
Abstract
The kidney is arguably the most important target of microvascular damage in diabetes. A substantial proportion of individuals with diabetes will develop kidney disease owing to their disease and/or other co-morbidity, including hypertension and ageing-related nephron loss. The presence and severity of chronic kidney disease (CKD) identify individuals who are at increased risk of adverse health outcomes and premature mortality. Consequently, preventing and managing CKD in patients with diabetes is now a key aim of their overall management. Intensive management of patients with diabetes includes controlling blood glucose levels and blood pressure as well as blockade of the renin–angiotensin–aldosterone system; these approaches will reduce the incidence of diabetic kidney disease and slow its progression. Indeed, the major decline in the incidence of diabetic kidney disease (DKD) over the past 30 years and improved patient prognosis are largely attributable to improved diabetes care. However, there remains an unmet need for innovative treatment strategies to prevent, arrest, treat and reverse DKD. In this Primer, we summarize what is now known about the molecular pathogenesis of CKD in patients with diabetes and the key pathways and targets implicated in its progression. In addition, we discuss the current evidence for the prevention and management of DKD as well as the many controversies. Finally, we explore the opportunities to develop new interventions through urgently needed investment in dedicated and focused research. For an illustrated summary of this Primer, visit: http://go.nature.com/NKHDzg
Similar content being viewed by others

Of the long-term complications of diabetes, chronic kidney disease (CKD) imposes the highest burden both in terms of financial cost and the effects on daily life. The presence and severity of CKD identify individuals who are at increased risk of adverse health outcomes — including frailty, reduced quality of life, end-stage renal disease (ESRD) and progressive end-organ damage at other sites — and premature mortality. Indeed, excess mortality associated with type 1 diabetes and type 2 diabetes is largely confined to those with CKD1–4. Consequently, preventing and managing CKD in patients with diabetes is a key aim of their overall management.

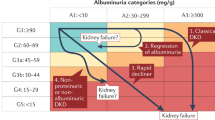
Approximately half of all patients with type 2 diabetes and one-third with type 1 diabetes will develop CKD, which is clinically defined by the presence of impaired renal function or elevated urinary albumin excretion, or both5,6 (Box 1). The percentage of these patients who can be considered to have CKD as a result of their diabetes is unclear. Invariably, other contributors to renal dysfunction are also present, including hypertension, dyslipidaemia, obesity, intrarenal vascular disease, acute kidney injury, glomerular atherosclerosis, renal ischaemia and ageing-related nephron loss. Accordingly, it is seldom possible to precisely define ‘diabetic kidney disease’ (DKD) or ‘diabetic nephropathy' in epidemiology or clinical practice, particularly in patients with type 2 diabetes. Consequently, it is more appropriate to identify patients with diabetes and CKD, and to undertake strategies for holistic renoprotection in patients with diabetes.
DKD was originally described by Mogensen7 in the 1980s as a progressive disease that began with the loss of small amounts of albumin into the urine (30–300 mg per day), known as microalbuminuria or occult or incipient nephropathy. As progressively larger amounts of albumin were lost in the urine, and albuminuria became detectable by the then standard dipstick urinalysis (>300 mg per day), the terms macroalbuminuria or overt nephropathy were used. This presentation was then classically followed by a relentless decline in kidney function, renal impairment and ultimately ESRD. This paradigm has proved useful in clinical studies, especially in type 1 diabetes, for identifying cohorts who are at increased risk of adverse health outcomes. However, any boundary between stages is artificial, and the relationship between urinary albumin excretion and adverse health outcomes is log-linear in clinical practice8. Moreover, many patients with type 1 diabetes, and most with type 2 diabetes, do not follow this classic course in modern clinical practice. For example, many patients with diabetes and renal impairment do not show excessive urinary albumin loss9,10. Indeed, of the 28% of the United Kingdom Prospective Diabetes Study (UKPDS) cohort who developed an estimated glomerular filtration rate (eGFR) of <60 ml/min/1.73 m2, half did not have preceding albuminuria11. Even in the Diabetes Control and Complications Trial (DCCT), of the 11% of patients with type 1 diabetes who developed an eGFR of <60 ml/min/1.73 m2 40% had never experienced overt nephropathy12. Similarly, most patients with microalbuminuria do not develop a progressive increase in their urinary albumin excretion as in the classic paradigm, and treatment-induced and spontaneous remission of albuminuria are commonly observed10,13.
Epidemiology
Although improvements in diabetes management have reduced the proportion of individuals with diabetes who develop CKD over any given time period14–16, their improved prognoses17,18 combined with the rising incidence of both type 1 and type 2 diabetes19 have seen the prevalence of CKD continue to grow20. Of the approximately 400 million people with type 2 diabetes worldwide19, approximately half will have evidence of CKD21. Approximately one in five adults with type 2 diabetes will have an eGFR of <60 ml/min/1.73 m2 and between 30% and 50% will have elevated urinary albumin excretion. In the UKPDS, for example, after a median 15 years of follow-up study, albuminuria was observed in 52% of participants and an eGFR of ≤60 ml/min/1.73 m2 in 28% of participants11.
The incidence of CKD in type 1 diabetes differs from that observed in type 2 diabetes. It is estimated that approximately one-third of all people with type 1 diabetes will develop CKD over the course of their lifetime15,22–24. This difference is mostly because subjects with type 1 diabetes are generally younger and healthier at diagnosis and carry fewer co-morbid conditions than those with type 2 diabetes. Consequently, the renal presentation in type 1 diabetes potentially better reflects DKD, rather than the mixed picture of CKD in type 2 diabetes that is confounded by omnipresent other contributors, such as ageing, vascular disease, insulin resistance and obesity.
The incidence, presentation and course of CKD in patients with diabetes vary considerably across countries and settings21 (Fig. 1). For example, African American, Middle Eastern, Hispanic, Asian and Polynesian patients with diabetes have a higher prevalence of elevated urinary albumin/creatinine ratio (ACR) than European populations25. Disadvantaged and minority populations also have a high prevalence of CKD and its subsequent progression. For example, the prevalence of albuminuria is nearly twice as common in Indigenous Australians in primary care compared with non-Indigenous Australian patients presenting to the same clinical practice26. The reasons for ethnic differences in CKD are complex27 and include economic, social or educational disadvantage, access to and uptake of care, lower achievement of treatment goals, lower screening rates, suboptimal early treatment of complications, diet and lifestyle factors, smoking, obesity, genetic factors and developmental programming. Another important feature is the younger age of onset of type 2 diabetes in these at-risk groups, which might be associated with a more malignant course, including accelerated β-cell loss in the pancreas, as well as renal and cardiovascular complications28.

Data from patients with type 2 diabetes surveyed in the US NHANES III4, the Australian NEFRON study5, the Italian RIACE study86,240 and the DEMAND study21. Yellow circles denote the percentage with an estimated glomerular filtration rate (eGFR) of <60 ml/min/1.73 m2. Blue circles denote patients with albuminuria. The percentage not included in either circle denotes patients without chronic kidney disease (CKD).
The cumulative risk of ESRD as a result of diabetes also differs considerably between populations both between and within countries, from <1% to as high as 13%25. This variability partly relates to the competing risk of premature mortality, chiefly owing to cardiovascular disease. Many (and probably most) patients with CKD will die before they develop ESRD13,17,29. Moreover, as most patients with diabetes now reside in developing countries19, the few that develop ESRD will seldom be able to access renal replacement therapy (RRT) programmes. However, the unparalleled number of patients with diabetes makes this disease the leading single cause of ESRD. In many countries, such as the United States, diabetes is present in more than half of all patients entering RRT programmes30.
Mechanisms/pathophysiology
DKD has been traditionally viewed as a microvascular disorder, clustered along with retinopathy and neuropathy, and separate from macrovascular disease that contributes to coronary heart disease, peripheral vascular disease and cerebrovascular disease. However, each disorder can be considered to be a tissue-specific manifestation of the same pathogenetic process, and DKD is the renal manifestation of the same glucose-driven process that occurs at susceptible sites elsewhere in the body31–34. Although all cells are chronically exposed to high plasma glucose levels in patients with diabetes, only some show progressive dysfunction, of which the endothelial cells lining the vasculature are a prime example. Specifically, the inability of endothelial cells to downregulate their glucose transport in response to high glucose levels35 leads to an overwhelming flux of intracellular glucose, which triggers the generation of pathogenetic mediators that contribute to the development of diabetic complications, including DKD.
Reactive oxygen species
Excessive glucose flux leads to the generation of toxic intermediates, the most important of which are thought to be reactive oxygen species (ROS). Excessive glucose flux can generate ROS in several different ways. Enhanced mitochondrial substrate oxidation with consequent enhanced mitochondrial membrane potential leads to the overproduction of superoxide. At the same time, increased glucose flux leads to the activation of NADPH oxidase and uncoupling of nitric oxide synthase36. ROS-mediated DNA strand breaks in the nucleus activate DNA repair mechanisms, including the enzyme poly(ADP ribose) polymerase 1 (PARP1), which inhibits the key glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH) by polyADP-ribosylation. Inhibition of GAPDH activity causes a bottleneck in glycolysis, resulting in the upstream accumulation of early glycolytic intermediates that are increasingly diverted into activating pathogenetic signalling pathways37,38 (Fig. 2). These diversions include increased polyol pathway flux, increased hexosamine pathway activity, increased formation of the highly reactive α-dicarbonyl methylglyoxal, increased expression of the receptor for advanced glycation end-products and its activating ligand S100A8/9, and activation of various protein kinase C (PKC) isoforms. Together, these diversions lead to cellular dysfunction, inflammation, apoptosis and fibrosis in cells exposed to excessive glucose flux. The central importance of ROS in initiating each of these processes is illustrated by the fact that each can be prevented when hyperglycaemia-mediated ROS generation is curtailed38.

Mitochondrial production of reactive oxygen species (ROS) accelerates in response to an increase in intracellular glucose. In addition, pathogenetic ROS are also generated through the ROS-induced uncoupling of nitric oxide synthase (eNOS) and inactivation of NADPH oxidases. ROS can mediate DNA damage, which in turn activates poly(ADP ribose) polymerase (PARP). PolyADP-ribosylation of glyceraldehyde-3-dehydrogenase (GAPDH) by PARP leads to the inhibition of this key glycolytic enzyme and a subsequent bottleneck in glycolysis. As a result, early glycolytic intermediates accumulate and are then diverted into pathogenetic signalling pathways. AGE, advanced glycation end-product; DAG, diacylglycerol; DHAP, dihydroxyacetone phosphate; GFAT, glutamine fructose-6-phosphate amidotransferase; NF-κB, nuclear factor-κB; PKC, protein kinase C; RAGE, receptor for AGE; UDP-GLcNAc, uridine diphosphate N-acetylglucosamine.
Nutrient-sensing pathways
Each cell has pathways that recognize and specifically respond to nutrient abundance to ensure efficient substrate use. The best known of these nutrient sensors include mammalian target of rapamycin (mTOR), 5′ AMP-activated protein kinase (AMPK) and the sirtuins. From the renal perspective, diabetes is sensed as a ‘bonanza state’ of nutrient surfeit that directly leads to changes in the expression and activity of AMPK, sirtuins and mTOR37 and downstream signalling effects on cellular homeostasis, including the downregulation of autophagy, regeneration, mitochondrial biogenesis and other cytoprotective responses that contribute to DKD39.
In addition, podocyte-specific activation of mTOR recapitulates many features of DKD, including mesangial expansion and proteinuria40,41. These findings have led to the concept of directed interventions to simulate energy depletion (associated with increased activity of AMPK and sirtuins and reduced mTOR activity) and promote efficient cellular function. Experimental data seem to support this strategy for renoprotection39–42, and agents such as metformin, peroxisome proliferator-activated receptor (PPAR) agonists37,38, phosphodiesterase inhibitors and resveratrol act on these pathways.
The multifactorial pathogenesis of DKD
Only one-third of patients with type 1 diabetes will develop overt nephropathy15,22–24, whereas almost all patients with type 1 diabetes eventually develop some degree of retinopathy. This suggests that additional risk factors beyond hyperglycaemia must also be involved in DKD. Indeed, although hyperglycaemia is an essential requirement for DKD, it is seldom the only contributor. Pathogenetic pathways initiated and sustained in the kidney by elevated glucose levels can be enhanced by several different factors. These include a range of metabolic factors, including excess fatty acids, carbonyl and oxidative stress, as well as haemodynamic factors, including shear stress induced by transmitted systemic hypertension, impaired autoregulation, hyperperfusion and hypoperfusion, and activation of the renin–angiotensin–aldosterone system (RAAS)43. On their own, these factors do not cause DKD but rather, in the presence of diabetes, feed into and enhance common pathogenetic mechanisms that include increased levels of growth factors, vasoactive hormones, cytokines and chemokines in the kidney. For example, glucose-induced endothelial dysfunction increases vascular susceptibility to shear stress, oxidative stress and other stressors. Endothelial dysfunction and subsequent microvascular rarefaction induced by hyperglycaemia also reduce blood flow while oxygen consumption is increased, leading to hypoxia. In turn, renal hypoxia induces compensatory — but ultimately maladaptive — changes in blood flow, metabolism and polar vasculosis (glomerular neoangiogenesis)44–46.
Key changes in the diabetic glomerulus
Despite the importance of the vascular endothelium in microvascular complications, many investigators propose that the early changes in renal glomeruli are critical for the subsequent development of glomerulosclerosis and nephron dropout (Fig. 3). Among these changes, the most important might be dysfunction of glomerular podocytes, which are highly specialized terminally differentiated cells that cover the urinary side of the glomerular basement membrane (GBM)47. Together with glomerular endothelial cells, podocytes are responsible for the maintenance of the GBM, its charge barrier and the shape and integrity of the glomerular capillary loop; all functions that are compromised in the diabetic glomerulus. The diabetic milieu induces ‘patho-adaptive’ changes in podocytes, including cytoskeletal rearrangement, de-differentiation, apoptosis and autophagy manifested by morphological widening, retraction and flattening (known as effacement), reduced motility, increased formation of intercellular tight junctions, a decrease in slit diaphragm length, glomerular hypertrophy, detachment and dropout48–50 (Fig. 3). Experimental models demonstrate that podocyte-specific injury can recapitulate a diabetes-like phenotype of glomerulosclerosis and tubulointerstitial fibrosis, even in the absence of hyperglycaemia51. Moreover, protecting podocytes from hyperglycaemia with a podocyte-specific deletion of the glucose transporter solute carrier family 2, facilitated glucose transporter member 4 (SLC2A4; also known as GLUT4)51 or from the resulting oxidative stress52 can prevent diabetes-associated albuminuria without restoring normal levels of glucose. Such data place podocytes, and more particularly the dysregulation of their growth and differentiation, at the very centre of the pathogenesis of DKD. Some studies suggest that a reduction in podocyte density might be a useful predictor for DKD and its progression53,54.

Morphological and functional alterations to renal glomeruli are one of the hallmarks of diabetic kidney disease. GBM, glomerular basement membrane.
One of the earliest and most characteristic of all glomerular changes in diabetes is a homogenous thickening of the GBM53,55. Thickening of the GBM is present in almost all patients with diabetes within a few years of diagnosis, although more pronounced changes are observed in DKD56. Whether GBM thickening is a marker of podocyte or endothelial dysfunction or a mediator of progressive DKD is unclear. Certainly, changes in the composition, charge or architecture of the GBM associated with thickening could contribute to albuminuria. Stiffening of the GBM might also reduce distensibility of the pericapillary wall and compromise the subpodocyte space, facilitating glomerular injury through haemodynamic mechanisms57.
Mesangial cells are also substantially altered by diabetes, undergoing proliferation and hypertrophy while increasing their production of matrix proteins. These changes lead to some of the unique structural features of diabetic glomerulopathy (Fig. 3), including an increase in the fractional volume of the glomerulus occupied by the mesangium (mesangial expansion), focal degeneration of mesangial cells and the mesangial matrix (mesangiolysis)58, and ultimately glomerulosclerosis59. There is a strong link between mesangial matrix expansion and progression of DKD53,55,60. However, unlike podocytes, upregulation of glucose transport into mesangial cells does not recapitulate a diabetic phenotype61, suggesting that crosstalk among podocytes, endothelial and inflammatory cells mediates mesangial matrix expansion rather than it being a direct effect of glucose exposure on mesangial cells. Although the molecular details of how diabetes alters mesangial cells are not completely understood, the importance of mesangial matrix expansion in the development and progression of diabetes-associated glomerulosclerosis is clear. For example, the resulting reduction in capillary surface area as a result of the expansion of the mesangium contributes to glomerular hypertension, proteinuria and reduced glomerular filtration62.
Inflammatory cell recruitment
Diabetes is also associated with the recruitment of activated leukocytes, particularly T cells and macrophages, into the glomerulus and tubulointerstitium, even in the early stages of DKD. Markers of renal and systemic inflammation correlate with albuminuria, matrix deposition and progressive decline in the GFR47,63. The influx of inflammatory cells into the diabetic kidney is partly in response to tissue injury but can also act as a mediator of DKD64,65, as inflammatory cells and their products (for example, cytokines, chemokines, activated complement and ROS) transform the renal microenvironment. In experimental models, inhibition of leukocyte recruitment and accumulation in the diabetic kidney protects against the development of albuminuria and progressive renal damage66,67. Indeed, Rag1-knockout mice, which are deficient in both T and B cells, fail to develop albuminuria associated with diabetes, although renal fibrosis and hyperfiltration still occur65.
Renal tubular dysfunction and fibrogenesis
The renal tubule is also adversely affected by diabetes. Early in diabetes, the increased glucose load delivered to the proximal tubule triggers maladaptive hypertrophy and hyperplasia of the cortical tubuli68 together with upregulation of glucose transport69, possibly to facilitate glucose reabsorption and reduce glucose wasting. However, as a consequence, sodium delivery to the macula densa is reduced and tubulo-glomerular feedback is activated, leading to increased intraglomerular pressure and hyperfiltration70–72. Chronic hyperglycaemia and other metabolic disturbances associated with diabetes also lead to progressive and cumulative atrophy of tubular epithelial cells. In DKD, up to half of the glomeruli are attached to dilated and atrophic tubules, and up to 17% of glomeruli may be atubular73. Such tubular dysfunction results in defective uptake, transcytosis and/or lysosomal processing of filtered protein, alterations that also contribute to albuminuria74.
Tubulointerstitial fibrosis is widely considered to be the final common pathway for loss of renal function in DKD75. Indeed, renal function and prognosis in DKD might ultimately correlate better with tubulointerstitial fibrosis than with classic and early glomerular changes75. It is generally thought that the accumulation of activated myofibroblasts is the major contributor to progressive renal scarring in diabetes. These fibrogenic cells might be derived from several different sources, including transformation of resident fibroblasts and mesenchymal stem cells, recruitment of fibroblasts from the bone marrow, and tubuloepithelial to mesenchymal trans-differentiation76 (Fig. 4).

The myofibroblasts responsible for the matrix deposition that leads to tubulointerstitial fibrosis in diabetic kidney disease (DKD) are derived from various sources. Transformation of local resident fibroblasts, mesenchymal stem cells and bone marrow-derived fibrocytes and the induction of endothelial to mesenchymal and tubuloepithelial to mesenchymal transitions are the main contributors241.
Complex histopathology of DKD
The same clinical presentation of DKD can be associated with a heterogeneous range of pathological features, including nodular or diffuse glomerulosclerosis, tubulointerstitial fibrosis, tubular atrophy and renal arteriolar hyalinosis, alone or in combination. The presence and severity of each of these features are independently associated with the risk of progressive renal disease, but not always with each other77. A histopathological staging system for glomerular lesions has been proposed77 (Box 2). However, its predictive utility remains to be established. Routine renal biopsy is not feasible or clinically appropriate beyond a research setting, and DKD remains a clinical diagnosis in most patients with diabetes.
Diagnosis, screening and prevention
Risk factors for DKD
Several different factors contribute to the development of CKD in patients with diabetes (Box 3). Some of these factors, including hyperglycaemia, hypertension, weight gain and dyslipidaemia, are potentially modifiable through optimized diabetes care. Moreover, robust clinical data show that intensive diabetes management significantly reduces the cumulative incidence of albuminuria, renal impairment and ESRD. Indeed, the major decline in the incidence of CKD over the past 30 years is considered to be largely attributable to improved diabetes care14,15.
Elevated blood glucose
The most important risk factor for CKD is hyperglycaemia. Although there are some structural similarities to other renal diseases, fundamentally, the phenotype of DKD is only observed in the context of elevated glucose levels. Elliot Joslin first hypothesized a relationship between glucose and diabetic complications78. However, the defining prospective clinical study by Jean Pirart and his Belgian colleagues unequivocally demonstrated that the degree and duration of hyperglycaemia were associated with microvascular complications, including CKD79. Subsequently, randomized controlled trials have validated this causal link in both type 1 diabetes80 and type 2 diabetes81,82. Nevertheless, although conventional markers of glucose levels, such as glycated haemoglobin (HbA1c), are associated with the incidence of microalbuminuria, it is also clear that many patients with poor glycaemic control do not develop renal complications, whereas others do despite intensive interventions and dedicated compliance (Fig. 5). This discordance might be because markers such as HbA1c fail to capture the dynamic dysglycaemia associated with diabetes. Indeed, even in the absence of chronic hyperglycaemia, transient hyperglycaemia, transient hypoglycaemia or increased glycaemic variability around a normal mean might have long-lasting and long-term effects on the development and progression of complications related to diabetes, including renal disease83–87.

The risk of development of an albumin excretion rate of >30 mg per day in adults from the FinnDiane study of individuals with type 1 diabetes and no chronic kidney disease (CKD) (orange), and the distribution of glycaemic control (histogram) in those patients with type 1 diabetes who develop microalbuminuria (bars) (P.-H.G. and M.C.T., unpublished observations).
Alternatively, past periods of poor glucose control, even before diagnosis, could also have a long-lasting legacy in the kidney, and therefore the risk for DKD might not be represented by current or recent HbA1c levels. This phenomenon has become known as ‘metabolic memory’ (Ref. 88), ‘metabolic karma’ (Ref. 89) or the ‘legacy effect’ (Ref. 83) and has been used to explain many clinical observations relating to diabetes and its management, including the persistent renal benefits observed as a result of intensive control during the DCCT80 and UKPDS trials81,90 as well as the apparent lack of benefits observed in many short-term and intermediate-term trials (as patient outcomes may be significantly determined by glucose exposure before the commencement of the trials91). The physiological mechanism or mechanisms responsible for metabolic karma remain poorly defined but might include epigenetic programming, remodelling and persistent post-translational modifications, such as advanced glycation end-products89. Further understanding the molecular basis of a metabolic legacy in diabetes will certainly provide new targets for intervention to reduce the burden of CKD in patients with diabetes.
High blood pressure
Elevated blood pressure is an important risk factor for the development CKD in both type 1 and type 2 diabetes92–94. In individuals with type 1 diabetes, blood pressure levels are usually normal at diagnosis, but become elevated proximate to the onset of microalbuminuria95. In type 2 diabetes, other factors contribute to the presence and severity of hypertension, which may precede CKD by many years or follow in its wake. This importance of hypertension to the pathogenesis of renal damage can be partly explained by the loss of renal autoregulation in diabetes, whereby systemic pressure is directly transmitted to vulnerable glomerular capillaries96,97. Consequently, there is no specific cut-off above which the specific risk for CKD can be denoted or below which the therapeutic impact of blood pressure control on the development of albuminuria can be ignored in patients with diabetes.
Blood lipid abnormalities
Dyslipidaemia is another important risk factor for the development of CKD in diabetes. In particular, elevated triglycerides, non-low-density lipoprotein cholesterol, apolipoprotein-B-100 or low high-density lipoprotein (HDL) cholesterol levels are independently associated with the development of CKD in both type 1 and type 2 diabetes98,99. However, conventional lipids and lipoprotein measurements do not fully account for the complex lipid and lipoprotein changes associated with diabetes and/or CKD. For example, HDL might not only lose its vasoprotective, antioxidant and anti-inflammatory properties in CKD, but dysfunctional HDL can be directly pathogenetic100. Detailed analyses of lipid sub-fractions have suggested that HDL3-cholesterol, sphingomyelin, apolipoprotein(a), apolipoprotein A-I and apolipoprotein A-II, apolipoprotein C-I and triglyceride enrichment might all be independently associated with progressive DKD101,102. Attempts have been made using lipidomics to establish a ‘lipid fingerprint’ associated with complications in diabetes103,104. However, exactly which lipids or lipoproteins are the most important in the pathogenesis of CKD in diabetes remains unclear.
Insulin resistance
Insulin resistance is also independently associated with CKD beyond its indirect links with glucose, blood pressure, body weight and lipid control105–107. Insulin-sensitizing interventions (for example, thiazolidinedione therapy, exercise and weight loss) all reduce albuminuria beyond their actions on metabolic control. In podocytes, resistance to insulin signalling, arising from deletion of the insulin receptor or its downstream effectors mTOR or RAC-beta serine/threonine protein kinase (also known as protein kinase AKT2), leads to progressive glomerular damage similar to that observed in diabetes51. Impaired insulin sensitivity also results in altered renal cell glucose metabolism105. At the same time, increased insulin signalling as a result of compensatory hyperinsulinaemia in the setting of pathway-selective insulin resistance might also contribute to abnormal vasoreactivity, angiogenesis, fibrogenesis and other pathways implicated in progressive renal disease108 as well as atherogenesis109.
Obesity
CKD is more prevalent and develops more rapidly in people with diabetes who are obese than their normal-weight counterparts28. This is one major reason why the cumulative incidence of CKD is greater in type 2 diabetes than type 1 diabetes110. Obesity negatively influences the major risk factors associated with CKD, including lipid, blood pressure and glucose control, as well as promoting insulin resistance. Obesity also has direct effects on the kidney, including changes in intraglomerular haemodynamics, increased sympathetic activity, hypertension, systemic inflammation, endothelial dysfunction, altered expression of growth factors and renal compression associated with visceral adiposity. Indeed, even in the absence of diabetes, obesity may be associated with an increased frequency and severity of albuminuria111, and obesity-related glomerulopathy has been extensively described112.
Programming for DKD
The majority of the variability in incident CKD remains unaccounted for by conventional risk factors. Indeed, the long-term survival of some of the very first patients to be treated with insulin without the advantages or intensity of modern treatment regimens suggest that some individuals are ‘protected'. This cannot be explained as simply having the ‘right’ genes. Although an inherited predisposition for DKD is evident113 and several potential loci have been reproducibly associated with CKD (Table 1), most genetic variants associated with CKD lie in non-coding regions. Overall, current evidence suggests that the genetic code explains only a small amount of why some individuals develop CKD and some do not114. Furthermore, any role for these genes, alone or in combination, in the molecular pathobiology of CKD remains to be established114.
Although the genetic programming for CKD remains elusive, risk can be imprinted through other means. In particular, epigenetics has emerged as an increasingly powerful paradigm to understand complex non-Mendelian diseases, including CKD. Persistent epigenetic changes can be acquired during development or in adaptations following environmental exposure (the so-called environmental footprint), including metabolic fluctuations associated with diabetes83,115–117. These epigenetic modifications — including changes in DNA methylation, histone modification and chromatin structure — store, retain and recall past experiences in a way that can shape the transcription of specific genes and, therefore, cellular functions118. Technological advances now make it possible to initiate epigenome-wide association studies119 to identify epigenetic marks associated with disease across the whole genome119,120, with comparable resolution and throughput to genome-wide studies. For example, some studies have identified differentially methylated regions in individuals with diabetes with CKD compared to those without CKD121,122. Many of the genes identified were also differentially expressed, including some that had been previously linked to CKD in genome-association studies. However, the broader utility of epigenetic markers to identify imprinted risk in individual patients beyond conventional risk factors remains to be established.
Some ‘risk’ programming also occurs during gestation and early development and is determined by the intrauterine environment, as well as the pre-conception nutrition and health of both the mother and father123. Cells are more sensitive to this epigenetic programming during development and differentiation, when gene regulatory regions are established. However, programming can also include constitutional or structural endowment. For example, reduction in nephron mass and filtration area associated with intrauterine growth retardation, maternal diabetes or vitamin A deficiency can increase the risk of CKD124–127. At present, intrauterine growth retardation affects one-quarter of live births in developing countries, the same countries in which the risk of diabetes and CKD are also the greatest128.
Estimating the GFR
CKD is a clinical diagnosis made in a patient with a reduction in their eGFR to <60 ml/min/1.73 m2, a persistently elevated urinary albumin excretion or both5,6 (Box 1). The eGFR is a measure of the flux of plasma fluid filtered from the glomerular capillaries into the Bowman's capsule per unit time (Fig. 3). An eGFR <60 ml/min/1.73 m2 is used to denote a moderate to severe renal impairment, and approximates an eGFR more than two standard deviations below the mean eGFR of healthy individuals aged 20–35 years. The eGFR can be inexpensively estimated using an appropriate mathematical formula from the serum creatinine levels, and patient age, gender and ethnicity. This calculation is often performed automatically by clinical pathology services. The importance of this initiative is illustrated by the fact that at least half of all individuals who currently have a reported eGFR of <60 ml/min/1.73 m2 still have a serum creatinine concentration in the normal range, meaning that until recently, renal impairment was frequently undetected in patients with diabetes until late in the course of their disease. However, serum creatinine is notably variable within individuals and is modified by several different factors (such as hydration status, physical activity and muscle mass), meaning that repeat testing is important to verify any abnormal results.
The best formula to accurately estimate GFR remains contentious129, although all widely used formulae will identify the majority of patients who have eGFR values of <60 ml/min/1.73 m2. Newer methods for estimating the GFR, including cystatin C-based formulae, have some advantages130, especially in the high to normal range of GFR for which serial monitoring using cystatin C can be used to accurately identify individuals with rapidly declining renal function (so-called progressors) well before the eGFR declines to <60 ml/min/1.73 m2 (Ref. 131). There is no place for the formal measurement of GFR using inulin, iothalamate or other substrates in the routine clinical assessment of renal function in patients with DKD132,133.
Estimating urinary albumin excretion
The second element used to identify individuals with diabetes and CKD is to detect those with persistently elevated urinary albumin excretion5,6 (Box 1). When the kidneys are healthy, little or no intact albumin enters the urine, meaning that the presence of albumin in the urine can be used to denote abnormal kidney function. Urinary albumin excretion can also be estimated in several different ways. The preferred method measures the concentration of albumin in a urine sample using a sensitive assay, adjusting the result for the urinary creatinine concentration. This metric is known as the ACR and is considered the most practical way to adjust for the void volume and urine concentration5,6. The ACR is best determined in urine collected at the first void in the morning, but can also be performed in a random manner; for example, at the time of a medical visit. Timed urine collections (for example, 4-hourly, overnight or 24-hourly urine collections) are also used but are time-consuming and seldom adequately performed outside hospital settings. Spot tests of urinary albumin concentration are not recommended as the concentration of urine varies considerably from void to void. A positive urinary dipstick test or elevated urine albumin concentration is almost always associated with an abnormal ACR134. However, fewer than half of adults with both type 2 diabetes and an abnormal ACR have an elevated urinary albumin concentration or a positive dipstick test134.
Owing to substantial day-to-day variability in urinary albumin excretion in any one individual (approximately 40%), any abnormal results should always be confirmed in at least one out of two additional samples collected over a 3–6-month period. If albumin excretion is within the normal range in all three initial tests, further screening is repeated on an annual basis. Any negative result, in an individual with previously negative tests, can simply be repeated annually as part of routine assessment for complications, as it is unlikely that significant CKD has been missed. However, any de novo abnormal results should be confirmed with an additional two tests during the 3–6 months5,6.
Cut-off values for defining what constitutes elevated urinary albumin excretion vary from guideline to guideline. The American Diabetes Association and the National Kidney Foundation Kidney Disease Outcomes Quality Initiative (NKF KDOQI) guidelines recommend that in patients with diabetes, the presence of albuminuria is defined by a persistent ACR of ≥30 mg g−1 in either men or women135,136. Other guidelines adjust for gender differences in urinary creatinine arising from differences in muscle mass between men and women (for example, defining albuminuria as persistent ACR of >22 mg g−1 in men and >31 mg g−1 in women), which might more accurately approximate clinical risk in patients with diabetes137. Formulae to estimate urinary albumin excretion using a single sample are also available, and as for GFR estimation, these might better adjust for demographic confounders138.
Screening for CKD in diabetes
All patients with type 2 diabetes should have their renal function screened at least annually post diagnosis, using both ACR and eGFR136, as both criteria are independently as well as synergistically associated with mortality and progression to ESRD139. In adults with type 1 diabetes, annual screening should begin at most 5 years after diagnosis. More frequent monitoring is appropriate for individuals with established renal impairment and those at increased risk of progressive kidney disease (for example, those with proteinuria of >1 g per day). Critically, such screening enables the identification of susceptible individuals so that appropriate preventive actions can be taken. Indeed, identification of risk through screening must be followed by intensification of and/or changes in management, such as those detailed in the next section140.
Other biomarkers of the risk for CKD
Although screening for albuminuria and renal impairment will identify most patients who are at risk of CKD, advanced and irreparable structural damage might already be present by the time CKD is diagnosed. Indeed, an eGFR of <60 ml/min/1.73 m2 denotes a loss of renal function of >50%. At the same time, an adverse prognosis is not inevitable in patients with overt nephropathy and/or a reduced eGFR131. Developing practical ways to identify patients with good prognoses from those with poor prognoses remain important for the management of patients with diabetes and CKD, especially in the primary care setting. Some researchers have developed models incorporating additional clinical variables such as age, ethnicity and retinopathy status for risk stratification141,142, although most of the variability in these models can be predicted on the basis of eGFR and albumin excretion alone143. Nevertheless, incorporating some of these additional patient variables adds to their predictive utility. An unmet clinical need is to identify novel biomarkers that have the potential to both diagnose and risk stratify CKD in patients with diabetes earlier than current techniques. Indeed, a number of individual biomarkers have been proposed (Box 4). Other studies have attempted to more broadly identify at-risk profiles using urine proteomics144, metabolomics145 and analysis of urinary exosomes (for microRNA)146. However, none of these techniques is currently applicable to the hundreds of millions of people with diabetes worldwide.
Management
Diabetes control in patients with established CKD
Intensive management of diabetes, including concurrent control of glucose, lipids and blood pressure as well as diet and lifestyle modifications, can slow the progression of established DKD17,18,147–149. Indeed, some data suggest such approaches can even reverse early glomerulopathy. For example, pancreatic transplantation, which restores normal glucose levels in patients with type 1 diabetes, is able to ameliorate the renal histological changes associated with diabetes150. However, it takes at least 10 years to observe any regression150, and metabolic control with standard therapy can seldom achieve that observed following pancreatic transplantation. Even with intensive management in the robust setting of clinical trials detailed below, many patients with diabetes still experience a progressive decline in renal function. This finding has led to the suggestion that, at best, current therapy simply delays the inevitable. Nonetheless, in the clinical setting, any delay in CKD has potentially profound effects on patient health.
Intensive glucose control
Whether preventing hyperglycaemia is sufficient on its own to treat progressive CKD once it is established is uncertain. Significant reductions in albuminuria and its progression are certainly observed following intensification of glucose control using standard therapies in both type 1 and type 2 diabetes151,152. However, within the limited confines of clinical trials, no significant effect has been observed on other renal outcomes, including doubling of the serum creatinine level, ESRD or death from renal disease151. Nonetheless, 6.5 years of intensive diabetes therapy in the DCCT study was ultimately associated with a 50% reduction in the risk of renal impairment in its follow-up Epidemiology of Diabetes Interventions and Complications (EDIC) study and a modestly lower rate of decline in renal function153. Moreover, this effect seemed to be entirely attributable to improved glucose control153. In addition, over the course of the EDIC study, RRT (haemodialysis, peritoneal dialysis or renal transplantation) was needed in only 8 participants in the intensive-therapy group, whereas 16 patients in the conventional-therapy group required RRT. Furthermore, the Action in Diabetes and Vascular Disease: Preterax and Diamicron MR Controlled Evaluation (ADVANCE) study of 11,140 patients with type 2 diabetes also reported that fewer patients required RRT following intensification of glucose control154 compared with a control group. Moreover, as with the EDIC study, a recent 5-year follow-up of the ADVANCE study confirmed this renal benefit155. However, total ESRD events (RRT plus deaths from renal disease) (Fig. 6) and doubling in serum creatinine were not significantly changed. Whether intensive glycaemic control has any influence on cardiovascular or mortality outcomes when initiated late, that is, after patients have established DKD or cardiovascular disease, also remains controversial91.

Incidence of end-stage renal disease (ESRD) stratified according to intervention group, whereby patients were subjected to either blood pressure (BP) lowering or glucose lowering treatments. Data are presented for sites (n = 144) that were able to follow the majority (≥85%) of patients surviving to participate in post-trial follow-ups, outlining the number of patients who progressed to ESRD and, of these, the number who were receiving renal replacement therapy (RRT) and the number who had died as a result of kidney disease (renal death). Neither BP-lowering nor glucose-lowering treatment significantly reduced the incidence of ESRD155.
Similarly, intensification of glucose control in patients with diabetes and CKD can be problematic as the multiple agents and high doses that are often required exposes patients to an increased risk of adverse drug reactions. Each class of glucose-lowering agent has some limitations (Box 5). In particular, the risk of severe hypoglycaemia is independently associated with a reduced eGFR and elevated urinary albumin excretion156. The increased risk of hypoglycaemia in patients with CKD can be explained by several different factors that include prescribing practices in this setting, altered insulin and drug pharmacology (including drug and metabolite accumulation, inadequate compensatory gluconeogenesis in CKD and flattening of the relationship between mean glucose control and HbA1c). Thus, careful individualized targeting, prescribing, patient education, planning and vigilance for hypoglycaemia are all important components in the management of CKD. Where possible, glucose-lowering agents not associated with hypoglycaemia are preferred, especially those not limited by renal impairment or associated co-morbid conditions such as heart failure. In some patients with CKD, less-intensive glycaemic control might be appropriate. Indeed, there may be a U-shape relationship between HbA1c and adverse outcomes, including hospitalization and mortality in patients with diabetes who have an eGFR of <60 ml/min/1.73 m2 (Ref. 157).
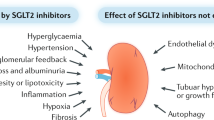
Several glucose-lowering agents are purported to have pleiotropic renoprotective actions in patients with diabetes and CKD beyond glucose lowering158. These drugs include metformin, dipeptidyl peptidase 4 (DPP4) inhibitors159, glucagon-like peptide 1 (GLP1) analogues160, thiazolidinediones161 and sodium/glucose co-transporter 2 (SGLT2) inhibitors162. These putative renal benefits are suggested from studies in which these agents reduced or prevented albuminuria in experimental models or in which renal benefits (such as reduced albuminuria) were observed in patients with DKD. Although plausible mechanisms can explain why such agents are renoprotective, these actions remain to be established by comprehensive clinical trials with a renal focus, although some are currently in progress163,164.
Blood pressure control
Lowering blood pressure is widely regarded the most efficacious treatment for CKD in diabetes, with many clinical trials demonstrating significant reductions in the risk of progression and increased rate of regression of albuminuria following interventions to lower systolic blood pressure165. For example, in the UKPDS trial, a reduction in systolic blood pressure from 154 mmHg to 144 mmHg was associated with a 30% reduction in microalbuminuria166. This benefit seems to occur regardless of whether patients had an elevated blood pressure to begin with167, and no evidence of a threshold for loss of efficacy or J-curve168 has been noted; the risk for albuminuria continues to decrease as the achieved blood pressure falls. Although this relationship might not be the same for mortality in patients with diabetes169, such data provide a renoprotective rationale for aggressively treating all patients with diabetes and CKD with antihypertensive agents, regardless of blood pressure.
Although a target blood pressure of <130/80 mmHg has been previously recommended for patients with diabetes and CKD, when achievable and tolerated, data from the ADVANCE and Action to Control Cardiovascular Risk in Diabetes (ACCORD) trials showed that intensification of blood pressure control failed to consistently improve ESRD, cardiovascular disease or other hard outcomes with the exception of stroke165. Overall, treatment of hypertension in patients with CKD at best only modestly reduces the risk of ESRD170, but exposes patients to increased drug costs, orthostatic symptomatology and potentially hypoperfusion in the setting of impaired autoregulation. Indeed, an increased risk of declining renal function and incident acute kidney injury has also been reported in some studies, which may itself contribute to a progressive decline in renal function in diabetes171. Moreover, the cost and challenges of achieving this level of blood pressure control in many patients has contributed to the 2014 Joint National Council 8 guidelines recommending a relaxed unified target of <140/90 mmHg172. However, stroke risk is also greatest in patients with CKD, and the most appropriate blood pressure target continues to be the subject of avid debate.
Although results from large observational studies suggest that the risk of albuminuria can be reduced by blood pressure reduction, regardless of modality173, the renoprotective efficacy of blockade of the RAAS using angiotensin converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs) seems to be greater than that achieved by other agents with a similar degree of blood pressure reduction167,174. For example, in the Irbesartan in Diabetic Nephropathy Trial (IDNT), fewer patients receiving irbesartan (an ARB) required RRT compared with those receiving amlodipine (a calcium channel blocker)175. However, despite these data, RAAS blockade (any and/or in adequate doses) in patients with diabetes and CKD continues to be underused in routine clinical care176. No differences in the clinical efficacy of ACE inhibitors versus ARBs with respect to reduction in blood pressure are evident, although tolerability and compliance might be greater with ARBs. The combination of ACE inhibitors and ARBs is not recommended in DKD, partly because of the increased risk of acute-on-chronic renal impairment and hyperkalaemia177. The addition of the direct renin inhibitor aliskiren to conventional RAAS blockade in patients with diabetes was also associated with adverse outcomes and had no effect on ESRD, although albuminuria was modestly reduced along with blood pressure levels178. Mineralocorticoid receptor antagonists also significantly reduce albuminuria when added to conventional RAAS blockade179, but are limited by anti-androgenic adverse effects and hyperkalaemia, especially in patients with renal impairment. Newer mineralocorticoid receptor antagonists that reduce these adverse effects are being actively explored for the management of DKD180.
Ultimately, any decision as to which blood-pressure-lowering agent is best is largely academic. Even in a trial setting, most patients require three to four different antihypertensive agents to achieve acceptable blood pressure targets181. Establishing the optimal combination is perhaps a more appropriate clinical question. For example, in the Avoiding Cardiovascular Events through Combination Therapy in Patients Living with Systolic Hypertension (ACCOMPLISH) trial, better renal outcomes (with respect to doubling of serum creatinine, starting dialysis or death) were observed in patients receiving benazepril (an ACE inhibitor) plus amlodipine (a calcium channel blocker) than in those receiving benazepril and hydrochlorothiazide (a diuretic), despite equivalent blood pressure control182.
Blood lipid lowering
Lipid-lowering treatment is widely recommended in all patients with CKD183 to reduce the risk of cardiovascular disease and associated mortality183. Whether lipid lowering also protects the kidneys remains controversial. No clear renoprotective effect of statins in patients with diabetes is evident184,185, and some potential risks have been recently identified186. By contrast, fibrate drugs reduce albuminuria187; whether this effect is mediated by lipid lowering, pleiotropic effects mediated by the activation of PPARα or trans-repression of other targets is unclear188–190. Fenofibrate is also associated with a rapid increase in serum creatinine (∼10–15%), leading to a fall in the eGFR, although the true GFR might be unaffected191. Nonetheless, an agent that increases serum creatinine makes its use in patients with established renal impairment challenging.
Diet and lifestyle interventions
Intensive diet and lifestyle interventions that are frequently recommended to patients with diabetes and CKD include weight loss, increased physical activity, smoking cessation, Mediterranean diet and sodium restriction. Limited research supports the ability of such interventions to reduce risk factors for progressive renal disease and albuminuria192–195. Indeed, the LOOK-AHEAD study reported a significant reduction in incident albuminuria following a multifactorial diet and lifestyle intervention196. However, the ability to truly modify renal progression or co-morbid vascular outcomes remains controversial, and the restrictions imposed by adherence might be associated with a reduced quality of life in precisely those patients who have the shortest life expectancy. Moreover, the beneficial impacts of multifactorial lifestyle intervention on hospitalizations and cost in the LOOK-AHEAD study were not evident among individuals with a history of cardiovascular disease (which is often typical in DKD). Certainly, significant weight loss is associated with reduced incidence of progressive CKD in diabetes, and regression of albuminuria has been observed following bariatric surgery196,197. Avoiding high levels of protein intake (that is, less than 1.3 g of protein per kilogram body weight per day or more than 20% of food energy from protein) is also appropriate for individuals with CKD198. However, formal protein restriction (<0.8 g per kg body weight per day) is not generally recommended as it is difficult to apply and enjoy and might be associated with clinically important risks, including malnutrition and bone remodelling199. Some studies have suggested that a dietary intake of omega-3 polyunsaturated fatty acids200 or omega-3 supplementation201 might also have beneficial effects on albuminuria in CKD.
Managing co-morbidity in patients with CKD
Patients with diabetes and CKD experience an increased risk and severity of other diabetic complications, including retinopathy, neuropathy, gastroparesis, sexual dysfunction, cognitive decline, sleep and mood disorders, heart failure, atrial fibrillation, cardiovascular disease and foot disease. The presence of CKD in a patient with diabetes can be considered a risk marker for each of these conditions33 but it is also often an aggravating factor. The more severe the renal impairment, or the greater the albuminuria, the greater the risk of cardiovascular as well as other complications. For example, myocardial infarction and stroke are approximately twice as common in those with diabetes and CKD than in those with diabetes but without renal disease202,203, and patients with ESRD carry a cardiovascular risk that is at least ten times greater again.
Such is the complexity of the management in CKD, it is common for other diabetic complications (for example, eye or foot disease) to go undiagnosed or to be relatively neglected, even though the risk of non-renal calamity can be very high. The presence of CKD in diabetes necessitates intensive prevention, monitoring and screening and early aggressive treatment of co-morbid disease. Indeed, aggressive multifactorial intervention specifically in patients with CKD has sustained beneficial effects with respect to their other vascular complications and reduces their mortality204. Moreover, the application of such treatments and improved control of risk factors has largely been responsible for the halving of age-standardized mortality in patients with CKD over the past 20 years17. As cardiovascular and cerebrovascular diseases are the major preventable causes of death in patients with diabetes and CKD, particular emphasis should be placed on reducing cardiovascular risk, including lowering lipid levels, treatment of hypertension, smoking cessation and lifestyle modification. Indeed, the absolute benefit from aggressive lipid lowering seems to be greatest in patients with CKD205,206. Low-dose aspirin can also be appropriate for the primary prevention of cardiovascular disease in patients with CKD, as most have a 10-year risk of cardiovascular events of more than 10%207. However, paradoxically harmful effects from antiplatelet therapy have also been reported for aspirin208 and clopidrogel in patients with CKD209. Patients at high risk of CKD can also be considered appropriate for screening for asymptomatic heart disease because early management can improve outcomes, although many of these patients are already maximally medically treated and the utility of cardiac screening beyond risk stratification remains unclear.
Similarly, the multifactorial interventions needed for the management of CKD in diabetes and its associated burden of co-morbid disease frequently exposes patients to iatrogenic complications. In particular, adverse drug reactions are more commonly observed in patients with CKD, which reflects the pill burden, altered pharmacokinetics, interactions with abnormal physiology and other medications, as well as frequently inadequate dose-adjustments in this setting. Appropriate targeting, cautious prescribing, judicious dosing and close monitoring are necessary for all therapies in patients with CKD, especially when multiple practitioners are involved and renal disease is not the primary focus. Given the sheer complexity of multifactorial management in patients with CKD, optimal care is best delivered by comprehensive multidisciplinary teams focused on individual patient needs. Such coordinated care is often limited and challenging to implement in routine clinical practice, although if only one subset of patients with diabetes could be targeted for such an intensive approach, it should be those with CKD.
Managing advanced CKD
Advanced-stage CKD is also associated with a range of complications that require specific additional management, including anaemia, fluid retention, itch, electrolyte disturbances, calciphylaxis and bone demineralization. In each case, these complications are more common, have greater severity and are less well tolerated in patients with diabetes than those without diabetes who have a similar degree of renal impairment210. Patients with CKD are also more vulnerable to episodes of acute kidney injury, including contrast nephropathy, renal ischaemia, hypovolaemia, sepsis, surgery and non-steroidal anti-inflammatory drug-induced acute kidney injury, all of which can be avoided by vigilance, education, close follow-up monitoring and assiduous early management, including stopping RAAS blockade, diuretic use and metformin treatment when appropriate. Ultimately, progressive renal decline requires timely referral to specialist services and, when appropriate, advanced care planning for some form of RRT or conservative care before their renal impairment becomes symptomatic211,212. The optimal timing for any RRT should be determined by individual circumstances, but generally dialysis should be considered when there are signs or symptoms of uraemia, inability to control hydration status or blood pressure or a progressive deterioration in nutritional status. Such individuals are usually identified when the eGFR falls to between 6–9 ml/min/1.73m2. Earlier asymptomatic initiation of dialysis specifically because of diabetes is not warranted, unless uraemic symptoms are difficult to detect and/or close supervision is not feasible.
Quality of life
The presence and severity of CKD in any individual with diabetes is also strongly associated with their health-related quality of life (HRQOL)213,214. The HRQOL in these patients is partly mediated by the presence and severity of co-morbid disease (Fig. 7) and associated risk factors. In parallel, CKD can also affect HRQOL through the burden of multifactorial interventions necessitated by the increased risk for or presence of co-morbid disease, which often leads to a costly time-consuming round of clinical appointments with multiple practitioners across different specialities, contributing to patient confusion, poly-pharmacy and an increased risk of iatrogenic complications156. Clinically relevant improvements in HRQOL in patients with diabetes and CKD can be obtained from structured management programmes that incorporate different specialties. Specific education and support programmes targeting at-risk patients with CKD can also vastly improve diabetes care; such care can be individualized or community-based care215. Formalized education of primary care physicians and other health care providers, as well as systematic management and decision-support programmes, can also improve outcomes for their patients, including HRQOL216–218.

The increased risk of diabetic complications for patients with chronic kidney disease (CKD) means that the management of CKD in diabetes is never only focused on the kidney, but must also involve the pro-active prevention, early detection and effective treatment of all diabetic complications.
Beyond its association with co-morbid disease, CKD can also directly affect HRQOL indices in individuals with diabetes through its negative effects on physical performance, fatigability, appetite, nutrition, immune function, bone mineralization, cognitive function, pruritus and fluid retention. Some of these complications are mediated by the retention of so-called uraemic toxins, which are highest when HRQOL is at its lowest. Renal anaemia might also play an important part in some patients. By the time the eGFR declines to <60 ml/min/1.73 m2, up to one in three individuals with diabetes will have anaemia219. Palliative correction of anaemia using erythropoietin receptor agonists can improve performance and quality of life, but not without considerable cost in terms of the financial cost of the medications themselves, potential for adverse effects220, and the systematic management and follow-up programme they require. Abnormal calcium phosphate homeostasis is also common in patients with CKD, as well as those with reduced HRQOL, but no clear evidence has shown that vitamin D, phosphate binders or calcimimetics improve HRQOL221. Limitations and restriction of certain foods and fluid in patients with advanced-stage CKD also places an additional burden.
However, by far the most important consideration for HRQOL in advanced CKD relates to the initiation of RRT, its appropriateness, its timing, modality and setting. It is beyond the scope of this Primer to discuss the enormous challenges of RRT in patients with diabetes. Importantly, even the finest RRT will at best achieve much less than a naturally functioning kidney, reinforcing the primary importance of renoprotection in the management of diabetes. In addition, RRT will not be appropriate for some patients with diabetes and CKD, because of co-morbidity, frailty, symptomatology and the anticipated excessive burden of therapy.
Outlook
The health implications of the diabetes epidemic are of unparalleled proportions, both in terms of morbidity and mortality as well as the vast health resources that they currently demand and will need in the future. The majority of these resources will be directed towards the prevention and management of diabetic complications, including CKD. A portent of the coming storm can be demonstrated by the Pima Indian population, among whom an ‘outbreak’ of type 2 diabetes began in the late 1950s. Inevitably, in the 1970s, an epidemic of DKD followed that has continued into this century, with a steadily increasing burden of ESRD222. Without effective prevention and treatment, the current global epidemic of diabetes combined with improved survival from heart disease may well lead to a similar crisis of CKD, with overwhelming requirements for RRT and health care systems, particularly in developing counties that carry the greatest burden of type 2 diabetes19. Even in the past 10 years, the number of people with diabetes in RRT programmes has more than doubled30.
Intensive management of diabetes, including control of glucose and blood pressure and blockade of the RAAS, will reduce the incidence of CKD and slow its progression. Indeed, a decline in the incidence of CKD over the past 30 years14,15 and recent plateau in the number of patients with diabetes who develop ESRD is considered to be attributable to improved diabetes care223,224. The prognosis of patients with DKD has also dramatically improved17,18. However, there remain deficiencies in implementation that need to be bridged through pragmatic guidelines and clinical pathways, Phase IV studies and audits, provision of adequate resources, and appropriate targeting of education and support.
An unmet need also remains for innovative treatment strategies for preventing, arresting, treating and reversing CKD in diabetes. Despite initially positive findings, clinical trials of new agents have frustratingly failed to live up to their promise225. Even the failure of early RAAS blockade to reduce the development of CKD226,227 in patients with diabetes has undermined what was widely viewed to be the best means of renoprotection. However, each failure has led to an evolution of our understanding of CKD, and led to newer agents, strategies and designs of clinical trials. Several novel therapies are currently in development225 (Box 6). However, at present, even when used in an optimal combination with standard medical care, renal complications seem to be only modestly reduced at best, and treatment often comes at the considerable expense of additional pill burden, cost and exposure to off-target effects. Given the primacy of CKD in clinical outcomes for those with diabetes, and the current absence of specific treatment, increased investment in CKD research is urgently required.
To provide evidence of efficacy in CKD it is necessary to target robust clinical end points. However, because the progression of renal disease is usually a slow process, over many years or even decades, clinical trials with defined end points such as ESRD are impractical. Although surrogate end points such as change in albuminuria and/or change in eGFR are useful indicators, individually or in combination they might not reflect the true renoprotective potential of interventions. Robust surrogate end points for progressive renal injury in diabetes are urgently needed to facilitate the testing of different strategies. Given the heterogeneous nature of CKD, a systematic panel of biomarkers and molecular phenotypes rather than a one-size-fits-all surrogate approach will probably be essential for the development of new treatments. Improving the design of clinical trials will also be important, including larger risk-enriched cohorts, early selection of responders and early exclusion of those intolerant to therapy, and appropriate surrogate and end point definition.
References
Groop, P. H. et al. The presence and severity of chronic kidney disease predicts all-cause mortality in type 1 diabetes. Diabetes 58, 1651–1658 (2009). A key paper showing the clustering of adverse outcomes in type 1 diabetes in individuals with CKD from the FinnDiane cohort.
Orchard, T. J., Secrest, A. M., Miller, R. G. & Costacou, T. In the absence of renal disease, 20 year mortality risk in type 1 diabetes is comparable to that of the general population: a report from the Pittsburgh Epidemiology of Diabetes Complications Study. Diabetologia 53, 2312–2319 (2010).
Bruno, G. et al. Estimated glomerular filtration rate, albuminuria and mortality in type 2 diabetes: the Casale Monferrato study. Diabetologia 50, 941–948 (2007).
Afkarian, M. et al. Kidney disease and increased mortality risk in type 2 diabetes. J. Am. Soc. Nephrol. 24, 302–308 (2013). This important study shows the clustering of adverse outcomes in type 2 diabetes in individuals with CKD from the NHANES cohort.
Thomas, M. C., Weekes, A. J., Broadley, O. J., Cooper, M. E. & Mathew, T. H. The burden of chronic kidney disease in Australian patients with type 2 diabetes (the NEFRON study). Med. J. Australia 185, 140–144 (2006).
Dwyer, J. P. et al. Renal dysfunction in the presence of normoalbuminuria in type 2 diabetes: results from the DEMAND study. Cardiorenal Med. 2, 1–10 (2012).
Mogensen, C. E., Christensen, C. K. & Vittinghus, E. The stages in diabetic renal disease. With emphasis on the stage of incipient diabetic nephropathy. Diabetes 32 (Suppl. 2), 64–78 (1983).
de Zeeuw, D., Parving, H. H. & Henning, R. H. Microalbuminuria as an early marker for cardiovascular disease. J. Am. Soc. Nephrol. 17, 2100–2105 (2006).
Thomas, M. C. et al. Nonalbuminuric renal impairment in type 2 diabetic patients and in the general population (national evaluation of the frequency of renal impairment co-existing with NIDDM [NEFRON] 11). Diabetes Care 32, 1497–1502 (2009).
Perkins, B. A. et al. Regression of microalbuminuria in type 1 diabetes. N. Engl. J. Med. 348, 2285–2293 (2003).
Retnakaran, R. et al. Risk factors for renal dysfunction in type 2 diabetes: U. K. Prospective Diabetes Study 74. Diabetes 55, 1832–1839 (2006).
Molitch, M. E. et al. Development and progression of renal insufficiency with and without albuminuria in adults with type 1 diabetes in the diabetes control and complications trial and the epidemiology of diabetes interventions and complications study. Diabetes Care 33, 1536–1543 (2010).
Adler, A. I. et al. Development and progression of nephropathy in type 2 diabetes: the United Kingdom Prospective Diabetes Study (UKPDS 64). Kidney Int. 63, 225–232 (2003). This paper has become the seminal natural history study for CKD in type 2 diabetes.
Bojestig, M., Arnqvist, H. J., Hermansson, G., Karlberg, B. E. & Ludvigsson, J. Declining incidence of nephropathy in insulin-dependent diabetes mellitus. N. Engl. J. Med. 330, 15–18 (1994).
Hovind, P. et al. Decreasing incidence of severe diabetic microangiopathy in type 1 diabetes. Diabetes Care 26, 1258–1264 (2003).
Gregg, E. W. et al. Changes in diabetes-related complications in the United States, 1990–2010. N. Engl. J. Med. 370, 1514–1523 (2014).
Andresdottir, G. et al. Improved survival and renal prognosis of patients with type 2 diabetes and nephropathy with improved control of risk factors. Diabetes Care 37, 1660–1667 (2014).
Andresdottir, G. et al. Improved prognosis of diabetic nephropathy in type 1 diabetes. Kidney Int. 87, 417–426 (2015).
International Diabetes Federation (IDF). IDF Diabetes Atlas 6th edn. IDF[online], (2013).
de Boer, I. H. et al. Temporal trends in the prevalence of diabetic kidney disease in the United States. JAMA 305, 2532–2539 (2011). This article explores the implications of the rising diabetes prevalence and changing management on the prevalence of CKD in the United States.
Parving, H. H. et al. Prevalence and risk factors for microalbuminuria in a referred cohort of type II diabetic patients: a global perspective. Kidney Int. 69, 2057–2063 (2006). A global survey documenting the high frequency of CKD in patients with type 2 diabetes worldwide.
Pambianco, G. et al. The 30-year natural history of type 1 diabetes complications: the Pittsburgh Epidemiology of Diabetes Complications Study experience. Diabetes 55, 1463–1469 (2006).
Krolewski, A. S., Warram, J. H., Christlieb, A. R., Busick, E. J. & Kahn, C. R. The changing natural history of nephropathy in type I diabetes. Am. J. Med. 78, 785–794 (1985).
Rossing, P., Rossing, K., Jacobsen, P. & Parving, H. H. Unchanged incidence of diabetic nephropathy in IDDM patients. Diabetes 44, 739–743 (1995).
Sheen, Y. J. & Sheu, W. H. Risks of rapid decline renal function in patients with type 2 diabetes. World J. Diabetes 5, 835–846 (2014).
Thomas, M., Weekes, A. J. & Thomas, M. C. The management of diabetes in indigenous Australians from primary care. BMC Public Health 7, 303 (2007).
Lanting, L. C., Joung, I. M., Mackenbach, J. P., Lamberts, S. W. & Bootsma, A. H. Ethnic differences in mortality, end-stage complications, and quality of care among diabetic patients: a review. Diabetes Care 28, 2280–2288 (2005).
TODAY Study Group et al. A clinical trial to maintain glycemic control in youth with type 2 diabetes. N. Engl. J. Med. 366, 2247–2256 (2012).
Forsblom, C. et al. Competing-risk analysis of ESRD and death among patients with type 1 diabetes and macroalbuminuria. J. Am. Soc. Nephrol. 22, 537–544 (2011).
Jha, V. et al. Chronic kidney disease: global dimension and perspectives. Lancet 382, 260–272 (2013). A detailed review of the current global burden and anticipated future impact of CKD.
Lundbaek, K. Diabetic angiopathy: a specific vascular disease. Lancet 266, 377–379 (1954).
Root, H. F., Pote, W. H. Jr & Frehner, H. Triopathy of diabetes; sequence of neuropathy, retinopathy, and nephropathy in one hundred fifty-five patients. AMA Arch. Intern. Med. 94, 931–941 (1954).
Deckert, T., Feldt-Rasmussen, B., Borch-Johnsen, K., Jensen, T. & Kofoed-Enevoldsen, A. Albuminuria reflects widespread vascular damage. The Steno hypothesis. Diabetologia 32, 219–226 (1989).
Stehouwer, C. D. Endothelial dysfunction in diabetic nephropathy: state of the art and potential significance for non-diabetic renal disease. Nephrol. Dial. Transplant. 19, 778–781 (2004).
Kaiser, N. et al. Differential regulation of glucose transport and transporters by glucose in vascular endothelial and smooth muscle cells. Diabetes 42, 80–89 (1993).
Inoguchi, T. et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 49, 1939–1945 (2000).
Schaffer, S. W., Jong, C. J. & Mozaffari, M. Role of oxidative stress in diabetes-mediated vascular dysfunction: unifying hypothesis of diabetes revisited. Vascul. Pharmacol. 57, 139–149 (2012).
Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 414, 813–820 (2001). A seminal paper describing the mechanics of glucose-dependent toxicity in endothelial cells and their role in microvascular complications.
Dugan, L. L. et al. AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J. Clin. Invest. 123, 4888–4899 (2013).
Inoki, K. et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J. Clin. Invest. 121, 2181–2196 (2011).
Godel, M. et al. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J. Clin. Invest. 121, 2197–2209 (2011).
Tikoo, K., Tripathi, D. N., Kabra, D. G., Sharma, V. & Gaikwad, A. B. Intermittent fasting prevents the progression of type I diabetic nephropathy in rats and changes the expression of Sir2 and p53. FEBS Lett. 581, 1071–1078 (2007).
Cooper, M. E. Pathogenesis, prevention, and treatment of diabetic nephropathy. Lancet 352, 213–219 (1998).
Blantz, R. C. Phenotypic characteristics of diabetic kidney involvement. Kidney Int. 86, 7–9 (2014).
Takiyama, Y. & Haneda, M. Hypoxia in diabetic kidneys. BioMed Res. Int. 2014, 837421 (2014).
Advani, A. & Gilbert, R. E. The endothelium in diabetic nephropathy. Semin. Nephrol. 32, 199–207 (2012).
Reidy, K., Kang, H. M., Hostetter, T. & Susztak, K. Molecular mechanisms of diabetic kidney disease. J. Clin. Invest. 124, 2333–2340 (2014). A detailed review of the pathobiology of DKD.
Hartleben, B. et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J. Clin. Invest. 120, 1084–1096 (2010).
Herman-Edelstein, M. et al. Dedifferentiation of immortalized human podocytes in response to transforming growth factor-β: a model for diabetic podocytopathy. Diabetes 60, 1779–1788 (2011).
Kato, H. et al. Wnt/-catenin pathway in podocytes integrates cell adhesion, differentiation, and survival. J. Biol. Chem. 286, 26003–26015 (2011).
Coward, R. & Fornoni, A. Insulin signaling: implications for podocyte biology in diabetic kidney disease. Curr. Opin. Nephrol. Hypertens. 24, 104–110 (2015).
Susztak, K., Raff, A. C., Schiffer, M. & Bottinger, E. P. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 55, 225–233 (2006).
Harindhanavudhi, T., Parks, A., Mauer, M. & Caramori, M. L. Podocyte structural parameters do not predict progression to diabetic nephropathy in normoalbuminuric type 1 diabetic patients. Am. J. Nephrol. 41, 277–283 (2015).
Meyer, T. W., Bennett, P. H. & Nelson, R. G. Podocyte number predicts long-term urinary albumin excretion in Pima Indians with type II diabetes and microalbuminuria. Diabetologia 42, 1341–1344 (1999).
Ponchiardi, C., Mauer, M. & Najafian, B. Temporal profile of diabetic nephropathy pathologic changes. Curr. Diabetes Rep. 13, 592–599 (2013).
Dalla Vestra, M., Saller, A., Bortoloso, E., Mauer, M. & Fioretto, P. Structural involvement in type 1 and type 2 diabetic nephropathy. Diabetes Metab. 26 (Suppl. 4), 8–14 (2000).
Lewko, B. & Stepinski, J. Hyperglycemia and mechanical stress: targeting the renal podocyte. J. Cell. Physiol. 221, 288–295 (2009).
Wada, T. et al. Nodular lesions and mesangiolysis in diabetic nephropathy. Clin. Exp. Nephrol. 17, 3–9 (2013).
Qian, Y., Feldman, E., Pennathur, S., Kretzler, M. & Brosius, F. C. 3rd. From fibrosis to sclerosis: mechanisms of glomerulosclerosis in diabetic nephropathy. Diabetes 57, 1439–1445 (2008).
Mauer, S. M. et al. Structural–functional relationships in diabetic nephropathy. J. Clin. Invest. 74, 1143–1155 (1984).
Weigert, C. et al. Evidence for a novel TGF-β1-independent mechanism of fibronectin production in mesangial cells overexpressing glucose transporters. Diabetes 52, 527–535 (2003).
Lemley, K. V. et al. Evolution of incipient nephropathy in type 2 diabetes mellitus. Kidney Int. 58, 1228–1237 (2000).
Chow, F., Ozols, E., Nikolic-Paterson, D. J., Atkins, R. C. & Tesch, G. H. Macrophages in mouse type 2 diabetic nephropathy: correlation with diabetic state and progressive renal injury. Kidney Int. 65, 116–128 (2004).
Chow, F. Y. et al. Monocyte chemoattractant protein-1 promotes the development of diabetic renal injury in streptozotocin-treated mice. Kidney Int. 69, 73–80 (2006).
Lim, A. K. & Tesch, G. H. Inflammation in diabetic nephropathy. Mediators Inflamm. 2012, 146154 (2012).
Chow, F. Y., Nikolic-Paterson, D. J., Ozols, E., Atkins, R. C. & Tesch, G. H. Intercellular adhesion molecule-1 deficiency is protective against nephropathy in type 2 diabetic db/db mice. J. Am. Soc. Nephrol. 16, 1711–1722 (2005).
Kanamori, H. et al. Inhibition of MCP-1/CCR2 pathway ameliorates the development of diabetic nephropathy. Biochem. Biophys. Res. Commun. 360, 772–777 (2007).
Osterby, R. & Gundersen, H. J. Glomerular size and structure in diabetes mellitus. I. Early abnormalities. Diabetologia 11, 225–229 (1975).
Rahmoune, H. et al. Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes. Diabetes 54, 3427–3434 (2005).
Vallon, V. et al. Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus. Am. J. Renal Physiol. 304, F156–F167 (2013).
Thomson, S. C. et al. Acute and chronic effects of SGLT2 blockade on glomerular and tubular function in the early diabetic rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 302, R75–R83 (2012).
Vallon, V. & Thomson, S. C. Renal function in diabetic disease models: the tubular system in the pathophysiology of the diabetic kidney. Annu. Rev. Physiol. 74, 351–375 (2012).
Najafian, B., Kim, Y., Crosson, J. T. & Mauer, M. Atubular glomeruli and glomerulotubular junction abnormalities in diabetic nephropathy. J. Am. Soc. Nephrol. 14, 908–917 (2003).
Russo, L. M. et al. Impaired tubular uptake explains albuminuria in early diabetic nephropathy. J. Am. Soc. Nephrol. 20, 489–494 (2009).
Bohle, A. et al. The consequences of tubulo-interstitial changes for renal function in glomerulopathies. A morphometric and cytological analysis. Pathol. Res. Pract. 186, 135–144 (1990).
Wang, B. et al. E-cadherin expression is regulated by miR-192/215 by a mechanism that is independent of the profibrotic effects of transforming growth factor-beta. Diabetes 59, 1794–1802 (2010).
Tervaert, T. W. et al. Pathologic classification of diabetic nephropathy. J. Am. Soc. Nephrol. 21, 556–563 (2010).
Joslin, E. P. Arteriosclerosis in diabetes. Ann. Int. Med. 4, 54–66 (1930).
Pirart, J. Glycaemic control and development of diabetic nephropathy. Acta Endocrinol. Suppl (Copenh.) 242, 41–42 (1981).
de Boer, I. H. & DCCT/EDIC Research Group. Kidney disease and related findings in the diabetes control and complications trial/epidemiology of diabetes interventions and complications study. Diabetes Care 37, 24–30 (2014). A pivotal study following the renal outcomes in the DCCT cohort of participants with type 1 diabetes.
Chalmers, J. & Cooper, M. E. UKPDS and the legacy effect. N. Engl. J. Med. 359, 1618–1620 (2008).
Patel, A. et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 358, 2560–2572 (2008).
Cooper, M. E. & El-Osta, A. Epigenetics: mechanisms and implications for diabetic complications. Circul. Res. 107, 1403–1413 (2010).
Kowluru, R. A. Mitochondria damage in the pathogenesis of diabetic retinopathy and in the metabolic memory associated with its continued progression. Curr. Med. Chem. 20, 3226–3233 (2013).
Pop-Busui, R. et al. DCCT and EDIC studies in type 1 diabetes: lessons for diabetic neuropathy regarding metabolic memory and natural history. Curr. Diabetes Rep. 10, 276–282 (2010).
Penno, G. et al. HbA1c variability as an independent correlate of nephropathy, but not retinopathy, in patients with type 2 diabetes: the Renal Insufficiency and Cardiovascular Events (RIACE) Italian multicenter study. Diabetes Care 36, 2301–2310 (2013).
Waden, J. et al. A1C variability predicts incident cardiovascular events, microalbuminuria, and overt diabetic nephropathy in patients with type 1 diabetes. Diabetes 58, 2649–2655 (2009).
Roy, S., Sala, R., Cagliero, E. & Lorenzi, M. Overexpression of fibronectin induced by diabetes or high glucose: phenomenon with a memory. Proc. Natl Acad. Sci. USA 87, 404–408 (1990).
Thomas, M. C. Glycemic exposure, glycemic control, and metabolic karma in diabetic complications. Adv. Chronic Kidney Dis. 21, 311–317 (2014).
Holman, R. R. 10-year follow-up of intensive glucose control in type 2 diabetes N. Engl. J. Med. 359, 1577–1589 (2008).
Bianchi, C. & Del Prato, S. Metabolic memory and individual treatment aims in type 2 diabetes — outcome-lessons learned from large clinical trials. Rev. Diabet. Stud. 8, 432–440 (2011).
Derby, L., Warram, J. H., Laffel, L. M. & Krolewski, A. S. Elevated blood pressure predicts the development of persistent proteinuria in the presence of poor glycemic control, in patients with type I diabetes. Diabete Metab. 15, 320–326 (1989).
Schmitz, A., Vaeth, M. & Mogensen, C. E. Systolic blood pressure relates to the rate of progression of albuminuria in NIDDM. Diabetologia 37, 1251–1258 (1994).
Tanaka, Y. et al. Role of glycemic control and blood pressure in the development and progression of nephropathy in elderly Japanese NIDDM patients. Diabetes Care 21, 116–120 (1998).
Ayodele, O. E., Alebiosu, C. O. & Salako, B. L. Diabetic nephropathy — a review of the natural history, burden, risk factors and treatment. J. Natl Med. Assoc. 96, 1445–1454 (2004).
Parving, H. H. et al. Impaired autoregulation of glomerular filtration rate in type 1 (insulin-dependent) diabetic patients with nephropathy. Diabetologia 27, 547–552 (1984).
Christensen, P. K., Hansen, H. P. & Parving, H. H. Impaired autoregulation of GFR in hypertensive non-insulin dependent diabetic patients. Kidney Int. 52, 1369–1374 (1997).
Tolonen, N. et al. Lipid abnormalities predict progression of renal disease in patients with type 1 diabetes. Diabetologia 52, 2522–2530 (2009).
Thomas, M. C. et al. Serum lipids and the progression of nephropathy in type 1 diabetes. Diabetes Care 29, 317–322 (2006).
Moore, K. J. & Fisher, E. A. Dysfunctional HDL takes its toll in chronic kidney disease. Immunity 38, 628–630 (2013).
Jenkins, A. J. et al. Lipoproteins in the DCCT/EDIC cohort: associations with diabetic nephropathy. Kidney Int. 64, 817–828 (2003).
Makinen, V. P. et al. Triglyceride–cholesterol imbalance across lipoprotein subclasses predicts diabetic kidney disease and mortality in type 1 diabetes: the FinnDiane Study. J. Intern. Med. 273, 383–395 (2013).
Reis, A. et al. Top-down lipidomics of low density lipoprotein reveal altered lipid profiles in advanced chronic kidney disease. J. Lipid Res. 56, 13–22 (2014).
Meikle, P. J., Wong, G., Barlow, C. K. & Kingwell, B. A. Lipidomics: potential role in risk prediction and therapeutic monitoring for diabetes and cardiovascular disease. Pharmacol. Ther. 143, 12–23 (2014).
Parvanova, A. I. et al. Insulin resistance and microalbuminuria: a cross-sectional, case-control study of 158 patients with type 2 diabetes and different degrees of urinary albumin excretion. Diabetes 55, 1456–1462 (2006).
Thorn, L. M. et al. Metabolic syndrome in type 1 diabetes: association with diabetic nephropathy and glycemic control (the FinnDiane study). Diabetes Care 28, 2019–2024 (2005).
Groop, L. et al. Insulin resistance, hypertension and microalbuminuria in patients with type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia 36, 642–647 (1993).
Groop, P. H., Forsblom, C. & Thomas, M. C. Mechanisms of disease: pathway-selective insulin resistance and microvascular complications of diabetes. Nat. Clin. Pract. Endocrinol. Metab. 1, 100–110 (2005).
Brown, M. S. & Goldstein, J. L. Selective versus total insulin resistance: a pathogenic paradox. Cell Metabolism 7, 95–96 (2008).
Yokoyama, H. et al. Higher incidence of diabetic nephropathy in type 2 than in type 1 diabetes in early-onset diabetes in Japan. Kidney Int. 58, 302–311 (2000).
Sharma, K. The link between obesity and albuminuria: adiponectin and podocyte dysfunction. Kidney Int. 76, 145–148 (2009).
Tsuboi, N., Utsunomiya, Y. & Hosoya, T. Obesity-related glomerulopathy and the nephron complement. Nephrol. Dial. Transplant. 28 (Suppl. 4), iv108–iv113 (2013).
The Diabetes Control and Complications Trial Research Group. Clustering of long-term complications in families with diabetes in the diabetes control and complications trial. Diabetes 46, 1829–1839 (1997).
Thomas, M. C., Groop, P. H. & Tryggvason, K. Towards understanding the inherited susceptibility for nephropathy in diabetes. Curr. Opin. Nephrol. Hypertens. 21, 195–202 (2012). A detailed review of the role of genetics in determining the risk for DKD.
Okabe, J. et al. Distinguishing hyperglycemic changes by Set7 in vascular endothelial cells. Circul. Res. 110, 1067–1076 (2012).
Brasacchio, D. et al. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes 58, 1229–1236 (2009).
El-Osta, A. et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J. Exp. Med. 205, 2409–2417 (2008).
Bird, A. Perceptions of epigenetics. Nature 447, 396–398 (2007).
Rakyan, V. K., Down, T. A., Balding, D. J. & Beck, S. Epigenome-wide association studies for common human diseases. Nat. Rev. Genet. 12, 529–541 (2011).
Guerrero-Bosagna, C. & Skinner, M. K. Environmentally induced epigenetic transgenerational inheritance of phenotype and disease. Mol. Cell. Endocrinol. 3–8 (2011).
Ko, Y. A. et al. Cytosine methylation changes in enhancer regions of core pro-fibrotic genes characterize kidney fibrosis development. Genome Biol. 14, R108 (2013).
Miao, F. et al. Evaluating the role of epigenetic histone modifications in the metabolic memory of type 1 diabetes. Diabetes 63, 1748–1762 (2014).
Perera, F. & Herbstman, J. Prenatal environmental exposures, epigenetics, and disease. Reprod. Toxicol. 31, 363–373 (2011).
Nelson, R. G., Morgenstern, H. & Bennett, P. H. Intrauterine diabetes exposure and the risk of renal disease in diabetic Pima Indians. Diabetes 47, 1489–1493 (1998).
Lemley, K. V. A basis for accelerated progression of diabetic nephropathy in Pima Indians. Kidney Int. 63, S38–S42 (2003).
Nelson, R. G., Morgenstern, H. & Bennett, P. H. Birth weight and renal disease in Pima Indians with type 2 diabetes mellitus. Am. J. Epidemiol. 148, 650–656 (1998).
Singh, G. R. & Hoy, W. E. Kidney volume, blood pressure, and albuminuria: findings in an Australian aboriginal community. Am. J. Kidney Dis. 43, 254–259 (2004).
Weil, E. J., Curtis, J. M., Hanson, R. L., Knowler, W. C. & Nelson, R. G. The impact of disadvantage on the development and progression of diabetic kidney disease. Clin. Nephrol. 74 (Suppl. 1), S32–S38 (2010).
Rognant, N., Lemoine, S., Laville, M., Hadj-Aissa, A. & Dubourg, L. Performance of the chronic kidney disease epidemiology collaboration equation to estimate glomerular filtration rate in diabetic patients. Diabetes Care 34, 1320–1322 (2011).
Shlipak, M. G. et al. Cystatin C versus creatinine in determining risk based on kidney function. N. Engl. J. Med. 369, 932–943 (2013).
Krolewski, A. S., Gohda, T. & Niewczas, M. A. Progressive renal decline as the major feature of diabetic nephropathy in type 1 diabetes. Clin. Exp. Nephrol. 18, 571–583 (2014). This paper highlights the importance of identifying and targeting renal decline when managing diabetes and DKD.
Hsu, C. Y. & Bansal, N. Measured GFR as “gold standard”—all that glitters is not gold? Clin. J. Am. Soc. Nephrol. 6, 1813–1814 (2011).
Nori, U. S., Pesavento, T. E. & Hebert, L. A. Measured GFR has limited clinical utility. Am. J. Kidney Dis. 57, 180; discussion 180–181 (2011).
Thomas, M. C. The assessment and management of albuminuria in primary care. Diabetes Res. Clin. Pract. 80, 83–88 (2008).
American Diabetes Association. (9). Microvascular complications and foot care. Diabetes Care 38, S58–S66 (2015).
KDOQI. KDOQI clinical practice guidelines and clinical practice recommendations for diabetes and chronic kidney disease. Am. J. Kidney Dis. 49, S12–S154 (2007).
Lambers Heerspink, H. J. et al. Comparison of different measures of urinary protein excretion for prediction of renal events. J. Am. Soc. Nephrol. 21, 1355–1360 (2010).
Fotheringham, J., Campbell, M. J., Fogarty, D. G., El Nahas, M. & Ellam, T. Estimated albumin excretion rate versus urine albumin–creatinine ratio for the estimation of measured albumin excretion rate: derivation and validation of an estimated albumin excretion rate equation. Am. J. Kidney Dis. 63, 405–414 (2014).
Amin, A. P. et al. The synergistic relationship between estimated GFR and microalbuminuria in predicting long-term progression to ESRD or death in patients with diabetes: results from the Kidney Early Evaluation Program (KEEP). Am. J. Kidney Dis. 61, S12–S23 (2013).
Thomas, M. C., Viberti, G. & Groop, P. H. Screening for chronic kidney disease in patients with diabetes: are we missing the point? Nat. Clin. Pract. Nephrol. 4, 2–3 (2008).
Elley, C. R. et al. Derivation and validation of a renal risk score for people with type 2 diabetes. Diabetes Care 36, 3113–3120 (2013).
Jardine, M. J. et al. Prediction of kidney-related outcomes in patients with type 2 diabetes. Am. J. Kidney Dis. 60, 770–778 (2012).
Thomas, M. C. & Groop, P. H. Diabetes: Assessing renal risk in patients with type 2 diabetes. Nat. Rev. Nephrol. 9, 559–560 (2013).
Roscioni, S. S. et al. A urinary peptide biomarker set predicts worsening of albuminuria in type 2 diabetes mellitus. Diabetologia 56, 259–267 (2013).
Sharma, K. et al. Metabolomics reveals signature of mitochondrial dysfunction in diabetic kidney disease. J. Am. Soc. Nephrol. 24, 1901–1912 (2013).
McClelland, A., Hagiwara, S. & Kantharidis, P. Where are we in diabetic nephropathy: microRNAs and biomarkers? Curr. Opin. Nephrol. Hypertens. 23, 80–86 (2014).
Gæ de, P., Vedel, P., Parving, H.-H. & Pedersen, O. Intensified multifactorial intervention in patients with type 2 diabetes mellitus and microalbuminuria: the Steno type 2 randomised study. Lancet 353, 617–622 (1999).
Gaede, P. et al. Multifactorial intervention and cardiovascular disease in patients with type 2 diabetes. N. Engl. J. Med. 348, 383–393 (2003). A seminal study demonstrating the utility of multifactorial intervention in patients with type 2 diabetes and established CKD.
Zhang, Z. et al. Renoprotective role of the vitamin D receptor in diabetic nephropathy. Kidney Int. 73, 163–171 (2008).
Fioretto, P., Steffes, M. W., Sutherland, D. E. R., Goetz, F. C. & Mauer, M. Reversal of lesions of diabetic nephropathy after pancreas transplantation. N. Engl. J. Med. 339, 69–75 (1998).
Coca, S. G., Ismail-Beigi, F., Haq, N., Krumholz, H. M. & Parikh, C. R. Role of intensive glucose control in development of renal end points in type 2 diabetes mellitus: systematic review and meta-analysis intensive glucose control in type 2 diabetes. Arch. Intern. Med. 172, 761–769 (2012). A meta-analysis exploring the utility of glucose control on renal outcomes in type 2 diabetes.
Fullerton, B. et al. Intensive glucose control versus conventional glucose control for type 1 diabetes mellitus. Cochrane Database Syst. Rev. 2, CD009122 (2014).
DCCT/EDIC Research Group. Intensive diabetes therapy and glomerular filtration rate in type 1 diabetes. N. Engl. J. Med. 365, 2366–2376 (2011).
ADVANCE Collaborative Group. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 358, 2560–2572 (2008).
Zoungas, S. et al. Follow-up of blood-pressure lowering and glucose control in type 2 diabetes. N. Engl. J. Med. 371, 1392–1406 (2014).
Zoungas, S. et al. Severe hypoglycemia and risks of vascular events and death. N. Engl. J. Med. 363, 1410–1418 (2010).
Shurraw, S. et al. Association between glycemic control and adverse outcomes in people with diabetes mellitus and chronic kidney disease: a population-based cohort study. Arch. Intern. Med. 171, 1920–1927 (2011).
Schernthaner, G., Mogensen, C. E. & Schernthaner, G. H. The effects of GLP-1 analogues, DPP-4 inhibitors and SGLT2 inhibitors on the renal system. Diab. Vasc. Dis. Res. 11, 306–323 (2014).
Groop, P. H. et al. Linagliptin lowers albuminuria on top of recommended standard treatment in patients with type 2 diabetes and renal dysfunction. Diabetes Care 36, 3460–3468 (2013).
Tanaka, T., Higashijima, Y., Wada, T. & Nangaku, M. The potential for renoprotection with incretin-based drugs. Kidney Int. 86, 701–711 (2014).
Sarafidis, P. A., Stafylas, P. C., Georgianos, P. I., Saratzis, A. N. & Lasaridis, A. N. Effect of thiazolidinediones on albuminuria and proteinuria in diabetes: a meta-analysis. Am. J. Kidney Dis. 55, 835–847 (2010).
Kohan, D. E., Fioretto, P., Tang, W. & List, J. F. Long-term study of patients with type 2 diabetes and moderate renal impairment shows that dapagliflozin reduces weight and blood pressure but does not improve glycemic control. Kidney Int. 85, 962–971 (2014).
Boehringer Ingelheim. MARLINA — T2D: Efficacy, Safety & Modification of Albuminuria in Type 2 Diabetes Subjects With Renal Disease With LINAgliptin. NCT01792518. ClinicalTrials.gov[online], (2015).
Janssen Research & Development. Evaluation of the Effects of Canagliflozin on Renal and Cardiovascular Outcomes in Participants With Diabetic Nephropathy (CREDENCE).NCT02065791. ClinicalTrials.gov[online], (2015).
Emdin, C. A. et al. Blood pressure lowering in type 2 diabetes: a systematic review and meta-analysis. JAMA 313, 603–615 (2015). A meta-analysis exploring the utility of blood pressure control on renal outcomes in type 2 diabetes.
UK Prospective Diabetes Study Group. Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38. BMJ 317, 703–713 (1998).
Strippoli, G. F., Craig, M., Deeks, J. J., Schena, F. P. & Craig, J. C. Effects of angiotensin converting enzyme inhibitors and angiotensin II receptor antagonists on mortality and renal outcomes in diabetic nephropathy: systematic review. BMJ 329, 828 (2004).
de Galan, B. E. et al. Lowering blood pressure reduces renal events in type 2 diabetes. J. Am. Soc. Nephrol. 20, 883–892 (2009).
Tsika, E. P., Poulimenos, L. E., Boudoulas, K. D. & Manolis, A. J. The J-curve in arterial hypertension: fact or fallacy? Cardiology 129, 126–135 (2014).
Lv, J. et al. Effects of intensive blood pressure lowering on cardiovascular and renal outcomes: a systematic review and meta-analysis. PLoS Med. 9, e1001293 (2012).
Van Buren, P. N. & Toto, R. D. The pathogenesis and management of hypertension in diabetic kidney disease. Med. Clin. North Amer. 97, 31–51 (2013).
James, P. A. et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 311, 507–520 (2014).
Casas, J. P. et al. Effect of inhibitors of the rennin–angiotensin system and other antihypertensive drugs on renal outcomes: systematic review and meta-analysis. Lancet 366, 2026–2033 (2005).
Lewis, E. J., Hunsicker, L. G., Bain, R. P. & Rohde, R. D. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N. Engl. J. Med. 329, 1456–1462 (1993).
Atkins, R. C. et al. Proteinuria reduction and progression to renal failure in patients with type 2 diabetes mellitus and overt nephropathy. Am. J. Kidney Dis. 45, 281–287 (2005).
United States Renal Data System. USRDS 2013 Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States National Institutes of Health (National Institute of Diabetes and Digestive and Kidney Diseases, 2013).
Tobe, S. W. et al. Cardiovascular and renal outcomes with telmisartan, ramipril, or both in people at high renal risk: results from the ONTARGET and TRANSCEND studies. Circulation 123, 1098–1107 (2011).
Parving, H. H. et al. Cardiorenal end points in a trial of aliskiren for type 2 diabetes. N. Engl. J. Med. 367, 2204–2213 (2012).
Bomback, A. S., Kshirsagar, A. V., Amamoo, M. A. & Klemmer, P. J. Change in proteinuria after adding aldosterone blockers to ACE inhibitors or angiotensin receptor blockers in CKD: a systematic review. Am. J. Kidney Dis. 51, 199–211 (2008).
Ruilope, L. M. et al. Rationale, design, and baseline characteristics of ARTS-DN: a randomized study to assess the safety and efficacy of finerenone in patients with type 2 diabetes mellitus and a clinical diagnosis of diabetic nephropathy. Am. J. Nephrol. 40, 572–581 (2015).
Bakris, G. L. A practical approach to achieving recommended blood pressure goals in diabetic patients. Arch. Intern. Med. 161, 2661–2667 (2001).
Bakris, G. L. et al. Renal outcomes with different fixed-dose combination therapies in patients with hypertension at high risk for cardiovascular events (ACCOMPLISH): a prespecified secondary analysis of a randomised controlled trial. Lancet 375, 1173–1181 (2010).
American Diabetes Association. Standards of medical care in diabetes — 2014. Diabetes Care 37, S14–S80 (2014).
Griffin, S. J. et al. Effect of early intensive multifactorial therapy on 5-year cardiovascular outcomes in individuals with type 2 diabetes detected by screening (ADDITION-Europe): a cluster-randomised trial. Lancet 378, 156–167 (2011).
Haynes, R. et al. Effects of lowering LDL cholesterol on progression of kidney disease. J. Am. Soc. Nephrol. 25, 1825–1833 (2014).
de Zeeuw, D. et al. Renal effects of atorvastatin and rosuvastatin in patients with diabetes who have progressive renal disease (PLANET I): a randomised clinical trial. Lancet Diabetes Endocrinol. 3, 181–190 (2015).
Jun, M. et al. Effects of fibrates in kidney disease: a systematic review and meta-analysis. J. Am. College Cardiol. 60, 2061–2071 (2012).
Park, C. W. et al. PPARα agonist fenofibrate improves diabetic nephropathy in db/db mice. Kidney Int. 69, 1511–1517 (2006).
Ren, S. et al. PPAR activation upregulates nephrin expression in human embryonic kidney epithelial cells and podocytes by a dual mechanism. Biochem. Biophys. Res. Commun. 338, 1818–1824 (2005).
Zhao, X. & Li, L. Y. PPAR-α agonist fenofibrate induces renal CYP enzymes and reduces blood pressure and glomerular hypertrophy in Zucker diabetic fatty rats. Am. J. Nephrol. 28, 598–606 (2008).
Ansquer, J. C. et al. Effect of fenofibrate on kidney function: a 6-week randomized crossover trial in healthy people. Am. J. Kidney Dis. 51, 904–913 (2008).
Hansen, H. P., Tauber-Lassen, E., Jensen, B. R. & Parving, H. H. Effect of dietary protein restriction on prognosis in patients with diabetic nephropathy. Kidney Int. 62, 220–228 (2002).
Chadban, S. et al. The CARI guidelines. Prevention and management of chronic kidney disease in type 2 diabetes. Nephrology 15 (Suppl. 1), S162–S194 (2010).
Bello, A. K. et al. Impact of weight change on albuminuria in the general population. Nephrol. Dial. Transplant. 22, 1619–1627 (2007).
Van Huffel, L. et al. Dietary restriction and exercise for diabetic patients with chronic kidney disease: a systematic review. PLoS ONE 9, e113667 (2014).
The Look AHEAD Research Group Effect of a long-term behavioural weight loss intervention on nephropathy in overweight or obese adults with type 2 diabetes: a secondary analysis of the Look AHEAD randomised clinical trial. Lancet. Diabetes Endocrinol. 2, 801–809 (2014).
Carlsson, L. M. et al. The incidence of albuminuria after bariatric surgery and usual care in Swedish obese subjects (SOS): a prospective controlled intervention trial. Int. J. Obes. 39, 169–175 (2015).
Evert, A. B. et al. Nutrition therapy recommendations for the management of adults with diabetes. Diabetes Care 37 (Suppl. 1), S120–S143 (2014).
Stevens, P. E., Levin, A. & Kidney Disease: Improving Global Outcomes Chronic Kidney Disease Guideline Development Work Group Members. Evaluation and management of chronic kidney disease: synopsis of the kidney disease: improving global outcomes 2012 clinical practice guideline. Ann. Intern. Med. 158, 825–830 (2013).
Lee, C. C., Sharp, S. J., Wexler, D. J. & Adler, A. I. Dietary intake of eicosapentaenoic and docosahexaenoic acid and diabetic nephropathy: cohort analysis of the diabetes control and complications trial. Diabetes Care 33, 1454–1456 (2010).
Miller, E. R. 3rdet al. The effects of n-3 long-chain polyunsaturated fatty acid supplementation on biomarkers of kidney injury in adults with diabetes: results of the GO-FISH trial. Diabetes Care 36, 1462–1469 (2013).
de Ferranti, S. D. et al. Type 1 diabetes mellitus and cardiovascular disease: a scientific statement from the American Heart Association and American Diabetes Association. Circulation 130, 1110–1130 (2014).
Newman, D. J. et al. Systematic review on urine albumin testing for early detection of diabetic complications. Health Technol. Assess. 9, iii–vi,xiii–163 (2005).
Gaede, P. Intensive glucose control and cardiovascular disease in type 2 diabetes — should we change the recommended target for glycated hemoglobin? Commentary to ACCORD and ADVANCE trials. Pol. Arch. Med. Wewn. 118, 619–621 (2008).
Shepherd, J. et al. Intensive lipid lowering with atorvastatin in patients with coronary artery disease, diabetes, and chronic kidney disease. Mayo Clin. Proc. 83, 870–879 (2008).
Palmer, S. C. et al. Benefits and harms of statin therapy for persons with chronic kidney disease: a systematic review and meta-analysis. Ann. Intern. Med. 157, 263–275 (2012).
Pignone, M. et al. Aspirin for primary prevention of cardiovascular events in people with diabetes: a position statement of the American Diabetes Association, a scientific statement of the American Heart Association, and an expert consensus document of the American College of Cardiology Foundation. Circulation 121, 2694–2701 (2010).
Kim, A. J. et al. Low-dose aspirin for prevention of cardiovascular disease in patients with chronic kidney disease. PLoS ONE 9, e104179 (2014).
Dasgupta, A. et al. Clinical outcomes of patients with diabetic nephropathy randomized to clopidogrel plus aspirin versus aspirin alone (a post hoc analysis of the clopidogrel for high atherothrombotic risk and ischemic stabilization, management, and avoidance [CHARISMA] trial). Am. J. Cardiol. 103, 1359–1363 (2009).
Johnson, R. J., Freehally, J. & Floege, J. Comprehensive Clinical Nephrology, (Elsevier Health Sciences, 2014).
National Institute for Health and Care Excellence (NICE). Chronic Kidney Disease: Early Identification and Management of Chronic Kidney Disease in Adults in Primary and Secondary Care. NICE Clinical Guideline 73 (NICE, 2008).
KDIGO. Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease KDIGO [online], (2012).
Dukes, J. L., Seelam, S., Lentine, K. L., Schnitzler, M. A. & Neri, L. Health-related quality of life in kidney transplant patients with diabetes. Clin. Transplant. 27, E554–E562 (2013).
Campbell, K. H. et al. Association between estimated GFR, health-related quality of life, and depression among older adults with diabetes: the Diabetes and Aging study. Am. J. Kidney Dis. 62, 541–548 (2013). A key paper exploring the impact of CKD on psychological health and quality of life of affected patients.
Hotu, C. et al. A community-based model of care improves blood pressure control and delays progression of proteinuria, left ventricular hypertrophy and diastolic dysfunction in Maori and Pacific patients with type 2 diabetes and chronic kidney disease: a randomized controlled trial. Nephrol. Dial. Transplant. 25, 3260–3266 (2010).
Rayner, H. C. et al. Does community-wide chronic kidney disease management improve patient outcomes? Nephrol. Dial. Transplant. 29, 644–649 (2014).
Thomas, B. Improving blood pressure control among adults with CKD and diabetes: provider-focused quality improvement using electronic health records. Adv. Chronic Kidney Dis. 18, 406–411 (2011).
Cortes-Sanabria, L. et al. Improving care of patients with diabetes and CKD: a pilot study for a cluster-randomized trial. Am. J. Kidney Dis. 51, 777–788 (2008).
Thomas, M. C., MacIsaac, R. J., Tsalamandris, C., Power, D. & Jerums, G. Unrecognized anemia in patients with diabetes: a cross-sectional survey. Diabetes Care 26, 1164–1169 (2003).
Parfrey, P. S. Critical appraisal of randomized controlled trials of anemia correction in patients with renal failure. Curr. Opin. Nephrol. Hypertens. 20, 177–181 (2011). A review of the utility of correcting anaemia in patients with CKD.
Block, G. A. et al. Phosphate homeostasis in CKD: report of a scientific symposium sponsored by the National Kidney Foundation. Am. J. Kidney Dis. 62, 457–473 (2013).
Nelson, R. G. et al. Development and progression of renal disease in Pima Indians with non-insulin-dependent diabetes mellitus. Diabetic Renal Disease Study Group. N. Engl. J. Med. 335, 1636–1642 (1996).
Burrows, N. R., Li, Y. & Geiss, L. S. Incidence of treatment for end-stage renal disease among individuals with diabetes in the U. S. continues to decline. Diabetes Care 33, 73–77 (2010).
Zoccali, C., Kramer, A. & Jager, K. The databases: renal replacement therapy since 1989 — the European Renal Association and European Dialysis and Transplant Association (ERA-EDTA). Clin. J. Am. Soc. Nephrol. 4 (Suppl. 1), S18–S22 (2009).
Thomas, M. C. Emerging drugs for managing kidney disease in patients with diabetes. Expert Opin. Emerg. Drugs 18, 55–70 (2013). A detailed review of the new treatments being developed for DKD.
Mauer, M. et al. Renal and retinal effects of enalapril and losartan in type 1 diabetes. N. Engl. J. Med. 361, 40–51 (2009).
Bilous, R. et al. Effect of candesartan on microalbuminuria and albumin excretion rate in diabetes: three randomized trials. Ann. Intern. Med. 151, 11–20 (2009).
Chakkarwar, V. A. Smoking in diabetic nephropathy: sparks in the fuel tank? World J. Diabetes 3, 186–195 (2012).
Hoy, W. E. et al. The multidimensional nature of renal disease: rates and associations of albuminuria in an Australian Aboriginal community. Kidney Int. 54, 1296–1304 (1998).
Nymark, M. et al. Serum lipopolysaccharide activity is associated with the progression of kidney disease in Finnish patients with type 1 diabetes. Diabetes Care 32, 1689–1693 (2009).
Thakar, C. V., Christianson, A., Himmelfarb, J. & Leonard, A. C. Acute kidney injury episodes and chronic kidney disease risk in diabetes mellitus. Clin. J. Am. Soc. Nephrol. 6, 2567–2572 (2011).
Forbes, J. M. & Cooper, M. E. Mechanisms of diabetic complications. Physiol. Rev. 93, 137–188 (2013). A detailed review of the molecular biology of diabetic complications.
Ahmed, S. B. et al. Oral contraceptives, angiotensin-dependent renal vasoconstriction, and risk of diabetic nephropathy. Diabetes Care 28, 1988–1994 (2005).
Hovind, P., Rossing, P., Johnson, R. J. & Parving, H. H. Serum uric acid as a new player in the development of diabetic nephropathy. J. Renal Nutr. 21, 124–127 (2011).
Zoppini, G. et al. Lower levels of 25-hydroxyvitamin D3 are associated with a higher prevalence of microvascular complications in patients with type 2 diabetes. BMJ Open Diab. Res. Care 3, e000058 (2015).
Forsblom, C. et al. Added value of soluble tumor necrosis factor-α receptor 1 as a biomarker of ESRD risk in patients with type 1 diabetes. Diabetes Care 37, 2334–2342 (2014).
Jha, J. C., Jandeleit-Dahm, K. A. & Cooper, M. E. New insights into the use of biomarkers of diabetic nephropathy. Adv. Chronic Kidney Dis. 21, 318–326 (2014).
Fufaa, G. D. et al. Association of urinary KIM-1, L-FABP, NAG and NGAL with incident end-stage renal disease and mortality in American Indians with type 2 diabetes mellitus. Diabetologia 58, 188–198 (2015).
Wong, M. G. et al. Circulating bone morphogenetic protein-7 and transforming growth factor-β1 are better predictors of renal end points in patients with type 2 diabetes mellitus. Kidney Int. 83, 278–284 (2013).
Pugliese, G. et al. Reproducibility of albuminuria in type 2 diabetic subjects. Findings from the Renal Insufficiency And Cardiovascular Events (RIACE) study Nephrol. Dial. Transplant. 26, 3950–3954 (2011).
LeBleu, V. S. et al. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 19, 1047–1053 (2013).
Author information
Authors and Affiliations
Contributions
Introduction (K.A.M.J-.D. and M.C.T.); Epidemiology (P-.H.G. and M.C.T.); Mechanisms/pathophysiology (M.B., K. Sustak and M.C.T.); Diagnosis, screening and prevention (K. Sharma and M.C.T.); Management (P.R., S.Z. and M.C.T.); Quality of Life (M.C.T.); Outlook (M.E.C. and M.C.T.); overview of Primer (M.E.C.).
Corresponding author
Ethics declarations
Competing interests
M.C.T. has received honoraria for educational meetings conducted on behalf of AbbVie, Boehringer Ingelheim, Eli Lilly and Company, Merck Sharpe & Dohme, Servier, Novartis, Takeda, Abbott, Allergan and AstraZeneca. M.B. declares no competing interests. K. Susztak has received research support from Boehringer Ingelheim and Biogen Idec for projects not related to this publication, and is on the advisory board of AbbVie. She has received research support from the US National Institutes of Health (NIH), the Juvenile Diabetes Research Foundation (JDRF) and the American Diabetes Association (ADA). K. Sharma has received research support from AbbVie, Boehringer Ingelheim and Stealth Peptides for projects not related to this publication, and is on the scientific advisory board of Merck and Astellas. He is founder of Clinical Metabolomics, and has received research support from the NIH, the JDRF and the ADA. K.A.M.J-.D. has received research grants from Genkyotex and Boehringer Ingelheim. S.Z. has served on the advisory board for Amgen, AstraZeneca, Bristol-Myers Squibb, Merck Sharp & Dohme, Novartis, Sanofi and Takeda Pharmaceuticals. S.Z. has received consultancy fees and honoraria from AstraZeneca, Bristol-Myers Squibb, Janssen-Cilag, Merck Sharp & Dohme and Servier Laboratories. She has received grants from the National Health and Medical Research Council and the Heart Foundation of Australia, and has undertaken institutional contract work for Bristol-Myers Squibb and the Commonwealth Department of Health. P.R. has received consultancy and/or speaking fees (to his institution) from AstraZeneca, Bristol-Myers Squibb, Boehringer Ingelheim, Eli Lilly and Company, Novo Nordisk, Sanofi Aventis, Astellas, AbbVie and Merck Sharp & Dohme. He has received research grants from AbbVie, Novo Nordisk and Astra Zeneca. P.R. has shares in Novo Nordisk. P-.H.G. has received lecture honoraria from Boehringer Ingelheim, AstraZeneca, Genzyme, Novartis, Novo Nordisk, Merck Sharp & Dohme, Eli Lilly and Company and Medscape. M.E.C. has received honoraria and consulting fees from AbbVie, Bayer, Boehringer Ingelheim, Eli Lilly and Company, Merck Sharpe and Dohme, Servier, Takeda, Novo Nordisk and AstraZeneca, as well as research grants from Novo Nordisk and AbbVie.
Rights and permissions
About this article

Cite this article
Thomas, M., Brownlee, M., Susztak, K. et al. Diabetic kidney disease. Nat Rev Dis Primers 1, 15018 (2015). https://doi.org/10.1038/nrdp.2015.18
Published:
DOI: https://doi.org/10.1038/nrdp.2015.18
This article is cited by
-
(Pro)renin receptor mediates tubular epithelial cell pyroptosis in diabetic kidney disease via DPP4-JNK pathway
Journal of Translational Medicine (2024)
-
Prevalence of chronic kidney disease in Tunisian diabetics: the TUN-CKDD survey
BMC Nephrology (2024)
-
DNI-MDCAP: improvement of causal MiRNA-disease association prediction based on deep network imputation
BMC Bioinformatics (2024)
-
Angiotensin II type 1 receptor-associated protein deletion combined with angiotensin II stimulation accelerates the development of diabetic kidney disease in mice on a C57BL/6 strain
Hypertension Research (2024)
-
Prevalence of diabetic kidney disease and the associated factors among patients with type 2 diabetes in a multi-ethnic Asian country
Scientific Reports (2024)
