
Abstract
Hepatic stellate cells (HSCs) are major contributors to liver fibrosis, as hepatic injuries may cause their transdifferentiation into myofibroblast-like cells capable of producing excessive extracellular matrix proteins. Also, HSCs can modulate engraftment of transplanted hepatocytes and contribute to liver regeneration. Therefore, understanding the biology of human HSCs (hHSCs) is important, but effective methods have not been available to address their fate in vivo. To investigate whether HSCs could engraft and repopulate the liver, we transplanted GFP-transduced immortalized hHSCs into immunodeficient NOD/SCID mice. Biodistribution analysis with radiolabeled hHSCs showed that after intrasplenic injection, the majority of transplanted cells rapidly translocated to the liver. GFP-immunohistochemistry demonstrated that transplanted hHSCs engrafted alongside hepatic sinusoids. Prior permeabilization of the sinusoidal endothelial layer with monocrotaline enhanced engraftment of hHSCs. Transplanted hHSCs remained engrafted without relevant proliferation in the healthy liver. However, after CCl4 or bile duct ligation-induced liver damage, transplanted hHSCs expanded and contributed to extracellular matrix production, formation of bridging cell-septae and cirrhosis-like hepatic pseudolobules. CCl4-induced injury recruited hHSCs mainly to zone 3, whereas after bile duct ligation, hHSCs were mainly in zone 1 of the liver lobule. Transplanted hHSCs neither transdifferentiated into other cell types nor formed tumors in these settings. In conclusion, a humanized mouse model was generated by transplanting hHSCs, which proliferated during hepatic injury and inflammation, and contributed to liver fibrosis. The ability to repopulate the liver with transplanted hHSCs will be particularly significant for mechanistic studies of cell-cell interactions and fibrogenesis within the liver.
Similar content being viewed by others

Introduction
Repopulation of the liver with transplanted cells is of much interest for biological studies and for therapeutic applications. Experimental transplantation of mature hepatocytes and liver sinusoidal endothelial cells (LSECs) has improved the understanding of how hepatic and endothelial cell compartments could be reconstituted, including for disease corrections [1, 2]. Other studies demonstrated the roles of cell-cell interactions, e.g., signaling from LSECs was found to be critical in liver regeneration [3]. Similarly, hepatic stellate cells (HSCs) may contribute to liver regeneration [4], although the mechanisms—especially within the intact organ in vivo—are incompletely defined. Therefore, availability of cell transplantation models, particularly in animals with human cells, may be helpful for translational studies. This requires consideration of the complexities involved in repopulation of the liver by transplanted cells. For instance, immediately after cell transplantation in the liver, transplanted hepatocytes cause hepatic ischemia with deleterious activation of inflammatory cells [5], which needs to be controlled for improving cell engraftment. Similarly, prior disruption of the endothelial barrier advanced entry of transplanted hepatocytes into the space of Disse, which was necessary for their subsequent integration in liver parenchyma [2]. Also, transplanted cell engraftment required hepatotrophic factors and matrix remodeling, which directly involved HSCs [6]. This role of HSCs in promoting engraftment of transplanted cells seemed distinct from their ability to transdifferentiate into profibrogenic myofibroblast-like cells expressing α-smooth muscle actin (α-SMA) with secretion of cytokines, chemokines or receptors, as well as extracellular matrix (ECM) components [7,8,9,10,11]. Nonetheless, mechanisms driving hepatic fibrogenesis are complex, with interactions between HSCs, other non-parenchymal cells, and hepatocytes through cell-cell contacts and soluble factors [12,13,14,15], which have been difficult to extrapolate from studies in vitro.
There is general agreement that HSCs are the major contributor to fibrogenesis in the liver. Following liver injury, HSCs migrate to sites of damage and undergo activation with excessive synthesis of ECM components. Even though the important role of HSCs in hepatic fibrogenesis is well documented, specific antifibrotic, HSC-directed therapies have yet to be established. One reason for this difficulty is that experimental modulation of HSCs in vivo is extremely challenging; until recently there was no established HSC-specific Cre-transgenic mouse model to study this cell compartment [16]. Therefore, we hypothesized that generation of animal models with transplantation of human HSCs (hHSCs) will be valuable for studying the contributions of HSCs in liver injury and fibrosis.
This project aimed to evaluate whether hHSCs could be successfully transplanted into the liver for studying their fate along with activation and migration to sites of liver injury. To avoid potential variables related to donor-specific differences in the properties of primary hHSCs, we utilized human HSCs that had been immortalized by the catalytic subunit of human telomerase reverse transcriptase (hTERT) and retained most aspects of primary HSCs, including typical morphology and gene expression profiles [17]. To avoid rejection, we transplanted hHSCs into xenograft-tolerant mice lacking T and B cells with non-obese diabetic-severe combined immunodeficiency (NOD/SCID). Marking of hHSCs with radiolabels or a lentivirally-introduced transgene to express green fluorescent protein (GFP) allowed tracking of transplanted cells over short- and long-term, respectively. This led to successful studies of the biodistributions, engraftment, proliferation and fate of transplanted hHSCs with or without fibrogenic damage in the liver.
Materials and methods
Animals
NOD.CB17-Prkdcscid/J mice were from Jackson Laboratories (Bar Harbor, ME), or from the Special Animal Core Facility, Hamburg University Medical Center. Animal Care and Use Committees at Albert Einstein College of Medicine and Hamburg University approved animal use in conformity with National Research Council’s Guide for the Care and Use of Laboratory Animals (United States Public Health Service Publication, revised 1996) and German regulations.
Cells
hTERT-hHSCs were from Dr. David Brenner and generation of these cells was described previously [17]. These hHSCs were cultured in 5% CO2 atmosphere on plastic dishes in Dulbecco’s minimum essential medium (DMEM) with antibiotics and 10% fetal bovine serum (FBS) (Atlanta Biologicals, Norcross, GA). Cells were split 1:3 every 4 to 5 d. The morphology of cells was evaluated by light microscopy. Staining for α-SMA was as described previously [6].
Lentiviral vector (LV) transduction of hHSCs with GFP
Vesicular stomatitis virus-pseudotyped LV stocks were obtained by pCCL.PPT.hPGK.GFP.Wpre transfer construct with packaging constructs, pMDLg/pRRE and pRSV-REV, and pMD2.G envelope construct, which were transfected into 293T cells to produce LV, as described previously [18]. For LV transduction, 4 × 106 hHSCs were incubated with vector for 90 min at 37 °C in 1 ml final volume, and transduced cells were then expanded in culture.
Cell transplantation
Mice were anesthetized with i.p. 90 mg/kg ketamine plus 8 mg/kg xylazine. Spleen was isolated by laparotomy and 1 × 106 hHSCs in DMEM with 2% FBS were injected into splenic pulp. Control animals received vehicle (DMEM/FBS) alone.
Radiolabeling of cells and biodistribution analysis
For radiolabeling, 3–6 × 106 hHSCs were incubated for 30 min at 37 °C in normal saline with 18.5 MBq Indium-111 (111In)-oxine or 370 MBq technetium-99m (99mTc) pertechnetate (UltraTag® kit, Mallinckrodt Inc, Hazelwood, MO) or 370 MBq99mTc-exametazime (Ceretec™, Amersham Biosciences, Piscataway, NJ). Cells were washed twice with PBS and pelleted under 300xg for 5 min. For labeling efficiency, radioactivity was measured in cell pellets and supernatants. Cell viability after radiolabeling was confirmed by trypan blue dye exclusion. For organ biodistributions, 1 × 106 radiolabeled hHSCs were transplanted into NOD/SCID mice followed 1 h later by gamma imaging (Argus; ADAC Laboratories, Milpitas, CA). Energy discrimination used a 15% window centered on 174- and 247-keV peaks of 111In. Dorsal images were acquired for 5 min on 64 × 64 × 16 matrix. After imaging, 111In activity was counted in excised spleen, liver, lungs, and kidneys (Cobra II counter; Packard Instrument Co., Meridien, CT), and expressed as fraction of total radioactivity in these organs.
Enhancement of hHSC engraftment and proliferation
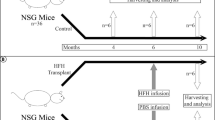
For endothelial injury, 200 mg/kg monocrotaline (MCT) was given i.p. in phosphate buffered saline, pH 7.4 (PBS), 16–24 h before cell transplantation. Control mice received PBS alone. For hepatic injury, the experimental design is shown in Fig. 1. To induce toxin-mediated liver fibrosis, 1 ml/kg carbon tetrachloride (CCl4) mixed 1:10 (v/v) in corn oil was given i.p. twice weekly for up to 6 weeks, starting 10 d after cell transplantation. Controls received corn oil alone. To induce biliary fibrosis, bile duct ligation (BDL) was performed 10 d after transplantation of hHSCs, as described previously [14].

Experimental design for elucidating proliferation and fate of transplanted hHSCs. After allowing 10 d for intrasplenically transplanted hHSCs to engraft, mice were given CCl4 injections twice weekly or subjected to BDL with analysis at various time points as indicated
Polymerase chain reaction (PCR)
Qualitative and quantitative PCR was performed for the human Charcot-Marie-Tooth disease type 1A repeat element (CMT1A) as well as murine c-mos sequence as described previously [19]. Quantitative PCR for human beta-globin gene was performed as described previously [20].
Western blot
Total proteins were from culture medium and extracted from hHSCs, blotted and analyzed for human collagen type I as described previously [21].
Tissue staining and in situ hybridization
For GFP-staining, liver tissue was fixed in 4% paraformaldehyde for 4 h with anti-GFP (1:300 Molecular Probes) staining, essentially as described previously [18]. Sections were incubated with secondary peroxidase-conjugated mouse or rabbit-specific goat IgG (1:300, Dako, Real Envision, HRP Rabbit/Mouse) for 30 min and developed with diaminobenzidine at room temperature. To distinguish GFP + transplanted cells from other cells or matrix components, tissues were co-stained with anti-GFP plus primary antibodies against following: anti-mouse F4/80 (eBioscience, Frankfurt, Germany), anti-human albumin, anti-mouse LSEC (CD146, Miltenyi Biotech, Bergisch-Gladbach, Germany), anti-human collagen type I (Southern Biotech, Birmingham, Alabama), collagen type IV (1:500, Sigma-Aldrich, Munich, Germany), anti-mouse cytokeratin (1:500, Dako, Hamburg, Germany), anti-human laminin (1:50, Dako). Alexa Fluor 488 and Alexa Fluor 555 conjugated secondary antibodies (Invitrogen) were used for fluorescence microscopy. Nuclei were stained with Hoechst 33258 (Invitrogen). Fibrillary tissue collagen was identified and quantitated by picrosirius red staining as described previously [14]. In situ hybridization for primate-specific alphoid satellite sequences was performed as described previously [19].
Statistical methods
Data are shown as mean ± SD. Differences were analyzed by t-tests with SigmaStat 3.1 software (Systat Inc., Point Richmond, CA). P < 0.05 was considered significant.
Results
Deposition and engraftment of transplanted hHSCs in mouse liver
In culture, hHSCs proliferated well with myofibroblast-like morphology, and showed expression of α-SMA by immunostaining (Fig. 2a) and collagen by western blotting (Fig. 2b), which are characteristics of HSCs. Also, virtually all LV-transduced hHSCs expressed GFP (Fig. 2c). Consistent with an activated phenotype, hHSCs displayed neither vitamin A autofluorescence nor lipid droplets (not shown). The mean diameter of trypsinized hHSCs (11.1 ± 3.5 µm) was greater than that of distal hepatic sinusoids (3–5 µm) but less than that of adult hepatocytes (20–35 µm) (Figs. 2d, e), suggesting that transplanted hHSCs would be entrapped within the liver after deposition into the portal circulation. To confirm this possibility, we radiolabeled hHSCs for biodistribution studies. The hHSCs incorporated 111In-oxine (to 62 ± 8% efficiency) but neither99mTc-exametazime nor 99mTc-pertechnetate. Gamma imaging of mice receiving 111In-labeled-hHSCs showed transplanted cells translocated rapidly from the spleen to the liver without entering extrahepatic organs, e.g., lungs (Fig. 2f), thereby confirming their entrapment in the liver. Measurement of 111In activity in excised organs established that the majority of transplanted hHSCs were in the liver (Fig. 2g). Some transplanted hHSCs remained in the spleen. Entry of hHSCs in lungs was negligible. Therefore, intrasplenic injection of hHSCs was successful in depositing cells to liver sinusoids, as was desired.

Analysis of hHSC characteristics and biodistribution studies with radiolabeled cells. a Immunohistochemistry showing α-SMA was expressed in all hHSCs. Note typical spindle shape or elongated morphology of cells in culture conditions. b Western blot of cell lysates and culture supernatants demonstrated collagen expression (α1 and α2 chains) by cultured hHSCs. c Lentivirally transduced hHSCs stably expressed GFP in all cultured cells supporting its use as a reporter in cell transplantation studies. d Comparison of the size of hHSCs released by trypsin-EDTA from culture dishes indicated these were substantially smaller than mouse hepatocytes (e) but were still much larger than the 3–5 μm diameter of hepatic sinusoids. f Gamma imaging of a mouse 1 h after intrasplenic transplantation of 111In labeled hHSCs showing tracer activity in spleen and liver. g Organ distributions of transplanted hHSCs as fractions of total 111In activity indicated that most hHSCs were deposited in the liver. Original magnifications A,C: 400x, D,E: 200x
We then studied engraftment of GFP-labeled hHSCs in mouse liver. PCR for human-specific CMT1 element identified transplanted cells in every mouse (n = 3 per time-point) (Fig. 3a). Transplanted cells were detected throughout a 7 days period, although the abundance of CMT1 PCR products decreased over time. Persistence of transplanted hHSCs in the liver was directly established by GFP staining, as well as by in situ hybridization for primate-specific alphoid satellite sequences identifying every centromere (Figs. 3b, c). hHSCs were localized in sinusoids or portal vein radicles immediately after transplantation. The abundance of transplanted hHSCs decreased after 7 days, which was in agreement with PCR results. It was noteworthy that transplanted hHSCs were distinct from sinusoidal liver cells, such as LSECs and hepatic macrophages, as shown by double immunofluorescence for GFP + transplanted cells and endothelial or F4/80 macrophage markers, respectively (Figs. 3d, e). Similarly, hHSCs were distinct from hepatocytes expressing albumin or cytokeratin (CK)-18 (not shown).

Engraftment of hHSCs in mouse liver. a PCR of livers from transplanted mice for human specific CMT1 indicated presence of hHSCs in recipients. b Immunohistochemistry for GFP reporter showing survival and distribution of transplanted cells in the liver after 7 d. c In situ hybridization for human alphoid sequences 1 h after transplantation demonstrated hHSCs in the space of Disse or within hepatic sinusoids (arrows). Combined staining for GFP (green) and anti-mouse LSEC (CD146) (d) or F4/80 (e) (red) after 7 d indicated that transplanted hHSC were within proximity to other non-parenchymal cells in the liver but were distinct from those other cell types. Original magnifications: B, D, E: 200x, C: 400x
Induction of hHSC proliferation in the mouse liver
We determined whether fibrogenic liver injury induced by CCl4 would promote proliferation of transplanted hHSCs. For this purpose, we analyzed proliferation of transplanted cells over 6 weeks in groups of mice given vehicle alone, MCT pretreatment alone, or MCT pretreatment plus CCl4 twice weekly post-transplant (n = 6 each) (Fig. 4). GFP-immunostaining identified few transplanted hHSCs in mouse livers without either MCT or CCl4. In these mice, hHSCs were observed in small groups, most likely representing accumulations in areas of ischemic necrosis induced by cell transplantation (Fig. 4a). In mice pretreated with MCT alone, hHSCs engrafted better with greater abundances (Fig. 4b). However, transplanted hHSCs expanded most in mice given MCT followed by repeated CCl4 (Fig. 4c). Transplanted hHSCs accumulated along fibrotic bands encircling liver nodules in this condition. To quantitate these differences in engraftment and proliferation of transplanted hHSCs we performed qRT-PCR for human beta-globin in mouse livers. The extent of liver repopulation with hHSCs approached 1.0 ± 2.5% in mice receiving MCT alone (n = 4 with 2 liver lobes per mouse) vs. 8.7 ± 4.9% in mice receiving MCT plus CCl4 (n = 6 with 2 lobes per mouse), p < 0.05 (Fig. 4d).

Induction of proliferation in transplanted hHSCs. Immunohistochemistry for GFP reporter 6 weeks after cell transplantation showing little proliferation of hHSCs in liver of otherwise untreated mouse. Transplanted cells mostly formed small clusters (a). By contrast, sinusoidal endothelial disruption with MCT prior to cell transplantation produced superior engraftment and proliferation of hHSCs (b). The greatest extent of hHSC expansion was observed in mice pretreated with MCT followed by CCl4 twice weekly (c). This greater proliferation of transplanted hHSCs was confirmed by qRT-PCR for human beta-globin (d). Original magnification A: 200x, B, C: 100x
Functional activity of transplanted hHSCs in mouse liver
We examined the ability of transplanted hHSCs to produce ECM components. In MCT plus CCl4-treated mice, immunostaining studies showed presence of substantial human collagen I in hHSC recipient livers (Fig. 5a). This was not the case in control mouse livers (Fig. 5b). Double staining for GFP and human collagen I demonstrated transplanted hHSCs were in close proximity to human collagen deposits, suggesting that this ECM component originated from transplanted hHSCs (Fig. 5c). Similarly, transplanted cells co-localized with collagen IV and laminin (Figs. 5d, e), which was also in agreement with the origin of these ECM components from transplanted hHSCs—a caveat being that the antibodies used to identify these components were not human-specific. Next, to determine the extent of liver fibrosis that could be assigned to transplanted hHSCs, we studied additional NOD/SCID mice with or without hHSC transplantation followed by CCl4 for six consecutive weeks. Quantitative analysis of Sirius Red-stained areas, as described previously [22], demonstrated that CCl4 caused greater fibrosis in mice with hHSCs vs. control mice without hHSC transplantation (Figs. 5f, g). The fractions of Sirius Red-stained areas constituted 7.2 ± 3.5% in hHSC transplant group (n = 10) vs. 1.0 ± 0.5% in controls with CCl4 alone (n = 8), p < 0.01 (Fig. 5h).

Transplanted hHSCs produced ECM components in mouse liver. (a, b) Immunohistochemistry showed presence of human collagen I in hHSC-transplanted (a) vs. untransplanted mouse livers (b) after 6 weeks of CCl4. Co-staining for GFP reporter (green) in transplanted hHSC and collagen I (c), collagen IV (d) and laminin (e) (red) showed ECM components within proximity to transplanted hHSCs. Sirius Red staining of vehicle control (f) and hHSC recipient livers (g) allowed quantification of fibrillary collagen (h) irrespective of its source in transplanted hHSCs or liver resident murine HSCs. Original magnifications A-G: 200x
Injury-related distributions of hHSCs in mouse liver
Transplantation of hHSCs in MCT-pretreated mice followed 10 d later by either hepatocellular injury with CCl4 or cholestatic injury with BDL demonstrated whether or not transplanted hHSCs would migrate to sites of injury. Control mice received vehicle alone or underwent sham surgery. After CCl4 injury, transplanted hHSCs accumulated in fibrotic septae with predominantly pericentral and also porto-central distribution, which was typical for liver fibrosis induced by CCl4. By contrast, BDL resulted in predominantly periportal accumulation of transplanted hHSCs (Figs. 6a, b). This distribution of hHSCs was confirmed by double staining for GFP and pan-cytokeratin as a marker of bile duct cells (Figs. 6c, d).

Injury-related distributions of transplanted hHSCs in mouse liver. Immunohistochemistry for GFP (a–b) as well as double-immunofluorescence for GFP (green) and biliary marker pan-cytokeratin (red) (c–d) showed pericentral distribution of transplanted hHSCs after CCl4-induced liver injury (a and c) and periportal accumulation after BDL (b and d). CV central vein, PF portal field, BD bile duct. Original magnifications 200x
To visualize migration of hHSCs within the injured liver, transplanted hHSCs were localized at various time-points. Transplanted hHSCs were present throughout the liver lobule after 10 d (Figs. 7a, b). However, 1 and 2 weeks after CCl4 hHSCs were predominantly in pericentrally located necrotic areas (Figs. 7c, d). During further CCl4-induced liver damage, hHSCs were in fibrotic septae joining adjacent central veins and occasionally extending from portal to central areas, indicating transplanted cells had migrated during fibrogenesis (Figs. 7e, f). In vehicle-treated controls, this pattern of hHSC distribution in the liver lobule was not observed at time points up to 4 weeks (Figs. 7g, h).

Migration of transplanted hHSCs to sites of liver injury. Immunohistochemistry for GFP visualized time-dependent migration of transplanted hHSCs to the site of CCl4-induced injury (a–f). In untreated control mice, hHSC distribution was without such a specific pattern (g–h). Original magnifications: 200x
To additionally evaluate whether Kupffer cells (KCs) may have contributed in recruitment of hHSCs through cell-cell interactions activated by tissue inflammation, we analyzed whether these cell types preceded one another or were simultaneously engaged at sites of injury. We found KCs and transplanted hHSCs co-localized in both, the CCl4 and BDL injury model, although this was more pronounced in CCl4-treated mice (Figs. 8a, b). In controls with MCT pretreatment only, we did not observe distinct co-localization of KCs and hHSCs (Fig. 8c). No GFP-positive cells were observed in mice without hHSC transplantation (Fig. 8d). Therefore, this hHSC transplantation model allowed identification of additional cell-cell interactions within the liver.

Co-localization of transplanted hHSCs and KCs. Combined immunofluorescence for GFP reporter (green) in transplanted hHSCs and F4/80 marker (red) of monocytes/macrophages showed close proximity of transplanted hHSCs and KCs. This was most pronounced in mice with CCl4-induced liver injury (a) and was also observed in mice after BDL (b). In mice with MCT pretreatment only, KCs and hHSCs were restricted to necrotic areas (c) Controls without transplantation of hHSCs were negative for GFP staining (d) Original magnifications: 200x
Discussion
In the liver, HSCs represent the major fibrogenic cell-type and are deemed crucial for progression of cirrhosis and related complications, such as portal hypertension. Therefore, understanding HSC pathophysiology will be important for therapeutic strategies aimed at delaying or reversing fibrosis in chronic liver diseases. Much current knowledge of HSC biology has been derived from studies with HSCs in vitro. However, we and others previously established that the widely used model of HSC activation by culturing isolated HSCs on a plastic surface does not accurately reflect changes of activated HSCs in vivo [23]. Obviously, interaction with the hepatic tissue environment is required for physiological HSC activation. This is in line with our finding that presence of liver macrophages during HSC culture shifted the gene expression pattern of in vitro activated HSCs towards the pattern observed in vivo [24]. Similarly, culture activation of primary human HSCs does not accurately reflect the biological phenotype of activated HSCs isolated from cirrhotic livers [25]. Whereas several existing mouse models of liver fibrosis provided valuable insight into fibrogenesis, the species-specific differences between murine and human HSCs do restrict their value as models for human fibrosis. Here, we successfully established a mouse model that allowed targeting of human HSCs to the liver along with analysis of their biological behavior and fate during hepatic fibrogenesis in vivo. Specifically, this study provided insights into how the liver may be repopulated with transplanted hHSCs. First, we found hHSCs were efficiently targeted to the liver via the intrasplenic route. Second, transplanted hHSCs engrafted in the liver, especially when the hepatic endothelium was injured beforehand to open up the space of Disse where HSCs are normally located. Third, transplanted hHSCs proliferated and migrated to the sites of liver damage and interacted with other cell types serving roles in hepatic injury and repair. Fourth, transplanted hHSCs displayed fibrogenic potential by generating myofibroblast-like cells and producing ECM components. In these ways, transplantation of hHSCs was effective for generating this humanized mouse model of liver fibrosis.
Radiolabeling of hHSCs was convenient for cell biodistribution analysis and this approach should similarly be useful in future transplantation studies, e.g., of primary hHSCs. The incorporation of 111In but not of 99mTc in hHSCs was recapitulated in primary mouse HSCs (Benten et al., unpublished data), indicating this was specific to this cell type. Previously, we showed that LSECs as well as KCs incorporated 111In and not 99mTc [18, 26], whereas hepatocytes incorporated both of these tracers [27], highlighting cell type-specific differences in radioligand uptake or transport mechanisms in parenchymal vs. non-parenchymal liver cells.
The restriction of transplanted hHSCs to the liver indicated that for achieving superior cell engraftment mechanical factors should be less important than cellular alterations, such as endothelial disruption by MCT. This was in accordance with previous studies demonstrating that endothelial damage improved engraftment of transplanted hepatocytes as well as LSECs [1, 18, 28]. Similarly, prior damage in native KCs with gadolinium chloride facilitated engraftment of transplanted KCs in the liver [26].
Remarkably, extensive proliferation of transplanted hHSCs required profibrogenic stimuli in vivo, since repeated CCl4 administration produced 8-fold greater liver repopulation with these cells. Transplanted hHSCs themselves were not cleared by this progressive liver injury. On the other hand, as hHSCs accumulated during CCl4-induced liver injury in fibrotic septae bridging pericentral or portovenous areas, this model identified for the first time injury-related migration of intrahepatic HSC-derived myofibroblasts. Although this migration of HSCs is considered to be central in fibrogenesis [29,30,31], it has not been possible previously to directly analyze this mechanism in vivo. Recently, Mederacke et al. generated a Cre-transgenic mouse for unequivocal identification of almost all endogenous HSCs in vivo, thereby allowing substantiation of these cells as major contributors in toxic, metabolic or biliary injuries in the liver [16]. Nonetheless, immunohistochemical identification of HSCs can be unreliable for tracking HSC migration in tissues since expression of cellular markers used for this purpose, e.g., α-SMA, desmin, glial fibrillary acidic protein, etc., may be regulated variably based on the functional state of the cells [30]. Moreover, lineage tracing for demonstrating migration of HSCs into areas of liver damage may not be possible due to difficulties in separating cells arising locally via transdifferentiation or changes induced by epithelial-mesenchymal transitions, cell proliferation, etc. By contrast, use of genetically marked transplanted hHSCs permitted identification of their recruitment to sites of injury and also functionality, i.e., production of ECM components, including collagens and laminin, in response to profibrogenic stimuli.
Co-localization of transplanted hHSCs and KCs after CCl4 and BDL-induced liver injury demonstrated these cell types were in close proximity. This was in agreement with our previous findings indicating that functional interactions between HSCs and KCs are significant in hepatic fibrogenesis [24]. As model systems have been successfully developed to identify the fate of transplanted KCs and their roles in regulating inflammatory events and processes through cytokine expression [26], exciting experimental studies to address interactions between hHSCs and KCs could now be launched. The biological behavior of transplanted hHSCs reproduced that of resident HSCs in vivo, and allowed for the first time delineation of how HSCs may migrate toward sites of injury, which has again been hypothesized to be a key mechanism in liver fibrogenesis [32]. In contrast with previous difficulties in determining whether HSCs originate locally or migrate to sites of liver injury, our study unequivocally established the ability of HSCs to migrate and to accumulate during liver injury. This finding should allow experimental studies of HSC-specific interventions, e.g., ex vivo genetic modification of HSCs prior to transplantation for demonstrating potential of candidate genes in fibrogenesis. Indeed, HSCs were readily and efficiently transduced by LV demonstrating the feasibility of introducing genes for interventions, such as gene constructs able to limit fibrogenesis and alter other processes ranging from inflammation to immunomodulation in the liver.
We found transplanted hHSCs did not transdifferentiate into alternative cell types. Whether HSCs possess stem/progenitor cell potential to generate additional cell types has been controversial [33,34,35]. However, we observed neither albumin nor CK-18 expression as hepatocyte markers in transplanted hHSCs, including after CCl4-induced hepatic injury. Although it may be that immortalized hHSCs studied here were no longer able to reproduce properties of primary hHSCs and species-specific barriers may have affected transdifferentiation capacity of these cells in mice, it should be noted that fetal human hepatocytes immortalized by hTERT retained their transdifferentiation potential, including after transplantation in immunodeficient mice [36].
We previously showed that hepatocyte transplantation activated native HSCs, which contributed in turn to transplanted cell engraftment in the liver [6]. In other experimental settings, co-transplantation of HSCs with pancreatic islet cells under the renal capsule improved parenchymal cell survival via immunoregulatory events and HSC-mediated generation of myeloid-derived suppressor cells [37, 38]. Since human hepatocytes tend not to survive and proliferate well in the mouse liver, our model could allow studies to determine whether co-transplantation of hHSCs would benefit engraftment and function of these or other human liver cell types. This should accelerate progress in other relevant translational research areas.
In summary, we demonstrated that immortalized hHSCs were successfully transplanted to generate a humanized mouse liver model of liver fibrosis and HSC migration in vivo. This animal model may offer new opportunities to study human fibrogenesis and to modulate liver fibrosis in a hHSC-specific manner in vivo.
References
Follenzi A, Benten D, Novikoff P, et al. Transplanted endothelial cells repopulate the liver endothelium and correct the phenotype of hemophilia A mice. J Clin Invest. 2008;118:935–45.
Gupta S, Inada M, Joseph B, et al. Emerging insights into liver-directed cell therapy for genetic and acquired disorders. Transpl Immunol. 2004;12:289–302.
Ding BS, Nolan DJ, Butler JM, et al. Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nature. 2010;468:310–5.
Omenetti A, Porrello A, Jung Y, et al. Hedgehog signaling regulates epithelial-mesenchymal transition during biliary fibrosis in rodents and humans. J Clin Invest. 2008;118:3331–42.
Gupta S, Bhargava KK, Novikoff PM. Mechanisms of cell engraftment during liver repopulation with hepatocyte transplantation. Semin Liver Dis. 1999;19:15–26.
Benten D, Kumaran V, Joseph B, et al. Hepatocyte transplantation activates hepatic stellate cells with beneficial modulation of cell engraftment in the rat. Hepatology. 2005;42:1072–81.
Brenner DA, Waterboer T, Choi SK, et al. New aspects of hepatic fibrosis. J Hepatol. 2000;32:32–8.
Murphy FR, Issa R, Zhou X, et al. Inhibition of apoptosis of activated hepatic stellate cells by tissue inhibitor of metalloproteinase-1 is mediated via effects on matrix metalloproteinase inhibition: implications for reversibility of liver fibrosis. J Biol Chem. 2002;277:11069–76.
Pinzani M, Marra F. Cytokine receptors and signaling in hepatic stellate cells. Semin Liver Dis. 2001;21:397–416.
Sanz S, Pucilowska JB, Liu S, et al. Expression of insulin-like growth factor I by activated hepatic stellate cells reduces fibrogenesis and enhances regeneration after liver injury. Gut. 2005;54:134–41.
Schaefer B, Rivas-Estilla AM, Meraz-Cruz N, et al. Reciprocal modulation of matrix metalloproteinase-13 and type I collagen genes in rat hepatic stellate cells. Am J Pathol. 2003;162:1771–80.
Benyon RC, Arthur MJ. Extracellular matrix degradation and the role of hepatic stellate cells. Semin Liver Dis. 2001;21:373–84.
Issa R, Zhou X, Constandinou CM, et al. Spontaneous recovery from micronodular cirrhosis: evidence for incomplete resolution associated with matrix cross-linking. Gastroenterology. 2004;126:1795–808.
Kluwe J, Pradere JP, Gwak GY, et al. Modulation of hepatic fibrosis by c-Jun-N-terminal kinase inhibition. Gastroenterology. 2010;138:347–59.
Maher JJ. Interactions between hepatic stellate cells and the immune system. Semin Liver Dis. 2001;21:417–26.
Mederacke I, Hsu CC, Troeger JS, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. 2013;4:2823.
Schnabl B, Choi YH, Olsen JC, et al. Immortal activated human hepatic stellate cells generated by ectopic telomerase expression. Lab Invest. 2002;82:323–33.
Benten D, Follenzi A, Bhargava KK, et al. Hepatic targeting of transplanted liver sinusoidal endothelial cells in intact mice. Hepatology. 2005;42:140–8.
Benten D, Cheng K, Gupta S. Identification of transplanted human cells in animal tissues. Methods Mol Biol. 2006;326:189–201.
Volz T, Lutgehetmann M, Wachtler P, et al. Impaired intrahepatic hepatitis B virus productivity contributes to low viremia in most HBeAg-negative patients. Gastroenterology. 2007;133:843–52.
Berna MJ, Seiz O, Nast JF, et al. CCK1 and CCK2 receptors are expressed on pancreatic stellate cells and induce collagen production. J Biol Chem. 2010;285:38905–14.
Kluwe J, Wongsiriroj N, Troeger JS, et al. Absence of hepatic stellate cell retinoid lipid droplets does not enhance hepatic fibrosis but decreases hepatic carcinogenesis. Gut. 2011;60:1260–8.
De Minicis S, Seki E, Uchinami H, et al. Gene expression profiles during hepatic stellate cell activation in culture and in vivo. Gastroenterology. 2007;132:1937–46.
Pradere JP, Kluwe J, De Minicis S, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology. 2013;58:1461–73.
Sancho-Bru P, Bataller R, Gasull X, et al. Genomic and functional characterization of stellate cells isolated from human cirrhotic livers. J Hepatol. 2005;43:272–82.
Merlin S, Bhargava KK, Ranaldo G, et al. Kupffer Cell Transplantation in Mice for Elucidating Monocyte/Macrophage Biology and for Potential in Cell or Gene Therapy. Am J Pathol. 2016;186:539–51.
Gupta S, Lee CD, Vemuru RP, et al. 111Indium labeling of hepatocytes for analysis of short-term biodistribution of transplanted cells. Hepatology. 1994;19:750–7.
Joseph B, Kumaran V, Berishvili E, et al. Monocrotaline promotes transplanted cell engraftment and advances liver repopulation in rats via liver conditioning. Hepatology. 2006;44:1411–20.
Bonacchi A, Romagnani P, Romanelli RG, et al. Signal transduction by the chemokine receptor CXCR3: activation of Ras/ERK, Src, and phosphatidylinositol 3-kinase/Akt controls cell migration and proliferation in human vascular pericytes. J Biol Chem. 2001;276:9945–54.
Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–69.
Marra F, Romanelli RG, Giannini C, et al. Monocyte chemotactic protein-1 as a chemoattractant for human hepatic stellate cells. Hepatology. 1999;29:140–8.
Seki E, De Minicis S, Gwak GY, et al. CCR1 and CCR5 promote hepatic fibrosis in mice. J Clin Invest. 2009;119:1858–70.
Kordes C, Sawitza I, Gotze S, et al. Hepatic stellate cells contribute to progenitor cells and liver regeneration. J Clin Invest. 2014;124:5503–15.
Choi SS, Omenetti A, Witek RP, et al. Hedgehog pathway activation and epithelial-to-mesenchymal transitions during myofibroblastic transformation of rat hepatic cells in culture and cirrhosis. Am J Physiol Gastrointest Liver Physiol. 2009;297:G1093–106.
Roskams T. Relationships among stellate cell activation, progenitor cells, and hepatic regeneration. Clin Liver Dis. 2008;12:853–60.
Zalzman M, Gupta S, Giri RK, et al. Reversal of hyperglycemia in mice by using human expandable insulin-producing cells differentiated from fetal liver progenitor cells. Proc Natl Acad Sci Usa. 2003;100:7253–8.
Chen CH, Kuo LM, Chang Y, et al. In vivo immune modulatory activity of hepatic stellate cells in mice. Hepatology. 2006;44:1171–81.
Chou HS, Hsieh CC, Yang HR, et al. Hepatic stellate cells regulate immune response by way of induction of myeloid suppressor cells in mice. Hepatology. 2011;53:1007–19.
Acknowledgements
This work was supported in part by NIH grants R01 DK071111 and P30-DK41296. DB received support from Deutsche Forschungsgemeinschaft, grants BE 2559/2–1 and BE 2559/2–2 and SFB 841. JK received support from the Deutsche Forschungsgemeinschaft, grant KL2140/2–1 and SFB 841. JK and DB received support from Forschungsförderungsfonds Medizin, Hamburg University.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Daniel Benten and Johannes Kluwe contributed equally to this work.
Rights and permissions
About this article

Cite this article
Benten, D., Kluwe, J., Wirth, J.W. et al. A humanized mouse model of liver fibrosis following expansion of transplanted hepatic stellate cells. Lab Invest 98, 525–536 (2018). https://doi.org/10.1038/s41374-017-0010-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-017-0010-7
This article is cited by
-
Modeling drug-induced liver injury and screening for anti-hepatofibrotic compounds using human PSC-derived organoids
Cell Regeneration (2023)
-
Computational simulation of liver fibrosis dynamics
Scientific Reports (2022)
-
The prognostic and clinicopathological significance of PD-L1 expression in patients with diffuse large B-cell lymphoma: a meta-analysis
BMC Cancer (2019)
