mitochondria (Fig. 5e, f). Naive PD-MSC coculture showed an increasing trend in the mitochondrial volume fraction but was not significantly different from the H2O2 treatment. Importantly, PD-MSCsPEDF completely prevented H2O2-injured reduction in the mitochondrial volume fraction. The volume fraction values can be affected by alterations in the numbers and/or size of the mitochondria.
Therefore, we next performed an analysis on the number of mitochondria per unit of cytoplasmic area. Intriguingly, the mitochondrial densities per individual group were not significantly different, indicating that the changes in the mitochondrial volume fraction were due to alterations in the size of mitochondria (Fig. 5g). In addition, the mitochondrial membrane potential of PD-MSCsPEDF cocultured with H2O2-treated RPE cells was significantly improved (Fig. 5h). The cristae density in the inner membrane of mitochondria by PD-MSCPEDF coculture was recovered, whereas its density by H2O2 treatment was decreased (Fig. 5i). These data demonstrate that PD-MSCsPEDF strongly promote antioxidant activity and mitochondrial function in OS-induced RPE cells.
PD-MSCsPEDF promote antiapoptotic activities and cell survival in oxidative stress-induced RPE cells through the JNK/ERK signaling pathway
OS can trigger the activation of multiple signaling pathways that influence cell death and survival [29]. To confirm the effects of PD-MSCsPEDF on OS-induced RPE apoptosis, we first determined the protein expression of caspase-3 and Bax, which are associated with apoptosis, and the antiapoptotic factor Bcl2 in H2O2-treated RPE cells. The expression levels of cleaved caspase-3 and Bax were significantly increased, while the expression level of Bcl2 in H2O2-treated RPE cells was decreased compared with that in the untreated control. In contrast, cleaved caspase-3 and Bax were hardly detected, but Bcl2 in H2O2-treated RPE cells cocultured with PD-MSCsPEDF was markedly increased compared to that in naive PD-MSCs (Fig. 6a, b). As another assay for cell death, the LDH concentration was analyzed using a colorimetric kit assay. As shown in Fig. 6c, the elevated level of LDH in H2O2-treated RPE cells was dramatically decreased in coculture with naive PD-MSCs and PD-MSCsPEDF. These results provide evidence that PD-MSCPEDF coculture promotes antiapoptotic effects in OS-induced RPE cells. In addition, PEDF displays retinal survival activity by interacting with the PEDF receptor (PEDF-R) on retinal cell surfaces [30]. We thus analyzed the expression level of PEDF-R in RPE cells. PD-MSCPEDF coculture induced a high expression level of PEDF-R in H2O2-treated RPE cells (Fig. 6d).

a, b Western blotting and quantification analysis of proapoptotic (e.g., procaspase-3, cleaved caspase-3, and Bax) and antiapoptotic (e.g., Bcl2) markers in H2O2-treated RPE cells cocultured with naive PD-MSCs and PD-MSCsPEDF. c Cytotoxicity of H2O2-treated RPE cells cocultured with naive PD-MSCs and PD-MSCsPEDF by an LDH assay. d, e Western blotting and quantification analysis of PEDF-R, total and phosphorylated forms (e.g., AKT, JNK, and ERK), f Proliferation activity by a CCK-8 assay in H2O2-treated RPE cells cocultured with naive PD-MSCs and PD-MSCsPEDF. *p < 0.05 vs. naive PD-MSC or PD-MSCPEDF coculture groups, **p < 0.05 vs. naive. Independent experiments were analyzed at least in triplicate. Data from each group are expressed as the mean ± SD and were determined by Student’s t test.
To further identify the signaling mechanisms activated by PD-MSCsPEDF in H2O2-treated RPE cells, we analyzed the phosphorylated protein expression of AKT, JNK, and ERK by western blotting. PD-MSCPEDF coculture resulted in significant increases in the phosphorylation of AKT, JNK, and ERK without affecting the total protein expression levels (Fig. 6d, e). Cell proliferation was also investigated using CCK-8 assays (Fig. 6f). The results indicated that PD-MSCPEDF coculture significantly protected RPE cells from H2O2-induced injury. Collectively, these results suggest that PEDF-overexpressing PD-MSCs promote antiapoptotic activities and cell survival in H2O2-treated RPE cells through the AKT/JNK/ERK signaling pathway.

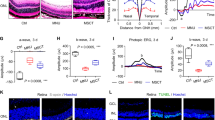
a Serum levels of inflammatory cytokines (e.g., INF-γ, IL-6, and IL-10) in H2O2-injured rats according to naïve PD-MSC or PD-MSCPEDF transplantation (n = 4–6). b qRT-PCR analysis of Vegf and Pedf expression in rat retinas. c Weight (g) of each eyeball from an individual rat (n = 6). d RT-PCR analysis of RPE-specific markers in rat retinas. e Histological images of ten retinal layers (inner limiting membrane (ILM), optic nerve fiber (ONF), ganglion cell layer (GCL), inner plexiform layer (IPL), inner nuclear layer (INL), outer plexiform layer (OPL), outer nuclear layer (ONL), outer limiting membrane (OLM), photoreceptor layer (PL), pigmented epithelial cells (PEC)) from each group by H&E. Scale bar = 50 μm. f Measurement of each total retina layer, INL, ONL, and IPL in rat retinas (n = 6). #p < 0.05 vs. Sham, *p < 0.05 vs. H2O2 injection groups, **p < 0.05 vs. naïve. Independent experiments were analyzed at least in triplicate. Data from each group are expressed as the mean ± SD.
Administration of PD-MSCsPEDF restores retinal functions by alleviating inflammation in an oxidative stress-induced animal model
To further analyze the therapeutic effects of PD-MSCsPEDF in an animal model, we induced retinal degeneration in rats using H2O2 (10 µg/μl) for 1 and 2 weeks as well as BSS-injected sham controls. One week after H2O2 injection, naive PD-MSCs and PD-MSCsPEDF were intravitreally transplanted into rats. To confirm the effects of PD-MSCsPEDF on H2O2-injured rats, we determined the serum levels of proinflammatory cytokines (e.g., IFN-γ and IL-6) and the anti-inflammatory marker IL-10. As shown in Fig. 7a, increased levels of proinflammatory cytokines (e.g., IFN-γ and IL-6) in H2O2-injured rats were dramatically decreased by PD-MSCPEDF transplantation, while the decreased anti-inflammatory marker IL-10 was elevated by PD-MSCPEDF transplantation. In particular, the effect of anti-inflammatory responses in the PD-MSCPEDF-transplanted group was significantly higher than that in the naive PD-MSC-transplanted group. H2O2-injured rat retina induced upregulated Vegf and downregulated Pedf expression, while PD-MSCPEDF transplantation recovered the Vegf and Pedf expression levels to levels similar to those of the sham control (Fig. 7b). In addition, endogenous Vegf levels by immunohistology in the naive PD-MSCs- or PD-MSCPEDF-transplanted group were reduced compared to those in the H2O2-injured rat group, even though there was no difference between the naive and genetically altered PD-MSC transplantations (Supplementary Fig. 3). Moreover, unlike naive PD-MSC transplantation, PD-MSCSPEDF transplantation restored the decreased weight of eyeballs in the H2O2-injured group to a similar level to that in the sham group (Fig. 7c), and all four RPE-specific markers (e.g., Rpe65, Best1, Rgr, and Rrh), except Rlbp1, in the H2O2-injured group were significantly upregulated (Fig. 7d).

a mRNA expression of antioxidant enzymes in H2O2-injured rat retinas according to naive PD-MSC or PD-MSCPEDF transplantation (n = 6). b MitoSOX-stained images in rat retinas. Scale bar = 100 μm. c mRNA expression of mitochondrial biogenesis markers in rat retinas. #p < 0.05 vs. Sham, ##p < 0.05 vs. H2O2 1 w, *p < 0.05 vs. H2O2 injection groups, **p < 0.05 vs. naive. Independent experiments were analyzed at least in triplicate. Data from each group are expressed as the mean ± SD.
We evaluated retinal structural changes induced by OS. The ten retinal layers were as follows: inner limiting membrane, optic nerve fiber, ganglion cell layer, inner plexiform layer (IPL), inner nuclear layer (INL), outer plexiform layer, outer nuclear layer (ONL), outer limiting membrane, photoreceptor layer, and pigmented epithelial cells (Fig. 7e). In particular, the ONL, which contains the cell bodies of the rod and cone photoreceptors, has important roles in the visual cycles in the retina. In addition, the INL and IPL regulate synaptic connections with neuron dendrites. To verify the in vivo retinal structural changes, images of the total retina, INL, ONL, and IPL were evaluated. Following intravitreal injection of H2O2, induced damage was assessed in each layer. Naive PD-MSC or PD-MSCPEDF transplantation rearranged the retinal layers. In particular, a PD-MSCPEDF injection increased the total retina, INL, and IPL compared with a naive injection (Fig. 7f). These data suggest that PD-MSCsPEDF promote mitochondrial biogenesis by reducing OS in H2O2-injured rats.
PD-MSCsPEDF have a prominent effect on mitochondrial biogenesis to protect against oxidative stress in H2O2-injured rats
To investigate the effect of PD-MSCPEDF transplantation on the OS levels in H2O2-injured rat retinas, we analyzed the expression levels of antioxidant genes (e.g., Hmox1, Hmox2, Sod1, Sod2, and Gpx1) using qRT-PCR. As shown in Fig. 8a, all expression levels of antioxidant genes were markedly increased in the naive PD-MSC- or PD-MSCPEDF-transplanted group. In particular, the expression of Hmox2, Sod2, and Gpx1 was significantly higher in the PD-MSCPEDF-transplanted group than in the naive PD-MSC-transplanted group, which was consistent with our in vitro data. Next, to clarify the mitochondrial functions of oxidatively damaged rat retinas after PD-MSCPEDF transplantation, we analyzed the mitochondrial ROS levels and mitochondrial biogenesis markers by MitoSOX staining and qRT-PCR, respectively. The mitochondrial ROS levels in the rat retina after intravitreal injection of H2O2 were remarkably upregulated compared to those in the sham control. In contrast, the naive PD-MSC- or PD-MSCPEDF-transplanted group showed improved mitochondrial ROS levels, similar to those of the sham control (Fig. 8b). Moreover, compared to their naive counterparts, PD-MSCsPEDF promoted the mRNA expression of mitochondrial biogenesis markers (e.g., Nrf1, Ppargc1a, and Tfam) in H2O2-injured rat retinas (Fig. 8c). These data demonstrate that intravitreal administration of PD-MSCsPEDF protects retinal degeneration against OS through increased antioxidant capacity and mitochondrial biogenesis.

PD-MSCsPEDF enhance the mitochondrial dynamics for antioxidant capacity and promote cell survival and proliferation by activating the AKT, ERK, and JNK signaling pathways for RPE regeneration.
Discussion
MSCs have been widely employed in various degenerative diseases. According to the results of preclinical studies and clinical trials, MSCs can effectively restore the visual system through several therapeutic mechanisms. PD-MSCs have more attractive properties with specific immunomodulatory functions and paracrine effects [1]. Several studies have shown that MSCs can significantly restore the visual system through MSC-mediated paracrine action by secreting various kinds of growth factors and cytokines [22]. Representatively, VEGF secretion from MSCs influences the differentiation from endothelial progenitor cells to endothelial cells [25]. As stem cell therapy, the paracrine secretion of VEGF from bone marrow-derived MSCs (BM-MSCs) mediates wound healing, promoting endothelial cell angiogenesis. Based on previous verification, gene modification of MSCs with specific factors can amplify their therapeutic ability for regenerative medicine.
The methods of transfection can generally be divided into viral- and nonviral-based vehicles. Viral vectors have emerged as the more popular transfection system [31]. This method is suitable for stability and integrates transfected DNA into the cell genome of the host. Previous studies have indicated that compared to nontransfected BM-MSCs, engineered BM-MSCs using a lentiviral vector to overexpress VEGF have beneficial effects, such as blood flow restoration for the treatment of critical limb ischemia [32]. Moreover, adenovirus-mediated transfection of modified PEDF into PD-MSCs inhibits angiogenesis and microvessels [33]. Similarly, the effect of PEDF-transfected umbilical cord MSCs using a lentiviral system induces increased PEDF and decreased VEGF expression (optical density) in diabetic retinopathy [34]. However, these methods suffer from critical limitations, such as cytotoxicity and safety concerns [35]. For useful means in gene therapy, a nonviral-based gene delivery system, including electroporation, is adequate for medical products. With electroporation, there is a decreased risk of genome integration in the host and immunogenicity. This procedure is safer for patients than other procedures [36]. Overexpression of PEDF in pigment epithelial cells using an electroporation system was performed to functionally rehabilitate neurodegenerative diseases and inhibit endothelial cell sprouting [37, 38]. In our previous report, TERT-overexpressing PD-MSCs using an AMAXA 2D electroporation method were successfully generated and enhanced the self-renewal ability of MSCs [39]. Recently, the updated transfection system by an AMAXA 4D Nucleofector™ device provided increased transfection efficiency and cell viability [40]. In the present study, we successfully constructed PEDF-overexpressing PD-MSCs (PD-MSCsPEDF) using a nonviral gene delivery system known as AMAXA 4D Nucleofector™. Compared to naive PD-MSCs, gene-modified PD-MSCs significantly secreted PEDF and retained the phenotypic characteristics.
Generally, OS has been identified as a major cause of RPE injury, which eventually could contribute to the development of retinal diseases, such as AMD, diabetic retinopathy, and glaucoma [41]. To support retina survival and maintenance during persistent OS and ROS production, RPE cells secrete a variety of regulatory factors, such as PEDF, which is a multifunctional protein with antiangiogenic, neuroprotective, and antioxidant properties [42]. PEDF directly regulates OS levels and controls mitochondrial structure and function in aging RPE cells through the PI3K/AKT signaling pathway [43]. In addition, PEDF expression induced antioxidant effects in RPE cells against oxidative injury by upregulating uncoupling protein 2 [17]. Our results demonstrate that overexpression of PEDF in PD-MSCs increases the expression of multiple antioxidant enzymes, including HMOXs, SODs, GPX1, and catalase. Moreover, PEDF promotes mitochondrial biogenesis and function.
Based on these data, we determined the effects of PD-MSCsPEDF on OS-induced RPE cell function under coculture conditions. PD-MSCsPEDF induced increased secretion of PEDF, antioxidant genes and RPE functional protein in H2O2-treated RPE cells. In addition, in the H2O2-injured retinal degeneration rat model, antioxidative effects were confirmed based on PD-MSCPEDF transplantation as well as the anti-inflammatory response. A previous study [44] found that iPSC-MSC transplantation downregulated proinflammatory cytokines in a retinal degeneration mouse model. PD-MSCsPEDF also significantly reduced VEGF expression. PEDF counteracts the angiogenic potential of VEGF. A recent report has shown that in an inherited retinal degeneration rat model, PEDF expression was remarkably upregulated in retinas by human umbilical cord MSC transplantation [45].
Under OS, decreased PEDF expression contributes to RPE dysfunction and AMD pathogenesis [46]. Of note, PD-MSCsPEDF can effectively protect RPE cells from oxidative damage and induce cell survival. Further, our data demonstrate that PD-MSCsPEDF dramatically reduced cytosolic and mitochondrial ROS production and induced mitochondrial biogenesis-related gene expression, including PPARGC1A and TFAM, in H2O2-treated RPE cells. In the same manner, administration of PD-MSCsPEDF in the H2O2-injured retinal degeneration model remarkably improved mitochondrial biogenesis as well as reduced mitochondrial ROS levels. PPARGC1A is a powerful activator of mitochondrial biogenesis and plays an important role in defending against the ROS generated by mitochondrial activity [47]. As described above, PEDF overexpression enhanced mitochondrial biogenesis-related gene expression, especially PPARGC1A, in PD-MSCs, and this expression might be affected as a paracrine action for of RPE cell regeneration under OS.
OS can trigger the activation of multiple signaling pathways that influence cell death and survival. PD-MSCPEDF coculture promoted antiapoptotic effects through decreased cleaved caspase-3 and Bax and increased antiapoptotic protein Bcl2 in H2O2-treated RPE cells. A recent study reported that the antiapoptotic effects of PEDF-R and PEDF were critical for PEDF-mediated retina survival activities [30].
In this study, we found that PD-MSCPEDF coculture increased the expression of PEDF-R, which is probably consistent with previously reported requirements for cell survival activity. Furthermore, we investigated whether PEDF signals the AKT, ERK, and JNK signaling pathways, which were proposed to play survival roles in retinal function [48]. Under OS, AKT, ERK, and JNK pathway activation declined in RPE cells. However, PD-MSCPEDF coculture with RPE cells promoted cell survival by activating these signaling pathways (Fig. 9).
In conclusion, the present study demonstrates that PEDF overexpression in PD-MSCs improves the mitochondrial activities of PD-MSCs, thereby promoting the reversal of OS-induced declines in RPE function by enhancing the antioxidative activity and stimulating the survival signaling pathway. Genetic modification of PEDF in PD-MSCs may provide a new MSC-based cell therapy for retinal degeneration diseases.
Data availability
All data analyzed for this study are included in this article.
References
Ding SLS, Kumar S, Mok PL. Cellular reparative mechanisms of mesenchymal stem cells for retinal diseases. Int J Mol Sci. 2017;18:1406.
Chichagova V, Hallam D, Collin J, Zerti D, Dorgau B, Felemban M, et al. Cellular regeneration strategies for macular degeneration: past, present and future. Eye. 2018;32:946–71.
Shaw PX, Stiles T, Douglas C, Ho D, Fan W, Du H, et al. Oxidative stress, innate immunity, and age-related macular degeneration. AIMS Mol Sci. 2016;3:196–221.
Athanasiou D, Aguila M, Bevilacqua D, Novoselov SS, Parfitt DA, Cheetham ME. The cell stress machinery and retinal degeneration. FEBS Lett. 2013;587:2008–17.
Fuhrmann S, Zou C, Levine EM. Retinal pigment epithelium development, plasticity, and tissue homeostasis. Exp Eye Res. 2014;123:141–50.
Bellezza I. Oxidative stress in age-related macular degeneration: Nrf2 as therapeutic target. Front Pharmacol. 2018;9:1280.
He Y, Zhang Y, Liu X, Ghazaryan E, Li Y, Xie J, et al. Recent advances of stem cell therapy for retinitis pigmentosa. Int J Mol Sci. 2014;15:14456–74.
Schwartz SD, Regillo CD, Lam BL, Eliott D, Rosenfeld PJ, Gregori NZ, et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: follow-up of two open-label phase 1/2 studies. Lancet. 2015;385:509–16.
Inoue Y, Iriyama A, Ueno S, Takahashi H, Kondo M, Tamaki Y, et al. Subretinal transplantation of bone marrow mesenchymal stem cells delays retinal degeneration in the RCS rat model of retinal degeneration. Exp Eye Res. 2007;85:234–41.
Garg A, Yang J, Lee W, Tsang SH. Stem cell therapies in retinal disorders. Cells. 2017;6:4.
Tang Z, Zhang Y, Wang Y, Zhang D, Shen B, Luo M, et al. Progress of stem/progenitor cell-based therapy for retinal degeneration. J Transl Med. 2017;15:99.
Fukuchi Y, Nakajima H, Sugiyama D, Hirose I, Kitamura T, Tsuji K. Human placenta-derived cells have mesenchymal stem/progenitor cell potential. Stem Cells. 2004;22:649–58.
Lee JM, Jung J, Lee HJ, Jeong SJ, Cho KJ, Hwang SG, et al. Comparison of immunomodulatory effects of placenta mesenchymal stem cells with bone marrow and adipose mesenchymal stem cells. Int Immunopharmacol. 2012;13:219–24.
Machalinska A, Kawa M, Pius-Sadowska E, Stepniewski J, Nowak W, Roginska D, et al. Long-term neuroprotective effects of NT-4-engineered mesenchymal stem cells injected intravitreally in a mouse model of acute retinal injury. Investig Ophthalmol Vis Sci. 2013;54:8292–305.
Tombran-Tink J, Shivaram SM, Chader GJ, Johnson LV, Bok D. Expression, secretion, and age-related downregulation of pigment epithelium-derived factor, a serpin with neurotrophic activity. J Neurosci. 1995;15:4992–5003.
Pang IH, Zeng H, Fleenor DL, Clark AF. Pigment epithelium-derived factor protects retinal ganglion cells. BMC Neurosci. 2007;8:11.
Wang X, Liu X, Ren Y, Liu Y, Han S, Zhao J, et al. PEDF protects human retinal pigment epithelial cells against oxidative stress via upregulation of UCP2 expression. Mol Med Rep. 2019;19:59–74.
Cayouette M, Smith SB, Becerra SP, Gravel C. Pigment epithelium-derived factor delays the death of photoreceptors in mouse models of inherited retinal degenerations. Neurobiol Dis. 1999;6:523–32.
Hsu YC, Wu YT, Yu TH, Wei YH. Mitochondria in mesenchymal stem cell biology and cell therapy: from cellular differentiation to mitochondrial transfer. Semin Cell Dev Biol. 2016;52:119–31.
Paliwal S, Chaudhuri R, Agrawal A, Mohanty S. Regenerative abilities of mesenchymal stem cells through mitochondrial transfer. J Biomed Sci. 2018;25:31.
Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005;1:361–70.
Kim SH, Jung J, Cho KJ, Choi JH, Lee HS, Kim GJ, et al. Immunomodulatory effects of placenta-derived mesenchymal stem cells on T cells by regulation of FoxP3 expression. Int J Stem Cells. 2018;11:196–204.
Huang B, Liang JJ, Zhuang X, Chen SW, Ng TK, Chen H. Intravitreal injection of hydrogen peroxide induces acute retinal degeneration, apoptosis, and oxidative stress in mice. Oxid Med Cell Longev. 2018;2018:5489476.
Kim KY, Rios LC, Le H, Perez AJ, Phan S, Bushong EA, et al. Synaptic specializations of melanopsin-retinal ganglion cells in multiple brain regions revealed by genetic label for light and electron microscopy. Cell Rep. 2019;29:628–44.
Ge Q, Zhang H, Hou J, Wan L, Cheng W, Wang X, et al. VEGF secreted by mesenchymal stem cells mediates the differentiation of endothelial progenitor cells into endothelial cells via paracrine mechanisms. Mol Med Rep. 2018;17:1667–75.
Ding DC, Wen YT, Tsai RK. Pigment epithelium-derived factor from ARPE19 promotes proliferation and inhibits apoptosis of human umbilical mesenchymal stem cells in serum-free medium. Exp Mol Med. 2017;49:e411.
He L, He T, Farrar S, Ji L, Liu T, Ma X. Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cell Physiol Biochem. 2017;44:532–53.
Ohno-Matsui K, Morita I, Tombran-Tink J, Mrazek D, Onodera M, Uetama T, et al. Novel mechanism for age-related macular degeneration: an equilibrium shift between the angiogenesis factors VEGF and PEDF. J Cell Physiol. 2001;189:323–33.
Zhou Y, Wang Q, Evers BM, Chung DH. Signal transduction pathways involved in oxidative stress-induced intestinal epithelial cell apoptosis. Pediatr Res. 2005;58:1192–7.
Subramanian P, Locatelli-Hoops S, Kenealey J, DesJardin J, Notari L, Becerra SP. Pigment epithelium-derived factor (PEDF) prevents retinal cell death via PEDF Receptor (PEDF-R): identification of a functional ligand binding site. J Biol Chem. 2013;288:23928–42.
Schnodt M, Buning H. Improving the quality of adeno-associated viral vector preparations: the challenge of product-related impurities. Hum Gene Ther Methods. 2017;28:101–8.
Beegle JR, Magner NL, Kalomoiris S, Harding A, Zhou P, Nacey C, et al. Preclinical evaluation of mesenchymal stem cells overexpressing VEGF to treat critical limb ischemia. Mol Ther Methods Clin Dev. 2016;3:16053.
Chen Q, Cheng P, Song N, Yin T, He H, Yang L, et al. Antitumor activity of placenta-derived mesenchymal stem cells producing pigment epithelium-derived factor in a mouse melanoma model. Oncol Lett. 2012;4:413–8.
Zhang W, Duan HT, Chen S, Wang YX, Kong JH, Dong M. et al. The protective effect of pigment epithelial-derived factor modified human umbilical cord mesenchymal stem cells on rats with diabetic retinopathy. Zhonghua Yan Ke Za Zhi. 2017;53:540–7.
Slanina H, Schmutzler M, Christodoulides M, Kim KS, Schubert-Unkmeir A. Effective plasmid DNA and small interfering RNA delivery to diseased human brain microvascular endothelial cells. J Mol Microbiol Biotechnol. 2012;22:245–57.
Ding XF, Fan M. Nonviral gene therapy of the nervous system: electroporation. Methods Mol Biol. 2016;1382:297–305.
Johnen S, Kazanskaya O, Armogan N, Stickelmann C, Stocker M, Walter P, et al. Endogenic regulation of proliferation and zinc transporters by pigment epithelial cells nonvirally transfected with PEDF. Investig Ophthalmol Vis Sci. 2011;52:5400–7.
Thumann G, Stocker M, Maltusch C, Salz AK, Barth S, Walter P, et al. High efficiency non-viral transfection of retinal and iris pigment epithelial cells with pigment epithelium-derived factor. Gene Ther. 2010;17:181–9.
Lee HJ, Choi JH, Jung J, Kim JK, Lee SS, Kim GJ. Changes in PTTG1 by human TERT gene expression modulate the self-renewal of placenta-derived mesenchymal stem cells. Cell Tissue Res. 2014;357:145–57.
Liu M, Asada M, Cao S, Adjou Moumouni PF, Vudriko P, Efstratiou A, et al. Transient transfection of intraerythrocytic Babesia gibsoni using elongation factor-1 alpha promoter. Mol Biochem Parasitol. 2017;216:56–59.
Ferrington DA, Ebeling MC, Kapphahn RJ, Terluk MR, Fisher CR, Polanco JR, et al. Altered bioenergetics and enhanced resistance to oxidative stress in human retinal pigment epithelial cells from donors with age-related macular degeneration. Redox Biol. 2017;13:255–65.
Ho TC, Yang YC, Cheng HC, Wu AC, Chen SL, Tsao YP. Pigment epithelium-derived factor protects retinal pigment epithelium from oxidant-mediated barrier dysfunction. Biochem Biophys Res Commun. 2006;342:372–8.
He Y, Leung KW, Ren Y, Pei J, Ge J, Tombran-Tink J. PEDF improves mitochondrial function in RPE cells during oxidative stress. Investig Ophthalmol Vis Sci. 2014;55:6742–55.
Jiang D, Xiong G, Feng H, Zhang Z, Chen P, Yan B, et al. Donation of mitochondria by iPSC-derived mesenchymal stem cells protects retinal ganglion cells against mitochondrial complex I defect-induced degeneration. Theranostics. 2019;9:2395–410.
Wang L, Li P, Tian Y, Li Z, Lian C, Ou Q, et al. Human umbilical cord mesenchymal stem cells: subpopulations and their difference in cell biology and effects on retinal degeneration in RCS rats. Curr Mol Med. 2017;17:421–35.
Bhutto IA, McLeod DS, Hasegawa T, Kim SY, Merges C, Tong P, et al. Pigment epithelium-derived factor (PEDF) and vascular endothelial growth factor (VEGF) in aged human choroid and eyes with age-related macular degeneration. Exp Eye Res. 2006;82:99–110.
Iacovelli J, Rowe GC, Khadka A, Diaz-Aguilar D, Spencer C, Arany Z, et al. PGC-1alpha induces human RPE oxidative metabolism and antioxidant capacity. Investig Ophthalmol Vis Sci. 2016;57:1038–51.
Wang X, Zhao T, Huang W, Wang T, Qian J, Xu M, et al. Hsp20-engineered mesenchymal stem cells are resistant to oxidative stress via enhanced activation of Akt and increased secretion of growth factors. Stem Cells. 2009;27:3021–31.
Acknowledgements
This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute, funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI16C1559) and by Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education (grant number: 2019R1l1A1A01057255). We thank Ho Jeong Kim (CHA University) for helping us with the graphical abstract.
Author information
Authors and Affiliations
Contributions
JYK: analysis of data, data interpretation, manuscript drafting; SP: data interpretation and manuscript drafting; SHP: animal experiment and data interpretation; DL, GHK, and JEN: data interpretation and analysis of data; KJL: critical discussion; GJK: conception and design, manuscript drafting, financial support, and final approval of manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Kim, J.Y., Park, S., Park, S.H. et al. Overexpression of pigment epithelium-derived factor in placenta-derived mesenchymal stem cells promotes mitochondrial biogenesis in retinal cells. Lab Invest 101, 51–69 (2021). https://doi.org/10.1038/s41374-020-0470-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-020-0470-z
This article is cited by
-
Stem cells and diabetic retinopathy: From models to treatment
Molecular Biology Reports (2023)
-
A Review on Mesenchymal Stem Cells for Treatment of Retinal Diseases
Stem Cell Reviews and Reports (2021)
