
Abstract
Parkinson’s disease (PD) is the second most common neurodegenerative disorder and is characterized by severe neuronal loss. Necroptosis, or programmed cell necrosis, is mediated by the receptor interacting protein kinase-1 and -3/mixed lineage kinase domain-like protein (RIP1/RIP3/MLKL) pathway, and is involved in several neurodegenerative diseases. Here we aimed to explore the involvement of necroptosis in 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine hydrochloride (MPTP)-induced PD and determine the potential mechanisms. We found that the protein levels of RIP1, RIP3, and MLKL increased significantly in a MPTP-induced mouse PD model. High expression of RIP1/RIP3/MLKL was associated with severe loss of dopaminergic neurons. Pretreatment with necrostatin-1 or the knockout of the RIP3/MLKL gene to block necroptosis pathway dramatically ameliorated PD by increasing dopamine levels and rescuing the loss of dopaminergic neurons, independent of the apoptotic pathway. Moreover, upregulation of inflammatory cytokines in MPTP-treated mice was partially inhibited by deletion of RIP3 or MLKL gene, indicating that a positive feedback loop exists between these genes and inflammatory cytokines. Our data indicate that RIP1/RIP3/MLKL-mediated necroptosis is involved in the pathogenesis of MPTP-induced PD. Downregulating the expression of RIP1, RIP3, or MLKL can significantly attenuate MPTP-induced PD. Future therapy targeting necroptosis may be a promising new option.
Similar content being viewed by others

Introduction
Parkinson disease (PD) is the second most common neurodegenerative disorder, surpassed only by Alzheimer’s disease [1, 2]. Loss of dopaminergic neurons (DA neurons) in the substantia nigra pars compacta (SNpc) is the critical neuropathological change, which contributes to the impaired movement seen in PD patients [3, 4]. Until recently, the pathophysiological mechanism underlying the loss of DA neurons has not been fully elucidated and the clinical outcome for PD patients is still poor [4, 5]. Therefore, identifying the potential mechanism underlying the loss of DA neurons in SNpc is needed to improve the management of PD patients.
Necroptosis, a newly recognized type of programmed cell death, has been found to contribute to inflammation and several diseases, such as cancer, stroke, and renal disease [6, 7]. When triggered by various stimuli, necroptosis is initiated by the activation of receptor interacting protein kinase-1 (RIP1). Activated RIP1 interacts with RIP3 via their RIP homotypic interaction motifs, phosphorylates RIP3 and forms a RIP1/RIP3 complex, termed the necrosome [8, 9]. Then, mixed lineage kinase domain-like protein (MLKL) is recruited and phosphorylated by RIP3 in the necrosome [10]. The phosphorylated MLKL monomers aggregate to form oligomers and translocate to the plasma membrane to execute necroptosis [6, 10, 11].
Although multiple cell death pathways are involved in PD, including apoptosis and autophagy, the effect of necroptosis remains largely unknown [12, 13]. Recently, Dionisio et al. reported that loss of microglial parkin inhibited necroptosis of BV-2 cells and contributed to neuroinflammation, however, whether necroptosis contributed to loss of DA neurons remains undefined [14]. Iannielli et al. found that pharmacological inhibition of necroptosis protected from dopaminergic neuronal cell death in Parkinson’s disease models, but they mainly focused on an inherited form of PD caused by mutations in the OPA1 gene and did not explore the role of the key components of necroptosis, RIP1/RIP3/MLKL [15]. Therefore, in this study, we aimed to explore the activation of necroptosis in PD and identify the role of RIP1/RIP3/MLKL-mediated necroptosis in an MPTP-induced mouse PD model. We found that necroptosis was involved in the pathogenesis of MPTP-induced PD. The loss of DA neurons was significantly reversed by administration of necrostatin-1 (Nec-1) or gene deletion of RIP3 or MLKL. Moreover, we also illustrated that the necroptosis pathway promoted the expression of pro-inflammatory genes, which may initiate neuroinflammation and in turn aggravate neuron necroptosis in MPTP-induced PD.
Materials and methods
Animals and treatment
RIP3−/− and MLKL−/− C57BL/6 mice were gifts from Jiahuai Han, School of Life Sciences, Xiamen University, China. Mice were kept in a specific pathogen-free room with constant temperature and humidity under a 12-h light/dark cycle and raised with unrestricted access to water and food. All experiments were approved by the Animal Care and Use Committee of Fujian Medical University, in accordance with the Chinese guidelines for the Care and Use of Laboratory Animals.
Each mouse genotype (wild type, RIP3−/−, or MLKL−/−, male, 8–10 weeks old)) was randomly divided into control and experiment groups. Each group included six mice. Mice were excluded from the study if they died before the indicated time to extract sample. The observers were blinded to the allocation of mice. The experimental mice were treated with one intraperitoneal injection of 1-Methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine hydrochloride (MPTP, 20 mg Kg−1 per dose, Sigma, St. Louis, MO, USA) every 2 h for a total of four doses over an 8 h in 1 day to induce an acute intoxication according to the previous reported protocol [16]. Control mice received saline only. Animals were killed at different time points: 1, 4, 7, and 21 days after the last MPTP injection. The brains were immediately stripped and cut sagittally into two identical hemispheres. One hemisphere was fixed in 4% formaldehyde and then embedded in paraffin for further histopathology studies. The other hemisphere was quickly dissected into SNpc and striatum and stored at −80 °C for further use as previously reported [17].
Necrostatin-1 (Nec-1, Sigma, St. Louis, MO, USA) was dissolved in dimethyl sulfoxide (DMSO, Sigma, St. Louis, MO, USA). All mice in which Nec-1 was administrated undertook intraperitoneal injection of Nec-1 (1.65 mg/kg, dissolved in 250 μl saline) 12 h before the application of MPTP as previously described [6, 18]. Repeated Nec-1 injections were ordered once per day until sacrifice. The control group received only injections of 250 μl saline with DMSO.
Reagents and antibodies
Rabbit polyclonal anti-RIP1 antibody (1:1000, ab106393, Abcam, MA), rabbit polyclonal anti-RIP3 antibody (1:1000, ab56164, Abcam, MA), rabbit monoclonal anti-RIP3 (phosphor T231 + S232) antibody (1:1000, ab222320, Abcam, MA), rabbit polyclonal anti-MLKL antibody (1:1000, ab172868, Abcam, MA), rabbit monoclonal anti-MLKL (phosphor S345) antibody (1:1000, ab196436, Abcam, MA), rabbit polyclonal anti-active caspase-3 antibody (1:1000, ab214430, Abcam, MA), and rabbit polyclonal anti-GAPDH antibody (1:1000, 10494-1-AP, Proteintech, Wuhan, China) were used for western blot. Rabbit polyclonal anti-tyrosine hydroxylase (TH) antibody (1:750, ab112, Abcam, MA) was used to conduct immunohistochemistry. Chicken polyclonal anti-TH primary antibody (1:1000, ab76442, Abcam, MA), rabbit polyclonal anti-RIP1 primary antibody (1:100, ab106393, Abcam, MA), rabbit polyclonal anti-RIP3 primary antibody (1:100, ab56164, Abcam, MA), rabbit polyclonal anti-MLKL primary antibody (1:100, 21066-1-AP, Proteintech, Wuhan, China), rabbit polyclonal anti-GFAP antibody (1:200, ab7260, Abcam, MA), rat anti-CD68 antibody (1:200, ab53444, Abcam, MA), Alexa 488-conjugated goat anti-chicken secondary antibody (1:200, ab150173, Abcam, MA) and Alexa 594-conjugated goat anti-rabbit secondary antibody (1:200, SA00006-3, Proteintech, Wuhan, China) were used to perform double immunofluorescence staining. In Situ Cell Death Detection Kit (Roche, 11684817910, Mannheim, Germany) was used to carried out TUNEL staining.
Western blot
Western blot analysis was performed as previously described [12]. Briefly, total protein samples were extracted from selected mouse midbrain by homogenization in radio immunoprecipitation assay lysis buffer (Beyotime Biotechnology, Shanghai, China). Protein concentration was assessed using a BCA kit (Beyotime Biotechnology, Shanghai, China). Equal amounts of protein were used to run on a SDS-PAGE gel, and then transferred to a polyvinylidene fluoride membrane. After blocking with 5% nonfat dry milk in PBS at room temperature for 1 h, the membranes were incubated at 4 °C overnight with appropriate primary antibodies. Following incubating with the secondary antibody, blot bands were examined by enhanced chemiluminescence (Beyotime, Biotechnology, Shanghai, China) and quantified with Image J software (NIH).
High-performance liquid chromatography (HPLC)
HPLC was carried out as previously described [13]. Briefly, striatum tissue sample was weighed and homogenized on ice in 0.1 mol/l perchloric acid, then centrifuged at 13000 rpm at 4 °C for 15 min. The supernatant was mixed with the HPLC mobile phase and injected into the HPLC column under analytical conditions. Data are expressed as ng dopamine per mg wet tissue.
Immunohistochemistry
Immunohistochemistry was performed as previously described [19]. Briefly, after incubating in 3% H2O2 for 20 min and then in 10% goat serum for 1 h, 4 μm-thick-paraffin sections were incubated overnight at 4 °C with appropriate anti-TH antibody. Then a biotinylated goat anti-rabbit secondary antibody (PV-9001, ZSGB-BIO, China) and diaminobenzidine (ZLI-9017, ZSGB-BIO, China) were used to assess immunoreactivity. The total number of TH-positive neurons was measured as previously described [13]. Briefly, 20 consecutive sections were collected from each brain for evaluation. Unbiased stereology was used to calculate the number of TH-positive neurons of each section in SNpc at ×40 magnification using a microscope (Olympus IX51) and Stereo Investigator software (MBF Bioscience, Williston, USA).
Double immunofluorescence staining
Double immunofluorescence staining was carried out as described previously [20]. Briefly, after blocking in 10% goat serum for 60 min, the sections were incubated at 4 °C overnight with indicated primary antibodies. After rinsed with PBS, the sections were then incubated at room temperature with a mix of proper secondary fluorescent antibodies for 1 h. Nuclei were then counterstained with DAPI for 5 min. The fluorescent images were acquired with a fluorescent microscope (Olympus OX51).
TUNEL staining
TUNEL staining was carried out according to the manufacturer’s protocol. Briefly, after perforating with proteinase K, brain sections were incubated with the TUNEL reaction mixture at 37 °C for 1 h and then counterstained with DAPI for 5 min. Samples were visualized using a fluorescent microscope (Olympus OX51). The number of apoptotic neurons was calculated in six different visual fields by Image J software (NIH).
Quantitative real time PCR
Total RNA was extracted from selected mouse brain tissue using Trizol reagent (Invitrogen, USA) and transcribed into cDNA using TakaRa PrimeScriptTM RT reagent Kit (TakaRa, Japan) as previously described. Real time PCR was carried out in quadruplicate on a 96-well plate using TaKaRa SYBR Premix Ex TaqTM Kit (TaKaRa, Japan). The level of mRNA was normalized to the expression of β-actin. The primer sequences used were listed as follows: TNF-α (Mouse), Forward 5′-CTTCTCATTCCTGCTTGTGG-3′, Reverse 5′-ATGAGAGGGAGGCCATTTG-3′; IL-1β (Mouse), Forward 5′-CGACAAAATACCTGTGGCCT-3′, Reverse 5′-TTCTTTGGGTATTGCTTGGG-3′; IL-6 (Mouse), Forward 5′-GCTACCAAACTGGATATAATCAGGA-3′, Reverse 5′-CCAGGTAGCTATGGTACTCCAGAA-3′; β-actin (Mouse), Forward 5′-ATGTGGATCAGCAAGCAGGA-3′, Reverse 5′-AAAGGGTGTAAAACGCAGCTC-3′.
Statistical analysis
Data were presented as mean ± SD. The statistical analysis was conducted using GraphPad Prism 5 software (GraphPad Inc., CA, USA). Differences between groups were evaluated by one-way analysis of variance followed by the Tukey multiple comparisons test. P < 0.05 was considered statistically significant.
Results
RIP3-mediated necroptosis contributed to MPTP-induced PD
As RIP3 was reported to be the determinant for cellular necroptosis, we first investigated the correlation between the expression of RIP3 and DA neuron degeneration in MPTP-treated mice [21, 22]. We found that the protein levels of RIP3 increased significantly and reached its peak at day 7 after MPTP induction (Fig. 1a). We also found that the striatal level of dopamine and the number of DA neurons in SNpc decreased dramatically, which was in consistent with the change of RIP3 expression (Fig. 1b, c). Since the expression of RIP3 reached its peak at day 7 after MPTP induction, all the following parameters were investigated at day 7 after MPTP induction. Moreover, phosphorylation of RIP3 at Thr231/Ser232 (P-RIP3) and phosphorylation of MLKL at Ser345 were also elevated after MPTP treatment (Fig. 1d).

RIP3-mediated necroptosis may be involved in MPTP-induced PD. a RIP3 expression in the midbrain was assessed by western blot after MPTP application at each indicated time points. b Striatal dopamine level was measured by HPLC. c TH-positive neurons in SNpc were assessed and quantified by immunohistochemistry. d Phosphorylation of RIP3 at Thr231/Ser232 and phosphorylation of MLKL at Ser345 were evaluated by western blot after MPTP treatment. n = 6 mice per group; RIP3 receptor interacting protein kinase-3, MPTP 1-Methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine hydrochloride, PD Parkinson disease, HPLC high-performance liquid chromatography, TH tyrosine hydroxylase, SNpc substantia nigra pars compacta. *P < 0.05 vs. control. Data were represented as mean ± SD. Scale bar: 200 μm
Downregulation of necroptosis signals by Nec-1 attenuates MPTP-induced PD
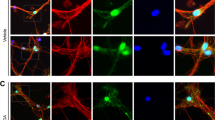
Nec-1, an effective small-molecule inhibitor of necroptosis, blocks a pivotal step in necroptosis [18]. Accordingly, we assessed the effect of Nec-1 on necroptosis signals in MPTP-induced PD. Our data show that MPTP application increased the expression of RIP1, RIP3, and MLKL, and were dramatically reversed by Nec-1 treatment (Fig. 2a–d). Moreover, the HPLC and immunohistochemistry data revealed that Nec-1 treatment significantly rescued the decreased level of dopamine and the loss of TH-positive neurons in SNpc (Fig. 2e, f). Double immunofluorescence staining further indicated that RIP1, RIP3, and MLKL were colocated with TH-positive DA neurons and highly expressed after MPTP induction (Fig. 3).

Nec-1 attenuated MPTP-induced PD. a–d Representative western blots and quantifications of the protein level of RIP1/RIP3/MLKL in the selected midbrain tissue. e Striatal dopamine level was measured by HPLC. f TH-positive neurons in SNpc were measured. n = 6 mice per group; Nec-1 necrostatin-1, RIP1 receptor interacting protein kinase-1, MLKL mixed lineage kinase domain-like protein. *P < 0.05 vs. control. #P < 0.05 vs. MPTP; Data were represented as mean ± SD

Double immunofluorescence staining showed colocalization and high expression of RIP1/RIP3/MLKL in SNpc at day 7 after MPTP treatment. a Immunostaining analysis of RIP1 in SNpc. b Immunostaining analysis of RIP3 in SNpc. c Immunostaining analysis of MLKL in SNpc. a–c Scale bar = 20 μm. n = 6 mice per group
Deletion of RIP3 or MLKL can attenuate MPTP-induced PD
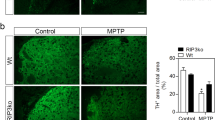
Both RIP3-knockout (RIP3-KO) and MLKL-KO mice were viable, with no detectable defects in normal development [6, 23]. Western blots of the selected midbrain tissue of RIP3-KO or MLKL-KO mice showed the absence of RIP3 or MLKL proteins, respectively (Fig. 4a, d). HPLC demonstrated that the levels of dopamine were much higher in the RIP3−/− and MLKL−/− littermates than that in the WT mice (Fig. 4b, e). Moreover, immunohistochemistry analysis indicated that the loss of TH-positive neurons was dramatically ameliorated in RIP3−/− mice and MLKL−/− mice (Fig. 4c, f).

Deletion of RIP3 or MLKL genes ameliorated MPTP-induced PD. a Western blot showed the absence of RIP3 protein in selected midbrain in RIP3−/− mice. b Striatal dopamine level was measured by HPLC. c TH-positive neurons in SNpc were measured. d Western blot showed the absence of MLKL protein in selected midbrain in MLKL−/− mice. e Striatal dopamine level was measured by HPLC. f TH-positive neurons in SNpc were measured. b–e, c–f n = 6 mice per group. WT wild type; *P < 0.05; Data were represented as mean ± SD
Because there is crosstalk between necroptosis and other cell death types [24], we then explored whether RIP3 or MLKL deficiency affected MPTP-induced apoptosis in SNpc. Although MPTP treatment also induced neuronal apoptosis, the number of TUNEL-positive neurons were similarly increased in both RIP3+/+ and RIP3−/− mice and in both MLKL+/+ and MLKL−/− mice (Fig. 5a, c). Cleaved caspase-3 was also increased similarly in both RIP3+/+ and RIP3−/− mice and in both MLKL+/+ and MLKL−/− mice (Fig. 5b, d). Thus, these finding indicated that the attenuation of MPTP-induced PD by the knockout of RIP3 or MLKL was independent of neuronal apoptosis.

Attenuation of MPTP-induced PD by the knockout of RIP3 or MLKL was independent of neuronal apoptosis. a Representative microphotograph of TUNEL staining showed the neuron apoptosis in SNpc after MPTP injection in both RIP3+/+ and RIP3−/− mice. TUNEL-positive cells were counted and averaged in six randomly selected fields per mouse. Scale bar = 50 μm. b Representative western blot (left panel) and quantification (right panel) of active caspase-3 are shown. c Representative microphotograph of TUNEL staining showed the neuron apoptosis in SNpc after MPTP injection in both MLKL+/+ and MLKL−/− mice. TUNEL-positive cells were counted and averaged in six randomly selected fields per mouse. Scale bar = 50 μm. d Representative western blot (left panel) and quantification (right panel) of active caspase-3 are shown. a–d n = 6 mice per group. *P < 0.05 vs. control. Data were represented as mean ± SD
RIP3/MLKL-mediated neuroinflammation may contribute to neuron necroptosis
Necrosis can cause inflammation, which in turn will enhance necrosis [25]. Accordingly, we further assessed the role of RIP3/MLKL pathway on neuroinflammation in SNpc. We found that astrocytes and microglia were significantly activated after MPTP induction (Fig. 6a–h). The mRNA levels of TNF-α, IL-1β, and IL-6 increased significantly after MPTP injection (Fig. 7a–f), which are consistent with previous reports [26, 27]. Notably, deletion of RIP3 or MLKL genes significantly ameliorated neuroinflammation by downregulating the expression of TNF-α, IL-1β, and IL-6 (Fig. 7a–f).

Astrocyte and microglia were activated by MPTP induction. a Immunostaining analysis of CD68-labeled microglia and GFAP-labeled astrocyte in control group. b Immunostaining analysis of CD68-labeled microglia and GFAP-labeled astrocyte in MPTP-treated group. Scale bar = 25 μm. n = 6 mice per group

RIP3/MLKL-mediated neuroinflammation might be involved in DA neuron necroptosis. a–c Relative mRNA levels of TNF-α, IL-1β, and IL-6 were measured by qRT-PCR in SNpc after MPTP injection in both RIP3+/+ and RIP3−/− mice. d–f Relative mRNA levels of TNF-α, IL-1β, and IL-6 were measured by qRT-PCR in SNpc after MPTP injection in both MLKL+/+ and MLKL−/− mice. a–f n = 6 mice per group; *P < 0.05 vs. control. #P < 0.05 vs. RIP3+/+ (a–c) or MLKL+/+ (d–f); Data were represented as mean ± SD
Discussion
In the present study, we found that RIP1/RIP3/MLKL-mediated necroptosis was activated in the MPTP-induced mouse PD model. Blockade of necroptosis through pharmacological intervention by Nec-1 or deletion of RIP3/MLKL gene can significantly restore the decreased level of dopamine and increase the number of DA neurons in mice after MPTP treatment. Moreover, we also revealed that necroptosis enhanced the expression of pro-inflammatory genes, which might initiate neuroinflammation and in turn aggravate DA neuron necroptosis in the MPTP-induced mouse PD model (Fig. 8).

Schematic representation of potential mechanism of RIP1/RIP3/MLKL-mediated DA neuron degeneration via regulation of necroptosis and neuroinflammation in SNpc after MPTP treatment in mice. DA neurons, dopaminergic neurons
Classically, cell death is classified into three main types: apoptosis, autophagy, and necrosis [24]. Necrosis has previously been described as uncontrolled cell lysis [24]. However, growing evidence has shown that necroptosis is a programmed form of necrosis [6, 10, 11]. Necroptosis was first detected in inflammation induced by TNF-α [18]. More recently, several studies have shown that numerous microenvironmental factors can initiate this pathway [6, 28]. RIP1, RIP3, and MLKL are the core components that execute necroptosis [11, 21]. In line with these findings, our data consistently show that MPTP treatment significantly elevated the expression of RIP1, RIP3, and MLKL in SNpc, which indicates that RIP1/RIP3/MLKL-mediated necroptosis might also contribute to PD. However, the mechanisms underlying the initiation of necroptosis remain undefined. One possible interpretation is that TNF-α, a feature of PD, modulated the RIP1/RIP3/MLKL pathway via conducting neuroinflammation in SNpc [28, 29]. Chronic activation of RIP1/RIP3/MLKL by TNF-α may cause enduring neuron necroptosis activation, which is consistent with a previous report [28].
To further identify the potential mechanism, we employed Nec-1, a specific RIP1 kinase inhibitor, to downregulate the necroptosis signals to investigate the role of necroptosis in MPTP-induced PD [6]. Consistently, our data clearly demonstrated that Nec-1 significantly decreased the expression of RIP1, and following the expression of RIP3 and MLKL also declined. In addition, a series of pathological changes that contributed to MPTP-induced PD, including decreased levels of dopamine and loss of DA neurons, were also dramatically rescued by Nec-1 treatment. Taken together, these data suggest a promising neuroprotective role of Nec-1 by inhibiting necroptosis pathway in MPTP-induced PD.
Because RIP3/MLKL activation is sufficient to initiate necroptosis through a RIP1-independent pathway, it is essential to identify the potential role of RIP3 and MLKL in mediating DA neuron necroptosis in the MPTP-induced mice PD model [30, 31]. In this study, we used RIP3−/− and MLKL−/− mice to address the problem, and we found that deletion of RIP3 or MLKL gene significantly increases the level of dopamine and rescued the loss of DA neurons in SNpc. Moreover, consistent with the previous study, we noted that the small number of necrotic neurons was detected in MPTP-induced RIP3−/− or MLKL−/− mice, suggesting that other types of cell death also occurred in PD [6].
Necroptosis is ultimately executed by oligomerization of MLKL, which will induce membrane perforation and subsequently release damage-associated molecular patterns into the extracellular environment and trigger necroinflammation [6, 11]. Accordingly, we found that MPTP treatment significantly promoted the activation of astrocytes and microglia, and increased the mRNA levels of several inflammatory cytokines in WT mice, and that the response can be partially rescued by deletion of RIP3 or MLKL gene, indicating that RIP3/MLKL-mediated necroptosis was involved in the formation of neuroinflammation. In addition, the inflammatory cytokines might in turn aggravate neuron necroptosis as previously reported [6, 11].
There are several limitations to this study. First, the use of only male mice is a limitation since PD affects males and females. Second, we did not carry out power calculations to ascertain optimal mice numbers. However, six animals per group were previously considered to properly power the simulated study in exploratory preclinical study [32]. Third, we did not study the effect of increasing RIP1/RIP3/MLKL-induced necroptosis nor did we assess the consequences on MPTP-treated PD mice. Further study to address this issue is warranted.
In summary, the present study demonstrates that RIP1/RIP3/MLKL-mediated necroptosis was involved in the pathogenesis of MPTP-induced PD. Downregulation of necroptosis signals by pharmacological intervention (Nec-1) or by RIP3 or MLKL gene knockout can attenuate MPTP-induced PD via increasing the striatal level of dopamine and the number of DA neurons in SNpc. Therapy targeting DA neuron necroptosis may be a potential protocol for treating PD.
References
de Lau LM, Breteler MM. Epidemiology of Parkinson’s disease. The Lancet Neurology. 2006;5:525–35.
Latourelle JC, Beste MT, Hadzi TC, Miller RE, Oppenheim JN, Valko MP, et al. Large-scale identification of clinical and genetic predictors of motor progression in patients with newly diagnosed Parkinson’s disease: a longitudinal cohort study and validation. Lancet Neurol. 2017;16:908–16.
Soldner F, Stelzer Y, Shivalila CS, Abraham BJ, Latourelle JC, Barrasa MI, et al. Parkinson-associated risk variant in distal enhancer of alpha-synuclein modulates target gene expression. Nature. 2016;533:95–9.
Zhao WZ, Wang HT, Huang HJ, Lo YL, Lin AM. Neuroprotective effects of baicalein on acrolein-induced neurotoxicity in the nigrostriatal dopaminergic system of rat brain. Mol Neurobiol. 2017;55:130–7.
Zhou Y, Lu M, Du R-H, Qiao C, Jiang C-Y, Zhang K-Z, et al. MicroRNA-7 targets nod-like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson’s disease. Mol Neurodegener. 2016;11. https://doi.org/10.1186/s13024-016-0094-3.
Xu Y, Ma H, Shao J, Wu J, Zhou L, Zhang Z, et al. A role for tubular necroptosis in cisplatin-induced AKI. J Am Soc Nephrol. 2015;26:2647–58.
Wallach D, Kang TB, Dillon CP, Green DR. Programmed necrosis in inflammation: toward identification of the effector molecules. Science. 2016;352:aaf2154.
Rickard JA, O’Donnell JA, Evans JM, Lalaoui N, Poh AR, Rogers T, et al. RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell. 2014;157:1175–88.
Zhang T, Zhang Y, Cui M, Jin L, Wang Y, Lv F, et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat Med. 2016;22:175–82.
Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54:133–46.
Ito Y, Ofengeim D, Najafov A, Das S, Saberi S, Li Y, et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science. 2016;353:603–8.
Wang H, Ye Y, Zhu Z, Mo L, Lin C, Wang Q, et al. MiR-124 regulates apoptosis and autophagy process in MPTP model of Parkinson’s disease by targeting to bim. Brain Pathol. 2016;26:167–76.
Liu J, Liu W, Lu Y, Tian H, Duan C, Lu L, et al. Piperlongumine restores the balance of autophagy and apoptosis by increasing BCL2 phosphorylation in rotenone-induced Parkinson disease models. Autophagy. 2018;14:845–61.
Dionisio PEA, Oliveira SR, Amaral J, Rodrigues CMP. Loss of microglial Parkin inhibits necroptosis and contributes to neuroinflammation. Mol Neurobiol. 2018;56:2990–3004.
Iannielli A, Bido S, Folladori L, Segnali A, Cancellieri C, Maresca A, et al. Pharmacological inhibition of necroptosis protects from dopaminergic neuronal cell death in Parkinson’s disease models. Cell Rep. 2018;22:2066–79.
Jackson-Lewis V, Przedborski S. Protocol for the MPTP mouse model of Parkinson’s disease. Nat Protoc. 2007;2:141–51.
Peng S, Wang C, Ma J, Jiang K, Jiang Y, Gu X, et al. Achyranthes bidentata polypeptide protects dopaminergic neurons from apoptosis in Parkinson’s disease models both in vitro and in vivo. Br J Pharmacol. 2017;175:631–43.
Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–9.
Hasegawa K, Yasuda T, Shiraishi C, Fujiwara K, Przedborski S, Mochizuki H, et al. Promotion of mitochondrial biogenesis by necdin protects neurons against mitochondrial insults. Nat Commun. 2016;7:10943.
Zhou K, Shi L, Wang Z, Zhou J, Manaenko A, Reis C, et al. RIP1-RIP3-DRP1 pathway regulates NLRP3 inflammasome activation following subarachnoid hemorrhage. Exp Neurol. 2017;295:116–24.
He S, Wang L, Miao L, Wang T, Du F, Zhao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–11.
Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–6.
Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y, et al. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. 2013;23:994–1006.
Lalaoui N, Lindqvist LM, Sandow JJ, Ekert PG. The molecular relationships between apoptosis, autophagy and necroptosis. Semin Cell Dev Biol. 2015;39:63–9.
Daniels BP, Snyder AG, Olsen TM, Orozco S, Oguin TH 3rd, Tait SWG, et al. RIPK3 restricts viral pathogenesis via cell death-independent neuroinflammation. Cell. 2017;169:301–13 e11.
Sathe K, Maetzler W, Lang JD, Mounsey RB, Fleckenstein C, Martin HL, et al. S100B is increased in Parkinson’s disease and ablation protects against MPTP-induced toxicity through the RAGE and TNF-alpha pathway. Brain. 2012;135:3336–47.
Iravani MM, Sadeghian M, Leung CC, Jenner P, Rose S. Lipopolysaccharide-induced nigral inflammation leads to increased IL-1beta tissue content and expression of astrocytic glial cell line-derived neurotrophic factor. Neurosci Lett. 2012;510:138–42.
Caccamo A, Branca C, Piras IS, Ferreira E, Huentelman MJ, Liang WS, et al. Necroptosis activation in Alzheimer’s disease. Nat Neurosci. 2017;20:1236–46.
Niranjan R. The role of inflammatory and oxidative stress mechanisms in the pathogenesis of Parkinson’s disease: focus on astrocytes. Mol Neurobiol. 2014;49:28–38.
Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. 2014;343:1357–60.
He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. PNAS. 2011;108:20054–9.
Selimkhanov J, Thompson WC, Guo J, Hall KD, Musante CJ. A quantitative analysis of statistical power identifies obesity end points for improved in vivo preclinical study design. Int J Obes. 2017;41:1306–9.
Acknowledgements
We would like to thank Institute of Neurology and Central laboratory of First Affiliated Hospital of Fujian Medical University for their kind support and technical guidance in conducting this study. This study was sponsored by key clinical specialty discipline construction program of Fujian, P.R.C. (No. 2014YZ0103 to De-Zhi Kang), major project of Fujian provincial department of science and technology (No. 2014YZ01 to DZK) and basic research and university production cooperation program of Fujian provincial department of science and technology, P.R.C. (No. 2018J01167 to PC).
Author information
Authors and Affiliations
Contributions
LQS, CP, YLH, LYX, and KDZ conceived and designed the experiments and wrote the manuscript. LQS, CP, WWX, LCC, and ZSY performed and analyzed most experiments, including established PD model, neurobehavior tests, western blot, immunofluorescence staining, and qRT-PCR. ZY assisted in experiments. WWX performed preliminary experiments with no data used. XXF assisted in the design of the experiments. The study was supervised by KDZ.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Lin, QS., Chen, P., Wang, WX. et al. RIP1/RIP3/MLKL mediates dopaminergic neuron necroptosis in a mouse model of Parkinson disease. Lab Invest 100, 503–511 (2020). https://doi.org/10.1038/s41374-019-0319-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-019-0319-5
This article is cited by
-
RIP3 in Necroptosis: Underlying Contributions to Traumatic Brain Injury
Neurochemical Research (2024)
-
MLKL deficiency alleviates neuroinflammation and motor deficits in the α-synuclein transgenic mouse model of Parkinson’s disease
Molecular Neurodegeneration (2023)
-
NeuroD1 administration ameliorated neuroinflammation and boosted neurogenesis in a mouse model of subarachnoid hemorrhage
Journal of Neuroinflammation (2023)
-
Necrosulfonamide exerts neuroprotective effect by inhibiting necroptosis, neuroinflammation, and α-synuclein oligomerization in a subacute MPTP mouse model of Parkinson’s disease
Scientific Reports (2023)
-
The role of ZBP1 in eccentric exercise-induced skeletal muscle necroptosis
Journal of Muscle Research and Cell Motility (2023)
