
Abstract
Renal cell carcinoma represents a model for contemporary classification of solid tumors; however, unusual and unclassifiable cases exist and are not rare in children and young adults. The anaplastic lymphoma kinase (ALK) gene has recently been implicated in subsets of pulmonary, esophageal, breast, and colon cancers. These findings strengthen the importance of molecular classification of carcinomas across different organ sites, especially considering the evolving targeted anticancer therapies with ALK inhibitors. In the current study of six pediatric renal cell carcinomas, two cases exhibited structural karyotypic abnormalities involving the ALK locus on chromosomal band 2p23. Fluorescence in situ hybridization (FISH) studies were positive for an ALK rearrangement in one case, and subsequent 5′ rapid amplification of cDNA ends analysis of this tumor revealed that the 3′ portion of the ALK transcript encoding for the kinase domain was fused in frame to the 5′ portion of vinculin (VCL, NM_003373). The new fusion gene is predicted to have an open reading frame of 4122 bp encoding for a 1374-aa oncoprotein; its expression was shown by immunoblotting with anti-VCL and anti-ALK antibodies in tumor tissue lysates. Immunohistochemistry with the same antibodies demonstrated cytoplasmic and subplasmalemmal localization of the oncoprotein determined by its N-terminal VCL portion. FISH with a custom-designed VCL–ALK dual-fusion probe set confirmed the presence of the fusion in neoplastic cells and demonstrated the potential clinical utility of this approach for detecting VCL–ALK in routinely processed tissue. The five remaining pediatric renal cell carcinomas did not show ALK rearrangement by FISH or ALK expression by immunohistochemistry. The data identify the kidney as a new organ site for ALK-associated carcinomas and VCL as a novel ALK fusion partner. The results should prompt further studies to advance the molecular classification of renal cell carcinoma and help to select patients who would benefit from appropriate targeted therapies.
Similar content being viewed by others
Main
Renal cell carcinoma has an incidence of 209 000 cases per year and is responsible for 102 000 deaths worldwide annually.1, 2 Approximately 80% of all renal carcinoma cases are currently classified as clear cell, papillary, or chromophobe subtypes with discrete molecular abnormalities characteristic for each group.2, 3, 4, 5, 6, 7 Rare subtypes have also been defined by the 2004 World Health Organization classification and include collecting duct, multilocular cystic, mucinous tubular and spindle cell, medullary, and Xp11.2 translocation- and neuroblastoma-associated carcinomas; each of these subtypes constitutes ∼1% of all primary kidney epithelial tumors and the three latter subtypes are diagnosed predominantly in children.8, 9 Additional rare morphologic variants have been proposed;10 however, ∼5% of all renal cell carcinomas do not fit into any of the currently defined categories. Despite the overall low incidence of pediatric renal carcinomas, their pathomorphological spectrum is different, and the fraction of unclassifiable tumors is higher, comprising ∼32% according to a recent series;9 this likely reflects a greater heterogeneity of molecular mechanisms underlying renal carcinomas in children and young adults.
The anaplastic lymphoma kinase (ALK) gene, named after anaplastic large cell lymphoma, was originally identified as a part of a fusion oncogene that resulted from a recurrent t(2;5)(p23;q35) in this malignancy.11 Subsequently, ALK rearrangements were found in several phenotypically different hematologic neoplasms12, 13, 14 and in histogenetically unrelated inflammatory myofibroblastic tumor.15, 16, 17, 18 Recently, ALK rearrangements and EML4-ALK and KIF5B-ALK fusions were discovered in a subset of non-small cell lung carcinomas;19, 20 proteomics reports suggested a possible TPM4-ALK fusion in esophageal carcinoma;21, 22 and an exon array profiling study demonstrated the EML4-ALK fusion in 2.5% of breast and colon cancers.23 ALK is also activated in ∼5 to 35% of neuroblastomas and neuroblastoma cell lines by a different mechanism involving point mutations and/or gene locus amplifications.24, 25
ALK belongs to the insulin receptor superfamily of receptor tyrosine kinases and is ontogenetically silenced in mature human tissues with the exception of certain areas of the brain; however, full-length ALK is expressed in some pediatric sarcomas.26 A total of 16 ALK fusion oncogenes have been reported; all include an obligatory 3′ portion encoding for the kinase domain. The various 5′ partners differ in their function and subcellular localization but are similar in their ubiquitous expression and homopolymerization properties of respective products that lead to an ectopic expression and constitutive activation of the ALK. Oncogenic activation in multiple tumor types makes ALK an attractive therapeutic target, and clinical trials of two kinase inhibitors have recently opened to enroll patients with either hematopoietic or solid tumor malignancies showing ALK alterations.27
In the current study of six pediatric renal cell carcinomas, we identified two cases with chromosomal rearrangements involving the ALK locus on chromosomal band 2p23 and discovered a novel VCL–ALK fusion expressed in one of the cases. Thus, we introduce the kidney as a new organ site of ALK-aberrant carcinoma.
Materials and methods
Clinical Specimens
In all, six cases of renal cell carcinoma were included in this study: cases 1 and 2 were the subjects of recent diagnostic pathology and clinical cytogenetic practices, and cases 3 through 6 from the archives of St Jude Children's Research Hospital (St Jude) were reviewed retrospectively. Clinicopathological information summarized in Table 1 was collected in accord with the National Institute of Health guidelines on human subject research and with the appropriate approval of the institutional review boards of St Jude and the University of Nebraska Medical Center. The cases were re-reviewed by two pathologists, and additional immunohistochemical analyses were performed.
Immunohistochemistry
Immunohistochemistry was performed on 4-μm sections of formalin-fixed, paraffin-embedded tumor tissue using Benchmark XT (Ventana Medical, Tucson, AZ, USA) and BondMax (Leica Microsystems, Bannockburn, IL, USA) automated stainers with the reagents supplied by the manufacturers. The primary antibodies were used according to the recommendations of suppliers, as presented in Table 2. Appropriate positive and negative controls were employed. The two anti-ALK antibodies used were a rabbit polyclonal ALK-11 developed and characterized in detail for clinical utility in anaplastic large cell lymphoma by Steven Morris's laboratory28 and a mouse monoclonal (clone 5A4) ALK/p80 (Thermo Fisher Scientific, Fremont, CA, USA) previously used by the authors to demonstrate an ectopic ALK expression in inflammatory myofibroblastic tumor with CARS-ALK fusion.18 An anaplastic large cell lymphoma with NPM-ALK fusion was used as a positive control for both anti-ALK antibodies utilized in the study. The immunohistochemistry results were scored by two pathologists and recorded by consensus as either positive, focally positive, focally weakly positive, or negative (Table 3).
Cytogenetic Analysis
Conventional cytogenetic analysis was performed on primary renal tumor samples of cases 1 and 2 and a lymph node metastasis sample of case 2 using standard tissue culture and harvesting procedures at the University of Nebraska Medical Center. Briefly, the tumor samples were disaggregated mechanically and enzymatically, and then cultured in RPMI-1640 media supplemented with 20% fetal bovine serum and 1% penicillin/streptomycin-L-glutamine (Irvine Scientific, Santa Ana, CA, USA) for 3–8 days (primary renal tumors) and for 24–48 h (lymph node). Following exposure to colcemid (0.02 μg/ml for 4 h for the primary tumor specimens and 0.2 μg/ml for 35 min for the lymph node specimen) and subsequent hypotonic treatment (0.8% sodium citrate for 30 min for the primary tumor specimens and 0.4% potassium chloride for 20 min for the lymph node specimen), the preparations were fixed with methanol:glacial acetic acid (3:1). Metaphase cells were banded with Giemsa trypsin and the karyotypes were described according to the International System for Human Cytogenetic Nomenclature (ISCN, 2009).29
Fluorescence In Situ Hybridization (FISH)
For analysis of the ALK (2p23) locus, dual-color FISH studies were conducted on destained metaphase cells and cytologic touch preparations of case 1, and 4-μm-thick formalin-fixed paraffin-embedded tissue sections of all six cases using the Vysis LSI ALK break-apart rearrangement probe (Abbott Molecular, Abbott Park, IL, USA). In addition, a dual-color, dual-fusion probe set was custom designed to span the vinculin (VCL; 10q22.1-23) and ALK loci using bacterial artificial chromosome clones RP11-836A7, CTD-2025L2, RP11-1073B3, RP11-664B23, RP11-610M22 spanning VCL and RP11-1062I24, CTD-2245E6, RP11-100C1, RP11-328L16, RP11-983I21, RP11-992E13, CTD-2544I11, RP11-684O3 spanning ALK (Invitrogen, Carlsbad, CA, USA); the clones were identified from the National Center for Biotechnology Information Map Viewer (http://www.ncbi.nlm.nih.gov/mapview) and the University of California, Santa Cruz Genome Browser (http://genome.ucsc.edu). Each bacterial artificial chromosome probe was directly labeled by nick translation with either Spectrum Green- or Spectrum Orange-dUTP per the manufacturer's protocol (Abbott Molecular) and hybridized to metaphase cell preparations of a karyotypically normal donor to confirm correct mapping, optimal signal strength, and lack of cross-hybridization, before proceeding with analysis of the patients' samples. This probe set was used to confirm or assess for the presence of a VCL–ALK fusion in cases 1 and 2, respectively, and to further characterize the cytogenetically complex chromosomal rearrangements observed in case 1. Whole chromosome paint and centromere-specific probes for chromosomes 9 and 10 (Cytocell, Cambridge, UK and Abbott Molecular) were also used to further define the karyotype of case 1.
Hybridization was conducted as previously described.16 Hybridization signals were assessed in 1–3 metaphase cells for each FISH probe combination (case 1) or in 200 interphase nuclei (cases 1–6); strong, well-delineated signals were independently scored by two individuals. An interphase cell specimen was interpreted as abnormal if split ALK break-apart probe signals were detected in >5% of the cells or juxtaposed VCL–ALK probe set signals were detected in >10% of cells (>2 s.d. above the average false-positive rate). Normal peripheral blood lymphocytes served as negative controls. Images were acquired using the CytoVision Image Analysis System (Applied Imaging, Santa Clara, CA, USA).
5′ Rapid Amplification of cDNA Ends (RACE)
RNA was extracted from 0.5 g homogenized frozen tumor tissue using guanidium-isothiocyanate followed by phenol–chloroform extraction (TRIzol reagent; Invitrogen). The RNA pellet was resuspended in diethylpyrocarbonate-treated water and incubated with DNase I (Qiagen, Valencia, CA, USA) for 1 h at room temperature followed by precipitation with isopropyl alcohol.
RACE was performed using 0.5 μg total tumor RNA and the 5′ RACE system from Invitrogen according to the manufacturer's protocol and using the ALK-specific primers described previously.18 The 5′ RACE products were analyzed on 1.2% agarose gels and cloned onto PGEM-T Easy Vector Systems (Promega, Madison, WI, USA) according to the manufacturer's instructions. The cloned DNA was analyzed for inserts by restriction digestion with EcoR1 (Promega); three clones were selected and sequenced using an automated sequencer (Biosystems, Foster City, CA, USA) at the sequencing facility of Hartwell Center for Bioinformatics and Biotechnology (St Jude). The sequences were analyzed using Lasergene v8.0 software (DNASTAR, Madison, WI, USA) and Basic Local Alignment Search Tool–Nucleotide (BLASTN) (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
Reverse Transcription-PCR (RT-PCR)
To confirm the presence of the VCL–ALK fusion transcript in case 1, RT-PCR analysis was performed using first-strand cDNA synthesized from 0.5 μg tumor RNA with the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA) containing a mixture of oligo(dT) and random hexamer primers. The primers were placed in exon 16 of the VCL (forward, 5′-TGCCTCTGATGAATTGAGC-3′) and in exon 20 of the ALK (reverse, 5′-CGGAGCTTGCTCAGCTTGTACTC-3′) to amplify a predicted 159-bp product. Reactions were run on the PTC 200 DNA Engine (MJ Research, Waltham, MA, USA) using previously described protocols.18 Positive (glyceraldehyde 3-phosphate dehydrogenase (GAPDH)) and negative (no reverse transcriptase) controls were concurrently run.
Western Blotting
Protein lysates were obtained from ∼1.0 g homogenized frozen tumor tissue using radioimmunoprecipitation assay buffer (Cell Signaling, Danvers, MA, USA) containing protease inhibitor cocktail (Sigma-Aldrich, St Louis, MO, USA). The protein concentrations were measured using the protein assay dye reagent (Bio-Rad). Following denaturation with β-mercaptoethanol, the tumor lysates were run on 10% sodium dodecyl sulfate polyacrylamide gels (Criterion Precast Gel; Bio-Rad), transferred to polyvinylidene fluoride membranes (Bio-Rad), and incubated with the ALK/p80 monoclonal anti-ALK (Thermo Fisher Scientific) and hVIN-1 monoclonal anti-VCL (Sigma-Aldrich) antibodies according to the manufacturers' recommendations. After a brief washing and subsequent incubation with the goat-anti-mouse horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology), the blots were developed by enhanced chemiluminescence reaction (ECL Detection reagents; GE Healthcare Global, Piscataway, NJ, USA). The human neuroblastoma cell line IMR3230 with a recognized full-length ALK expression and normal kidney tissue were used as controls of ALK and VCL expression, respectively. Mouse monoclonal anti-β-actin primary antibody (Sigma-Aldrich) was used as the protein-loading control.
Results
Clinicopathological Findings
Case 1
A 16-year-old African-American boy with sickle cell trait presented with right flank pain and gross hematuria. The family history included prostate cancer in the maternal grandfather at the age of 46 and breast cancer in the maternal aunt at the age of 33 years. An abdominal ultrasound and computed tomography examination revealed a solid, 6.2 × 4.0-cm mass within the right renal collecting system causing mild hydronephrosis. A needle core biopsy was diagnosed as ‘malignant pleomorphic neoplasm consistent with renal medullary carcinoma of sickle cell trait’. The family refused definitive surgery initially, and a right nephrectomy with regional lymph node sampling was performed after a 5-month delay; preoperative abdominal computed tomography images showed only a few millimeters of increase in the tumor size during this period. Extensive imaging studies were negative for distant metastatic disease. On the basis of imaging and pathology results, the patient was assigned stage III (T1N1M0) and received no adjuvant treatment; he is alive without evidence of disease 4 months after surgery.
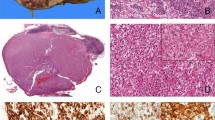
Gross pathological examination showed a 6.5-cm irregularly shaped solid tumor mass with infiltrative borders centered in the right renal medulla with extension into the collecting system, ureter, and renal cortex (Figure 1a). Microscopically, the tumor exhibited a diffuse sheet-like growth pattern with circular accumulations of collagen focally present at the tumor periphery, consistent with the original capsule that by the time of surgery was widely infiltrated. Round, oval, and polygonal tumor cells contained abundant vaguely granular eosinophilic cytoplasm characterized by frequent intracytoplasmic lumina and moderately polymorphic, predominantly vesicular nuclei with small nucleoli, occasional grooves, and rare vacuoles (Figure 1b). Secondary changes, which were likely caused by the previous needle biopsy and intermittent sickling, included thick vascular walls, hemosiderosis, focal poor cellular cohesion, and lymphoplasmacytic infiltrate. The tumor cells expressed keratins AE1/AE3, Cam 5.2, CK7, and EMA, retained INI1, showed TFE3 nuclear staining (Figure 1c), and were negative for CD10, renal antigen, S-100, HMB45, and WT1. Approximately 75% of tumor cells were p53 positive, but the Ki-67 proliferative index was relatively low (5%; Table 3). A small metastasis was identified in one of the ipsilateral paracaval lymph nodes. The tumor had some but not all features of renal medullary carcinoma, chromophobe carcinoma, and transitional cell carcinoma, but the overall pathology, immunohistochemistry, clinical presentation, and preoperative course did not strictly adhere to any of the categories defined by the latest World Health Organization classification, and the diagnosis of renal cell carcinoma, indeterminate subtype, was rendered.


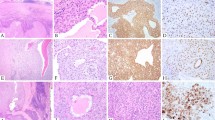
Pathology of six cases of pediatric renal cell carcinoma. (a) Case 1. Bivalved kidney showing an irregularly shaped 6.5-cm solid renal mass expanding to the collecting system and ureter with mild hydronephrosis. (b) Case 1. Hematoxylin and eosin (H&E) staining of a tumor with diffuse sheet-like growth of focally poorly adherent cells with large eosinophilic cytoplasm, intracytoplasmic lumina, mildly pleomorphic nuclei with open chromatin, small nucleoli, and occasional groves. Hemosiderosis and lymphoplasmacytic infiltrate are seen. (c) Case 1. Immunohistochemical analyses using anti-TFE3 antibody shows nuclear staining in tumor cells. (d) Case 2. H&E staining of the primary tumor shows diffuse growth of undifferentiated tumor cells with relatively small cytoplasm and round nuclei with prominent nucleoli. (e) Case 3. H&E staining of undifferentiated tumor cells shows small cytoplasm and large round nuclei with prominent nucleoli growing in sheet-like and cribriform patterns with intervening desmoplastic stroma. (f) Case 4. H&E staining shows eosinophilic and clear tumor cells with small uniform nuclei and papillary tumor architecture. (g) Case 5. H&E staining shows eosinophilic and clear tumor cells with large moderately polymorphic nuclei with open chromatin, small nucleoli, and papillary and acinar tumor architecture. (h) Case 6. H&E staining of predominantly clear tumor cells with mildly polymorphic nuclei with open chromatin, small nucleoli, and papillary and acinar tumor architecture with numerous psammoma bodies. Magnification (panels b–h) × 400.
Case 2
The patient was a 10-year-old African-American girl with sickle cell trait who presented with severe stomach cramps, vomiting, and fever. Radiographic studies demonstrated a mass in the right kidney, as well as mediastinal and cervical lymphadenopathy. A cervical lymph node biopsy showed replacement of the native lymphoid tissue by a poorly differentiated carcinoma arranged in sheets with focal necrosis and perinodal involvement. The tumor cells were immunoreactive for MAK-6 and EMA and negative for desmin, actin, and CD45. In light of the patient's clinical history of sickle cell trait and the presence of a large renal mass, the lymph node lesion was diagnosed as metastatic high-grade carcinoma consistent with renal medullary carcinoma. Following chemotherapy that dramatically shrunk the lymphadenopathy, the patient underwent a radical right nephrectomy. However, 7 months later, she succumbed to disease.
Gross pathological examination of the nephrectomy specimen revealed multiple, partially necrotic tumor nodules involving the superior pole (largest lesion measuring 3.0 × 2.3 cm), renal papillae, and ureteropelvic junction. Microscopically, the malignant cells were arranged in lobules and sheets with focal desmoplasia. Although regions of postchemotherapy necrosis and dystrophic calcifications were noted, significant viable tumor remained with evidence of vascular, lymphatic, and soft tissue invasion in the renal sinus. The neoplastic cells were large and polygonal with a small amount of cytoplasm and large nuclei with prominent eosinophilic nucleoli (Figure 1d). Mitotic rate was brisk (3 to 4 per high-power field) with scattered abnormal multipolar figures. Immunohistochemical analysis at initial examination showed results similar to that observed in the lymph node metastasis. Additional immunohistochemical assessment of the renal tumor cells revealed loss of INI1 and no ALK expression with two anti-ALK antibodies utilized in the study (Table 3).
Cases 3–6
Four additional renal cell carcinomas lacking cytogenetic studies were also subjected to ALK FISH and immunohistochemical analyses at St Jude. Three of the children (cases 3, 4, and 6) were of African-American descent and had documented sickle cell trait; 1 child (case 5) was of Hispanic descent (Table 1). Histopathologically, case 3 was classified as a renal medullary carcinoma (Figure 1e). Cases 4 through 6 exhibited papillary architecture and clear cytoplasm but differed in the nuclear expression of TFE3 (positive in cases 4 and 5 but negative in case 6). Thus, although tumor karyotypes were not available, cases 4 (Figure 1f) and 5 (Figure 1g), originally diagnosed as papillary renal carcinomas in 2002, could have in fact represented Xp11.2 translocation-associated carcinomas. Case 6 also demonstrated morphological (Figure 1h) and some immunohistochemical (Table 3) features assigned to the Xp11.2 translocation-associated carcinoma,31 but lacked expression of TFE3 and TFEB and was, therefore, diagnosed as a renal cell carcinoma, indeterminate subtype. Immunohistochemical analyses using two anti-VCL antibodies were positive in cases 1 through 5, which is consistent with a collecting duct origin of these neoplasms.32
Cytogenetic and FISH Data
Examination of case 1 by conventional cytogenetic analysis and multiple FISH studies on destained metaphase cells revealed the following abnormal complex karyotype: 46,XY,der(2)t(2;10)(p23;q22)t(10;?)(q23;?),−9,der(10)t(2;10)(p23;q22),+der(?)t(9;?)(?q13;?)[1]/47,idem,+der(?2)del(2)(q12)t(2;?)(p23;?)[4].ish der(2)t(2;10)(ALK+;VCL+),+der(?2)del(2)t(2;?)(5′ALK+,3′ALK-),der(10)t(2;10) (ALK+;VCL+),der(?)t(9;?)(wcp9+) (Figure 2a, b, and d). Eleven cells were karyotypically normal. Interphase FISH analysis conducted on cytologic touch preparations and formalin-fixed paraffin-embedded tissue sections with the ALK break-apart probe revealed either one fused (normal chromosome 2 homolog) and one split (indicative of a rearranged ALK locus) orange–green signal pattern (Figure 2c) or one fused and one split orange–green signal pattern with an extra green (5′ ALK) signal, the latter likely corresponding to the der(?2)del(2)t(2;?) in nearly all cells analyzed on the cytologic touch preparation and >50% of cells analyzed from the formalin-fixed paraffin-embedded tissue sections. Similarly, two signal patterns could be appreciated with the dual-color, dual-fusion VCL–ALK probe set. Specifically, a single orange signal (spans VCL) corresponding to the normal chromosome 10 homolog, one to two isolated green signals (spans ALK) representative of the normal chromosome 2 with or without the der(?2)del(2)t(2;?), and two orange–green fused signals indicative of VCL–ALK fusions on the der(2) and der(10) chromosomes of the t(2;10) were identified in metaphase cells and interphase nuclei of case 1 (Figure 2d and e).

Cytogenetic analyses of cases 1 and 2. (a) Representative karyotype of case 1: 47,XY,der(2)t(2;10)(p23;q22)t(10;?)(q23;?),+der(?2)del(2)(q12)t(2;?)(p23;?),−9,der(10)t(2;10)(p23;q22),+der(?)t(9;?)(?q13;?) (arrowheads indicate breakpoints). (b) FISH analysis of a destained metaphase cell from case 1 with the ALK break-apart probe demonstrating a translocation of the orange distal (3′) to ALK signal to chromosome 10 (orange arrowhead) with retention of the green proximal (5′) to ALK signal on the der(2)t(2;10), (green arrow). A green signal is also seen on the der(?2)del(2) (white arrowhead). A fused orange–green signal is visible on the normal chromosome 2 homolog (yellow arrowhead). (c) Rearrangement of the ALK locus in an interphase cell from case 1 represented by the split orange and green signals. (d) FISH analysis of a destained metaphase cell from case 1 with a chromosome 10 CEP probe (aqua) and the VCL–ALK dual-fusion probe set confirming the presence of an orange–green fusion signal on der(2)t(2;10) (yellow arrow) and der(10) (white arrow) chromosomes. The VCL (orange) and the ALK (green) probe signals can be seen on the normal chromosomes 10 and 2, respectively. (e) Interphase FISH with the VCL–ALK dual-fusion probe set in case 1 showing the presence of two VCL–ALK fusions (juxtaposed orange and green signals). (f) Partial karyotype and schematic illustration of the chromosome 2 pericentric inversion identified in the lymph node metastasis of case 2 [inv(2)(p23q11.2)]. (g) The interphase nuclei of the primary renal tumor of case 2 negative for an ALK rearrangement (orange and green signals remain fused).
Karyotypic analysis of case 2 revealed the following abnormal clone in all seven cells detected in the lymph node metastasis: 46,XX,inv(2)(p23q11.2) (Figure 2f). An abnormal clone was not detected in the postchemotherapy primary renal tumor nephrectomy specimen by conventional cytogenetic analysis. Subsequent interphase FISH studies conducted on formalin-fixed paraffin-embedded tissue sections of both the lymph node metastasis and the primary renal tumor were negative for a rearrangement of the ALK locus (Figure 2g) or the presence of a VCL–ALK fusion. Despite repeated attempts, FISH studies were unsuccessful on destained metaphase cells from the lymph node metastasis exhibiting the 2p23 rearrangement, most likely because of the advanced age (>15 years) of the cytogenetic slide preparations.
Cases 3 through 6 were negative for an ALK rearrangement by interphase FISH, a finding concordant with the immunohistochemical analysis (Table 3).
Identification of the 5′ ALK Fusion Partner and VCL–ALK Expression in Case 1
The first round of 5′ RACE did not yield products that could be visualized on the gel; however, a weak, ∼600-bp, band was apparent from the nested reaction. Sequencing analysis of the purified and cloned products of the nested reaction showed that it contained a portion of ALK cDNA (exon 20) immediately preceded by >562 bp of non-ALK sequence. BLASTN analysis revealed the non-ALK sequence to be 100% identical to the portion of VCL mRNA (NCBI reference number NM_3373) that contained exons 13 through 16 of the gene. Confirmatory RT-PCR performed using independently synthesized tumor cDNA and VCL exon 16 forward and ALK exon 20 reverse primers yielded a predicted 159-bp product (Figure 3a), the sequencing of which confirmed the breakpoint (Figure 3b).

Breakpoint of the VCL–ALK fusion gene and expression of the VCL–ALK chimeric protein in case 1. (a) Electrophoresis of products from RT-PCR analysis of the tumor cDNA with the VCL exon 16 forward and ALK exon 20 reverse primers shows an expected 159-bp product (column 2, arrow). In column 1, GAPDH was amplified as a positive control of tumor cDNA, and in column 3, a negative RT-PCR control reaction was run. (b) Sequence of the VCL–ALK fusion breakpoint with in-frame translation (medium line); the 811 residue on VCL (proline) is immediately followed by the 1058 residue on ALK (valine). The breakpoint (marked by a vertical black line in the center of the sequence) is located at the beginning of the ALK exon 20. (c) Western blot analysis of protein lysates. Column 1, neuroblastoma line IMR32; column 2, tumor tissue, column 3, normal kidney tissue. In line 1, immunoblotting with the anti-VCL antibody hVIN1 shows an ∼117-kDa product in all three samples, consistent with the wild-type VCL expression. An ∼150-kDa additional product is seen in the tumor (arrow), consistent with the expression of abnormally sized fusion protein. In line 2, immunoblotting with the ALK/p80 antibody shows an ∼180-kDa band consistent with full-length ALK in the neuroblastoma line IMR32. An aberrantly sized (∼150 kDa) product (arrow), consistent with the ALK-VCL fusion is seen in the tumor. No ALK expression is observed in normal renal tissue. In line 3, immunoblotting with the anti-β-actin antibody shows sample-loading control. (d) Immunohistochemistry with the anti-ALK antibody ALK11 shows cytoplasmic and subplasmalemmal staining of tumor cells. (e) Immunohistochemistry with the anti-VCL antibody hVIN1 shows subplasmalemmal and cytoplasmic staining identical to that observed with the ALK11 antibody (d), which is consistent with colocalization of VCL and ALK reaction products to the VCL subcellular domains. Magnification (panels d and e) × 600.
Western blot analysis of tumor protein lysates with anti-VCL (hVIN1) anti-ALK (ALK/p80) antibodies showed an abnormally shifted, ∼150 kDa band consistent with the predicted fusion protein (Figure 3c). Immunohistochemistry of the tumor with both anti-ALK antibodies showed cytoplasmic staining with subplasmalemmal augmentation (Figure 3d), consistent with an ectopic ALK expression. The immunohistochemical staining patterns with anti-ALK and anti-VCL antibodies were identical, thereby demonstrating the colocalization of both reaction products in tumor cells (Figure 3d and e).
ALK Immunohistochemical Analysis of Cases 2–6
Both anti-ALK antibodies utilized in the study showed identical negative results; lack of ALK expression by immunohistochemistry correlated with negative ALK FISH rearrangement data (Table 3).
Discussion
Of the six examined pediatric renal neoplasms in our series, two exhibited structural karyotypic abnormalities of the chromosomal band 2p23 harboring the ALK gene. The ALK gene rearrangement was originally discovered in anaplastic large cell lymphoma, and the NPM1 gene was its most common translocation partner.11 Subsequently, recurrent ALK chimeric oncogenes involving diverse partner genes (RanBP2, CLTC, CARS, and others) were identified in inflammatory myofibroblastic tumors.15, 16, 17, 18 More recently, the ALK gene has been shown to be involved in small subsets of epithelial malignancies, including carcinomas of pulmonary, esophageal, mammary gland, and gastrointestinal origin.19, 20, 21, 22
To the best of our knowledge, this is the first report of a recurrent ALK locus rearrangement in a primary kidney tumor. Although 2p23 chromosomal abnormalities have not been previously emphasized as recurrent in renal cell carcinoma, a review of the literature and search of the Mitelman database (http://www.ncbi.nlm.nih.gov/cancerchromosomes) yielded four additional primary renal carcinomas and one renal carcinoma cell line exhibiting karyotypic aberrations at or near the ALK gene5, 33, 34, 35, 36 (Table 4). Notably, in contrast to cytogenetic findings in the current two cases, the earlier reported 2p23–25 rearrangements in renal tumors were accompanied by multiple numerical and structural abnormalities that are common in renal carcinomas and include deletions/rearrangements of the short arm of chromosome 3, loss of chromosome Y, and hypodiploidy. The significance of 2p23–25 rearrangements in this latter backdrop of frequently occurring renal carcinoma-associated anomalies is not known.
Further characterization of the two renal cell carcinomas with structural chromosomal abnormalities of 2p23 revealed an ALK rearrangement by metaphase and interphase FISH in case 1, but not in case 2. Significantly, the VCL gene that spans the chromosomal region 10q22.1-23 was identified as the ALK fusion partner involved in the t(2;10) in case 1. Involvement of VCL by a molecular alteration has not been previously reported in any tumor type in humans or animals.
The VCL gene encodes for two transcript variants. The first is the ubiquitously expressed VCL, and the second is the muscle-specific isoform metavinculin that includes an additional 68-amino acid (aa) sequence encoded by an alternatively spliced exon 19.37 The VCL RNA encodes for a 1066-aa protein that has a molecular weight of ∼117 KDa. The structure of the protein is well studied; VCL has a head domain (residues 1–835) and a tail domain (residues 896–1066) that are connected by a flexible hinge region.38 VCL functions as an adhesion protein that couples extracellular matrix through integrins to the acto-myosin cytoskeleton. VCL binds talin at the head and actin at the tail; binding to talin opens hydrogen bonds, thereby allowing homodimerization of VCL molecules in their active conformational forms.39 Animal experiments showed the role of VCL in the normal development of muscle and nervous systems; even heterozygous inactivation of VCL (Vcl+/−) is sufficient to evoke heart problems in mice,40 and missense mutations of metavinculin have been demonstrated in humans with dilated and hypetrophic cardiomyopathies.41 VCL participates in the focal adhesion complexes located on the lateral surfaces of glandular epithelium; it is expressed in kidneys and VCL immunohistochemical studies demonstrated labeling of the collecting ducts, as well as tumors of collecting duct origin.32 The role of VCL in human malignancies has not been demonstrated; however, knockout VCL cells showed increased cellular migration speed and enhanced proliferation in vitro.42 These findings led to the hypothesis that cancer cells with decreased VCL expression might be highly metastatic.
The predicted VCL–ALK gene has an open reading frame of 4122 nucleotides and encodes for a 1374-aa fusion protein that contains the first 811-aa residues of VCL with most of its head domain and 563-aa residues of ALK to include the entire kinase domain. Similar to previously reported ALK fusion partners, VCL is ubiquitously expressed and self-polymerizes because of the multivalent binding of the head to talin, which anchors the VCL head to the inner plasmolemmal surfaces.43 In the current study, we demonstrated that VCL–ALK fusion-positive tumor cells immunoreacted with antibodies directed against the C-terminal ALK portion and full-length VCL. The observed colocalization of the signals in subplasmalemmal regions and cytoplasm is consistent with the predicted subcellular localization of the oncoprotein determined by the N-terminal head domain of VCL. The fusion product lacks the VCL tail; thus, it cannot bind F-actin and function as a part of the focal adhesion complexes. The spatial disruption of the VCL head and tail as a result of the rearrangement would preclude effective mechanical coupling of integrins to the cytoskeleton. The neoplastic cells would, therefore, experience functional consequences of the VCL haploinsufficiency that may influence the shape of tumor cells, as well as proliferative and metastatic potentials of the tumor. Consequently, the intracytoplasmic lumina and poor cellular cohesion observed in case 1 may be related to VCL deficiency, which reduces cellular stiffness and adhesion in vitro.44
Before chemotherapy, a clonal inversion involving 2p23 was identified in the lymph node metastasis of case 2. Cytogenetic analysis of the primary postchemotherapy, partially necrotic renal tumor was normal, which most likely reflected stromal overgrowth while the cells were maintained in culture. Neither an ALK rearrangement by interphase FISH nor ALK expression by immunohistochemistry could be identified in the primary tumor or lymph node metastasis; interphase FISH for VCL–ALK fusion was negative, and further molecular studies were precluded by lack of additional fresh or frozen tissue. Therefore, the matter of ALK involvement in case 2 remains open.
Small percentages (1.8–2.0%) of cells with an ALK rearrangement by FISH and lack of ALK expression by immunohistochemistry have been reported in some non-small cell lung carcinomas with EML4-ALK fusion transcripts detected by RT-PCR.45 These findings question the relevance of EML4-ALK transcript expression not accompanied by corresponding protein expression and/or definitive FISH confirmation, and also emphasize the need for continued refinement of ALK-testing algorithms with implementation of complementary, corroborating approaches when needed. In this sense, the developed dual-fusion VCL–ALK probe set may be useful in future clinical practice to diagnose malignancies carrying the t(2;10) VCL–ALK fusion.
The two tumors with cytogenetic abnormalities of the chromosomal locus harboring the ALK gene differed clinically and histopathologically. Case 1 showed an unusual histology featuring a solid growth of large polygonal cells with polymorphic clear nuclei and abundant eosinophilic cytoplasm with intracytoplasmic lumina and rigid cellular walls. The tumor metastasized to a regional lymph node but otherwise exhibited a slow growth over the 5-month preoperative period and lack of distant metastatic disease in 9 months since the initial presentation. Case 2 displayed a primitive cellular morphology with abundant necrosis, high proliferative rate, and rapid progressive clinical course with distant metastatic disease at presentation. These features, combined with the lack of INI1 expression, were diagnostic of a renal medullary carcinoma. The morphology of case 1 was also dissimilar to that of ALK-negative cases 3 through 6. In fact, the histopathological features of case 1 do not clearly fit within a predefined renal cell carcinoma subtype, although the solid growth pattern, abundant eosinophilic cytoplasm, and large intracytoplasmic lumina (cytoplasmic vacuoles) are somewhat reminiscent of a ‘vacuolated lipoid’ renal carcinoma reported by Bruder et al;9 unfortunately, no cytogenetic data on this tumor were presented. Outside the renal carcinoma spectrum, the morphology of case 1 also resembles the so-called ‘solid signet-ring’ non-small cell lung carcinomas reported as a predominate subtype among ALK-aberrant pulmonary carcinomas in the western world population.46 Interestingly, the neoplastic cells of case 1 also expressed TFE3 immunohistochemically, although an Xp11.2 rearrangement was not identified karyotypically. This might reflect a possible activation or upregulation of the MITF by a mechanism other than translocation and exemplifies a tumor with simultaneous ALK and MITF activation.
Of the six children in our series of renal cell carcinoma of various morphologies, five also had sickle cell trait. Epidemiologic studies have shown an excess frequency of sickle cell anemia, but not sickle cell trait, in African Americans with renal cancer.47 Suggested mechanisms for this association include a chronic renal injury from erythrocyte slugging and an immunosuppressive effect of multiple red blood cell transfusions. Although an association of sickle cell trait and pediatric renal cell carcinoma in a small series is not statistically significant, our data indicate that sickle cell trait is not exclusive to the renal medullary carcinoma subtype to which it was originally linked,48 but could be a risk factor for a broader spectrum of renal tumors, including ALK-associated carcinomas.
The discovery of a novel VCL–ALK fusion produced by a complex t(2;10)(p23;q22) in a pediatric renal cell carcinoma at the DNA, RNA, and protein levels highlights the kidney as a new organ site for ALK-induced malignancies and expands the carcinoma-associated ALK fusion gene partner list to four.19, 20, 21, 22 The predicted structure and function of VCL–ALK oncoprotein follows the archetypical pattern defined by the combined properties of the N-terminal partner (ubiquitous expression and homopolymerization) and the C-terminal kinase domain (constitutive activation). On the basis of the current data, we propose that ALK fusion genes represent an oncogenic driver in a subset of renal cell carcinomas, and that recognition of these cases may be useful in ascertaining a patient's eligibility for emerging tyrosine kinase-targeted therapies, the clinical efficacy of which has been recently reported in a trial of ALK-positive non-small cell lung carcinoma.49 Our findings should stimulate future studies to determine the frequency, morphologic spectrum, and clinical characteristics of ALK-aberrant carcinomas of the kidney.
References
Chow WH, Dong LM, Devesa SS . Epidemiology and risk factors for kidney cancer. Nat Rev Urol 2010;7:245–257.
Rini BI, Campbell SC, Escudier B . Renal cell carcinoma. Lancet 2009;373:1119–1132.
Delahunt B, Eble JN . History of the development of the classification of renal cell neoplasia. Clin Lab Med 2005;25:231–246.
Yan BC, Mackinnon AC, Al-Ahmadie HA . Recent developments in the pathology of renal tumors: morphology and molecular characteristics of select entities. Arch Pathol Lab Med 2009;133:1026–1032.
Rigola MA, Casadevall C, Bernués M, et al. Analysis of kidney tumors by comparative genomic hybridization and conventional cytogenetics. Cancer Genet Cytogenet 2002;137:49–53.
Arsanious A, Bjarnason GA, Yousef GM . From bench to bedside: current and future applications of molecular profiling in renal cell carcinoma. Mol Cancer 2009;8:1–20.
Polascik TJ, Bostwick DG, Cairns P . Molecular genetics and histopathologic features of adult distal nephron tumors. Urology 2002;60:941–946.
Renshaw AA, Granter SR, Fletcher JA, et al. Renal cell carcinomas in children and young adults: increased incidence of papillary architecture and unique subtypes. Am J Surg Pathol 1999;23:795–802.
Bruder E, Passera O, Harms D, et al. Morphologic and molecular characterization of renal cell carcinoma in children and young adults. Am J Surg Pathol 2004;28:1117–1132.
Srigley JR, Delahunt B . Uncommon and recently described renal carcinomas. Mod Pathol 2009;22:S2–S23.
Morris SW, Kirstein MN, Valentine MB, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science 1994;263:1281–1284.
Delsol G, Lamant L, Mariamé B, et al. A new subtype of large B-cell lymphoma expressing the ALK kinase and lacking the 2;5 translocation. Blood 1997;89:1483–1490.
Onciu M, Behm FG, Downing JR, et al. ALK-positive plasmablastic B-cell lymphoma with expression of the NPM-ALK fusion transcript: report of 2 cases. Blood 2003;102:2642–2644.
Chan JK, Lamant L, Algar E, et al. ALK+ histiocytosis: a novel type of systemic histiocytic proliferative disorder of early infancy. Blood 2008;112:2965–2968.
Lawrence B, Perez-Atayde A, Hibbard MK, et al. TPM3-ALK and TPM4-ALK oncogenes in inflammatory myofibroblastic tumors. Am J Pathol 2000;157:377–384.
Bridge JA, Kanamori M, Ma Z, et al. Fusion of the ALK gene to the clathrin heavy chain gene, CLTC, in inflammatory myofibroblastic tumor. Am J Pathol 2001;159:411–415.
Ma Z, Hill DA, Collins MH, et al. Fusion of ALK to the Ran-binding protein 2 (RANBP2) gene in inflammatory myofibroblastic tumor. Genes Chromosom Cancer 2003;37:98–105.
Debelenko LV, Arthur DC, Pack SD, et al. Identification of CARS-ALK fusion in primary and metastatic lesions of an inflammatory myofibroblastic tumor. Lab Invest 2003;83:1255–1265.
Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448:561–566.
Takeuchi K, Choi YL, Togashi Y, et al. KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin Cancer Res 2009;15:3143–3149.
Jazii FR, Najafi Z, Malekzadeh R, et al. Identification of squamous cell carcinoma associated proteins by proteomics and loss of beta tropomyosin expression in esophageal cancer. World J Gastroenterol 2006;12:7104–7112.
Du XL, Hu H, Lin DC, et al. Proteomic profiling of proteins dysregulted in Chinese esophageal squamous cell carcinoma. J Mol Med 2007;85:863–875.
Lin E, Li L, Guan Y, et al. Exon array profiling detects EML4-ALK fusion in breast, colorectal, and non-small cell lung cancers. Mol Cancer Res 2009;7:1466–1476.
Mossé YP, Laudenslager M, Longo L, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008;455:930–935.
Chen Y, Takita J, Choi YL, et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 2008;455:971–974.
Webb TR, Slavish J, George RE, et al. Anaplastic lymphoma kinase: role in cancer pathogenesis and small-molecule inhibitor development for therapy. Expert Rev Anticancer Ther 2009;9:331–356.
Garber K . ALK, lung cancer, and personalized therapy: portent of the future? J Natl Cancer Inst 2010;102:672–675.
Hutchison RE, Banki K, Shuster J, et al. Use of an anti-ALK antibody in the characterization of anaplastic large-cell lymphoma of childhood. Ann Oncol 1997;8 (Suppl 1):S37–S42.
Shaffer LG, Slovak ML, Campbell LJ, (eds). An International System for Human Cytogenetic Nomenclature. Karger, ISCN: Basel, 2009, pp 138.
Lamant L, Pulford K, Bischof D, et al. Expression of the ALK tyrosine kinase gene in neuroblastoma. Am J Pathol 2000;156:1711–1721.
Argani P, Olgac S, Tickoo SK, et al. Xp11 translocation renal cell carcinoma in adults: expanded clinical, pathologic, and genetic spectrum. Am J Surg Pathol 2007;31:1149–1160.
Kuroda N, Naruse K, Miyazaki E, et al. Vinculin: its possible use as a marker of normal collecting ducts and renal neoplasms with collecting duct system phenotype. Mod Pathol 2000;13:1109–1114.
Kovacs G, Szücs S, De Riese W, et al. Specific chromosome aberration in human renal cell carcinoma. Int J Cancer 1987;40:171–178.
Zhao WP, Gnarra JR, Liu S, et al. Renal cell carcinoma. Cytogenetic analysis of tumors and cell lines. Cancer Genet Cytogenet 1995;82:128–139.
Yoshida MA, Ohyashiki K, Ochi H, et al. Cytogenetic studies of tumor tissue from patients with nonfamilial renal cell carcinoma. Cancer Res 1986;46:2139–2147.
Pavlovich CP, Padilla-Nash H, Wangsa D, et al. Patterns of aneuploidy in stage IV clear cell renal cell carcinoma revealed by comparative genomic hybridization and spectral karyotyping. Genes Chromosomes Cancer 2003;37:252–260.
Weller PA, Ogryzko EP, Corben EB, et al. Complete sequence of human vinculin and assignment of the gene to chromosome 10. Proc Natl Acad Sci USA 1990;87:5667–5671.
Bakolitsa C, Cohen DM, Bankston LA, et al. Structural basis for vinculin activation at sites of cell adhesion. Nature 2004;430:583–586.
Bailly M . Connecting cell adhesion to the actin polymerization machinery: vinculin as the missing link? Trends Cell Biol 2003;13:163–165.
Zemljic-Harpf AE, Ponrartana S, Avalos RT, et al. Heterozygous inactivation of the vinculin gene predisposes to stress-induced cardiomyopathy. Am J Pathol 2004;165:1033–1044.
Vasile VC, Will ML, Ommen SR, et al. Identification of a metavinculin missense mutation, R975W, associated with both hypertrophic and dilated cardiomyopathy. Mol Genet Metab 2006;87:169–174.
Lifschitz-Mercer B, Czernobilsky B, Feldberg E, et al. Expression of the adherens junction protein vinculin in human basal and squamous cell tumors: relationship to invasiveness and metastatic potential. Hum Pathol 1997;28:1230–1236.
Johnson RP, Craig SW . F-actin binding site masked by the intramolecular association of vinculin head and tail domains. Nature 1995;373:261–264.
Goldmann WH . Mechanical aspects of cell shape regulation and signaling. Cell Biol Int 2002;26:313–317.
Martelli MP, Sozzi G, Hernandez L, et al. EML4-ALKrearrangement in non-small cell lung cancer and non-tumor lung tissues. Am J Pathol 2009;174:661–670.
Rodig SJ, Mino-Kenudson M, Dacic S, et al. Unique clinicopathologic features characterize ALK-rearranged lung adenocarcinoma in the western population. Clin Cancer Res 2009;15:5216–5223.
Baron BW, Mick R, Baron JM . Hematuria in sickle cell anemia—not always benign: evidence for excess frequency of sickle cell anemia in African Americans with renal cell carcinoma. Acta Haematol 1994;92:119–122.
Davis Jr CJ, Mostofi FK, Sesterhenn IA . Renal medullary carcinoma. The seventh sickle cell nephropathy. Am J Surg Pathol 1995;19:1–11.
Bang Y, Kwak EL, Shaw AT, et al. Clinical activity of the oral ALK inhibitor PF-02341066 in ALK-positive patients with non-small cell lung cancer (NSCLC). J Clin Oncol 2010;28 (18S):3.
Acknowledgements
We thank Elizabeth Perlman, Deborah Perry, Bruce Beckwith, and the Children's Oncology Group for sharing their diagnostic expertise on submitted cases. We also thank St Jude Tumor Bank and Department of Pathology for support of the study and, in particular, Charlene Henry for help with immunohistochemistry; Julia Wilbourne and Rashmi Sharma for help with interphase FISH; Sheila Shurtleaf, molecular pathology division, and Hartwell Center for Bioinformatics and Biotechnology for help with sequencing. Jill Lahti (St Jude Department of Genetics and Tumor Cell Biology) has generously provided the IMR32 cell line. We thank Angela McArthur for editing the manuscript. This work was supported in part by the Edna Ittner Pediatric Research Grant and the Eppley Pediatric Cancer Award (to JAB) and the American Lebanese Syrian Associated Charities (ALSAC).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Debelenko, L., Raimondi, S., Daw, N. et al. Renal cell carcinoma with novel VCL–ALK fusion: new representative of ALK-associated tumor spectrum. Mod Pathol 24, 430–442 (2011). https://doi.org/10.1038/modpathol.2010.213
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2010.213
Keywords
This article is cited by
-
ALK-rearranged renal cell carcinoma with TPM3::ALK gene fusion and review of the literature
Virchows Archiv (2023)
-
ALK rearrangement-associated renal cell carcinoma morphologically mimicking mucinous tubular and spindle cell carcinoma: a case report
Diagnostic Pathology (2022)
-
Epithelioid inflammatory myofibroblastic sarcoma with VCL–ALK fusion of central nervous system: case report and brief review of the literature
Brain Tumor Pathology (2022)
-
Histologische Subtypen des Nierenzellkarzinoms
Der Pathologe (2021)
-
Novel, emerging and provisional renal entities: The Genitourinary Pathology Society (GUPS) update on renal neoplasia
Modern Pathology (2021)
